
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn the nature of the triplet electronic states of naphthalene dimers†
L.
Martinez-Fernandez‡
a,
Peicong
Wu‡
b,
Lin-Tao
Bao‡
c,
Xueli
Wang
b,
Rui-Hua
Zhang
c,
Wei
Wang
 c,
Hai-Bo
Yang
c,
Jinquan
Chen
*bd and
R.
Improta
*e
c,
Hai-Bo
Yang
c,
Jinquan
Chen
*bd and
R.
Improta
*e
aDepartamento de Química Física de Materiales, Instituto de Química Física Blas Cabrera, CSIC, Calle Serrano 119, 28006, Madrid, Spain
bState Key Laboratory of Precision Spectroscopy, East China Normal University, Shanghai 200241, China. E-mail: jqchen@lps.ecnu.edu.cn
cShanghai Key Laboratory of Green Chemistry and Chemical Processes, Shanghai Frontiers Science Center of Molecule Intelligent Syntheses, Chang-Kung Chuang Institute, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
dCollaborative Innovation Center of Extreme Optics, Shanxi University, Taiyuan 030006, Shanxi, China
eIstituto di Biostrutture e Bioimmagini-CNR (IBB-CNR), Via De Amicis 95, I-80145 Napoli, Italy. E-mail: roberto.improta@cnr.it
First published on 28th January 2025
Abstract
Elucidating the photophysical mechanisms within multi-chromophore assembly (MCA) is essential for many key technological and biological processes. Although it has been established that one of the most important photoactivated applications of MCA is intimately linked to efficient intersystem crossing (ISC) to triplet states and the interplay between delocalized/localized triplet excited states, the underlying mechanism between such equilibrium and the observed optical properties remains elusive. Herein, four suitably designed dinaphthyl compounds, covalently bonded in a face-to-face configuration and encompassing the primary possible stacking geometries, were prepared and their triplet state properties investigated by combining transient absorption spectroscopy experiments with quantum chemistry calculations. Our results offer direct evidence of both localized and delocalized triplet states, with the most stable and long-lived triplet state consistently localized on a single naphthalene unit, irrespective of the stacking configuration. Moreover, depending on the stacking geometry, even if localized, the triplet transient absorption spectrum was demonstrated to be significantly different from that of an isolated naphthalene.
1 Introduction
The triplet electronic states of aggregates containing many interacting chromophores (hereafter, generically, multichromophore assembly, MCA) have been unequivocally demonstrated to hold paramount significance in a multitude of biological processes1–3 and technological advancements.4–8 In particular, these applications are attributed to the direct involvement and regulation of triplet states among different molecular processes in MCA, such as phosphorescence,9 triplet–triplet annihilation,10–12 delayed fluorescence,13,14 triplet energy transfer,15 or circularly polarized luminescence (CPL).16,17 Consequently, any enhancement in our comprehension of the nature and properties of the MCA triplet states is of critical importance. In this field, naphthalene dimers have emerged as an extremely interesting model system,18–39 as prototypes of a very important family of compounds, polyacenes,40–42 that are promising components for optoelectronic devices.43–46 In the last two decades the nature and the properties of the lowest energy triplet electronic states of the dimers composed of two units of naphthalene (or some of its derivatives) have been, indeed, quite intensively debated. Firstly, several experimental studies28,30–33,47 showed that the lowest energy triplet state in different dinaphthyl compounds has spectral properties (for example its phosphorescence energy) different from those of the naphthalene triplet and suggested that it was an ‘excimer’. Several computational studies20,24,36 proposed that the excimer triplet, characterized by a dimerization energy substantially larger than that of the ground state,48 is delocalized over the two moieties. The existence of a triplet excimer in dinaphthyl has been criticized, and the related red-shifted phosphorescence attributed to impurities.25 Secondly, additional controversies arose concerning the preferential geometries adopted by the triplet excimer (e.g. face-to-face, in the following f2f, bent, T-shape etc.).24,36 The scarcity of experimental results has been an important bottleneck, preventing a firm assessment of these issues. For example, the absence of ‘excimer-like’ phosphorescence does not allow their presence to be ruled out, since for a perfectly parallel face-to-face structure it is forbidden by symmetry.32 Moreover, the results obtained on covalently bonded dimers are not easily generalized, since not only their structural restraints can have an overwhelming effect on the nature of the triplet minima,31 but the substituents (e.g. carbonyl groups, as in the ‘Agosta’ dimers)31,49 can also affect the electronic properties of the naphthalene moieties.In particular, in the presence of a suitable electron donor bridge substituent, naphthalene50–52 and naphthalene derived molecules (as the naphthalene mono-imide (NI)53,54 and di-imide (NDI))55 have been shown to be involved in very interesting (symmetry breaking) charge separation processes.56–58
In order to advance the understanding of the effects ruling the behaviour of the triplet states of naphthalene dimers, we have here synthesized and characterized four different dinaphthyl compounds where two stacked naphthalenes are rigidly locked with a specific orientation by covalent bonds (Scheme 1(b)). We show that in all of them UV irradiation leads to triplet electronic states with a quantum yield (QY) ≥0.5, which we characterize by integrating steady state and time resolved spectra and quantum mechanical calculations.
 | ||
| Scheme 1 (a) Schematic diagram of triplet states involved in face-to-face naphthalene dimers. (b) Chemical structure of the dinaphthalene studied in this work. (c) Schematic drawing of the ground state optimized structures of the different dinaphthalene compounds. For each compound both the side (on the left) and the top (on the right) views are reported. η is the dihedral formed by the two rings, and d is the distance between the middle point of the inner C–C bonds. | ||
The four compounds studied all have an f2f arrangement, but they exhibit different stacking geometries and degrees of asymmetry (Scheme 1(b) and (c)), enabling a more detailed and general picture of the relationship between the structure of the dimers and the properties of their triplet electronic states to be obtained. In parallel, the same computational approach has been applied to study the interplay between the localized and delocalized triplet electronic states of ‘free’ naphthalene dimers in toluene solution. We show that for all the species the lowest energy triplet state is essentially localized on a single naphthalene moiety, although it can exhibit quite different spectral properties depending on the dimer, especially for the more symmetric 1515 compound. On the other hand, we find that delocalized triplet excimer minima are possible for all the species examined, i.e. also for stacking geometries different from the face-to-face one. Excimer minima are disfavoured by inclusion of vibrational and entropic effects with respect to the localized one, which are characterized by shallower potential energy surfaces (PES). Finally, we show that it cannot be taken for granted that ‘localized’ triplets in a dimer have exactly the same spectral behaviour as the monomer, since they are affected by the presence of a closely stacked partner. Since many of these conclusions are expected to be valid also for other organic multimers, this study, besides providing fresh insights on an “old problem”, provides new information on the effects that determine the properties of the triplet electronic states in closely stacked systems.
