
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGenerating the Pd-catalyzed δ C–H chalcogenation of aliphatic picolinamides: systematically decreasing the bias†
Soumya Kumar
Sinha
a,
Aniket
Gholap
b,
Yazhinimuthu
C M
a,
Anirban
Pal
a,
Anant R.
Kapdi
 *b and
Debabrata
Maiti
*a
*b and
Debabrata
Maiti
*a
aDepartment of Chemistry, Indian Institute of Technology Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@iitb.ac.in
bDepartment of Chemistry, Institute of Chemical Technology, Nathalal Parekh Road, Matunga, Mumbai-400019, India. E-mail: ar.kapdi@ictmumbai.edu.in
First published on 17th March 2025
Abstract
Reaching out to the distal sp3 C–H bonds remains a daunting challenge to synthetic organic chemists, primarily due to the relative inertness of the C–H bonds in alkanes. As such, most reports have envisaged the employment of sterically biased substrates, which render the other possible positions inaccessible for functionalization. Herein, we report a palladium-catalyzed highly selective δ-chalcogenation of aliphatic picolinamides, whereby both sterically biased and relatively unbiased substrates are made feasible for site-selective δ-C–H functionalization. The successful employment of the Thorpe–Ingold effect explains the reactive intermediates involved. The present protocol also provides direct access to the introduction of structural modifications on α-amino acid structural motifs, such as leucine, with high regioselectivity. Sequential hetero-di-functionalization has been carried out at δ-sp3 C–H bonds, resulting in the desymmetrization of quaternary centers. A thorough mechanistic investigation has been carried out, which provided evidence for the reaction pathway and the plausible mechanistic cycle involved.
Introduction
α-Amino acids and aliphatic amines have been widely explored, primarily due to their ubiquitous influence in biologically important compounds and as building blocks in organic synthesis.1,2 As such, the difficulty of selectively functionalizing any of the distal positions in this class of substrates remains not only a daunting challenge to synthetic organic chemists, but also an important one in terms of its application in drugs and natural products alike.3 C–H activation has enabled the easy generation of such complex molecules.4,5 Synthetic organic chemistry has seen innovative methodologies implemented in the past few decades, which have changed the outlook of chemists towards overcoming challenges, and C–H activation has remained at the forefront of this innovation.6 The main obstacle in developing the selective distal functionalization of aliphatic amines lies in the relative inertness of the C–H bonds in alkanes. Further, the aliphatic chains tend to be fluxional, making regioselectivity issues a significant cause of concern. Additionally, inhibiting the proximal C–H bond activation via a kinetically favoured five-membered metallacycle and driving the reaction towards a less-favoured six-membered metallacycle, thus enabling site-selective distal C–H functionalization, has proven to be a significant hurdle. To overcome these challenges, attempts have been made to employ a designated substrate structure or a suitable directing group that offers the advantage of bringing the transition metal closer to the required distal sp3 C–H bond.7 However, the variety of substrates to carry out this strategy has often been limited. Recently, various groups have used covalent as well as transient template moieties that place the metal catalyst closer to the target C–H bond to overcome the favoured proximal C–H bond activation.8,9 However, most reports to date have outlined the formation of C–C bonds from C–H bonds, with only a single report of site-selective δ C–B bond formation (Scheme 1B).10 Earlier this year, our group reported a ligand-enabled orthogonal strategy that can achieve orthogonal selectivity between the distal δ and γ positions of aliphatic amines based solely on the choice of ligand (Scheme 1C).11 The direct functionalization of C–H bonds to synthesize C–S bonds in aliphatic substrates opens up new avenues towards a vast number of pharmaceutical drugs that can be used as synthons towards many complex molecules (Scheme 1D). Hence, the investigation of site-selective C–S bond formation at the distal positions of aliphatic moieties is of immense importance. | ||
| Scheme 1 Site-selective δ-thioarylation of aliphatic motifs: a brief overview. (A) α-Amino acids prone to selective δ-functionalization. (B) Prior work in distal δ-functionalization. (C) Ligand-controlled orthogonal selectivity between the δ and γ positions: Previous work. (D) Importance of sulfur motifs in complex derivatives. (E) Site-selective δ C–H chalcogenation of α-amino acids and aliphatic amines: this work. | ||
Organosulfur compounds as functional groups are known for their prevalence in several natural products and pharmaceutical drugs.12,13 The incorporation of a C–S moiety at an unactivated aliphatic C–H bond is a highly promising pathway for the synthesis of a variety of such biologically complex molecules (Scheme 1D). However, the tendency of sulphur groups to bind metal ions and poison the metal catalyst is one of the most prominent obstacles towards the C–H functionalization pathway.14
This explains the incompatibility of our reaction protocol when thiols were employed as a sulphur source.15 This unsuccessful attempt led us to examine related reports in the literature in which various groups have employed dichalcogenides to minimize such catalyst-retarding effects, resulting in an effective and highly efficient C–H functionalization process.16 Consequently, we employed the simplest diphenyl disulfide to substrate 1 and observed 38% of our desired δ-thioarylated product.15 Buoyed by this observation and convinced of the need for a highly regioselective pathway that would result in the completely site-selective δ thioarylation of aliphatic amines and α-amino acids to provide synthons towards a wide variety of complex molecules, herein, we report a palladium-catalyzed site-selective δ C–H chalcogenation on various classes of aliphatic picolinamides (Scheme 1E).
