DOI:
10.1039/D4SC07743A
(Edge Article)
Chem. Sci., 2025,
16, 6089-6098
Cysteine-selective [188Re]Re(V) radiolabelling of a Nanobody® for targeted radionuclide therapy using a “chelate-then-click” approach†
Received
14th November 2024
, Accepted 25th February 2025
First published on 26th February 2025
Abstract
In this study, we present the first reported use of bioorthogonal click chemistry with rhenium-188 for radiolabelling of an anti-c-Met VHH Nanobody®. We employed a “chelate-then-click” strategy, wherein a bifunctional chelator was designed in two parts, which were subsequently joined post-labelling and post-conjugation via the strain-promoted azide–alkyne cycloaddition (SPAAC) reaction. Cysteine-selective conjugation of the VHH was achieved through thiol-Michael addition, forming a VHH-DBCO construct. Radiolabelling of the azide-functionalised chelator with [188Re]Re(V) was optimised to achieve a radiochemical conversion of ∼70%, despite challenges associated with maintaining the azide functionality under reducing conditions. The final product, [188Re]Re-VHH, demonstrated high radiochemical purity and good in vitro stability over 48 h. In vitro cell-binding studies against U87MG and BxPC3 cell lines proved the retention of c-Met binding post-labelling. In vivo biodistribution studies on mice bearing BxPC3 tumour xenografts, however, exhibited suboptimal tumour uptake, likely a result of the low molar activity (1.4–3.3 MBq nmol−1) of the radioconjugate. This work illustrates the potential of bioorthogonal click chemistry for radiolabelling biomolecules with 188Re, although further optimisation or alternative radiolabelling strategies to enhance the molar activity are necessary to improve pharmacokinetics.
1. Introduction
Rhenium-188, with its favourable physical properties and generator production, has often been recognised as an attractive radionuclide in the targeted treatment of cancer. In addition to its emission of high-energy β− particles (Emax = 2.12 MeV, Eav = 763 keV) and relatively short 16.9 h half-life, 188Re is readily available as no-carrier-added perrhenate ([188Re]ReO4−) from a 188W/188Re generator.1–3 The latter makes the development of 188Re-radiopharmaceuticals of particular interest, as generator-based radionuclides can be eluted on demand at reasonable costs and with high specific activity. 188Re is the group 7 congener of the widely used SPECT imaging radionuclide, 99mTc, sharing many chemical similarities and, therefore, chelator systems as well.4 Since the ability to reduce [188Re]ReO4− and prevent its reoxidation to the +7 oxidation state is more difficult than for [99mTc]TcO4−, harsher labelling conditions for 188Re than used with 99mTc (higher temperatures, more reducing agent, etc.) are often required.5,6 Additionally, effort needs to be put towards finding a chelator system that ensures a high level of chemical inertness and stability for 188Re, particularly in vivo. The tetradentate NxS4−x chelator system has been used extensively to coordinate Tc(V) and Re(V) to form well-defined, stable complexes.1,7,8 In particular, N2S2 monoamine-monoamide dithiol (MAMA) chelators are especially attractive, as MAMA derivatives complexed with 99mTc(V) and 186/188Re(V) are neutral and have been shown to be stable under physiological conditions.9–13 Additionally, the single amine on N2S2 MAMA chelators, as opposed to the two amines on N2S2 bis(aminoethanethiol) (BAT) chelators or no amines on N2S2 diamido-dithiol (DADS) chelators (Fig. 1), allows for the easy and unambiguous conjugation of a targeting vector such as a small molecule, peptide or biomolecule.13
 |
| | Fig. 1 Examples of N2S2 chelators for chelating 99mTc(V) and 186/188Re(V), and the [188Re]Re(V)-N2S2 complex used in this study. | |
While antibodies are the biomolecules most frequently used as targeting vectors in targeted radionuclide therapy (TRNT), the 16.9 h half-life of 188Re makes it more compatible with vectors that have shorter biological half-lives.14 There has been a growing interest in the use of Nanobodies® (VHH, variable domain of the heavy chain of heavy-chain only antibodies) as they have several favourable advantages, particularly when it comes to their in vivo pharmacokinetics.14–16 They are among the smallest known antigen-binding antibody fragments (∼15 kDa), which gives them excellent tumour penetration properties, as well as the ability to pass through the glomerular filtration barrier for rapid excretion.15,16 Their small size, however, often does not allow for sufficient residence time in the bloodstream, which is a requirement for adequate tumour accumulation.17 For this reason, the plasma half-life of a VHH is oftentimes increased by the addition of a biological half-life extending (HLE) group, such as one which binds to serum albumin,17,18 or tuned by PEGylation.19 This helps to provide a balance between excretion and unwanted radiation toxicity side-effects for successful therapy.
Herein, we describe the radiolabelling of a HLE anti-c-Met VHH with [188Re]Re(V)-N2S2 (Fig. 1). C-Met is a membrane receptor protein belonging to the receptor tyrosine kinase family and is commonly activated by hepatocyte growth factor.20 Once activated, it stimulates cell proliferation and motility, including the invasion and migration of cancer cells, making it a good target in the treatment of cancer.20 However, the radiolabelling of a heat-sensitive biomolecule, such as a VHH, with 188Re(V) involves several challenges. The harsh labelling conditions of the Re(V)-oxo core would effectively destroy any sensitive biomolecule in the process. Additionally, it is preferred to attach bifunctional chelators to biomolecules in a site-specific manner in order to produce well-defined conjugates with retained affinity.21 Since this usually involves a cysteine-mediated conjugation and the chelator of choice, N2S2, contains free thiols prior to radiolabelling, it seemed almost impossible to proceed with this method. Post-conjugation labelling would destroy the VHH, while pre-conjugation labelling would result in the free thiols of the bifunctional chelator reacting with the Michael-acceptor needed for bioconjugation. For this reason, we turned to the efficient method of bioorthogonal click chemistry. With the advent of bioorthogonal click chemistry, more possibilities than ever before are available in the fields of radioimaging and radiotherapy.22 In particular, the copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) and inverse electron demand Diels–Alder (IEDDA) reactions have shown promise in the development of radiopharmaceuticals because of their mild reaction conditions and relatively quick reaction times.23 In this work, we employed SPAAC in a “chelate-then-click” approach to effectively radiolabel a VHH with 188Re (Fig. 2). This method found success with Lodhi et al. in the labelling of human serum albumin with 99mTc.24 To our knowledge, this is the first reported use of bioorthogonal click chemistry with 188Re.