2 Results and discussion
2.1 Triplet state properties of 1515
Although the synthesis of 1515 has already been reported, its photophysical properties have never been examined in depth.59,60 The steady-state absorption spectrum of 1515 in dichloromethane (DCM) exhibits a low-energy absorption band centred at 285 nm (Fig. 1(a)). The emission band centered at 450 nm can be quenched by O2 and display a significant Stokes shift relative to the absorption peak (Fig. 1(a) and (b)). Nanosecond time-resolved transient absorption (TA) measurements with 285 nm excitation in N2-saturated DCM are depicted in Fig. 2. Excitation at 285 nm results in the generation of a very broad excited state absorption (ESA) band (Fig. 2(a)), with maxima at 425 nm, 475 nm and 600 nm. In the next ∼100 ns, a significant decay can be observed centered at 425 nm and 525 nm. When the time delay is 130 ns, two distinct ESA bands can be detected with maxima at 475 nm and 600 nm. Then the TA data decay to zero in ∼5 μs without an apparent change in spectral shape. The TA data can be best fit with a two-exponential decay function, yielding lifetimes of 32 ± 1 ns and 1.4 ± 0.1 μs, and the relative Decay Associated Difference Spectra (DADS) are shown in Fig. 2(b). DADS1(blue) shows a broad band covering the entire 360–600 nm region, the dip present at 460 nm being likely associated with stimulated emission, as suggested by the steady state emission spectrum (Fig. 1(b)), suggesting DADS1 is contributed by a triplet state. DADS2 shows two ESA bands peaking at 475 nm and 600 nm. To further study the two species that survive to the nanosecond time scale, the O2 concentrate dependent and triplet state sensitization experiments have been carried out. As shown in Fig. S1,† the lifetime of 1515 is quenched by the presence of dissolved oxygen in DCM, suggesting that the observed excited state species are likely contributed by triplet states. Moreover, triplet state sensitization experiments were carried out, employing benzophenone (Bp) as the triplet state energy donor. A wavelength of 355 nm was selected for excitation, which is specific to the excitation of Bp. As shown in Fig. S2a,† BP exhibits a triplet state spectrum with a maximum at 530 nm.61,62 After adding 1515, the lifetime of BP is quenched significantly in lockstep with the generation of several new bands that resemble the spectral shape of the measured TA spectra of 1515 (Fig. S2b and S3a†), confirming that these two components are associated with triplet states, which are formed with a total triplet state QY of 0.88 (Table S5†). | ||
| Fig. 1 (a) Steady-state absorption and emission spectra of 1515, 2626, 2615 and 1515cross in DCM. (b) The emission spectrum of 1515 under N2- and air-saturated conditions in DCM. | ||
 | ||
| Fig. 2 (a) Nanosecond time-resolved transient absorption spectra of 1515 excited at 285 nm in DCM under N2-saturated conditions. (b) Decay-associated difference spectra (DADS) and (c) kinetic traces probed at 430 nm and 600 nm. | ||
In Section 2.2 of the ESI† we report the TA spectra measured for 1515 in the more polar acetonitrile solvent. The DADS are very similar to those found in DCM, indicating that long-lived charge separated triplet states are not involved in the photoactivated dynamics.
In order to assign these spectra, we resorted to Quantum Mechanical (QM) calculations. We used different basis sets and density functionals (namely ωB97XD63 and M052X64), and the double hybrid B2LYPD365,66 (Section 4 in the ESI†), since the latter have recently emerged as very promising tools for the calculation of the excited state properties.67 In the following we shall use ωB97XD and B2LYPD3 as reference methods. We have performed our study both in the gas phase and in DCM solution, including a study on solvent effects using the Polarizable Continuum Model (PCM).68 In the ESI† we discuss our approach in more detail, which is also adopted in a parallel study on the naphthalene dimer in the gas phase69 and in the study of other closely stacked MCA.70
We succeeded in locating the minima of two different triplet electronic states, both in the gas phase and in DCM solution, with all the adopted computational methods. In one of them, the analysis of the Molecular Orbitals (MO) and Mulliken spin densities indicates that the triplet is essentially localized over a single naphthalene moiety. In the following we shall label this excited state as T1-loc (Fig. 3(a)). The other minimum corresponds to the ‘excimer’ introduced above, the triplet is equally delocalized over the two naphthalene rings (Fig. 3(b)), and we shall label it as T1-exc (and its minimum, T1-exc-min). In T1-exc-min, the bond lengths are intermediate between those of a naphthalene in its lowest energy singlet or triplet state. In T1-loc-min, instead, one of the moieties keeps the geometry of ground state singlet while the other adopts one close to that of a triplet naphthalene monomer. Moreover, T1-exc-min is characterized by a closer approach of the two rings, d ∼3 Å, with respect to the value of ∼3.2 Å, found for S0-min and T1-loc-min (Section 4.2. in the ESI†). As collected in Table 1, all the adopted computational approaches predict that T1-loc-min is the most stable minimum, both in the gas phase (Section 4 in the ESI†) and in DCM. T1-loc-min is also relatively favoured by the inclusion of vibrational and entropic effects. Confirming the indications obtained on stacked naphthalene dimers,69 standard density functionals underestimate the relative stability of T1-exc-min, whereas inclusion of the solvent effect has a negligible effect (Table S6†).