Results and discussion
Based on our previous investigations toward understanding the nature of substrates prone to site-selective δ-functionalization,9d our initial objective lay in decoding the kinetic effect for a range of aliphatic amines and α-amino acids. It was theoretically verified that the C–H activation step for an α-methyl substrate is 3.9 kcal mol−1 (ΔG‡) higher than that for the corresponding biased α-dimethyl substrate. This observation was attributed to the lack of Thorpe–Ingold effect in the α-methyl substrate, making the C–H activation step more difficult. The hypothesis can be further correlated with the bond distances between the metal centre and the atoms in the amine motif. The α-dimethyl amine substrate (1) has a Pd–N bond distance of 2.0494 Å, which is slightly longer than the Pd–N bond distance of 2.0065 Å for substrate 1′t (Scheme 2, Int A and Int C). Correspondingly, we wanted to determine whether we could still be successful in promoting site-selective distal δ-C–H functionalization in this class of substrates in which there is no assistance from the Thorpe–Ingold effect. To obtain a clear view of the nature of the organometallic intermediates, we attempted to isolate the C–H activation intermediates for substrates having varying substituents at aliphatic positions. Picolinamide-mediated 2,4,4-trimethylpentan-2-amine (1a), which has two methyl substituents at the α position, yielded a mononuclear six-membered palladacycle δ-C–H activated intermediate at room temperature (Scheme 2, Int A).10 However, attempts to isolate a δ-C–H activated intermediate starting from other substrates were met with less success. We tried to modify the substrate motif based on the substituents at the α- and γ-positions. The α substituent controls the rigidity of the cyclometallated intermediate, while the substituent at the γ position influences the statistical factor, rendering any other functionalization, apart from functionalization at the δ-position, improbable.11 Similar reactions were repeated with picolinamide-mediated pentan-2-amine (1′n) with one methyl substituent at the α position, which, upon reaction with palladium at low temperatures, resulted in a di-nuclear Pd(II) pre-catalyst (Scheme 2, Int B).11 However, it did not provide the δ-C–H activated palladacycle even at elevated temperatures. We observed that the δ-carbon shows a favourable orientation towards the metal catalyst as the bias at the α position increases. These observations indicate the significance of the α-substituents for isolating the δ-C–H activated intermediate. Correspondingly, the influence of the γ-substituents on the desired δ-C–H activated intermediate was studied. 3-Methylbutan-1-amine, with one γ-methyl substituent (1′t) and 3,3 dimethylbutan-1-amine, with two γ-methyl substituents (1′v), both of which were unsubstituted at the α-position, were used for the reaction with palladium. However, the corresponding pre-catalysts (Scheme 2, Int C and Int D) showed identical structures to that of Int B, indicating that the substituents at the γ-position played no role in lowering the C–H activation transition state. Consequently, the Thorpe–Ingold effect is likely to be operative, facilitating the C–H activation state by lowering the transition state for substrates having gem-dimethyl groups at the α-position. Our next goal was to determine whether these C–H activated and pre-C–H activated intermediates were catalytically effective towards our desired site-selective δ-thioarylation.15 Gratifyingly, we observed that all these intermediates were competent in synthesizing our desired δ-product. Hence, we hypothesize that aliphatic motifs having flexible substituents at the α- and γ-positions are unable to facilitate the formation of C–H activated intermediates.11 However, this kinetic effect does not hinder the applicability of our protocol, whereby the developed methodology steers the reaction towards site-selective δ-C–H chalcogenation. | ||
| Scheme 2 X-ray analysis of organometallic intermediates. | ||
We began our inspection with picolinamide-guided 2,4,4-trimethylpentan-2-amine (1). Various templates ranging from quinoline- to pyridine-based ones were screened, and 2-picolinic acid was found to serve as the optimal directing template for site-selective δ functionalization. The choice of ligands has had a very crucial role in controlling the efficiency of both C(sp2)–H and C(sp3)–H activation.17 In this regard, pyridine and quinoline motifs have been ubiquitous choices.17,18 This led us to screen such moieties extensively. Stringent screening led to 4-hydroxyquinoline being the optimum ligand for this class of amine substrates.15 Pd(OAc)2 was found to be the most effective transition metal catalyst, while the best oxidant and solvent were found to be Ag2CO3 and MeOH, respectively. Interestingly, the methodology worked better under an O2 atmosphere than under normal air conditions, and 90 °C was found to be the optimum temperature for this system, with further increases in temperature promoting the di-δ-chalcogenated product as a reaction side-product. Crucially, 1 eq. of disulphide was found to be suitable for promoting the desired site-selective δ-thioarylation. Control of the reaction parameters led us to understand the importance of each component. The reaction was unable to proceed in the absence of a metal catalyst, which is indicative of the importance of the transition metal catalyst in generating the metallacycle, and was considerably less effective in the absence of an Ag salt as an oxidant, which suggested the hindrance of the regeneration of the metal catalyst in the absence of an oxidant.15
Having obtained suitably compatible reaction conditions for site-selective δ-thioarylation, we proceeded to investigate the scope and limitations of the protocol. Gratifyingly, the electronic nature of the disulfides was found to have no effect on the compatibility of the protocol. Both electron-rich (Me) and electron-deficient (Cl, NO2) groups at the 4-position of disulfides were found to promote selective δ-functionalization with promising yields (Scheme 3, entry 2b–2d). Almost identically favourable outcomes could also be obtained with substrates having diverse groups (F, NO2) at the 2- or 3-position of the disulfide ring (Scheme 3, entry 2e–2h). Our next aim was to not limit our work to mono δ-functionalization, but extend it further towards the possibility of functionalizing all the available δ positions. Unfortunately, once a thioarylated group is inserted at the δ position, it inactivates other available δ positions towards any functionalization. Hypothetically, this can be correlated to the electronic nature of sulphur, which renders the metallacycle and correspondingly the substrate inactive for further functionalization. Consequently, our focus shifted from a homo-di-functionalization strategy towards a less-favourable hetero-di-functionalization strategy. Our strategy was centered around utilizing δ arylation9d and then establishing our δ-thioarylation protocol on the already-arylated amine, thus achieving hetero-di-δ-functionalization. As a proof of concept, using 2,4,4-trimethylpentan-2-amine, a –COMe substituted arylating group was inserted at one of the available δ-sites followed by a –NO2 substituted disulfide motif at the other available δ-site, enabling successful hetero-di-δ-functionalization (Scheme 3, entry 2i).