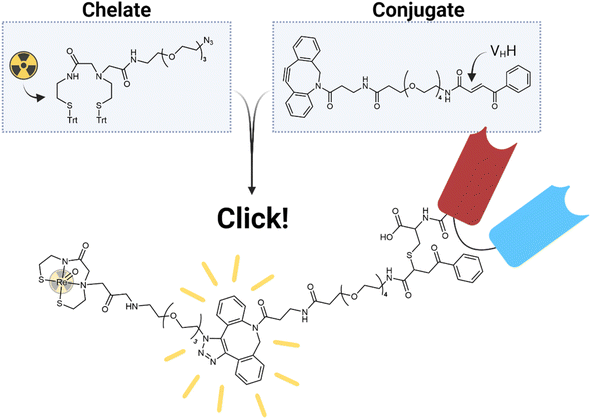 |
| | Fig. 2 The “chelate-then-click” labelling strategy employed in this study. | |
2. Results and discussion
2.1 Synthesis
The N2S2 MAMA ligand was chosen as a suitable chelator as it is known to form a stable neutral complex with Re(V), the most easily accessible oxidation state of Re from perrhenate.25 Additionally, to our knowledge, this chelator has not yet been investigated for the labelling of a heat-sensitive biomolecule such as a VHH with 188Re. The trityl-protected N2S2 MAMA chelator (1) was synthesised in two steps according to a literature procedure.10 Reaction of the chelator with ethyl bromoacetate and subsequent saponification of the ester (2) yielded the chelator with a pendant carboxylic acid (3), as described previously.26 This acid allowed for the amide coupling of H2N-PEG3-N3 to produce N2S2-PEG3-N3 (4) (Scheme 1A). The dibenzocyclooctyne (DBCO)-functionalised carbonylacrylic (CA) compound for cysteine VHH modification was synthesised via a series of amide couplings, resulting in DBCO-PEG4-CA (7) (Scheme 1C). Detailed spectra and characterisation data are provided in the ESI.†
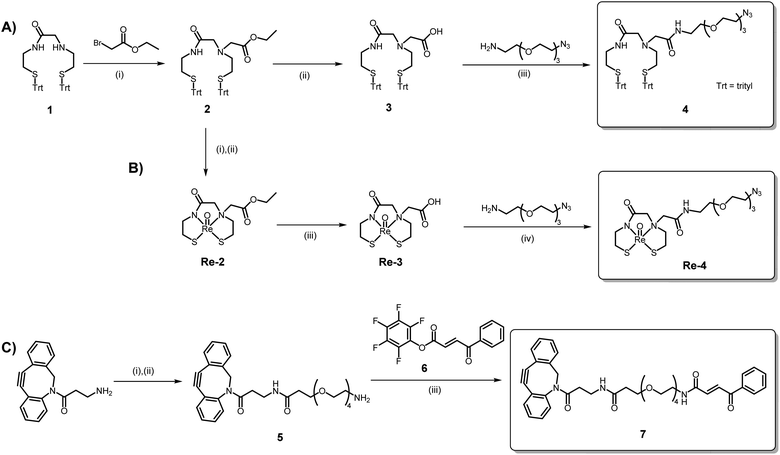 |
| | Scheme 1 (A) Synthesis of the N2S2-PEG3-N3 chelator, 4. Reagents and conditions: (i) DIPEA, MeCN, 85 °C, 4 h (84%); (ii) LiOH, THF/H2O, r.t., 20 h (91%); (iii) EDC, HOBt, DCM, r.t., 20 h (58%). (B) Synthesis of the natReO(N2S2-PEG3-N3) complex, Re-4. Reagents and conditions: (i) TFA, Et3SiH, 0 °C, 20 min. (ii) (PPh3)2ReOCl3, NaOAc, MeOH, 75 °C, 18 h (62%); (iii) LiOH, THF/H2O, r.t., 2 h (68%); (iv) EDC, HOBt, DMF, r.t., 20 h (30%). (C) Synthesis of the DBCO-PEG4-CA ligand, 7. Reagents and conditions: (i) Boc-NH-PEG4-COOH, DIPEA, DCM, r.t., 20 h; (ii) TFA, DCM, r.t., 1 h (81%); (iii) DCM, DIPEA, r.t., 20 h (80%). | |
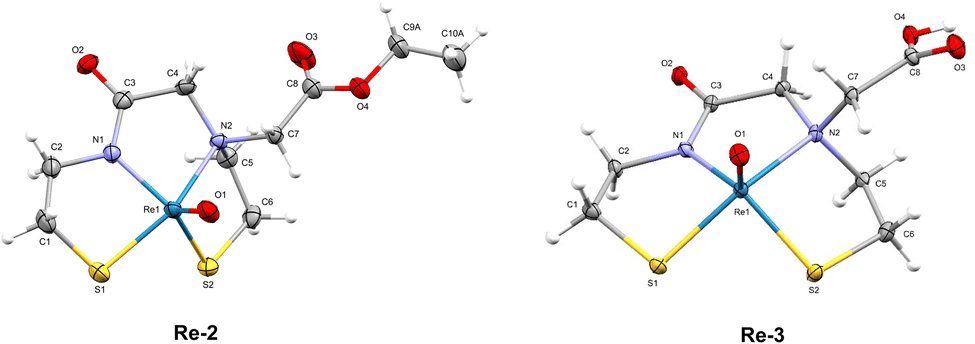
The “cold” natReO(N2S2-PEG3-N3) complex (Re-4) was synthesised in three steps, starting with the deprotection of the S-trityl groups on ester compound 2 with TFA and triethylsilane (Scheme 1B). The deprotected chelator 2 was then reacted with (Ph3P)2ReOCl3 in the presence of NaOAc to form complex Re-2. Subsequent saponification of the ester yielded Re-3 and amide coupling with H2N-PEG3-N3 thereafter produced the natReO(N2S2-PEG3-N3) complex, Re-4 (Scheme 1B). The synthesis of Re-4 had to be performed over three steps to prevent the Staudinger reduction of the azide by the PPh3 ligands of the (Ph3P)2ReOCl3 precursor. The 1H NMR spectra of all three rhenium complexes exhibit complex splitting patterns as a result of the chelator backbone protons becoming diastereotopic upon coordination to the Re(V) oxo core. Additionally, all chemical shifts of the chelator protons on Re-4 are present downfield upon complexation compared to the trityl-protected ligand, 4, with the exception of the protons on the carbon alpha to the amine (protons e, Fig. S17†). These protons experience a dramatic splitting where one proton shifts upfield (1.57 ppm) and one shifts downfield (3.95 ppm). This is indicative of the syn isomer of the complex being formed, as previously observed.13,26 The syn configuration of the isolated Re complexes was confirmed by the X-ray crystal structures obtained for Re-2 and Re-3 (Fig. 3).