 | ||
| Fig. 3 Spin density computed at the B2LYPD3/6-31+G(d,p) level in the gas phase for T1-loc-min (a) and T1-exc-min (b). Color code: red, negative spin density; green, positive spin density. | ||

|
Then, we simulated the absorption spectra of T1-loc-min and T1-exc-min, by computing the excitation energies to the lowest 30 excited states at the TDA/6-31+G(d,p) level and by broadening the stick transitions with a Gaussian with a half width at half maximum of 0.25 eV. This simple procedure reproduces well the vibrationally resolved spectrum of the monomer69 and the spectra obtained at the TD level, or by using larger basis sets, but for a weak blue-shift (10–20 nm, depending on the method) of the absorption maximum (Section 4.3.1 in the ESI†). The computed TDA/B2LYPD3/6-31+G(d,p) absorption spectra of T1-loc-min and T1-exc-min in the gas phase are shown in Fig. 4.
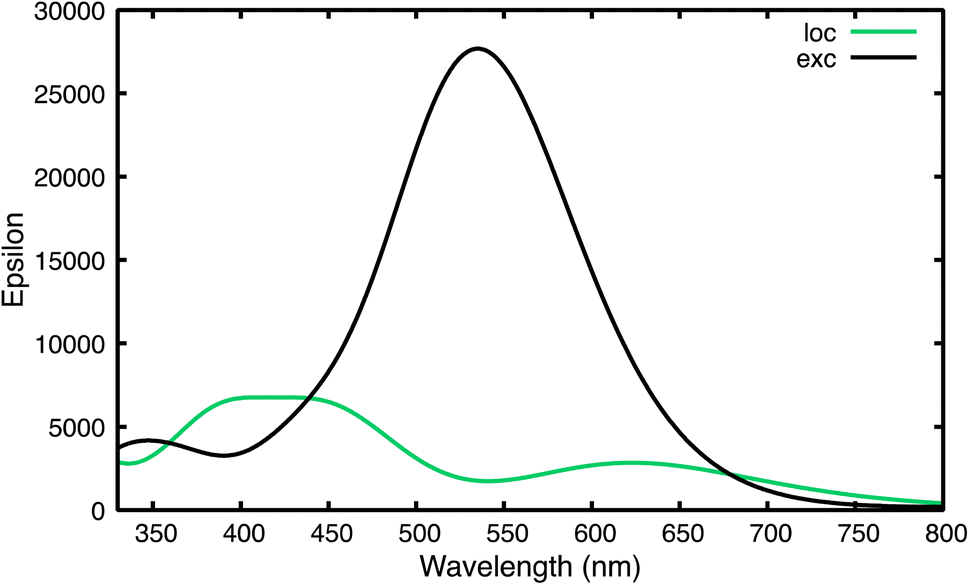 | ||
| Fig. 4 TDA-B2LYPD3/6-31+G(d,p) computed absorption spectra for the 1515 triplet minima: T1-loc-min (green) and T1-exc-min (black) in the gas phase. | ||
The spectrum of T1-loc-min is very similar to that of the long-lived DADS (DADS2), with a maximum above 450 nm and another shallow band at ∼600 nm. On the other hand, the spectrum of T1-exc-min exhibits a single, extremely intense band peaking at 540 nm. Moreover, ADC(2) calculations predict for the triplet excimer minimum of a stacked naphthalene dimer a very intense band at ∼500 nm, which moderately aligns with the ESA spectrum of DADS1 extracted from the TA experiment.23 The indications provided by TDA/B2LYPD3 double hybrid calculations are confirmed by TDA/ωB97XD and TDA/M052X methods, when applied in DCM solution, also when using large basis sets (e.g. aug-cc-pvtz) (Section 4.3.1 in the ESI†).
We do not find any indication of stable charge separated triplet states, in line with the experimental results in acetonitrile. Naphthalene is indeed a poorer electron acceptor of NI and NDI, and the ethylenic bridges are not suitable electron donors. Moreover, in our closely stacked arrangement, Frenkel excitonic couplings are very large, and the steric restraints of the short covalent bridges prevent substantial structural rearrangements in the excited state minima, reducing the reorganization energy, which is an important factor for the population of CT states in closely stacked MCA.70,71 Finally, calculations for stacked naphthalene dimers in the gas phase indicate that the contribution of CT states to triplet excitons is smaller than for singlet excited states20
2.2 Triplet state properties of 2626, 2615 and 1515cross
Different from 2626,59,60 to the best of our knowledge, the synthesis of 2615 has never been reported. As detailed in the ESI,† we produced it by using a suitable etherifying group. We have measured the TA spectra for these species following excitations at 285 nm, and in the presence of Bp (excited at 355 nm). The TA signals and the DADS obtained by their fitting are reported in Fig. 5. The three compounds exhibit quite similar spectral features, and, therefore we shall discuss them together. In all cases examined, there are indeed two distinct DADS species with lifetimes extending into the nanosecond time scale. The spectral shapes of these species are very similar to the corresponding feature present after sensitization with Bp (Fig. S3–S7†). Such observations allow for a confident assignment of these species to triplet excited states (Fig. S8–S10†). This DADS exhibits a maximum at ∼440 nm, with a long tail in the red wing, more prominent for 2615, where a shallow maximum at ∼550 nm can be recognized. As detailed in the ESI,† the triplet QY is large in all of these three compounds: it is >0.7 for 2615 and 2626, and >0.5 for 1515cross (Table S5†). | ||
| Fig. 5 Nanosecond time-resolved transient absorption spectra of (a) 2615, (c) 2626 and (e) 1515cross excited at 285 nm in DCM under N2-saturated conditions. Decay-associated difference spectra (DADS) of (b) 2615, (d) 2626 and (f) 1515cross. Excited state absorption kinetic traces probed at 430 nm and 600 nm of (a) 2615, and at 440 nm and 600 nm of (c) 2626 and (e) 1515cross. | ||
As reported in Table 1 we succeeded in localizing both the T1-loc and the T1-exc minima for all of these three species, with the T1-loc minimum always being the most stable, both in the gas phase and in DCM solution (Table S6†). For 2626, we succeeded in locating two localized minima, differing in the stacking geometry of the two rings (Fig. S14†). In the most stable one (T1-loc-min), they are rotated with respect to the other (η = −39.3°). In the other one, less stable by 0.12 eV, they are close enough to be face-to-face (η = −7.5°, T1-loc-f2f). For 2626, in T1-exc-min η = −39.7°, showing that delocalized minima do not necessarily require f2f arrangements. However, due to the structural restraints imposed by the inter-ring bonds, d is 3.30 Å, i.e. longer than what found for the 1515 excimer minimum. As a consequence, for 2626 T1-exc-min is much less stable than T1-loc-min, when compared to 1515.
2615 is the only compound among those investigated here where the two naphthalene rings are different, as one is substituted in 1,5 positions and the other one in 2,6 positions (Scheme 1). We succeeded in optimizing two different localized triplet minima, T1-loc (triplet localized on the 15 naphthalene moiety, see Fig. S15a†) and T1-loc1 (triplet localized on the 26 naphthalene moiety, see Fig. S15b†), with very similar stability (T1-loc-min is 0.01 more stable). Considering that the two naphthalene rings are different, for this compound T1-exc-min (0.40 eV less stable than T1-loc-min) is not perfectly symmetric, i.e. different from all the excimer minima considered until now, the structural parameters and the spin density are not the same in the two moieties. The long axes of the rings are almost perpendicular (η = 85.3°), while d is 3.13 Å, much shorter than that found for T1-loc-min (3.37 Å). For 1515cross, in T1-exc-min, significantly less stable than T1-loc-min, the rings are very close (d ∼3.0 Å) with η = 16.9°.
We have computed the TDA absorption spectra for all the minima (Fig. 6 and S19†) both in the gas phase and in DCM. The lineshapes of the B2LYPD3 computed spectra for T1-loc-minima (panel a in Fig. 6) are very similar to those of the experimental long-lived DADS2 (panel b in Fig. 6), but for a small, expected, uniform blue-shift (∼20 nm). The most significant differences between the different compounds are also well reproduced. For example, for 2615, a shallow band at ∼600 nm is present, and the main peaks (those above 400 nm) are slightly blue-shifted with respect to that of 1515. The spectra associated with T1-exc minima are reported in Fig. 6(c) and they are very different from the spectrum associated with the long-lived, most stable triplet state. In analogy with those reported above for 1515, at the B2LYPD3 level, they all exhibit an extremely intense peak in the red, whose maximum (in the range 450–550 nm) changes depending on the molecule considered. Moreover, the absorption intensity above 600 nm is always larger with respect to that found in the experiments (2626 and 2615 have strongly absorption until 800–900 nm). As reported in Fig. S17–S25,† the TDA spectra computed with ωB97XD and M052X functionals in DCM solution provide the same picture obtained with double hybrid TDA-DFT, where TD-ωB97XD spectra provide a similar qualitative picture. The signature of the excimer minimum is, thus, a very strong absorption band at 500–600 nm, whose intensity and position depends on the structural and electronic features of the minimum. The closer the approach of the bases, the more intense and red-shifted is the absorption band.
 | ||
| Fig. 6 (a) TDA-B2LYPD3/6-31+G(d,p) computed absorption spectra for the T1-loc-minima in the gas phase. (b) Experimental μs decay-associated difference spectra (DADS2) for all the dimers (the red traces in Fig. 2 and 5). (c) TDA-B2LYPD3/6-31+G(d,p) computed absorption spectra for the T1-exc-minima in the gas phase. | ||
2.3 Triplet state properties of the stacked naphthalene dimer in toluene
In the dinaphthalene species studied here, the covalent bonds connecting the two naphthalene moieties lead to some distortion of the ring planarity, and obviously induce some structural restraints, which could affect the relative stability of localized and delocalized minima. In order to assess this issue and to increase the generality of our conclusions, we have applied our computational approach to the study of naphthalene monomers and dimers in toluene solution, for which the experimental triplet spectrum is available. As shown in Fig. 7, it exhibits a strong band at ∼420 nm, but, as the concentration increases, a weak shoulder at longer wavelength appears, which has been attributed to the presence of a triplet excimer involving two stacked naphthalene molecules (i.e. the T1-exc species we have discussed in the previous Section).47 | ||
| Fig. 7 TDA-PCM/B2PLYBD3/6-31+G(d,p) computed absorption spectra in toluene. The experimental absorption spectrum of naphthalene in toluene47 is shown in the inset on the right. | ||
In addition to the triplet of the naphthalene monomer, we succeeded in locating both T1-loc and T1-exc for a dimer. For T1-loc different stationary points are possible. In the most stable one (hereafter T1-loc-min), the two monomers exhibit a slipped parallel arrangement (Fig. S26†), but a perfect ‘face-to-face’ arrangement is also possible. On the other hand, this latter structure (hereafter T1-loc-f2f) exhibits a small negative frequency (<50 cm−1) and it is not an absolute minimum of the T1-loc PES. In T1-exc-min, the monomers adopt a perfect ‘face-to-face’ parallel arrangement, and two moieties are much closer than in the ground state minimum, with d decreasing by 0.4–0.5 Å. As reported in Table 1, for a naphthalene dimer the relative stability of T1-exc strongly increases. From the electronic point of view it is slightly more stable than T1-loc, as predicted also by ADC(2) calculations in the gas phase. T1-loc-min, likely due to its much shallower PES than T1-exc, is however relatively stabilized (by ≥0.2 eV) when vibrational and entropic corrections are included, resulting in the most stable triplet minimum also at the B2LYPD3 level.