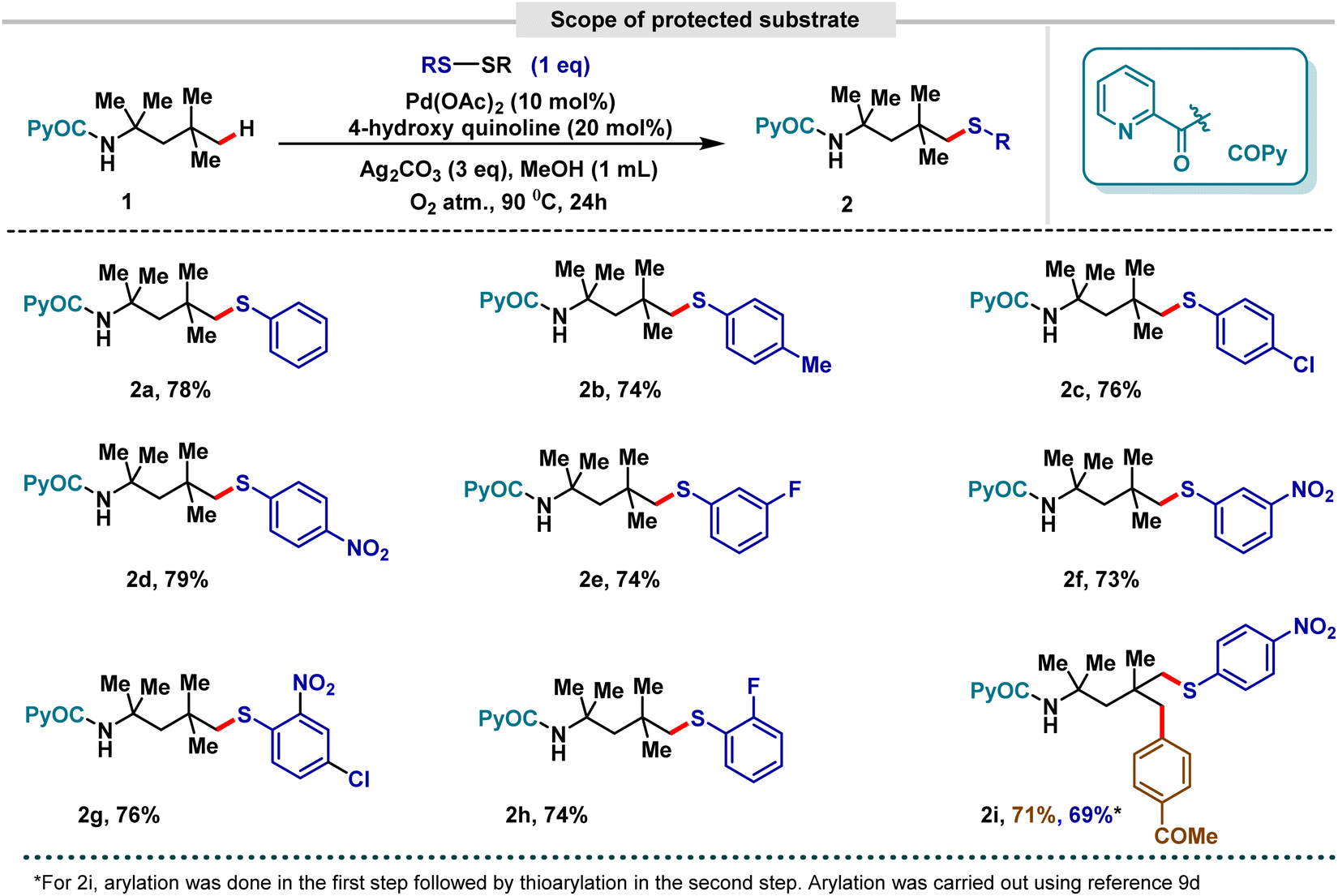 | ||
| Scheme 3 Scope of δ-thioarylation of protected aliphatic amines. | ||
Concurrently, we attempted to focus on extending our methodology to various classes of aliphatic amines and α-amino acids. To execute our protocol and with the aim of decreasing the bias in a systematic manner, we tried to implement α-amino acids that would have a compatible distal δ-position for functionalization. Our attempts centered around developing a protocol that would be suitable to site-selectively functionalize the desired distal δ-position over other positions that might be compatible for functionalization. Fortunately, we found that leucine fit our required model of substrate; however, applying the above optimized reaction conditions led to only 20% formation of the desired δ product, with the unreacted starting material being present in majority.15 To attempt to improve the efficiency of our reaction methodology, an extensive optimization of the reaction conditions was carried out. This led us to find that the combination of Pd(OPiv)2 and Ag2O in TFT solvent at 130 °C was optimal for site-selective δ-thioarylation. Completely site-selective δ-thioarylation was observed over all the other plausible aliphatic positions. This can be hypothesized as being due to the high activation energy for C–H activation at the tertiary γ-position, making it kinetically unfavourable. Consequently, the kinetically favoured δ C–H activation is observed, resulting in facile site-selective δ-thioarylation. An extensive screening of various pyridine and quinoline ligands was attempted, and 2-chloroquinoline was found to be the best ligand for the δ thioarylation of leucine, resulting in an 82% yield of our desired site-selective δ-chalcogenated product (Scheme 4B). 1.8 eq. of disulfide was found to be optimal for obtaining 82% yield. To investigate directing template structures that would enable site-selective δ-functionalization over other available aliphatic positions, we tested various pyridine, substituted pyridine, quinoline, pyrazine and isoquinoline motifs (Scheme 4A). Based on the initial forays into picolinamide templates by Daugulis and the developments of the Chen group in regioselective C(sp3)–H bond functionalization via the assistance of picolinic acid directing groups,19,2a we also tested a number of substituted picolinic acid templates. However, our previously employed 2-picolinic acid was found to be most suitable for promoting our desired product with a yield of 82%.
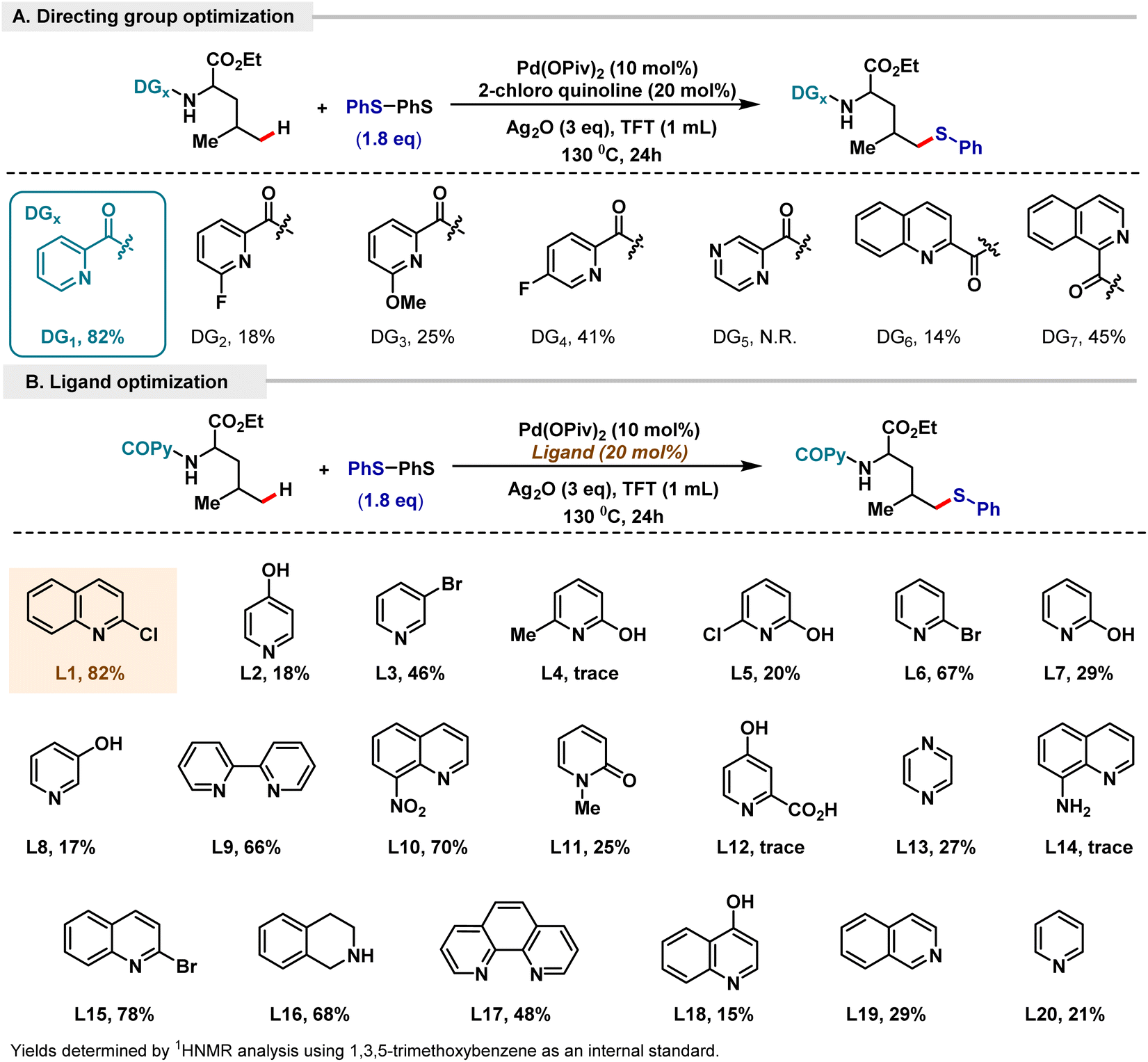 | ||
| Scheme 4 Optimization of the δ-thioarylation of leucine. (A) Directing group variation. (B) Ligand optimization. | ||
After developing the optimized reaction parameters, we proceeded to determine the feasibility and generality of our protocol. Considering the lower thermodynamic feasibility of a six-membered metallacycle over a five-membered one,9 an intriguing assessment of scope of the methodology was awaited. Our investigation began with investigating the compatibility of our optimized reaction conditions with the heavier congener of the chalcogen group, selenium. Sulphur is succeeded in the periodic table by selenium; consequently, its reactivity pattern is expected to be similar to that of sulphur. However, limited reports are available on C–Se bond formation from aliphatic C–H bonds.20,21,8b The protocol worked suitably, generating the δ-selenoarylated product in 82% yield by a simple tuning of the metal catalyst from Pd(OPiv)2 to Pd(OAc)2 (Scheme 5, entry 2′b). Concurrently, we proceeded to determine the feasibility of reacting leucine moieties with a variety of disulfides. Irrespective of the electronic nature of the substituent at the 4-position of the arene ring of the disulfides (Cl, Me, NO2), the reaction was found to be compatible, affording satisfactory yields of the δ-thioarylated products (Scheme 5, entry 2′c–2′e). Similar results were found for disulfides having various substituents at the 3-position of the arene ring, with the nature of substituents varying from highly electron-rich –OMe groups to highly electron-deficient –NO2 groups (Scheme 5, entry 2′f–2′h). The protocol was also applicable for disulfides with doubly substituted aromatic rings (Scheme 5, entry 2′i), thus demonstrating the generality of this transformation and its ability to diversify α-amino acids with a wide variety of disulfides and diselenides, promoting the formation of the site-selective δ-chalcogenated product in synthetically useful yields. To demonstrate the importance of our method for generating diastereoselective δ-chalcogenated products, the L-isomer of leucine was subjected to the above optimized reaction conditions. Delightfully, the L-isomer of leucine successfully reacted with diphenyl disulfide, providing the δ-thioarylated product in 84% yield with a moderate diastereoselectivity ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Scheme 5, entry 2′j). Similar results were found for the higher congener, selenium, with the protocol proving successful in synthesizing the δ-selenoarylated product with a similar diastereoselectivity ratio of 5:1 (Scheme 5, entry 2′k).
:1 (Scheme 5, entry 2′j). Similar results were found for the higher congener, selenium, with the protocol proving successful in synthesizing the δ-selenoarylated product with a similar diastereoselectivity ratio of 5:1 (Scheme 5, entry 2′k).
 | ||
| Scheme 5 Reaction scope towards site-selective δ-thioarylation of α-amino acids and aliphatic amines. | ||
Moving towards our primary objective of systematically decreasing the bias in the aliphatic amines while not altering the possibility of site-selective δ functionalization, we tried to apply our protocol to aliphatic picolinamides having multiple competitive reaction sites. Starting from our initially screened picolinic-acid-mediated 2,4,4-trimethylpentan-2-amine, we formulated other differently substituted amines by removing one substituent each from adjacent α and γ positions. As hypothesized earlier, the two substituents at the α-position are required to bring about the rigidity to hold the transition metal catalyst closer to the desired C–H bond and stabilize the metallacycle intermediate, while the γ-substitution is essential to overcome the kinetically and thermodynamically more favourable γ-functionalization and achieve the less-favourable δ functionalization via both statistical and steric factors combined.11 We predicted that aliphatic amines that are structurally analogous to leucine motifs should be applicable to our methodology. To our delight, treatment of picolinic-acid-mediated 4-methylpentan-2-amine (1′n) with Ph2S2 yielded our desired product in 78% yield (Scheme 5, entry 2′n). We could further extend the methodology to 4-methylhexan-2-amine (1′p), with diselenides as well as unsubstituted and substituted disulfides proving successful in generating the desired δ-chalcogenated products in synthetically viable yields (Scheme 5, entry 2′p–2′s). Interestingly, in spite of the presence of two inequivalent δ-C(sp3)–H sites, the primary C–H bond is preferentially thioarylated over the secondary δ-C–H bond. This can be attributed to two factors: (a) the more facile activation of the primary C–H compared to the secondary ones11,22 and (b) steric clashes between the ε-methyl group and the heterocyclic ring of the directing group and the distal methyl group on the α-carbon.9d Moving towards the next step of our objective, removing all the substituents from the α-position and generating fully selective δ functionalization even without the rigidity that holds the metallacycle together remained our priority. The first attempt was made using picolinic-acid-guided 3,3-dimethylbutan-1-amine (1′t), in which the γ position is sterically biased, but the α position remains free from any bias. Agreeably, the condition of maintaining the rigidity to hold the metallacycle proved unnecessary, as this substrate resulted in the formation of the δ-chalcogenated products with both disulfides and diselenides, emphasizing the feasibility and generality of our protocol for synthesizing site-selective δ-chalcogenated products from a wide variety of aliphatic picolinamides and chalcogenated derivatives (Scheme 5, entry 2′t and 2′u) in good yields. The final step was removing all the substituents from not only the α-position, but also from the γ-position, and then using our methodology to promote site-selective δ-chalcogenation. In this regard, picolinic-acid-mediated 3-methylbutan-1-amine (1′v) was examined with a range of unsubstituted and substituted disulfides and diselenides. All these were found to be compatible, giving the desired δ-chalcogenated products in synthetically viable yields (Scheme 5, entry 2′v–2′y).