 |
| | Fig. 3 ORTEP diagrams of the crystal structures of Re-2 and Re-3 (thermal ellipsoids are drawn at the 30% probability level for both). | |
2.2 X-ray crystallography
Crystals of Re-2 and Re-3 were analysed by X-ray diffraction (Fig. 3). The crystal data for both complexes are summarised in Table S1.† Both complexes have a distorted square-pyramidal geometry as described previously for similar N2S2 complexes.27–30 The complexes are “distorted” in the way that the rhenium(V) oxo group lies above the plane of the four basal nitrogen and sulphur atoms. Both structures have the pendant ester (Re-2) or carboxylic acid (Re-3) functionality on the same side as the apical Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, confirming the syn isomer was formed. Additionally, each of the crystals for Re-2 and Re-3 contain both enantiomers of the syn conformation in a 1
O group, confirming the syn isomer was formed. Additionally, each of the crystals for Re-2 and Re-3 contain both enantiomers of the syn conformation in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
2.3 Cysteine-selective VHH bioconjugation
The bivalent HLE anti-c-Met VHH provided by Sanofi® was engineered at its C-terminus with a terminal cysteine residue, allowing for cysteine-selective VHH bioconjugation. A carbonylacrylic (CA) moiety was chosen as the Michael-acceptor for this conjugation as it has been shown to be highly selective for cysteines in stoichiometric quantities, rapidly forming a stable product under biocompatible conditions.31,32 Most importantly, unlike with maleimides which have shown instability in vivo,33 the thioether product formed by addition of a cysteine onto a CA moiety has been reported to be irreversible.31,32
Since the free thiols of the VHH constructs are cross-reactive with each other, dimerisation happens readily in basic or neutral solutions. To free the thiol for bioconjugation, the VHH dimer (VHH-S–S-VHH) was cleaved using dithiothreitol (DTT) at room temperature to yield the monomer (VHH-SH). This reaction was monitored via size exclusion high performance liquid chromatography (SEC-HPLC), which showed only one peak corresponding to the monomer after 1 h (Fig. S24†). Removal of the excess DTT using a PD-10 desalting column and immediate reaction with 3 equivalents of DBCO-PEG4-CA (7) for 1.5 h yielded the desired DBCO-PEG4-VHH construct (Fig. 4). ESI-MS analysis of the conjugate after a final PD-10 purification showed a single mass after deconvolution corresponding to the expected mass of the desired VHH-DBCO construct (Fig. S26B†). The total protein recovery of the bioconjugation was 71 ± 3% and the purity of the conjugate was determined to be ∼97% by SEC-HPLC (Fig. S27†).
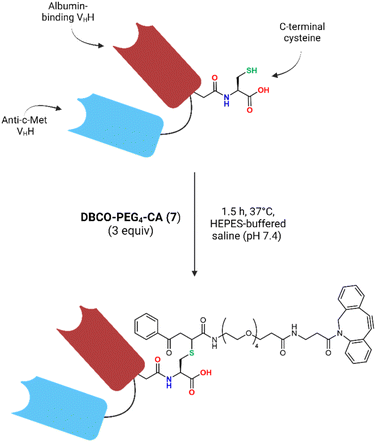 |
| | Fig. 4 Schematic overview of the conjugation of the VHH with DBCO-PEG4-CA (7). | |
2.4
188Re(V)-radiolabelling of N2S2-PEG3-N3 (4)
No-carrier-added [188Re]ReO4− was eluted from an in-house built 188W/188Re generator using saline. The perrhenate was concentrated after elution to provide a sufficient activity concentration for radiolabelling (∼500 MBq mL−1). A report on the construction of the generator and quality control of its eluate is referenced in the ESI.† To monitor the reaction process and optimise the radiolabelling effectively, [188Re]ReO(N2S2-PEG3-N3) was synthesised in two steps (Scheme S1†). The aim was first to produce a [188Re]Re(V)-citrate precursor by reducing [188Re]ReO4− in citrate buffer in the presence of the reducing agent, stannous chloride (SnCl2),34,35 and then add an aliquot of this to the freshly-deprotected N2S2 chelator (4) to afford the final labelled compound, [188Re]ReO(N2S2-PEG3-N3), after transchelation. Ascorbic acid was also included in both steps as an antioxidant to keep the 188Re-labelled compounds stable. Initial radiolabelling attempts with the N2S2-PEG3-N3 chelator (4), however, yielded three distinct peaks on the radio-HPLC chromatogram (Fig. 5). Upon further investigation and comparison to the HPLC-UV chromatogram of the natRe(V) analogue, Re-4, it was determined that the minor peak (retention time 7.4 min) in the radiochromatogram corresponds to the labelled chelator with the azide group intact. The two largest peaks (retention times 6.4 and 6.6 min), however, were attributed to reduction products resulting from an excess of SnCl2 in the labelling mixture. This was proven by treating Re-4 with SnCl2, which led to the formation of two additional distinct Re(V) complexes according to ESI-MS (Fig. S31 and S32†). HPLC analysis demonstrated that these products co-elute with the byproducts observed in the labelling mixture (Fig. 5 and S33–S35†). Increasing the amount of SnCl2 resulted in the exclusive formation of the product corresponding to the peak at 6.4 min (Fig. 5), and ESI-MS data enabled the assignment of this peak to the amine derivative formed via reduction of the azide. Further evaluation of the reduction of Re-4 under radiolabelling conditions at varying pH values (4, 5, and 5.7) indicated that the formation of the amine is slightly favoured at lower pH (Fig. S33–S35†). The identity of the middle peak (m/z = 628.3) at retention time 6.6 min remains undetermined, however, the available evidence strongly suggests that it represents another Re complex and a reduction product.