The TDA spectrum computed for the triplet monomer is in good agreement with the experimental one (Fig. S27†), but for the expected weak blue-shift of the maximum. In agreement with the ADC(2) calculations in the gas phase, an extremely strong band, with a maximum at ∼550 nm, is associated with the T1-exc minimum of the dimer. The spectrum of T1-loc, though its maximum falls at ∼400 nm, is different to that of the monomer. As detailed in the ESI,† in addition to a ‘monomer-like’ transition at ∼400, quite similar to that typical of isolated naphthalene, we find two additional weaker transitions, on the red-wing of the main absorption band, involving also the stacked naphthalene molecule. The position and the intensity of these transitions depend on the stacking geometry of the dimer. For T1-loc-min, they simply produce a weak red-tail in the spectrum. On the other hand, for T1-loc-f2f, an intense band appears just below 500 nm, extending until 600 nm.
3 Conclusions
We here reported a comparative study of different stacked naphthalene dimers in the triplet electronic states, where the two moieties are either covalently bonded or simply stabilized by van der Waals interactions. In the former case, we have explored different stacking geometries, from cases where the two ring molecular axes are almost parallel (as in 1515) to other molecules where they are almost perpendicular (as in 2615). Triplet electronic states are always formed in high yield (>0.5), and time-resolved absorption experiments always show the presence of two components with decay lifetimes extending into the nanosecond and microsecond time scales, whose shapes are the same recorded after photosensitization experiments, confirming their identification as triplet states. Our calculations (by using either standard hybrid or double hybrid DFT calculations) indicate that for all the molecules the most stable triplet is localized on one of the two rings (T1-loc). This assignment is confirmed by the experimental phosphorescence spectra and by the remarkable agreement between the shape of the experimental spectra and that computed for the localized minima. The absorption spectra of the localized triplet are similar to that of a triplet naphthalene monomer, with a strong peak slightly above 400 nm. However, a tail appears in the red (until 550–650 nm), which, depending on the stacking arrangement, can gain significant intensity. For 1515, where the rings are close to being f2f, a second intense band, peaking at 600 nm is indeed present both in experimental and in computed T1-loc spectra. The computed spectra for delocalized ‘excimer-like’ triplet minima (T1-exc) are instead not consistent with the experimental ones at longer times, exhibiting a very strong band at 500–550 nm, and extending until 800 nm. Interestingly, also the T1-exc spectrum strongly depends on the stacking geometry, with the main absorption band red-shifting and gaining intensity for f2f arrangements (as in 1515). According to our calculations, the relative stability of T1-exc is also larger for f2f stacking, which enables a larger decrease of the inter-ring distance, a major stabilizing factor for excimer minima.Charge separated triplet electronic states are not expected to play a significant role in the compounds here examined, also in more polar solvents, as confirmed by the test experiments performed in acetonitrile. Suitable modifications in the bridge (e.g. increasing its electron donor efficiency and reducing the steric hindrance) and in the naphthalene moieties (increasing their electron acceptor properties) are likely necessary to observe symmetric breaking charge separation in covalently bridged stacked naphthalene dimers.
For a free stacked dimer, which does not suffer from the small distortion of the ring planarity (necessary to reduce the steric hindrance) of covalently bonded dimers, the most accurate electronic methods (e.g. double hybrids density functionals or ADC(2))24 predict indeed that ‘excimer’ minima are more stable (by 0.1–0.2 eV). However, localized minima, for which a large number of almost isoenergetic minima are available, are strongly relatively favoured by entropic and vibrational effects, and, at least at the double hybrid level, are slightly more stable also in this latter case. Based on our calculations, it cannot be thus taken for granted that the shallow absorption band appearing above 500 nm when the naphthalene concentration in toluene increases is due to triplet excimers, as the f2f localized triplet would have a spectral signature consistent with the experimental features.
Based on our experimental and computational analysis, the possibility that for stacked naphthalene dimers the absolute triplet minimum is a delocalized excimer is expected to be more an exception than a rule. At the same time, our calculations show that ‘excimer’ minima are relative minima also when the axes of the rings are perpendicular, although they are relatively more stable for symmetric face-to-face arrangements, and when ring planarity is not distorted. In these cases, their stability is comparable to that of localized minima. On these grounds, excimer triplet minima can be transiently populated following light absorption and/or triplet sensitization of dinaphthalene and, therefore, their presence has to be considered when interpreting TR spectra. Moreover, when naphthalene dimers are trapped inside rigid hosts, which decreases the weight of entropic effects, the excimer can become the most stable triplet minima.
These conclusions are expected to be valid for several other MCAs, with the role played by delocalized excimers being more important for more symmetric stacking arrangements and when a strong decrease of the inter-ring distance in the triplet minimum is sterically feasible. Another general consideration concerns the expected spectral features of the localized and excimer minima, which, for closely stacked systems, can significantly depend on the stacking geometry of the chromophores, and, therefore, on their electronic interactions. In other words, we cannot expect that ‘localized’ minima have exactly the same spectra of the free monomer.
Data availability
The data that support the findings of this study are available in the ESI† of this article and are available on request from the corresponding authors.Author contributions
Conceptualization, L. M. F., X. W., W. W., H. Y., J. C. and R. I.; methodology, L. M. F., X. W., W. W., H. Y., J. C, and R. I.; formal analysis, all authors; investigation, all authors; writing – original draft, L. M. F., X. W., J. C. and R. I.; writing – review and editing, all authors; supervision, H. Y., J. C. and R. I.; funding acquisition, L. M. F., H. Y., J. C. and R. I. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research project is part of the activities of the National Center for Gene Therapy and Drugs Based on RNA Technology, funded by the European Union – Next Generation EU, Project CN00000041, CUP B93D21010860004. R. I. also thanks CNR, program “Progetti di Ricerca@cnr”, project UCATG4 and NUTRAGE funded by FOE-2021 DBA.AD005.225 for financial support. This research project was made possible through the access granted by the Galician Supercomputing Center (CESGA) to its supercomputing infrastructure. The supercomputer FinisTerrae III and its permanent data storage system have been funded by the Spanish Ministry of Science and Innovation, the Galician Government and the European Regional Development Fund (ERDF). This study was funded by the National Natural Science Foundation of China (No. 92156024 to J. Chen, No. 92056203 and 92356307 to H. Yang). We thank the Materials Characterization Center of East China Normal University for help with the measurement of the ns-TA spectra.References
- J. Ho, E. Kish, D. D. Méndez-Hernández, K. WongCarter, S. Pillai, G. Kodis, J. Niklas, O. G. Poluektov, D. Gust, T. A. Moore, A. L. Moore, V. S. Batista and B. Robert, Triplet–triplet energy transfer in artificial and natural photosynthetic antennas, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5513–E5521 CAS.