Most of the reports so far have involved acyclic amines. However, the generality of our methodology lies in its extension to cyclic aliphatic amines as well. We proceeded to investigate the compatibility of our developed protocol towards alicyclic amines having several competitive reaction sites. Picolinic-acid-protected cyclic hexyl amines (1′z) were hugely successful in providing the corresponding δ thioarylated and selenoarylated products (Scheme 5, entry 2′z–2′ac), and complex alicyclic amines such as pinamine (1′ad) reacted with a wide range of disulphides ranging from electron-rich –Me containing disulphides to electron-deficient –NO2 bearing ones to give the corresponding products in good-to-moderate yields (Scheme 5, entry 2′ad and 2′ae). In all these cases, site-selective δ C–H chalcogenation was observed over all other possible δ- and γ-reactive sites. This can be attributed to the unfavourable interactions in the C–H activation transition state.9d Having obtained a complete idea of the protocol for site-selective chalcogenation of one available δ-site, we proceeded to examine the feasibility of generating hetero-difunctionalized products via both the available C–H bonds at the δ-position of α-amino acids such as leucine, which would provide the first such example of truly selective δ-hetero-bi-functionalized leucine derivatives. Delightfully, initial δ-arylation9d followed by our developed δ-thioarylation methodology was successful in functionalizing both available δ positions of leucine (Scheme 5, entry 2′af), providing the first example of δ-hetero-bi-functionalized leucine motifs, which could be useful synthons for a wide variety of drug derivatives and complex molecules.
To gain insight into the mechanism of the overall reaction and the role of the synthesized organometallic intermediates, we embarked on various control experiments.15 The acetonitrile-coordinated [5,6]-organometallic complex Int. A10 was synthesized and found to be catalytically competent towards our reaction methodology.15 Additionally, stoichiometric reaction of Int. A with 1 eq. of diphenyl disulfide provided our desired δ-thioarylated product in 69%, hence implying the competency of the complex in the overall reaction mechanism. Similar analogies were drawn with Int. C,11 which was also found to be catalytically competent, thus demonstrating its importance in the methodology. Since our protocol provides easy access to the δ-chalcogenation of α-amino acids, our mechanistic attempts were centred around understanding the reaction pathway in such δ-selective leucine motifs (1′a). Firstly, we conducted kinetic studies to probe the role of individual components in the reaction methodology. We observed a drastic four-fold delay in the reaction performance as soon as we removed either or both the ligand and oxidant (Scheme 6B). This is consistent with our mechanistic hypothesis: (a) the absence of the ligand hinders the formation of the metal–ligand complex, thus impeding the reaction, while (b) the absence of the Ag oxidant inhibits the oxidative addition of disulfide into the C–H activated complex, demonstrating its importance in promoting the successful implementation of the reaction. Additionally, in accordance with literature precedents, Ag+ ions have been found to facilitate hetero-bimetallic cluster formation, facilitating product release and significantly enhancing the catalytic performance of the organometallic complex.23 Order-determination studies with substrate 1′a and disulfide showed an order of 1 and 0 respectively, demonstrating the presence of an amide substrate and the absence of disulfide in the overall rate-determining step of the reaction protocol (Scheme 6A).15 Additionally, no deuterium incorporation was observed even after two cycles in such amide substrates without a quaternary γ-center (1′a), indicating the C–H activation step to be irreversible for this class of substrate (Scheme 6C).
 | ||
| Scheme 6 Mechanistic insights. (A) Order determination studies of substrate 1′. (B) Role of individual components. (C) Reversibility experiment. (D) Plausible mechanistic cycle. | ||
Based on the kinetic studies and related reports in the literature, a plausible mechanistic design was crafted for α-amino acids such as leucine (Scheme 6D). Complexation of amide 1′a to Pd(OPiv)2 affords the metal–ligand complex A. The presence of complex A was demonstrated experimentally via X-ray crystallography. Intermediate A undergoes δ C–H activation to form the six-membered palladacycle B, which is irreversible for this class of substrate (2′a). Intermediate B then undergoes subsequent oxidative addition of diphenyl disulfide to form intermediate C, which undergoes reductive elimination to form the δ-thioarylated product 2′a, regenerating the metal catalyst for the next cycle.