 |
| | Fig. 5 Radio-HPLC chromatogram of the crude labelling mixture (black) compared to the UV chromatograms of the natRe(V) complexes, Re-4 (red) and natReO(N2S2-PEG3-NH2), produced by the reduction of Re-4 by excess SnCl2 in ethanol at room temperature (blue). | |
Unfortunately, having the azide reduced would prevent the SPAAC click reaction from taking place with the DBCO-PEG4-VHH construct in the subsequent step. Switching the click reagents (i.e. the DBCO and N3) between the two halves would also not work as the DBCO is unstable in the presence of the excess TFA required to remove the protecting groups of the chelator.36 Having the azide on the half with the CA is additionally unfavourable as azides have been reported to react with α,β-unsaturated carbonyl compounds.37 It was therefore decided to keep the azide on the chelator and optimise the radiolabelling. Careful optimisation of the radiolabelling conditions to maximise the ratio of azide to amine and perrhenate were performed, testing factors such as SnCl2 concentration (0.0–1.5 μg μL−1), chelator amount (0.01–0.5 μmol), reaction volume (0.1–1.0 mL), temperature (40 °C or 90 °C) and reaction time (15–60 min), as well as the influence of having a N2 atmosphere. The results of these optimisations are displayed in Fig. 6. The ultimate goal was to decrease the amount of SnCl2 to a concentration low enough to prevent azide reduction but high enough to reduce Re(VII) to Re(V).
 |
| | Fig. 6 Radiolabelling optimisation of [188Re]ReO(N2S2-PEG3-N3). Conditions tested: (A) influence of N2 atmosphere; (B) SnCl2 concentration; (C) chelator quantity; (D) reaction volume; (E) reaction temperature and time. The fixed values for each variable during the optimisations were as follows: activity (20 MBq), SnCl2 concentration (0.1 μg μL−1), volume (100 μL), chelator quantity (0.1 μmol), temperature (90 °C) and time (60 min). RCC = radiochemical conversion. | |
SnCl2 concentration and influence of N2.
It is known that in comparison to Tc(VII), a large excess of Sn(II) is usually required to sufficiently reduce Re(VII) to its lower oxidation states. This is as a result of the lower standard reduction potential of Re(VII) over Tc(VII).38 Performing the reduction in an acidic citrate solution significantly increases the ease of reduction of [188Re]ReO4− by an effect known as the “expansion of the coordination sphere,” leading to an increase in the standard reduction potential and thus a decrease in the concentration of Sn(II) required.39,40
Initially, our radiolabelling experiments were conducted in the presence of air with no N2 purging of the citrate buffer. These experiments showed that over 1.0 μg μL−1 of SnCl2 was necessary to achieve a radiochemical conversion (RCC) to [188Re]Re(V)-citrate greater than 95% (Fig. 6A, pink curve). However, when the resultant [188Re]Re(V)-citrate was reacted with the N2S2-PEG3-N3 chelator (4) in the presence of air, the RCC to [188Re]ReO(N2S2-PEG3-N3) was only about 50% (Fig. 6B, pink curve). This prompted further optimisation of the radiolabelling conditions to improve the yield. Subsequent experiments were performed in a N2 atmosphere using N2-purged citrate buffer while varying the concentration of SnCl2 (0.025–1.50 μg μL−1). The activity (20 MBq), volume (100 μL), chelator quantity (0.1 μmol), temperature (90 °C) and time (60 min) were kept constant. The results indicate that under a N2 atmosphere, the required SnCl2 concentration could be reduced by ten times (0.1 μg μL−1) to achieve a RCC greater than 95%, compared to the reaction in the presence of air (Fig. 6A). In fact, to our knowledge, we were able to reach a concentration of SnCl2 per MBq of activity lower than any recorded concentration in literature for the reduction of 188Re(VII) to 188Re(V). This was oftentimes 15 to 20 times lower (and sometimes up to 200 times lower) than published values,34,41–45 bringing it closer to the concentration range of SnCl2 used for the reduction of [99mTc]TcO4−. This was an important outcome for the labelling of our chelator bearing a reducible azide. Fig. 6B shows that by maintaining a constant chelator concentration and adjusting the SnCl2 concentration, the maximum RCC to [188Re]ReO(N2S2-PEG3-N3) was increased to over 70% using 0.1 μg μL−1 of SnCl2. At concentrations lower than this, more [188Re]ReO4− was present, while higher concentrations resulted in more azide reduction to amine, thereby lowering the yield. Ensuring an inert atmosphere is therefore crucial to achieving the highest possible RCC in this labelling process.
Chelator concentration and reaction volume.