- X. Wang, L. Martínez-Fernández, Y. Zhang, P. Wu, B. Kohler, R. Improta and J. Chen, Ultrafast Formation of a Delocalized Triplet-Excited State in an Epigenetically Modified DNA Duplex under Direct UV Excitation, J. Am. Chem. Soc., 2024, 146, 1839–1848 CrossRef CAS PubMed.
- M. A. Miranda and V. Lhiaubet-Vallet, in DNA Photodamage: From Light Absorption to Cellular Responses and Skin Cancer, ed. R. Improta and T. Douki, The Royal Society of Chemistry, 2021, 10.1039/9781839165580-00055.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S.-J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T.-Q. Nguyen, D. Beljonne and R. H. Friend, The role of charge recombination to triplet excitons in organic solar cells, Nature, 2021, 597, 666–671 CrossRef CAS PubMed.
- J. Grüne, G. Londi, A. J. Gillett, B. Stähly, S. Lulei, M. Kotova, Y. Olivier, V. Dyakonov and A. Sperlich, Triplet Excitons and Associated Efficiency-Limiting Pathways in Organic Solar Cell Blends Based on (Non-) Halogenated PBDB-T and Y-Series, Adv. Funct. Mater., 2023, 33, 2212640 CrossRef.
- A. Privitera, J. Grüne, A. Karki, W. K. Myers, V. Dyakonov, T.-Q. Nguyen, M. K. Riede, R. H. Friend, A. Sperlich and A. J. Gillett, Geminate and Nongeminate Pathways for Triplet Exciton Formation in Organic Solar Cells, Adv. Energy Mater., 2022, 12, 2103944 CrossRef CAS.
- E. Bassan, A. Gualandi, P. G. Cozzi and P. Ceroni, Design of BODIPY dyes as triplet photosensitizers: electronic properties tailored for solar energy conversion, photoredox catalysis and photodynamic therapy, Chem. Sci., 2021, 12, 6607–6628 RSC.
- L. R. Weiss, S. L. Bayliss, F. Kraffert, K. J. Thorley, J. E. Anthony, R. Bittl, R. H. Friend, A. Rao, N. C. Greenham and J. Behrends, Strongly exchange-coupled triplet pairs in an organic semiconductor, Nat. Phys., 2017, 13, 176–181 Search PubMed.
- W. Zhao, Z. He and B. Z. Tang, Room-temperature phosphorescence from organic aggregates, Nat. Rev. Mater., 2020, 5, 869–885 Search PubMed.
- A. Olesund, S. Ghasemi, K. Moth-Poulsen and B. Albinsson, Bulky Substituents Promote Triplet–Triplet Annihilation Over Triplet Excimer Formation in Naphthalene Derivatives, J. Am. Chem. Soc., 2023, 145, 22168–22175 CrossRef CAS PubMed.
- L. Huang and G. Han, Triplet–triplet annihilation photon upconversion-mediated photochemical reactions, Nat. Rev. Chem., 2024, 8, 238–255 CrossRef CAS PubMed.
- N. J. Turro, V. Ramamurthy and J. Scaiano, in Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, California, 2010 Search PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bässler, D. Beljonne, M. Buck, Y. Olivier, A. Köhler and E. Zysman-Colman, A Deep Blue B,N-Doped Heptacene Emitter That Shows Both Thermally Activated Delayed Fluorescence and Delayed Fluorescence by Triplet–Triplet Annihilation, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- H. Sternlicht, G. C. Nieman and G. W. Robinson, Triplet—Triplet Annihilation and Delayed Fluorescence in Molecular Aggregates, J. Chem. Phys., 1963, 38, 1326–1335 CrossRef CAS.
- B. Sk and S. Hirata, Förster resonance energy transfer involving the triplet state, Chem. Commun., 2023, 59, 6643–6659 RSC.
- D. Yang, J. Han, M. Liu and P. Duan, Photon Upconverted Circularly Polarized Luminescence via Triplet–Triplet Annihilation, Adv. Mater., 2019, 31, 1805683 CrossRef CAS PubMed.
- J. Han, P. Duan, X. Li and M. Liu, Amplification of Circularly Polarized Luminescence through Triplet–Triplet Annihilation-Based Photon Upconversion, J. Am. Chem. Soc., 2017, 139, 9783–9786 CrossRef CAS PubMed.
- H. Fliegl, Z.-Q. You, C.-P. Hsu and D. Sundholm, The Excitation Spectra of Naphthalene Dimers: Frenkel and Charge-transfer Excitons, J. Chin. Chem. Soc., 2016, 63, 20–32 CrossRef CAS.
- M. Gudem and M. Kowalewski, Triplet-triplet Annihilation Dynamics of Naphthalene, Chem.–Eur. J., 2022, 28, e202200781 CrossRef CAS PubMed.
- Y. Dai, A. Calzolari, M. Zubiria-Ulacia, D. Casanova and F. Negri, Intermolecular Interactions and Charge Resonance Contributions to Triplet and Singlet Exciton States of Oligoacene Aggregates, Molecules, 2023, 28, 119 CrossRef CAS PubMed.
- D. Kim, Effects of Intermolecular Interactions on the Singlet–Triplet Energy Difference: A Theoretical Study of the Formation of Excimers in Acene Molecules, J. Phys. Chem. C, 2015, 119, 12690–12697 CrossRef CAS.