Conclusion
In summary, this protocol provides easy access to a series of novel unnatural α-amino acids, which could be important building blocks towards complex molecules and drug derivatives alike, via site-selective δ-chalcogenation of the leucine motif. In addition, we have demonstrated that its ability is not limited to the δ-functionalization of substrates that are conformationally and sterically biased towards the distal δ-position, but extends to all classes of substrates. Various classes of aliphatic amines and α-amino acids have been screened, with the substrate bias being decreased systematically without compromising the fully exclusive δ site-selectivity. Through this approach, we have demonstrated the first example of the hetero-di-δ functionalization of α-amino acids (leucine). We have managed to replicate the nature of the intermediate associated with the removal of substrate bias and performed kinetic studies to obtain an idea of the overall mechanistic cycle. Through this approach, we have mostly shown the generality of performing site-selective distal δ-chalcogenation, thus opening the door for new avenues in the field of aliphatic C–H activation.Data availability
Data supporting the manuscript are provided in the ESI,† including the experimental methods and protocols for this study.Author contributions
S. K. S. and D. M. conceived the project. S. K. S., A. G., Y. C. M., A. P. performed the experiments under the supervision of D. M. and A. K. S. K. S. and D. M. co-wrote the paper.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Financial support received from SERB India (CRG/2022/004197) is gratefully acknowledged. Financial support received from UGC-India (Fellowship to S. K. S.) is gratefully acknowledged. We would like to thank Mr Tanay Pal (PhD student in DM Lab) for his help during the revision work.Notes and references
- Asymmetric Synthesis and Application of α-Amino acids, ed. V. A. Soloshonok and K. Izawa, American Chemical Society, Washington DC, 2009, vol. 1009 Search PubMed.
- (a) V. G. Zaitsev, D. Shabashov and O. J. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CAS; (b) M. Fan and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 12152–12155 CAS; (c) E. T. Nadres, G. I. F. Santos, D. Shabashov and O. J. Daugulis, J. Org. Chem., 2013, 78, 9689–9714 CrossRef CAS PubMed; (d) J. Calleja, D. Pla, T. W. Gorman, V. Domingo, B. Haffemayer and M. J. Gaunt, Nat. Chem., 2015, 7, 1009–1016 CAS; (e) C. He and M. J. Gaunt, Angew. Chem., Int. Ed., 2015, 54, 15840–15844 CrossRef CAS PubMed; (f) Z. Fan, S. Shu, J. Ni, Q. Yao and A. Zhang, ACS Catal., 2016, 6, 769–774 CAS; (g) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2018, 7, 1178–1192 CrossRef CAS; (h) S. K. Sinha, P. Ghosh, S. Jain, S. Maiti, S. A. Al-Thabati, A. A. Alsehri, M. Mokhtar and D. Maiti, Chem. Soc. Rev., 2023, 52, 7461–7503 RSC.
- A. Ambrogelly, S. Palioura and D. Söll, Nat. Chem. Biol., 2007, 3, 29–35 CrossRef CAS.
- (a) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (b) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (c) R. H. Crabtree and A. Lei, Chem. Rev., 2017, 117, 8481–8482 CrossRef CAS PubMed; (d) T. Dalton, T. Faber and F. Glorius, ACS Cent. Sci., 2021, 7, 245–261 CrossRef CAS PubMed.
- (a) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed; (b) U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, 701 CrossRef PubMed; (c) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS PubMed; (d) Z. Chen, M.-Y. Rong, J. Nie, X.-F. Zhu, B.-F. Shi and J.-A. Ma, Chem. Soc. Rev., 2019, 48, 4921–4942 RSC; (e) N. Y. S. Lam, Z. Fan, K. Wu, H. S. Park, S. Y. Shim, D. A. Strassfeld and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 2793–2803 CrossRef CAS PubMed; (f) S. Kaltenberger and M. van Gemmeren, Acc. Chem. Res., 2023, 56, 2459–2472 CrossRef CAS PubMed; (g) U. Dutta and D. Maiti, Acc. Chem. Res., 2022, 55, 354–372 CrossRef CAS PubMed.
- (a) Y. Feng and G. Chen, Angew. Chem., Int. Ed., 2010, 49, 958–961 CrossRef CAS PubMed; (b) D. Y.-K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452–9474 CrossRef CAS PubMed; (c) L. M. Chapman, J. C. Beck, L. Wu and S. E. Reisman, J. Am. Chem. Soc., 2016, 138, 9803–9806 CrossRef CAS PubMed; (d) P. Gandeepan, T. Muller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- G. He and G. Chen, Angew. Chem., Int. Ed., 2011, 50, 5192–5196 CrossRef CAS PubMed.
- (a) X. Ye, Z. He, T. Ahmed, K. Weise, N. G. Akhmedov, J. L. Peterson and X. Shi, Chem. Sci., 2013, 4, 3712–3716 RSC; (b) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664–2669 CrossRef CAS; (c) G. Xia, J. Weng, L. Liu, P. Verma, Z. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571–577 CrossRef CAS PubMed; (d) Y. Wu, Y.-Q. Chen, T. Liu, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14554–14557 CrossRef CAS PubMed; (e) J. Das, S. Guin and D. Maiti, Chem. Sci., 2020, 11, 10887–10909 RSC.
- (a) J.-W. Xu, Z.-Z. Zhang, W.-H. Rao and B.-F. Shi, J. Am. Chem. Soc., 2016, 138, 10750–10753 CrossRef CAS PubMed; (b) Y.-Q. Chen, Z. Wang, Y. Wu, S. R. Wisniewski, J. X. Qiao, W. R. Ewing, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2018, 140, 17884–17894 CrossRef CAS PubMed; (c) H. Lin, X. Pan, A. L. Barsamian, T. M. Kamenecka and T. D. Bannister, ACS Catal., 2019, 9, 4887–4891 CrossRef CAS; (d) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrasekhar, S. S. Anjana, R. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633–5638 CrossRef CAS PubMed; (e) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS PubMed; (f) T. Bhattacharya, P. K. Baroliya, S. A. Al-Thabaiti and D. Maiti, JACS Au, 2023, 3, 1975–1983 CrossRef CAS PubMed.
