
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComment on “Discovery of a polyketide carboxylate phytotoxin from a polyketide glycoside hybrid by β-glucosidase mediated ester bond hydrolysis” by X. Wang, D.-K. Kong, H.-R. Zhang, Y. Zou, Chem. Sci., 2024, 15, 17183
Seyed Amirhossein
Nasseri
 ab,
Rajneesh K.
Bains
ab,
Yuqing
Tian
b and
Stephen G.
Withers
ab
ab,
Rajneesh K.
Bains
ab,
Yuqing
Tian
b and
Stephen G.
Withers
ab
aDepartment of Chemistry, University of British Columbia, Vancouver, Canada. E-mail: withers@chem.ubc.ca
bMichael Smith Laboratories, University of British Columbia, Vancouver, Canada
First published on 28th January 2025
Abstract
We challenge the conclusion that the β-glucosidase in question directly catalyses hydrolysis of the substrate ester linkage. Rather we propose that this enzyme performs a normal glucoside hydrolysis and that the released aglycone undergoes rearrangement with formation of a quinone methide-like species through spontaneous cleavage of the ester.
The above paper by Wang et al. describes the pathway by which polyketide phytotoxins are biosynthesised in the fungus F. proliferatum.1 The work is, for the most part, well performed and convincing. However, the conclusion, which they highlight in the title and the abstract, that a β-glucosidase (ProL) from CAZy family GH3 directly catalyses the hydrolysis of an ester bond within the key substrate (1) to release the unsaturated fatty acid as shown below, is highly unlikely. It is unclear whether the authors believe that ProL cleaves both the glycosidic bond and the ester bond or whether they believe it is just an esterase with no glycosidic activity, as the fate of the pyrone aglycone (coloured blue in the scheme) is not discussed. In any case, it would be an unprecedented role for a glycosidase, with the classical active site residues, to act as an esterase.

A much simpler, and in many ways more interesting mechanism, is one in which enzymatic hydrolysis of the glycosidic bond by ProL is followed by spontaneous rearrangement of the α-pyrone moiety with release of the fatty acid, through the formation of a quinone methide (QM)-like intermediate. This intermediate in turn most likely becomes hydrated by addition of water to the terminal alkene carbon with regeneration of an α-pyrone, as shown below.
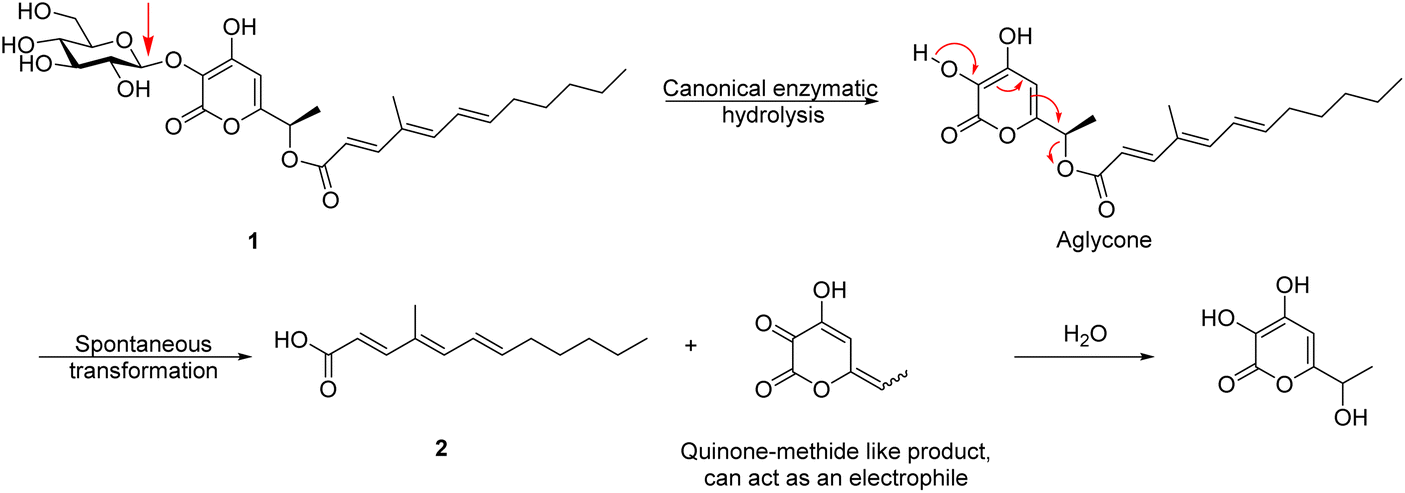
The key experiments cited to support the contention that ester hydrolysis is enzymatic were conducted with A. nidulans (AN), a commonly used heterologous expression system. Here, the authors showed production of (2) with the ANproL strain, whereas the ANWT showed minimal formation of (2), as also did mutants in which key active site residues had been substituted (ANproL D266A and ANproL E493A). These experimental data are all consistent with our proposed mechanism as the inactivation of the glucosidase activity will abolish the rearrangement and release. Indeed, even if ProL was responsible for the hydrolysis of the ester, it would be unlikely that the canonical glycosidase active site residues (D266 and E349) would be involved in an entirely different reaction.
The experiments using H218O shown in their Fig. 5b would, at first glance, appear to disqualify the quinone methide-mediated mechanism we propose, since according to our proposed mechanism, the carboxylate oxygens should both derive from the parent compound 1 and not from the water. However, careful reading of the experimental section makes it clear that the figure is (unintentionally) misleading. The experiment was performed by incubating the organism (Fusarium proliferatum) in H218O such that all steps in the formation of the finally formed and detected fatty acid take place in labelled water. Consequently, it is highly probable that the parent compound 1 carried 18O within its carboxylic acid moiety, especially since the ketone of intermediate 8 (the substrate for the Baeyer–Villiger reaction, as shown in their Fig. 4d) would undergo reversible hydration thus “wash-in” of 18O over that lengthy time period. The authors should really have reported the mass spectrum of a sample of 1 isolated from samples grown for four days in H218O. In addition, it is probable that the 18O “washed in” to the carboxylic acid product spontaneously, as suggested by the presence of doubly 18O-labelled material in the spectra of Fig. 5b. A control experiment in which the carboxylic acid 2 was incubated for 4 days in H218O under the same conditions would have confirmed whether such “wash-in” occurs.