The influence of the chelator quantity (0.01–0.5 μmol) and reaction volume (0.1–1.0 mL) on the RCC of [188Re]ReO(N2S2-PEG3-N3) was determined (Fig. 6C and D). The activity (20 MBq), SnCl2 concentration (0.1 μg μL−1), temperature (90 °C) and time (60 min) were kept constant. The maximum RCC (∼70%) was reached at a chelator quantity of 0.1 μmol, after which no further increase in conversion is observed. This equates to a relatively low apparent molar activity of 0.2 MBq nmol−1, meaning that a post-labelling purification step is necessary. Lowering the amount of chelator in this labelling results in an increase in the percentage of [188Re]ReO4− and [188Re]Re(V)-citrate in the product mixture, thus reducing the overall yield. The maximal conversion of ∼70% was also observed at the lowest reaction volume tested (0.1 mL), with an increase in the volume of citrate buffer leading to a steady decrease in RCC. This is as a result of the increase in the amount of [188Re]ReO4− present in the final product mixture as the volume increased.
Reaction temperature and time.
Finally, the reaction temperature (40 °C or 90 °C) and reaction time (15–60 min) were evaluated for the labelling of N2S2-PEG3-N3 (Fig. 6E). The activity (20 MBq), SnCl2 concentration (0.1 μg μL−1), volume (100 μL) and chelator quantity (0.1 μmol) were kept constant. At a temperature of 40 °C, the RCC was poor, only reaching ∼50% after 1 h. At a temperature of 90 °C, however, the maximum RCC of ∼70% was already reached after 15 min. These results confirm what is already known about the high temperatures required for producing 188Re(V)-labelled compounds.46
The final reaction conditions for the transchelation from [188Re]Re(v)-citrate (∼140 MBq) to [188Re]ReO(N2S2-PEG3-N3) are as follows: 0.1 μg μL−1 SnCl2·2H2O (from the first step), 0.1 μmol N2S2-PEG3-N3 (4), 100 μL total volume, 90 °C, 15 min. The pH of the reaction was kept around 5 using 1.0 M citrate buffer, as it has been shown that 188Re(V)-radiolabelling is most efficient at a slightly acidic pH, likely because it prevents hydrolysis of the SnCl2.47,48 The final RCC of [188Re]ReO(N2S2-PEG3-N3) was ∼70%, which was then purified by prep-HPLC to yield a single peak in the radio-HPLC chromatogram (Fig. 7). Breakthrough of the unlabelled chelator during prep-purification was determined to be unlikely, as shown by the separation between the chelator and natRe(V) complex, Re-4, when studied by HPLC (Fig. S36†). Additionally, the presence of a single peak and the fact that the reaction proceeds at a high temperature suggests there was complete conversion to the syn product with no anti diastereomer observed, a finding reported previously in literature.45
 |
| | Fig. 7 Radio-HPLC chromatograms of (A) the unoptimised radiolabelling; (B) the optimised radiolabelling; (C) the final purified radiolabelling product, [188Re]ReO(N2S2-PEG3-N3) and (D) UV chromatogram of natReO(N2S2-PEG3-N3) (358 nm). | |
2.5 SPAAC click reaction
After purification of the labelled chelator, [188Re]ReO(N2S2-PEG3-N3), and removal of the volatiles, the residue was redissolved in 25 μL of a 0.3 M sodium citrate solution (pH 8), which acted to neutralise any residual TFA and bring the final solution to a pH of 5.5. To this solution, the DBCO-PEG4-VHH was added in HEPES buffer (enough to have an apparent molar activity of ∼5 MBq nmol−1), and the reaction was allowed to proceed for 30 min, after which time the extent of the click reaction was evaluated by iTLC (Fig. S37†). The yield of SPAAC click reaction ranged between 30-65%, resulting in a final apparent molar activity of between 1.4–3.3 MBq nmol−1. Attempts at reaching a higher molar activity were not successful, likely due to the slower reaction kinetics of the SPAAC reaction. After 30 min, the [188Re]Re-VHH was purified from any non-VHH-associated activity using a PD MiniTrap™ column, yielding the pure 188Re-labelled VHH-construct with a radiochemical purity (RCP) >99%. In an effort to try to improve the yield of the click reaction between the 188Re-labelled N2S2 chelator and VHH conjugate, the inverse electron demand Diels–Alder (IEDDA) reaction was adopted (Page S49 in the ESI†). Since the chelator bearing a tetrazine was also easily reduced in the presence of excess SnCl2, this method was abandoned.
2.6
In vitro stability
When stored in saline without the addition of a quencher (i.e. ascorbate), the [188Re]Re-VHH radioconjugate exhibited a dramatic time-dependent decrease in radiochemical stability due to the release of 188Re from the complex, dropping to ∼8% RCP after 24 h (Fig. 8). This was shown to be prevented by the addition of ascorbate at a slightly acidic pH of 5.5, whereby >96% of the conjugate was still intact after 24 h and >93% after 48 h. Radio-HPLC chromatograms of this solution show the decrease in RCP is as a result of the release of perrhenate from the conjugate over time without signs of VHH or chelator degradation (Fig. S38†). Interestingly, the same concentration of ascorbate added at pH 7 did not keep the radioconjugate as stable as at pH 5.5, with the RCP gradually dropping to >80% and >71% after 24 h and 48 h, respectively. This indicates that having a slightly acidic environment aids in ensuring the stability of the [188Re]Re(V)-N2S2 complex. Finally, upon incubation of the radioconjugate in human serum at 37 °C with the addition of ascorbate, the results mirrored those seen with the test in saline at pH 7. After 24 h, a moderate RCP of ∼85% was observed.
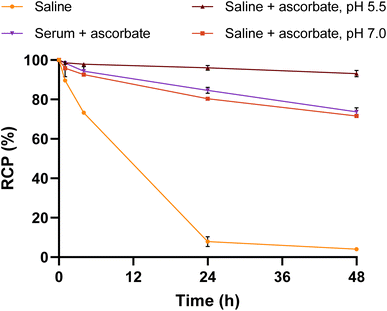 |
| | Fig. 8
In vitro stability of [188Re]Re-VHH in physiological saline (r.t.) or human serum (37 °C) over 48 h. Also included in the study is the influence of ascorbate and the pH of the solution. All solutions were monitored by iTLC and all saline solutions were also monitored by radio-HPLC over time. RCP = radiochemical purity. | |
2.7
In vitro cell-binding of [188Re]Re-VHH
To verify the c-Met binding affinity of the [188Re]Re-VHH construct, cell-binding experiments were performed with both a U87MG (glioblastoma) cell line and a BxPC3 (pancreatic cancer) cell line. Both cell lines are known to express c-Met.49–51 Owing to our low molar activity, this experiment was performed with the cells in suspension to provide a greater surface area for binding compared to surface-adhered cells. After 2 h of incubation at 37 °C, only 3.17 ± 0.06% and 12.37 ± 0.19% of the radiolabelled VHH was found to be associated with the U87MG and BxPC3 cells, respectively (Fig. 9). Receptor blocking studies to assess the specificity of the binding were performed by co-incubating cells with a 1000-fold excess of VHH conjugated to N-ethylmaleimide (NEM). This decreased the binding by over 90% and 96% for the U87MG and BxPC3 cell lines respectively, proving the binding specificity of the VHH.
 |
| | Fig. 9
In vitro cell-binding experiments with [188Re]Re-VHH against c-Met expressing U87MG and BxPC3 cell-lines. VHH-NEM was used as a blocking agent. | |
2.8
Ex vivo biodistribution of [188Re]Re-VHH
Biodistribution studies were performed in order to evaluate the pharmacokinetic profile of the radioconjugate by injecting BxPC3 xenograft mice intravenously with [188Re]Re-VHH. Full biodistribution results are provided in the ESI.† Overall, low standardised uptake values (SUV) were calculated for all tissues of interest, including the blood, consistent with the rapid pharmacokinetics associated with VHHs (Fig. 10). Moderate excretion from the body was observed over time with 32.98 ± 6.06% of the injected activity being excreted after 4 h, climbing to 98.47 ± 0.22% after 48 h (Fig. S39†).17Fig. 10 shows that a high uptake was observed in the kidneys (SUV4h = 1.32 ± 0.24%) and liver (SUV4h = 1.12 ± 0.34%), which slowly washed out of these organs by 48 h p. i. (SUV48h = 0.26 ± 0.04% and 0.083 ± 0.02%, respectively). The higher kidney uptake was expected, as unbound VHHs are small enough to pass through the glomerular filter (∼60 kDa) to be cleared from the bloodstream.17,52 High uptake in the liver and intestines (Fig. S39†) indicate that there is some hepatobiliary clearance in addition to renal excretion. This could be a result of the lipophilicity of the chelator and SPAAC linker, as similarly high intestinal and liver uptakes were observed with a 186Re-labelled N2S2 chelator13 as well as a 99mTc-labelled HSA containing a SPAAC linker.24 [188Re]Re-VHH exhibited minimal tumour uptake, which was at maximum at 4 h p. i. (SUV = 0.71 ± 0.27%) and slowly decreased over the subsequent timepoints. After 24 h p. i. however, only limited retention of the radioconjugate was observed in the tumour compared to other organs as the activity cleared from the bloodstream (tumour-to-blood ratio = 1.31 ± 0.51). Tumour-to-blood ratios for each timepoint can be found in Table S5.†
 |
| | Fig. 10
Ex vivo biodistribution over time expressed as a standard uptake value (SUV) of [188Re]Re-VHH in male NMRI nude mice bearing BxPC3 tumour xenografts (n = 4). p. i. = post-injection. | |
3. Conclusion
In what we believe to be the first reported use of bioorthogonal click chemistry with 188Re, we have successfully radiolabelled a HLE anti-c-Met VHH with 188Re(V) by employing a “chelate-then-click” approach. This was performed by designing and synthesising a bifunctional chelator in two halves which were joined post-labelling via SPAAC click chemistry. Cysteine-selective thiol-CA conjugation was achieved efficiently with the Michael-addition of the VHH-SH monomer onto the DBCO-PEG4-CA (7) half, resulting in the desired VHH-DBCO construct. The radiolabelling of the N2S2-PEG3-N3 (4) chelator half, however, proved to be challenging as the excess SnCl2 required to reduce the Re(VII) to Re(V) also reduced the azide on the chelator to an amine. Careful optimisation of the reactant quantities and conditions required for the labelling was performed, with the final optimised conditions resulting in a high RCC to [188Re]ReO(N2S2-PEG3-N3) of ∼70%. This was achieved by ensuring an inert atmosphere during the radiolabelling, thereby allowing the concentration of SnCl2 used to be lowered dramatically. Reaction of the purified labelled chelator, [188Re]ReO(N2S2-PEG3-N3), with the VHH-DBCO conjugate resulted in the final SPAAC product, [188Re]Re-VHH, in high radiochemical purity and which displayed good in vitro stability over 48 h. Proof-of-concept studies for [188Re]Re-VHH as a c-Met targeting agent demonstrate specific binding in vitro, though suboptimal tumour uptake in vivo. Overall, this research displays the potential for the labelling of a biomolecule, such as a VHH, with 188Re by making use of the approach of bioorthogonal click chemistry.
Data availability
The data supporting this article has been included as part of the ESI.† Crystallographic data has been deposited at the CCDC under 2384422 and 2384423.
Author contributions
The research study was conceptualised by MO, GG and TO. Experimental design was carried out by DM, CS, MO and TO. Production of 188W for development of a 188W/188Re generator was done by MVdV. Generator fabrication, elution and analysis was performed by DM, MVdV and TO. Synthesis, bioconjugation, radiolabelling optimisation and stability studies were performed by DM and TO. X-ray crystal structure analysis was performed by OB. In vitro cell binding assays were performed by DM and CS. Ex vivo biodistribution experiments were performed by DM, CS, JW and MO. The first draft of the manuscript was written by DM, with all authors having contributed to manuscript review and editing, and having approved the final submission.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
The authors would like to acknowledge the contributions of Peter Casteels (Large Molecule Research Innovation, Sanofi, 9052 Ghent, Belgium) and Melissa Crabbé, Sunay Rodriguez Pérez, Koen Vermeulen and Amelie Coolkens (SCK CEN) to the research reported. We also thank Andrew Burgoyne for his help in initiating the project. The authors would also like to thank the SCK CEN Radioprotection Officers, Job Cools, Stefanie de Rudder, Zjef Beelen and Marc Eysermans, for their willingness to help with last-minute transports and to work late days for this project. This publication was partially created within the project SECURE funded by the European Union under grant agreement No. 101061230. The graphical abstract, Fig. 2, 4 and the graphical abstract were created with http://BioRender.com.
References
- D. R. Melis, A. R. Burgoyne, M. Ooms and G. Gasser, RSC Med. Chem., 2022, 13, 217–245 RSC.
- F. F. Knapp, Int. J. Nuclear Med. Res., 2017, 3–15 Search PubMed.
- B. Ponsard, J. Hiltunen, P. Penttilla, H. Vera Ruiz, A. L. Beets, S. Mirzadeh and F. F. Knapp, J. Radioanal. Nucl. Chem., 2003, 257, 169–174 CrossRef.
- U. Abram and R. Alberto, J. Braz. Chem. Soc., 2006, 17, 1486–1500 CrossRef.
- P. S. Donnelly, Dalton Trans., 2011, 40, 999–1010 RSC.
- A. Boschi, L. Uccelli, M. Pasquali, A. Duatti, A. Taibi, G. Pupillo and J. Esposito, J. Chem., 2014, 2014, 1–14 CrossRef.
- S. S. Jurisson and J. D. Lydon, Chem. Rev., 1999, 99, 2205–2218 CrossRef.
-
J. R. Dilworth and S. I. Pascu, in The Chemistry of Molecular Imaging, Wiley, 2014, pp. 137–164 Search PubMed.
- M. Friebe, A. Mahmood, C. Bolzati, A. Drews, B. Johannsen, M. Eisenhut, D. Kraemer, A. Davison and A. G. Jones, J. Med. Chem., 2001, 44, 3132–3140 CrossRef PubMed.
- M. Ono, R. Ikeoka, H. Watanabe, H. Kimura, T. Fuchigami, M. Haratake, H. Saji and M. Nakayama, ACS Chem. Neurosci., 2010, 1, 598–607 CrossRef PubMed.
- K. Ogawa, T. Mukai, Y. Arano, H. Hanaoka, K. Hashimoto, H. Nishimura and H. Saji, J. Labelled Compd. Radiopharm., 2004, 47, 753–761 CrossRef.
- T.-Y. Luo, I.-C. Tang, Y.-L. Wu, K.-L. Hsu, S.-W. Liu, H.-C. Kung, P.-S. Lai and W.-J. Lin, Nucl. Med. Biol., 2009, 36, 81–88 CrossRef PubMed.
- D. W. Demoin, A. N. Dame, W. D. Minard, F. Gallazzi, G. L. Seickman, T. L. Rold, N. Bernskoetter, M. E. Fassbender, T. J. Hoffman, C. A. Deakyne and S. S. Jurisson, Nucl. Med. Biol., 2016, 43, 802–811 CrossRef PubMed.
- M. D'Huyvetter, C. Xavier, V. Caveliers, T. Lahoutte, S. Muyldermans and N. Devoogdt, Expert Opin. Drug Delivery, 2014, 11, 1939–1954 CrossRef.
- M. Kijanka, B. Dorresteijn, S. Oliveira and P. M. van Bergen en Henegouwen, Nanomedicine, 2015, 10, 161–174 Search PubMed.
- R. Chakravarty, S. Goel and W. Cai, Theranostics, 2014, 4, 386–398 CrossRef CAS PubMed.
- P. Bannas, J. Hambach and F. Koch-Nolte, Front. Immunol., 2017, 8, 1603 CrossRef.
- S. Hoefman, I. Ottevaere, J. Baumeister and M. Sargentini-Maier, Antibodies, 2015, 4, 141–156 CrossRef.
- V. Gupta, S. Bhavanasi, M. Quadir, K. Singh, G. Ghosh, K. Vasamreddy, A. Ghosh, T. J. Siahaan, S. Banerjee and S. K. Banerjee, J. Cell Commun. Signaling, 2019, 13, 319–330 CrossRef CAS.
- G. Floresta and V. Abbate, Med. Res. Rev., 2022, 42, 1588–1606 CrossRef.
- A. Sadiki, S. R. Vaidya, M. Abdollahi, G. Bhardwaj, M. E. Dolan, H. Turna, V. Arora, A. Sanjeev, T. D. Robinson, A. Koid, A. Amin and Z. S. Zhou, Antibiot. Ther., 2020, 3, 271–284 CAS.
- D. Bauer, S. M. Sarrett, J. S. Lewis and B. M. Zeglis, Nat. Protoc., 2023, 18, 1659–1668 CrossRef CAS.
- D. Zeng, B. M. Zeglis, J. S. Lewis and C. J. Anderson, J. Nucl. Med., 2013, 54, 829–832 CrossRef CAS.
- N. A. Lodhi, J. Y. Park, K. Kim, Y. J. Kim, J. H. Shin, Y.-S. Lee, H.-J. Im, J. M. Jeong, M. Khalid, G. J. Cheon, D. S. Lee and K. W. Kang, Mol. Pharm., 2019, 16, 1586–1595 Search PubMed.
- V. Carroll, D. W. Demoin, T. J. Hoffman and S. S. Jurisson, Radiochim. Acta, 2012, 100, 653–667 CrossRef CAS.
- L. Mei, Y. Wang and T. Chu, Eur. J. Med. Chem., 2012, 58, 50–63 Search PubMed.
- H. F. Kung, Y. Z. Guo, C. C. Yu, J. Billings, V. Subramanyam and J. C. Calabrese, J. Med. Chem., 1989, 32, 433–437 CrossRef CAS PubMed.
- C. S. John, L. C. Francesconi, H. F. Kung, S. Wehrli, G. Graczyk and P. Carroll, Polyhedron, 1992, 11, 1145–1155 CrossRef CAS.
- R. A. Bell, B. E. McCarry and J. F. Valliant, Inorg. Chem., 1998, 37, 3517–3520 CrossRef.
- L. G. Luyt, H. A. Jenkins and D. H. Hunter, Bioconjugate Chem., 1999, 10, 470–479 CrossRef PubMed.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef PubMed.
- B. Bernardim, M. J. Matos, X. Ferhati, I. Compañón, A. Guerreiro, P. Akkapeddi, A. C. B. Burtoloso, G. Jiménez-Osés, F. Corzana and G. J. L. Bernardes, Nat. Protoc., 2019, 14, 86–99 CrossRef PubMed.
- A. D. Baldwin and K. L. Kiick, Bioconjugate Chem., 2011, 22, 1946–1953 CrossRef.
- R. E. Nuttall, T. T. Pham, A. C. Chadwick, I. N. Hungnes, G. Firth, M. A. Heckenast, H. A. Sparkes, M. C. Galan, M. T. Ma and P. G. Pringle, Inorg. Chem., 2023, 62, 20582–20592 CrossRef PubMed.
- T. T. Pham, I. N. Hungnes, C. Rivas, J. Cleaver, G. Firth, P. J. Blower, J. Sosabowski, G. J. R. Cook, L. Livieratos, J. D. Young, P. G. Pringle and M. T. Ma, J. Nucl. Med., 2024, 65, 1087–1094 CrossRef PubMed.
- W. Shi, F. Tang, J. Ao, Q. Yu, J. Liu, Y. Tang, B. Jiang, X. Ren, H. Huang, W. Yang and W. Huang, Angew. Chem., 2020, 132, 20112–20116 CrossRef.
- H.-Z. Zhu, G. Wang, H.-L. Wei, H.-J. Chu and J. Zhu, Macromol. Res., 2016, 24, 793–799 CrossRef.
- J. O. Dickson, J. B. Harsh, W. W. Lukens and E. M. Pierce, Chem. Geol., 2015, 395, 138–143 CrossRef.
- J. J. Vajo, D. A. Aikens, L. Ashley, D. E. Poeltl, R. A. Bailey, H. M. Clark and S. C. Bunce, Inorg. Chem., 1981, 20, 3328–3333 CrossRef.
- J. Kleynhans, A. Duatti and C. Bolzati, Molecules, 2023, 28, 1487 CrossRef.
- S. Guhlke, A. Schaffland, P. O. Zamora, J. Sartor, D. Diekmann, H. Bender, F. F. Knapp and H.-J. Biersack, Nucl. Med. Biol., 1998, 25, 621–631 CrossRef PubMed.
- J. L. Crudo, M. M. Edreira, E. R. Obenaus, M. Chinol, G. Paganelli and S. G. de Castiglia, Int. J. Pharm., 2002, 248, 173–182 CrossRef PubMed.
- J. L. Crudo, M. M. Edreira, E. R. Obenaus and S. G. de Castiglia, J. Radioanal. Nucl. Chem., 2004, 261, 337–342 CrossRef.
- G. Liu, S. Dou, J. He, D. Yin, S. Gupta, S. Zhang, Y. Wang, M. Rusckowski and D. J. Hnatowich, Appl. Radiat. Isot., 2006, 64, 971–978 CrossRef PubMed.
- V. A. Sanders, D. Iskhakov, D. Abdel-Atti, M. Devany, M. C. Neary, K. R. Czerwinski and L. C. Francesconi, Nucl. Med. Biol., 2019, 68–69, 1–13 CrossRef PubMed.
- Y. Feng, T. E. Phelps, V. Carroll, F. Gallazzi, G. Sieckman, T. J. Hoffman, C. L. Barnes, A. R. Ketring, H. M. Hennkens and S. S. Jurisson, Dalton Trans., 2017, 46, 14677–14690 RSC.
- S. Prakash, M. J. Went and P. J. Blower, Nucl. Med. Biol., 1996, 23, 543–549 CrossRef PubMed.
- L. Meléndez-Alafort, G. Ferro-Flores, C. Arteaga-Murphy, M. Pedraza-López, M. A. González-Zavala, J. I. Tendilla and L. García-Salinas, Int. J. Pharm., 1999, 182, 165–172 CrossRef PubMed.
- H. Luo, H. Hong, M. R. Slater, S. A. Graves, S. Shi, Y. Yang, R. J. Nickles, F. Fan and W. Cai, J. Nucl. Med., 2015, 56, 758–763 CrossRef PubMed.
- M. J. W. D. Vosjan, J. Vercammen, J. A. Kolkman, M. Stigter-van Walsum, H. Revets and G. A. M. S. van Dongen, Mol. Cancer Ther., 2012, 11, 1017–1025 Search PubMed.
- F. E. Escorcia, J. L. Houghton, D. Abdel-Atti, P. R. Pereira, A. Cho, N. T. Gutsche, K. E. Baidoo and J. S. Lewis, Theranostics, 2020, 10, 151–165 CrossRef PubMed.
- G. Bao, M. Tang, J. Zhao and X. Zhu, EJNMMI Res., 2021, 11, 6 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental, characterisation and X-ray crystallographic details. Crystallographic data for Re-2 and Re-3. CCDC [2384422 and 2384423]. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07743a. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Charlotte
Segers
ab,
Charlotte
Segers