- H. B. Chandrashekar, P. Dolui, B. Li, A. Mandal, H. Liu, S. Guin, H. Ge and D. Maiti, Angew. Chem., Int. Ed., 2021, 60, 18194–18200 CrossRef CAS PubMed.
- S. K. Sinha, N. Goswami, Y. Li, S. Maji, D. Raja, A. S. Sarala, S. Guin, R. S. Paton and D. Maiti, ACS Catal., 2024, 14, 12681–12693 CrossRef CAS.
- (a) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133–5209 CAS; (b) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (c) C. Zhao, K. P. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed; (d) S. K. Sinha, S. Panja, J. Grover, P. S. Hazra, S. Pandit, Y. Bairagi, X. Zhang and D. Maiti, J. Am. Chem. Soc., 2022, 144, 12032–12042 CrossRef CAS PubMed; (e) A. L. Demain, Adv. Appl. Microbiol., 1959, 1, 23 CAS; (f) T. Bottiglieri, K. Hyland and E. H. Reynolds, Drugs, 1994, 48, 137–152 CrossRef CAS PubMed; (g) J. M. Williams, K. M. J. Brands, R. T. Skerji, R. B. Jobson, G. Marchesini, K. M. Conrad, B. Pipik, K. A. Savary, F.-R. Tsay, P. G. Houghton, D. R. Sidler, U.-H. Dolling, L. M. DiMichele and T. J. Novak, J. Org. Chem., 2005, 70, 7479–7487 CrossRef CAS PubMed.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed.
- L. L. Hegedus and R. W. McCabe, in Catalyst Poisoning, Marcel Dekker, New York, 1984 Search PubMed.
- See ESI† for further details.
- (a) X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Xin, Org. Lett., 2015, 17, 1970–1973 CrossRef CAS PubMed; (b) H.-Y. Xiong, T. Besset, D. Cahard and X. Pannecoucke, J. Org. Chem., 2015, 80, 4204–4212 CrossRef CAS PubMed; (c) S.-Y. Yan, Y.-J. Liu, B. Liu, Y.-H. Liu, Z.-Z. Zhang and B.-F. Shi, Chem. Commun., 2015, 51, 7341–7344 RSC; (d) X. Ye, J. L. Petersen and X. Shi, Chem. Commun., 2015, 51, 7863–7866 RSC.
- (a) Y.-H. Zhang, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 5072 CrossRef CAS PubMed; (b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118–1126 CrossRef; (c) A. D. Ryabov, I. K. Sakodinskya and A. K. Yatsimirsky, J. Chem. Soc., Dalton Trans., 1985, 1985, 2629–2638 RSC.
- (a) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833–851 CrossRef CAS PubMed; (b) M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS PubMed; (c) N. Goswami, N. Kumar, P. Gupta and D. Maiti, ACS Catal., 2024, 14, 3798–3811 CAS; (d) C. He, W. G. Whitehurst and M. J. Gaunt, Chem, 2019, 5, 1031–1058 CrossRef CAS.
- (a) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef CAS PubMed; (b) T. T. Nguyen and O. Daugulis, Chem. Commun., 2017, 53, 4609–4611 CAS; (c) G. He, B. Wang, W. A. Nack and G. Chen, Acc. Chem. Res., 2016, 49, 635–645 CAS; (d) H. Wang, H.-R. Tong, G. He and G. Chen, Angew. Chem., Int. Ed., 2016, 55, 15387–15391 CrossRef CAS PubMed; (e) B. Li, X. Li, Z. Chen, X. Zhang, G. He and G. Chen, J. Am. Chem. Soc., 2019, 141, 9401–9407 CAS; (f) Y. Huang, X. Lv, H.-R. Tong, W. He, Z. Bai, H. Wang, G. He and G. Chen, Org. Lett., 2024, 26, 94–99 CAS.
- X. Wang, R. Qiu, C. Yan, V. P. Reddy, L. Zhu, X. Xu and S.-F. Yin, Org. Lett., 2015, 17, 1970 CrossRef CAS PubMed.
- C. Lin, W. Yu, J. Yao, B. Wang, Z. Liu and Y. Zhang, Org. Lett., 2015, 17, 1340 CrossRef CAS PubMed.
- (a) J. D. Lawrence, M. Takahashi, C. Bae and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 15334–15335 CrossRef CAS PubMed; (b) R. Ge, F. Herrington, A. Mangawang, D. Maiti and H. Ge, Tetrahedron Chem, 2023, 7, 100046 CrossRef CAS; (c) Y.-H. Li, Y. Ouyang, N. Chekshin and J.-Q. Yu, J. Am. Chem. Soc., 2022, 144, 4727–4733 CrossRef CAS PubMed; (d) J. Das, W. Ali, A. Ghosh, T. Pal, A. Mandal, C. Teja, S. Dutta, R. Pothikumar, H. Ge, X. Zhang and D. Maiti, Nat. Chem., 2023, 15, 1626–1635 CrossRef CAS PubMed.
- T. Bhattacharya, S. Dutta and D. Maiti, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07897g |
| This journal is © The Royal Society of Chemistry 2025 |