- Y. Tian, Y. Li, B. Chen, R. Lai, S. He, X. Luo, Y. Han, Y. Wei and K. Wu, Sensitized Molecular Triplet and Triplet Excimer Emission in Two-Dimensional Hybrid Perovskites, J. Phys. Chem. Lett., 2020, 11, 2247–2255 CrossRef CAS PubMed.
- P. Bao, C. P. Hettich, Q. Shi and J. Gao, Block-Localized Excitation for Excimer Complex and Diabatic Coupling, J. Chem. Theory Comput., 2021, 17, 240–254 CrossRef CAS PubMed.
- M. Pabst, B. Lunkenheimer and A. Köhn, The Triplet Excimer of Naphthalene: A Model System for Triplet−Triplet Interactions and Its Spectral Properties, J. Phys. Chem. C, 2011, 115, 8335–8344 CrossRef CAS.
- B. Nickel and M. F. Rodriguez Prieto, Triplet Excimer Phosphorescence from Aromatic Compounds in Liquid Solutions: A Critical Review and New Negative Results, Z. Phys. Chem., 1986, 150, 31–67 CrossRef CAS.
- G. Huettmann and H. Staerk, Delayed luminescence of naphthalene in isooctane spectral, lifetime, and magnetic field studies of delayed fluorescence and monomer phosphorescence at 293 K, J. Phys. Chem., 1991, 95, 4951–4954 CrossRef CAS.
- R. J. Locke and E. C. Lim, Orientational effect on intramolecular triplet excimer formation in (1,n)-dinaphthylalkanes, J. Phys. Chem., 1989, 93, 6017–6019 CrossRef CAS.
- E. C. Lim, Molecular triplet excimers, Acc. Chem. Res., 1987, 20, 8–17 CrossRef CAS.
- J. Cai and E. C. Lim, Intramolecular photoassociation and photoinduced charge transfer in bridged diaryl compound. 5. Intramolecular triplet excimer of 1,1′-dinaphthylamine, J. Phys. Chem., 1993, 97, 6203–6207 CrossRef CAS.
- S. H. Modiano, J. Dresner, J. Cai and E. C. Lim, Intramolecular photoassociation and photoinduced charge transfer in bridged diaryl compounds. 4. Temporal studies of intramolecular triplet excimer formation in dinaphthylmethanes and dinaphthyl ethers, J. Phys. Chem., 1993, 97, 3480–3485 CrossRef CAS.
- M. Terazima, J. Cai and E. C. Lim, Time-Resolved EPR and Optical Studies of Intermoiety Interactions in the Lowest Triplet State of L-Shaped Dimers of Naphthalene: Conformation Dependence of Excitation Exchange Interaction, J. Phys. Chem. A, 2000, 104, 1662–1669 CrossRef CAS.
- M. Yamaji, H. Tsukada, J. Nishimura, H. Shizuka and S. Tobita, Investigations of triplet excimer of naphthalenophane by emission and transient absorption measurements in solution, Chem. Phys. Lett., 2002, 357, 137–142 CrossRef CAS.
- S. Hashimoto and M. Yamaji, Observation of intramolecular singlet and triplet excimers of tethered naphthalene moieties under the geometric constraints imposed by the host framework of zeolites, Phys. Chem. Chem. Phys., 2008, 10, 3124–3130 RSC.
- A. K. Chandra and E. C. Lim, Semiempirical Theory of Excimer Luminescence, J. Chem. Phys., 1968, 48, 2589–2595 CrossRef CAS.
- A. K. Chandra and E. C. Lim, Semiempirical Theory of Excimer Luminescence. II. Comparison with Previous Theories and Consideration of the Transition Probability and the Stability of the Excimer Triplet State, J. Chem. Phys., 1968, 49, 5066–5072 CrossRef CAS.
- A. L. L. East and E. C. Lim, Naphthalene dimer: Electronic states, excimers, and triplet decay, J. Chem. Phys., 2000, 113, 8981–8994 CrossRef CAS.
- A. C. Hancock and L. Goerigk, Noncovalently bound excited-state dimers: a perspective on current time-dependent density functional theory approaches applied to aromatic excimer models, RSC Adv., 2023, 13, 35964–35984 RSC.
- N. O. Dubinets, A. A. Safonov and A. A. Bagaturyants, Structures and Binding Energies of the Naphthalene Dimer in Its Ground and Excited States, J. Phys. Chem. A, 2016, 120, 2779–2782 CrossRef CAS PubMed.
- S. Shirai, Y. Kurashige and T. Yanai, Computational Evidence of Inversion of 1La and 1Lb-Derived Excited States in Naphthalene Excimer Formation from ab Initio Multireference Theory with Large Active Space: DMRG-CASPT2 Study, J. Chem. Theory Comput., 2016, 12, 2366–2372 CrossRef CAS PubMed.
- Y. C. Cheng, R. J. Silbey, D. A. da Silva Filho, J. P. Calbert, J. Cornil and J. L. Brédas, Three-dimensional band structure and bandlike mobility in oligoacene single crystals: A theoretical investigation, J. Chem. Phys., 2003, 118, 3764–3774 CrossRef CAS.
- J. E. Anthony, The Larger Acenes: Versatile Organic Semiconductors, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- K. Hummer and C. Ambrosch-Draxl, Electronic properties of oligoacenes from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 205205 CrossRef.
- G. Schweicher, G. Garbay, R. Jouclas, F. Vibert, F. Devaux and Y. H. Geerts, Molecular Semiconductors for Logic Operations: Dead-End or Bright Future?, Adv. Mater., 2020, 32, 1905909 CrossRef CAS PubMed.
- M. W. B. Wilson, A. Rao, J. Clark, R. S. S. Kumar, D. Brida, G. Cerullo and R. H. Friend, Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene, J. Am. Chem. Soc., 2011, 133, 11830–11833 CrossRef CAS PubMed.
- S. Bettis Homan, V. K. Sangwan, I. Balla, H. Bergeron, E. A. Weiss and M. C. Hersam, Ultrafast Exciton Dissociation and Long-Lived Charge Separation in a Photovoltaic Pentacene–MoS2 van der Waals Heterojunction, Nano Lett., 2017, 17, 164–169 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, External Quantum Efficiency Above 100% in a Singlet-Exciton-Fission–Based Organic Photovoltaic Cell, Science, 2013, 340, 334–337 CrossRef CAS PubMed.
- X. Wang, W. G. Kofron, S. Kong, C. S. Rajesh, D. A. Modarelli and E. C. Lim, Transient Absorption Probe of Intermolecular Triplet Excimer of Naphthalene in Fluid Solutions: Identification of the Species Based on Comparison to the Intramolecular Triplet Excimers of Covalently-Linked Dimers, J. Phys. Chem. A, 2000, 104, 1461–1465 CrossRef CAS.
- S. Tsuzuki, K. Honda, T. Uchimaru and M. Mikami, High-level ab initio computations of structures and interaction energies of naphthalene dimers: Origin of attraction and its directionality, J. Chem. Phys., 2004, 120, 647–659 CrossRef CAS PubMed.
- W. C. Agosta, Transannular and interannular effects in 1,8-(1,8)-naphthalylnaphthalene and related compounds, J. Am. Chem. Soc., 1967, 89, 3505–3510 CrossRef CAS.
- C. Climent, M. Barbatti, M. O. Wolf, C. J. Bardeen and D. Casanova, The photophysics of naphthalene dimers controlled by sulfur bridge oxidation, Chem. Sci., 2017, 8, 4941–4950 RSC.
- G. D. Scholes, G. O. Turner, K. P. Ghiggino, M. N. Paddon-Row, J. J. Piet, W. Schuddeboom and J. M. Warman, Electronic interactions in rigidly linked naphthalene dimers, Chem. Phys. Lett., 1998, 292, 601–606 CrossRef CAS.
- S. Tretiak, W. M. Zhang, V. Chernyak and S. Mukamel, Excitonic couplings and electronic coherence in bridged naphthalene dimers, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13003–13008 CrossRef CAS PubMed.
- X. Niu, K. Tajima, J. Kong, M. Tao, N. Fukui, Z. Kuang, H. Shinokubo and A. Xia, Symmetry-breaking charge separation in a nitrogen-bridged naphthalene monoimide dimer, Phys. Chem. Chem. Phys., 2022, 24, 14007–14015 RSC.
- R. Jing, Y. Li, K. Tajima, Y. Wan, N. Fukui, H. Shinokubo, Z. Kuang and A. Xia, Excimer Formation Driven by Excited-State Structural Relaxation in a Covalent Aminonaphthalimide Dimer, J. Phys. Chem. Lett., 2024, 15, 1469–1476 CrossRef CAS PubMed.
- N. Banerji, A. Fürstenberg, S. Bhosale, A. L. Sisson, N. Sakai, S. Matile and E. Vauthey, Ultrafast Photoinduced Charge Separation in Naphthalene Diimide Based Multichromophoric Systems in Liquid Solutions and in a Lipid Membrane, J. Phys. Chem. B, 2008, 112, 8912–8922 CrossRef CAS PubMed.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Ultrafast Elementary Photochemical Processes of Organic Molecules in Liquid Solution, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Tuning symmetry breaking charge separation in perylene bichromophores by conformational control, Chem. Sci., 2019, 10, 10629–10639 RSC.
- M. Madhu, R. Ramakrishnan, V. Vijay and M. Hariharan, Free Charge Carriers in Homo-Sorted π-Stacks of Donor–Acceptor Conjugates, Chem. Rev., 2021, 121, 8234–8284 CrossRef CAS PubMed.
- M. Haenel and H. A. Staab, Transanulare Wechselwirkungen bei [2.2]Phanen, III. [2,2](2,6)Naphthalinophan und [2.2](2,6)Naphthalinophan-1,11-dien, Chem. Ber., 1973, 106, 2203–2216 CrossRef CAS.
- M. W. Haenel, Transanulare Wechselwirkungen bei [2.2]Phanen, XII: Chirales und achirales [2.2](1,5)Naphthalinophan, Chem. Ber., 1978, 111, 1789–1797 CrossRef CAS.
- M. B. Ledger and G. Porter, Primary photochemical processes in aromatic molecules. Part 15. – The photochemistry of aromatic carbonyl compounds in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1972, 68, 539–553 RSC.
- E. J. Land and G. Porter, Extinction coefficients of triplet-triplet transitions, Pro. R. Soc. London, Ser. A, 1997, 305, 457–471 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- L. Goerigk and S. Grimme, Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2011, 7, 291–309 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. E. Sandoval-Salinas, E. Brémond, A. J. Pérez-Jiménez, C. Adamo and J. C. Sancho-García, Excitation energies of polycylic aromatic hydrocarbons by double-hybrid functionals: Assessing the PBE0-DH and PBE-QIDH models and their range-separated versions, J. Chem. Phys., 2023, 158, 044105 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Quantum Mechanical Continuum Solvation Models, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- L. Martinez-Fernandez and R. Improta, Localized and Excimer Triplet Electronic States of Naphthalene Dimers: A Computational Study, Molecules, 2025, 30, 298 CrossRef CAS PubMed.
- L. Martínez Fernández, F. Santoro and R. Improta, Nucleic Acids as a Playground for the Computational Study of the Photophysics and Photochemistry of Multichromophore Assemblies, Acc. Chem. Res., 2022, 55, 2077–2087 CrossRef PubMed.
- L. Martinez-Fernandez and R. Improta, in Nucleic Acid Photophysics and Photochemistry, ed. S. Matsika and A. H. Marcus, Springer Nature Switzerland, Cham, 2024, pp. 29–50, DOI:10.1007/978-3-031-68807-2_2.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2405198, 2405200, 2405201, and 2405218. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07982e |
| ‡ Equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2025 |