The authors comment that the “β-glucosidase-mediated hydrolysis of ester bonds” has been reported before.2,3 However, in both those cases the cleavage was via a normal glycosidase reaction in which the carboxylate is the leaving group, as shown above. It had never been implied that the glycosidases were acting as esterases. Indeed, Kiso et al.2 through 18O labelling experiments, proved that this was not the case for the β-glucosidase they studied. Thus, even in these cases, the glycosidases studied hydrolyze the substrates by acting on the glycosidic bond (shown with the red arrow in the figure below) and not the ester bond.

The authors could also identify all products formed upon incubation of ProL (crude mixture) with compound 1 and look at what else is formed besides the unsaturated acid 2. According to their hypothesis the other product should/might be the glucosyl α-pyrone. According to our mechanism this will be a mixture of glucose and the α-pyrone that we contend has formed via the QM-like intermediate. Depending on its stability, it might be possible to observe this QM-like intermediate directly, or else confirm its existence by conducting the reaction in the presence of highly reactive nucleophiles, such as the thiol β-mercaptoethanol as shown above, which is often used to trap QMs.
Additionally, the authors could incubate compound 1 with any other promiscuous β-glucosidase that is capable of hydrolyzing the glycosidic bond of 1 but that is unlikely to have evolved specific esterase activity. Our hypothesis predicts that this would yield compound 2 in all cases, as the cleavage of the glycosidic linkage should result in the spontaneous breakdown of the ester bond.
Interestingly, this likely constitutes an example of Nature employing a linker moiety that, as a consequence of cleavage of the bond at one end, spontaneously breaks a bond at the other end to expel a second molecule. Linkers with this property, referred to as “self-immolative linkers”, were first developed by organic chemists in 1981 (ref. 4) and have found a wide variety of applications in release of therapeutics or reporter molecules.5 Probably the most common self-immolative linkers involve the formation and release of QMs and related compounds, much like this. We are aware of a few other examples of ‘natural’ self-immolative linkers, with perhaps the most similar being that in the plant natural product salicortin. This phenolic glycoside is produced as an anti-feedant by plants of the willow family and has been shown to generate reactive QMs upon cleavage by β-glucosidases, as shown below.6 In this case, however, it is not clear whether its toxicity derives from the released QM or from the released cyclohexenoyl fragment, or oxidation products thereof. Another, more chemically modest example is that of the cyanogenic glycosides7 which degrade to release cyanide as part of the plants' defense systems, as shown below. The “pyrone-quinone-methide”-forming linker proposed here, might therefore be an additional example of a ‘natural’ self-immolative linker: the authors of the paper are well-placed to establish that.

Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors have no conflicts of interest to report.Notes and references
- X. Wang, D.-K. Kong, H.-R. Zhang and Y. Zou, Chem. Sci., 2024, 15, 17183–17192 RSC.
- T. Kiso, S. Kitahata, K. Okamoto, S. Miyoshi and H. Nakano, J. Biosci. Bioeng., 2000, 90, 614–618 CrossRef CAS PubMed.
- Y. Hua, S. Sansenya, C. Saetang, S. Wakuta and J. R. Ketudat Cairns, Arch. Biochem. Biophys., 2013, 537, 39–48 CrossRef CAS PubMed.
- P. L. Carl, P. K. Chakravarty and J. A. Katzenellenbogen, J. Med. Chem., 1981, 24, 479–480 CrossRef CAS PubMed.
- A. Abe and M. Kamiya, Bioorg. Med. Chem., 2021, 44, 116281 CrossRef CAS PubMed.
- J. Zhu, S. G. Withers, P. B. Reichardt, E. Tradwell and T. P. Clausen, Biochem. J., 1998, 332, 367–371 CrossRef CAS PubMed.
- R. M. Gleadow and B. L. Møller, Annu. Rev. Plant Biol., 2014, 65, 155–185 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |