
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultiligand-enabled, copper-catalyzed Hiyama coupling of arylsilanes with unactivated secondary alkyl halides: reaction development and mechanistic insights†
Jiajing
Zhou
a,
Zhiqiang
Zhang
a,
Yan
Cao
a and
Weilong
Xie
 *ab
*ab
aCollege of Chemistry and Chemical Engineering, Donghua University, Shanghai 201620, China. E-mail: weilongxie@dhu.edu.cn
bState Key Laboratory of Elemento-Organic Chemistry, Nankai University, Tianjin 300071, China
First published on 4th February 2025
Abstract
Construction of carbon–carbon bonds is the cornerstone in organic synthesis, and Hiyama coupling is the representative synthetic approach for realizing linkages between silyl compounds and organohalides. In previous literature, such couplings are mainly utilized for the bond formations of arylsilanes with sp2-aryl halides, yet Hiyama couplings with sp3-hybridized alkyl halides still remain scarce. Copper catalysis has recently been scrutinized in several important transformations of unactivated secondary alkyl halides, whereas their conversions with organosilanes are far less developed. Herein, we leverage a multiligand catalysis to offer a solution for efficient copper-catalyzed Hiyama couplings with such unactivated alkyl halides. Detailed mechanistic studies disclosed that the incorporation of an NHC ligand with a phenanthroline–copper system would dramatically enhance the reaction efficiency, where the copper species with NHC and phenanthroline-type ligands were most likely to account for the C(sp2)–Si bond activation and C(sp2)–C(sp3) bond formation process, respectively.
Introduction
Cross-coupling reactions allow the construction of various compounds from small to highly complex molecules by the utilization of simple and commercially available chemicals, and the exploration of efficient catalytic systems has witnessed a tremendous advancement in organic synthesis along with the lines of methodology development in past decades.1 In this context, Hiyama coupling is one important transformation for providing reliable synthetic approaches to the linkages between silyl compounds with organic (pseudo)halides.2 Considering the low toxicity, ready availability and high abundance of silicon chemicals, the exploration of such coupling systems would be highly valuable in synthesis.3The majority of the reported Hiyama couplings are focused on the reactions between aryl or vinyl silane nucleophiles with sp2-electrophiles.2,3 Recently, with the increasing demand for C(sp3)-rich scaffolds in numerous research fields, a surge of interest has been initiated in the conversions of alkyl halides.4 However, late transition-metal-catalyzed conversions of alkyl halides often encounter several problematic issues including sluggish oxidation of alkyl electrophiles and facile β-hydride elimination of alkyl-metal species.5 Thus, approaches for efficient couplings with alkyl halides are in high demand.
In this line, first-row transition metals were proven to be promising catalysts in coupling with sp3-alkyl halides via single-electron mechanisms, where nickel, cobalt and iron have recently been scrutinized in this realm.6 In comparison, copper catalysis utilized in this field is still lagging far behind,7 although copper-mediated Ullmann reactions are regarded as the earliest transformations in organic synthesis.8 Thus, further methodology development of copper systems would be valuable from the aspects of both important theoretical significance and practical advantages, including earth abundance, cost efficiency and environmental benignity of such catalytic systems.9
The last two decades have witnessed the significant achievement of copper-catalyzed sp2-aryl halide transformations,10–12 and a burgeoning interest has recently been aroused in the development of unactivated secondary sp3-alkyl halides (Scheme 1a).4–7 Fu et al., in a seminal report, disclosed a photo-enabled copper catalyzed amination of secondary alkyl halides with carbazoles.13 Later, Leonori et al. illustrated a halogen-atom-transfer (XAT) approach assisting copper-catalyzed amination of alkyl iodides.14 Recently, Lin and colleagues reported an NHC–copper system catalyzing the coupling of alkyl halides with amines under photo-catalysis.15 Cyanation16 and Sonogashira couplings17 of alkyl halides were also recently achieved by several research groups. Meanwhile, Suzuki couplings of alkyl halides with arylborons were also achieved. For instance, primary alkyl halides coupling with arylborons was previously disclosed by Liu et al.18 and unactivated secondary alkyl iodides were recently reported by Leonori and co-workers using preactivated boronates with the assistance of XAT approaches.19 Our group recently established a practical copper system catalyzing Suzuki couplings of various alkyl halides with arylborons.20a Motivated by the aforementioned reports, and also in continuation of our research interest in copper catalysis,20 we endeavor to develop Hiyama couplings of unactivated alkyl halides with copper systems.
 | ||
| Scheme 1 Copper-catalyzed cross-couplings of unactivated secondary alkyl halides. | ||
In fact, Hiyama reactions with sp3-alkyl electrophiles still remain scarce.21 Fu et al. reported palladium-catalyzed reactions of aryltrialkoxysilanes with primary alkyl halides.21a Later, the same group developed nickel-catalyzed cross-couplings of secondary alkyl halides with highly polarized trifluorosilylarenes.21b,c The problematic issues impeding the development might lie in the relatively lower propensity for bond activation of the inherent lower Lewis acidity C(sp2)–Si bonds, compared with arylborons in Suzuki coupling.
In the scenario for the enhancement of the catalytic performance of traditional catalytic systems, considerable efforts have been devoted to developing a variety of synthetic strategies recently. For instance, modification of the ligand shell of catalysts would provide efficient systems.22 However, tedious synthetic processes and capricious reactivity of catalysts often require massive efforts. Bimetallic catalysis with two distinct systems have also been proven to be successful by capitalizing on the advantages of different transition metal systems.23 On the other hand, the sole metal-based multiligand relay catalysis has also emerged as an attractive approach for efficient C–C bond formation.24 This strategy could trigger different reaction patterns by using two types of simple ligands rather than a complicated ligand design, thus providing a more efficient approach. Buchwald et al. demonstrated that palladium-mediated C–N cross-couplings could be accelerated by binary phosphine ligand mixtures.25a Recently, the Wang group successfully developed a two-ligand system involving assisted palladium catalysis for the polyfluoroarylation-allylation of diazocompounds.25b Sevov and co-workers established a nickel-catalyzed reductive coupling system to construct C(sp3)–C(sp2) bonds enabled by pyrazolylpyridine and phosphine binary ligands.25c Mauleón and colleagues reported that copper-catalyzed carboboration or hydroboration of alkynes could also be enhanced by mixed ligands.25d,e Herein, we first develop a multiligand-enabled copper catalysis for the efficient Hiyama couplings of unactivated secondary alkyl halides and arylsilanes with the assistance of phenanthroline and N-heterocyclic carbene (NHC) ligands (Scheme 1b). Detailed mechanistic studies revealed that the phenanthroline ligand was crucial for C–C bond formations, and the strong σ-electron-donating NHC ligands accounted for C–Si bond activation. Notably, the current protocol operated in conjunction with the different advantages of two-ligand/copper systems, thus eliminating complicated ligand design and providing a facile and straightforward synthetic route.
Results and discussion
Reaction optimization
We commenced the study by investigating the coupling of phenyltrimethoxysilane (1a) with bromocyclohexane (2) with the copper catalyst (CuBr·SMe2), nitrogen bidentate ligands and base at 80 °C (Fig. 1a, and also see Table S1–S6 in the ESI† for details). The desired coupling product 3 could be obtained in a moderate yield with the employment of phenanthroline ligand L1 (32%). Sterically congested ligand L2 having methyl groups adjacent to the nitrogen atoms could barely form the desired product (<1%). These sterically accessible ligands with electronically distinct groups including phenyl, methoxide, nitro and methyl groups did not enhance the coupling efficiency (L3–L6, <1–35%). Bipyridines were also found to be sluggish for the current C(sp2)–C(sp3) bond formation (L7–L9, 11–18%). Further efforts with other ligands including NHCs, phosphines and terpyridines were observed to be inefficient for the current alkylation reactions.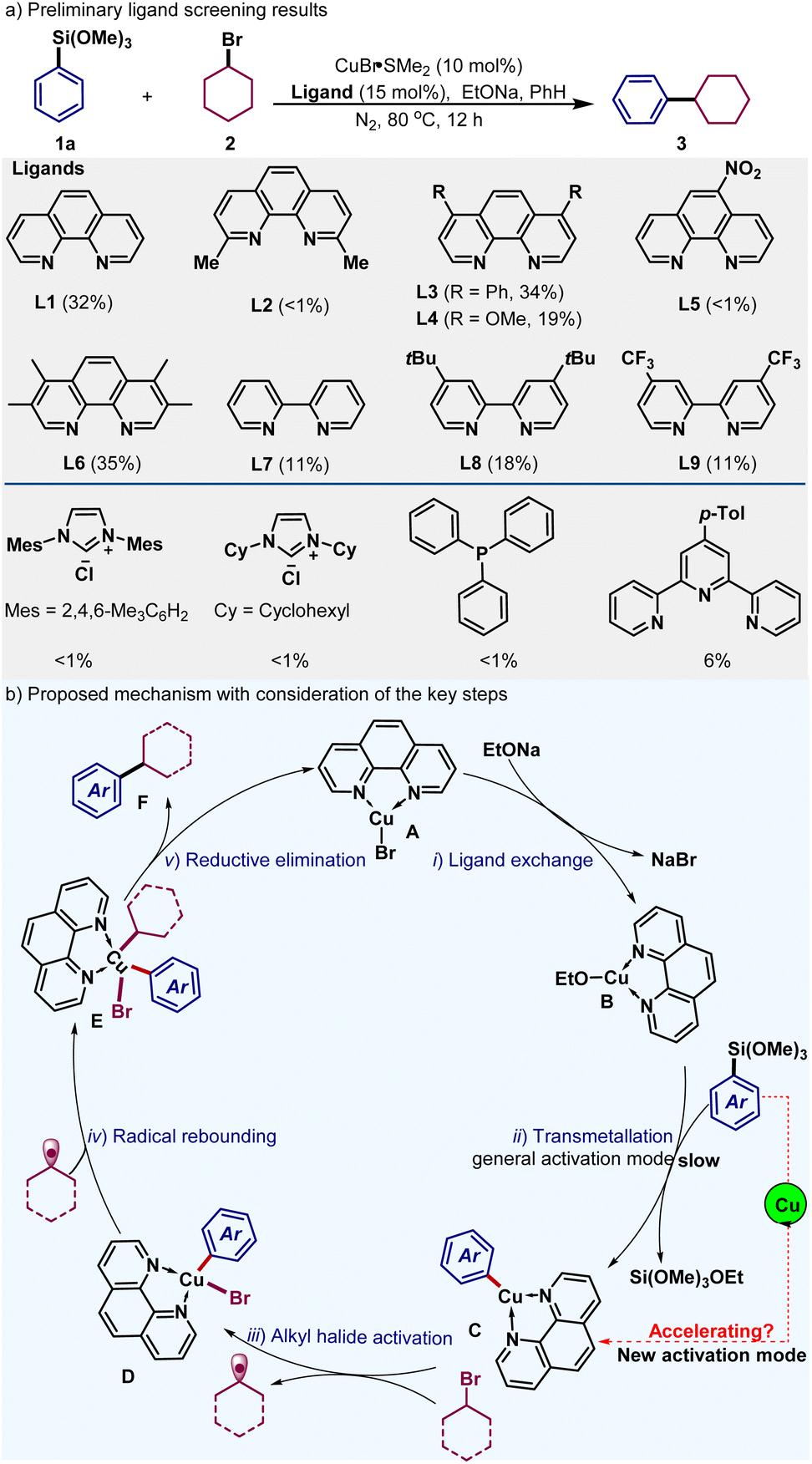 | ||
| Fig. 1 (a) Preliminary results for the ligand effect of copper-catalyzed sp2–sp3 Hiyama couplings. (b) Simplified mechanistic considerations of the key steps. | ||
We reasoned that the mediocre performance of the current coupling probably resulted from a certain rate-determining step of the reaction pathways. Thus, a proposed reaction mechanism is depicted in Fig. 1b to illustrate the possible key steps of the catalytic cycle for the phenanthroline ligand-enabled copper-catalyzed Hiyama coupling system. Based on prior literature,18,20a the catalytic cycle would consist of the following steps: the ligand exchange of the phenanthroline–copper bromide with alkoxide base, the transmetallation step of the arylsilane forming the copper-aryl species, the radical activation of alkyl halides, the radical rebounding process of the alkyl radicals to form high-valent copper species, and the final reductive elimination step to generate the cross-coupling product. Copper-aryl species have been reported to be capable of activation of alkyl halides,18,20a and the reductive elimination step from high-valent copper is well documented to be facile.9e Thus, we reasoned that the current Hiyama coupling was probably inhibited by the transmetallation process. In fact, the polarity of the C(sp2)–Si bond is much weaker than that of the C(sp2)–B bond in arylborons, which might lead to the sluggish bond activation of arylsilanes. In this circumstance, polar trifluorosilylarenes have been previously utilized in nickel-catalyzed Hiyama coupling.21b,c Thus, the exploration of an alternative activation pathway for the arylsilane reagents might address the problematic issues in the current system (dashed red arrow in Fig. 1b).
We envisioned that the leverage of an efficient copper catalyst for C(sp2)–Si bond activation might facilitate the current reactions effectively. In fact, strong σ-electron-donating NHC ligands have been utilized for the transmetallation of arylsilanes or arylborons in copper systems to form the corresponding copper-aryl species, reported by the Ball, Hou and other groups.26,27 Thus, we assumed that the incorporation of NHCs with the above phenanthroline–copper system might offer a solution for the development of efficient Hiyama couplings.
Toward this end, the model reaction was further investigated with copper, a phenanthroline-type ligand (L6) and a variety of NHC ligands (Table 1). Gratifyingly, the product yield was dramatically increased from 35% to 72% with the combination of NHC-1 (5.0 mol%), L6 (5.0 mol%) and the same loading of copper (CuBr·SMe2, 10 mol%). Encouraged by this observation, a series of NHC ligands with distinct steric environment substituents were subsequently examined. Sterically hindered NHC ligands were found to give high yields (NHC-1–NHC-3: 72–78%). An NHC ligand with a saturated backbone did not affect the efficiency (NHC-4, 73%). Notably, the cyclohexyl group-substituted carbene precursor NHC-5 gave an excellent yield of 93%. However, decreased yields were observed with sterically accessible ligands (NHC-6–NHC-8, 2–74% yields). Collectively, the steric environment of NHC ligands has a profound effect on the efficiency, and the bulkier ligands were comparably more effective than the small-size NHCs. This observation could be rationalized in that the unhindered NHCs were likely to form heteroleptic copper species, thus leading to a lower activity (vide infra). Redox-active ligand NHC-11 was found to be sluggish. Additionally, the use of NHC alone was inefficient (<1%; see Table S1 in the ESI† for details), indicating that the cooperation effect between the NHC and phenanthroline ligands probably accelerated the current multiple ligand-enabling coupling system.
|
|
||||
|---|---|---|---|---|
| Entry | [Cu] (X) | Solvent | Base | Yieldc (%) |
| a Reaction conditions: PhSi(OMe)3 (0.80 mmol), CyBr (0.40 mmol), copper catalyst (X mol%), L6 [(1/2)X mol%], NHC [(1/2)X mol%] and EtONa (0.60 mmol) in the indicated solvent (0.50 mL) at 80 °C for 12 h under nitrogen. b X = 10. c Yields were determined by GC analyses against a calibrated internal standard of n-hexadecane. | ||||
| 1 | CuBr·SMe2 (10) | PhH | EtONa | 93 |
| 2 | CuBr·SMe2 (5) | PhH | EtONa | 90 |
| 3 | CuBr (5) | PhH | EtONa | 78 |
| 4 | CuI (5) | PhH | EtONa | 57 |
| 5 | CuCl (5) | PhH | EtONa | 38 |
| 6 | CuTc (5) | PhH | EtONa | 75 |
| 7 | Cu(acac)2 (5) | PhH | EtONa | 85 |
| 8 | — | PhH | EtONa | <1 |
| 9 | CuBr·SMe2 (5) | Toluene | EtONa | 83 |
| 10 | CuBr·SMe2 (5) | CyH | EtONa | 80 |
| 11 | CuBr·SMe2 (5) | 1,4-Dioxane | EtONa | 64 |
| 12 | CuBr·SMe2 (5) | THF | EtONa | 23 |
| 13 | CuBr·SMe2 (5) | PhH | tBuONa | 72 |
| 14 | CuBr·SMe2 (5) | PhH | MeONa | 89 |
| 15 | CuBr·SMe2 (5) | PhH | MeOK | 36 |
| 16 | CuBr·SMe2 (5) | PhH | MeOLi | <1 |

Further investigation of other reaction parameters was performed. The reaction efficiency was slightly diminished with a decreased catalyst loading (entry 2, 90% yield with 5.0 mol% of copper). The evaluation of the copper catalysts disclosed that the efficiency was related to the counter anions. Copper bromide promoted the coupling in a yield of 78% (entry 3), and other copper salts with iodide and chloride anions enabled the reactions with decreased yields (entries 4 and 5, 57% and 38%, respectively). Copper catalysts with thiophene-2-carboxylate and acetylacetonate anions were also effective (entries 6 and 7, 75–85%). The coupling product could not be detected in the absence of copper, indicating the necessity of copper in the current reaction (entry 8, <1%). Solvent screening disclosed that the polarity has a pronounced effect on the efficiency, and nonpolar solvents were observed to be superior to polar solvents (entries 9–12). Further investigation of the base revealed that sodium bases were optimal (entries 13–16). The reaction could not proceed below 40 °C (see the ESI† for details).
Reaction scope
After the optimization of the current multiligand-enabled copper catalysis, we proceeded to evaluate the feasibility of substrate scope. An array of arylsilanes was firstly tested in the current system (Table 2). Phenyltrimethoxysilane was reacted with bromocyclohexane and afforded the desired product 3 in a high yield of 89%. Arylsilanes with carbon substituents including methyl, ethyl, tert-butyl and phenyl groups all generated the corresponding products smoothly (4–8, 64–90%). Reagents containing an electron-rich methoxide group gave the corresponding products 9 and 10 in good yields with increasing catalyst loading (10 mol% of copper; 78% and 72% yields, respectively). However, a highly electron-rich substrate with two methoxide groups failed to generate the alkylation product 11, which was probably due to the low polarity of the C(sp2)–Si bond induced by the electronic effect, thus leading to an ineffective transmetallation. Substrates with electron-deficient groups, such as chloro, fluoro, and trifluoromethyl moieties, were all able to afford the products in moderate to good yields (12–15, 50–77%). Low isolated yields of the fluorine-containing products could probably be ascribed to their volatility property.
A substrate containing the reactivity distinct trialkylsilyl and trialkoxysilyl moieties was tested, and the coupling proceeded selectively at the polarized carbon site of the trialkoxysilyl group, leading to the formation of product 16 in 74% yield. A π-extended arylsilane coupled with cyclohexyl bromide generated the alkylation product in a high yield (17, 82%). Furthermore, the steric influence on the coupling system was also tested. 2-Methylarylsilane formed the alkylation product in a moderate yield with increased catalyst loading (18, 45%; 10 mol% of copper). Unfortunately, sterically congested 2,6-disubstituted coupling reagents failed to generate the desired product (19, <1%).
The catalytic ability with various alkyl halides was further investigated (Table 3). The compound tert-butylphenyltrimethoxysilane was employed as the coupling partner due to the volatility of certain products. Cyclic alkyl bromides including seven-, six- and five-membered rings were all arylated without difficulty (6, 20 and 21, 78−82%). Interestingly, a substrate of the strained four-membered cyclobutyl bromide was subjected to the coupling system, and the target product 22 could be obtained in a high yield of 83%. However, a highly stained bromocyclopropane failed to generate the product.

Linear alkyl halides including 2-bromobutane and 2-bromopropane both gave the C(sp2)–C(sp3) coupling products in good isolated yields (23 and 24, 61% and 55%, respectively), and the lower isolated yields were probably due to the volatility of the products. Tetrahydropyranyl-4-bromide was coupled to afford the desired product 25 in 50% isolated yield. Subsequently, alkyl electrophiles containing aryl moieties were reacted, and both of the target products were obtained without the isomerization of the reactive sites to their benzylic positions (26 and 27, 70% and 66%, respectively). Next, the tolerance of other functional groups was investigated. The unsaturated alkynyl and vinyl moieties were both untouched during the coupling process, as exemplified by the products 28 and 29. A substrate of ether substituted alkyl bromide was also able to afford the corresponding product in a synthetically useful yield (30, 39%). Notably, excellent diastereoselectivities could be obtained for cyclic alkyl halides, as exemplified by the products 31–33 (d.r. >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), while the linear reagent afforded the target product 34 with a moderate diastereoselectivity (d.r. = 8:1). It deserves particular mention that the enhanced stereoselectivity from the alkyl halides to the coupling products provided strong evidence for the involvement of radicals in the current system. For instance, the diastereomeric ratio for the (3-bromocyclohexyl)benzene reagent was 9:1, and that value for the coupling product 32 was increased to 22:1 (see the ESI† for details). Unfortunately, the extension of the current protocol to sterically hindered tertiary sp3-hybridized alkyl halides was observed to be ineffective. For instance, no coupling product was detected for the reaction of 1a with tert-butyl bromide.
:1), while the linear reagent afforded the target product 34 with a moderate diastereoselectivity (d.r. = 8:1). It deserves particular mention that the enhanced stereoselectivity from the alkyl halides to the coupling products provided strong evidence for the involvement of radicals in the current system. For instance, the diastereomeric ratio for the (3-bromocyclohexyl)benzene reagent was 9:1, and that value for the coupling product 32 was increased to 22:1 (see the ESI† for details). Unfortunately, the extension of the current protocol to sterically hindered tertiary sp3-hybridized alkyl halides was observed to be ineffective. For instance, no coupling product was detected for the reaction of 1a with tert-butyl bromide.
Mechanistic studies
After the investigation of the reaction scope, a series of mechanistic relevant experiments were performed to shed light on the mechanism (Scheme 2). A stoichiometric amount of TEMPO [2,2,6,6-(tetramethylpiperidin-1-yl)oxyl] as radical scavenger was added to the standard conditions, and the reaction was observed to be fully inhibited, indicating that the coupling process probably occurred via radical pathways (Scheme 2a). The sp3-alkyl radical species was trapped to form the adduct 35 in 58% yield. Similarly, 1,1-diphenylethylene was also observed to largely dampen the reaction, and the cyclohexyl radical formed products 36 and 37 in 29% and 4% yields, respectively. Next, a radical clock experiment was performed with bromomethylcyclopropane 38 (Scheme 2b), and the reaction predominantly gave the ring-opening product 39 along with a minor product 40, and this observation further confirmed the radical species involved in the current system. | ||
| Scheme 2 Preliminary mechanistic studies. | ||
Next, comparative studies of the reaction courses with the monoligand and multiligand copper systems were performed side-by-side (Scheme 2c). Evidently, the catalytic ability of the multiligand copper system was remarkably increased compared to the monoligand system of phenanthroline ligand L6. Product 3 was obtained in 89% yield with a mixed NHC-5 and L6 ligand system in 4 h, while with L6 alone the product was only obtained in a low yield of 8% under otherwise identical conditions. The use of the sole NHC ligand could not promote the current coupling (see the ESI† for details), disclosing the crucial role of phenanthroline ligand for C(sp2)–C(sp3) bond formation, and the additional NHC ligand could promote the Hiyama couplings effectively.
Furthermore, we questioned the possibility of the heteroleptic copper complexes incorporating the phenanthroline-type ligand and NHC ligand catalyzing the current cross-coupling reactions. Thus, copper complex Cu-1 supported by NHC-2 and L6 was independently prepared and its structure was confirmed by NMR spectroscopic and X-ray crystallographic analyses (CCDC 2361575). The utilization of Cu-1 as the catalyst in the current cross-coupling system was observed to be ineffective for C(sp2)–C(sp3) bond formation (Scheme 3a), thus precluding the involvement of the heteroleptic copper species as catalytically active in the cross-coupling process.
 | ||
| Scheme 3 Evaluation of the activity of copper complexes and proposed reaction mechanism. | ||
Importantly, a combination of the NHC–copper complex Cu-2 and a phenanthroline-type ligand-supported complex Cu-3 was observed to afford the alkylation product 6 in 85% NMR yield. The efficiency was almost identical to that for conditions using premixed CuBr·SMe2, L6 and NHC-5 (Table 2, 81%). Especially noteworthy was that the Hiyama coupling product 6 was not detected with either Cu-3 or Cu-2 copper complex at 2.5 mol% catalyst loading, and the productive yield could be increased to 35% with 10 mol% loading of Cu-3 as the catalyst. As anticipated, the employment of 10 mol% of Cu-2 could not afford the coupling product (see the ESI† for details). The above observation strongly supported that the two individual copper complexes accounted for the highly efficient C(sp2)–C(sp3) coupling reactions.
Subsequently, a comparison of the catalytic performance for copper catalyst Cu-3 and the binary catalyst system of Cu-3 and Cu-2 was conducted with a variety of arylsilanes (Scheme 3b). Evidently, the experimental data disclosed that the additional NHC–copper complex Cu-2 showed a prominent effect on the efficiency enhancement. For instance, the mixed catalyst systems gave good to high yields in 3 h or 12 h, and using complex Cu-3 alone afforded the products in low yields. Moreover, arylsilanes with the electron-rich methoxide group required an increased catalyst loading to achieve high product yields (1g and 1h, 5.0 mol%), indicating that C(sp2)–Si bond activation probably has a crucial effect on the coupling system.
With the observation of catalytically active copper complexes, further mechanistic scrutiny was performed in due course. NHC–copper alkoxides have previously been reported to react with arylsilanes, forming the relevant copper-aryl species via the transmetallation process.27 Thus, a copper–aryl complex, Cu-4, was separately prepared, and employed as a cocatalyst with 2.5 mol% catalyst loading (Scheme 3c). The alkylation product 6 was obtained in 86% yield with the assistance of the copper-aryl catalyst Cu-4. From GC-MS and NMR spectral analyses of the reaction mixtures, the 4-methoxyphenyl group was not transferred from the NHC–copper complex to couple with alkyl halides even with a 10 mol% loading of Cu-4. However, increasing its loading to 20 mol% would allow the detection of the coupling product 9 in 7% NMR yield. This observation indicated that the current system was unlikely to be triggered either by the transfer of the aryl moieties from the NHC–copper catalyst to the phenanthroline–copper catalyst or by the dynamic ligand exchange process between the binary ligands.25 Furthermore, the Hiyama reaction was not workable with ligand L6 as the sole ligand in parallel controlled experiments. Increasing its loading to 10 mol% only afforded the product 6 in a low yield of 28%. In addition, the inability of Cu-4 to catalyze the cross-coupling reactions was confirmed experimentally, and this result indicated that the phenanthroline ligand was integral to C(sp2)–C(sp3) bond formation.
Next, a chemically stoichiometric reaction of complex Cu-4, bromocyclohexane and base even with a high loading of copper catalyst (CuBr·SMe2) and phenanthroline-type ligand L6 (25 mol%) did not generate the coupling product 9 (Scheme 3d). These experimental data further precluded the ligand exchange process in the current multiligand-enabled copper catalysis. Interestingly, a reaction of complex Cu-4 with an excess amount of phenyltrimethoxysilane (1a, 10 equiv) would generate an aryl group exchanged copper complex, Cu-5, which was probably formed by the reaction of copper-aryl species with the base-activated pentacoordinate silicate species. The concomitantly generated aryl anion would be in equilibrium with silyl ethers to form arylsilanes, and such transient naked and reactive aryl anions could probably account for the high efficiency of the current multiligand-enabled couplings. However, revealing the detailed pathways for the aryl group transfer might require further effort.
Based on the above experimental data and prior literature,10–12,18–20,27 a plausible mechanism is proposed in Scheme 3f. An in situ generated phenanthroline–copper complex A would react with a transient aryl anion, generating via the NHC-copper catalyst the aryl group release from arylsilanes, to form a copper-aryl species B. Subsequently, the copper-aryl species B would activate the sp3-alkyl halides via a single-electron process to afford the copper species C with concomitant formation of alkyl radical species, and a following rebounding step of the radical species would afford the high-valent copper species D. Finally, the ensuing reductive elimination of D would afford the Hiyama coupling product with the regeneration of copper species A. It should be mentioned that the coupling reactions also could be enabled by phenanthroline ligand alone in a slower fashion via the traditional transmetallation process (gray arrows).28
Conclusions
In conclusion, the current study first demonstrates multiligand-enabled copper-catalyzed Hiyama couplings of arylsilanes with unactivated secondary alkyl halides to forge C(sp2)–C(sp3) bonds, thus offering an efficient method for Hiyama couplings with a cost-effective copper system. A simple combination of phenanthroline and NHC ligands remarkably increased the reaction efficiency, and mechanistic studies disclosed that the two types of ligands supported copper catalysis shown by their respective abilities in the coupling system, where the NHC–copper catalyst accounted for the transmetallation process of arylsilanes and the phenanthroline–copper catalyst was responsible for carbon–carbon bond formation. This protocol offered a new solution for the development of Hiyama couplings. Further effort would be required to understand the mechanistic aspects and broaden the application range of the current multiligand-enabled copper system.Data availability
ESI† is available and includes the experimental procedures, characterization data and crystallographic data for Cu-1. Deposition number 2361575 (for Cu-1) contains the supplementary crystallographic data for this paper.29 The data are provided free of charge by the Cambridge Crystallographic Data Centre.Author contributions
W. X. conceived the project. J. Z., Z. Z. and Y. C. conducted the experiments. W. X., J. Z., Z. Z. and Y. C. analysed the data and W. X. wrote the manuscript with the assistance of all authors.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
We are grateful to the Natural Science Foundation of Shanghai (22ZR1402500) and the National Natural Science Foundation of China (22201034) for generous funding. We thank the staff from College of Materials Science and Engineering (Donghua University) for X-ray data collection using a Bruker D8 Venture, and the group of Prof. Dr Weimin Xuan (Donghua University) for their great support with the X-ray diffraction analysis.Notes and references
- (a) F. Diederich, P. J. Stang, Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH, 2004 Search PubMed; (b) J. C. H. Lee, D. J. Hall, Metal-Catalyzed Cross-Coupling Reactions and More, ed. A. de Meijere, S. Brase and M. Oestreich, Wiley-VCH, Weinheim, 2014, pp 65–132 Search PubMed; (c) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- For selected references, see: (a) Y. Hatanaka and T. Hiyama, J. Org. Chem., 1988, 53, 918–920 CrossRef CAS; (b) S. E. Denmark and J. Y. Choi, J. Am. Chem. Soc., 1999, 121, 5821–5822 CrossRef CAS; (c) S. E. Denmark and R. F. Sweis, J. Am. Chem. Soc., 2001, 123, 6439–6440 CrossRef CAS PubMed; (d) T. Komiyama, Y. Minami and T. Hiyama, ACS Catal., 2017, 7, 631–651 CrossRef CAS; (e) S. B. Pawley, A. M. Conner, H. M. Omer and D. A. Watson, ACS Catal., 2022, 12, 13108–13115 CrossRef CAS PubMed; (f) S. E. Denmark and C. S. Regens, Acc. Chem. Res., 2008, 41, 1486–1499 CrossRef CAS PubMed; (g) S. E. Denmark and J. H. C. Liu, Angew. Chem., Int. Ed., 2010, 49, 2978–2986 CrossRef CAS PubMed; (h) Y. Nakao and T. Hiyama, Chem. Soc. Rev., 2011, 40, 4893–4901 RSC; (i) H. F. Sore, W. R. J. D. Galloway and D. R. Spring, Chem. Soc. Rev., 2012, 41, 1845–1866 RSC; (j) S. E. Denmark and A. Ambrosi, Org. Process Res. Dev., 2015, 19, 982–994 CrossRef CAS PubMed.
- (a) Science of Synthesis, ed. I. Fleming, Thieme, Stuttgart, 2002, vol. 4, ch. 4.4 Search PubMed; (b) The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, New York, vol. 2, 1998 Search PubMed; (c) P. Nareddy, F. Jordan and M. Szostak, Chem. Sci., 2017, 8, 3204–3210 RSC; (d) K. Shin, Y. Park, M.-H. Baik and S. Chang, Nat. Chem., 2018, 10, 218–224 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) D. H. Liu, P. M. Pfluger, A. Outlaw, L. Luckemeier, F. Zhang, C. Regan, H. Rashidi Nodeh, T. Cernak, J. Ma and F. Glorius, J. Am. Chem. Soc., 2024, 146, 11866–11875 CrossRef CAS PubMed; (c) J. Choi and G. C. Fu, Science, 2017, 356, eaaf7230 CrossRef PubMed; (d) A. Kaga and S. Chiba, ACS Catal., 2017, 7, 4697–4706 CrossRef CAS.
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science, Sausalito, CA, 2010 Search PubMed.
- For the selected references on nickel systems, see: (a) T. Iwasaki and N. Kambe, Top. Curr. Chem., 2016, 374, 66 CrossRef PubMed; (b) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 1340–1341 CrossRef CAS PubMed; (c) P. Zhang, C. C. Le and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 8084–8808 CrossRef CAS PubMed ; for the selected references on cobalt systems, see: ; (d) A. Guérinot and J. Cossy, Acc. Chem. Res., 2020, 53, 1351–1363 CrossRef PubMed; (e) L. R. Mills, D. Gygi, J. R. Ludwig, E. M. Simmons, S. R. Wisniewski, J. Kim and P. J. Chirik, ACS Catal., 2022, 12, 1905–1918 CrossRef CAS PubMed; (f) L. R. Mills, D. Gygi, E. M. Simmons, S. R. Wisniewski, J. Kim and P. J. Chirik, J. Am. Chem. Soc., 2023, 145, 17029–17041 CrossRef CAS PubMed ; for the selected references on iron systems, see: ; (g) T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara, H. Seike, H. Takaya, Y. Tamada, T. Ono and M. Nakamura, J. Am. Chem. Soc., 2010, 132, 10674–10676 CrossRef CAS PubMed; (h) M. P. Crockett, A. S. Wong, B. Li and J. A. Byers, Angew. Chem., Int. Ed., 2020, 59, 5392–5397 CrossRef CAS PubMed; (i) P. O. Peterson, M. V. Joannou, E. M. Simmons, S. R. Wisniewski, J. Kim and P. J. Chirik, ACS Catal., 2023, 13, 2443–2448 CrossRef CAS; (j) D. Chen, C. Lepori, R. Guillot, R. Gil, S. Bezzenine and J. Hannedouche, Angew. Chem., Int. Ed., 2024, 63, e202408419 CrossRef CAS PubMed.
- L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC.
- F. Ullmann and J. Bielecki, Ber. Dtsch. Chem. Ges., 1901, 34, 2174–2185 CrossRef CAS.
- (a) S. Saranya, G. Anilkumar, Copper Catalysis in Organic Synthesis, John Wiley & Sons, Ltd, 2020 Search PubMed; (b) G. Evano, N. Blanchard, Copper-Mediated Cross-Coupling Reactions, John Wiley & Sons, 2013 CrossRef; (c) S. R. Chemler, Copper Catalysis in Organic Synthesis, Beilstein-Institut, 2015, vol. 11, pp 2252–2253 Search PubMed; (d) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed; (e) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed; (f) R. Trammell, K. Rajabimoghadam and I. Garcia-Bosch, Chem. Rev., 2019, 119, 2954–3031 CrossRef CAS PubMed; (g) Y. Luo, Y. Li, J. Wu, X.-S. Xue and J. F. Hartwig, Science, 2023, 381, 1072–1079 CrossRef CAS PubMed.
- For the selected reviews, see: (a) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337–2364 CrossRef CAS; (c) D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS PubMed; (d) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC; (e) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed; (f) Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- For the representative references on C–C bond formation, see: (a) H.-Q. Do and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 1128–1129 CrossRef CAS PubMed; (b) H.-Q. Do, R. M. Kashif Khan and O. Daugulis, J. Am. Chem. Soc., 2008, 130, 15185–15192 CrossRef CAS PubMed; (c) Y. Zhou, W. You, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2014, 53, 3475–3479 CrossRef CAS PubMed.
- For the selected references on carbon-heteroatom bond formation, see: (a) Z. Chen, Y. Jiang, L. Zhang, Y. Guo and D. Ma, J. Am. Chem. Soc., 2019, 141, 3541–3549 CrossRef CAS PubMed; (b) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS PubMed; (c) R. Ray and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 60, 8203–8211 CrossRef CAS PubMed; (d) M. Fan, W. Zhou, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2016, 55, 6211–6215 CrossRef CAS PubMed; (e) S.-T. Kim, M. J. Strauss, A. Cabré and S. L. Buchwald, J. Am. Chem. Soc., 2023, 145, 6966–6975 CrossRef CAS PubMed; (f) A. Tlili, N. Xia, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 8725–8728 CrossRef CAS PubMed; (g) S. Xia, L. Gan, K. Wang, Z. Li and D. Ma, J. Am. Chem. Soc., 2016, 138, 13493–13496 CrossRef CAS PubMed.
- (a) A. C. Bissember, R. J. Lundgren, S. E. Creutz, J. C. Peters and G. C. Fu, Angew. Chem., Int. Ed., 2013, 52, 5129–5133 CrossRef CAS PubMed; (b) Q. M. Kainz, C. D. Matier, A. Bartoszewicz, S. L. Zultanski, J. C. Peters and G. C. Fu, Science, 2016, 351, 681–684 CrossRef CAS PubMed.
- B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef.
- H. Luo, Y. Yang, Y. Fu, F. Yu, L. Gao, Y. Mao, Y. Li, K. Wu and L. Lin, Nat. Commun., 2024, 15, 5647 CrossRef CAS PubMed.
- T. S. Ratani, S. Bachman, G. C. Fu and J. C. Peters, J. Am. Chem. Soc., 2015, 137, 13902–13907 CrossRef CAS PubMed.
- (a) A. Hazra, M. T. Lee, J. F. Chiu and G. Lalic, Angew. Chem., Int. Ed., 2018, 57, 5492–5496 CrossRef CAS PubMed; (b) X. Zeng, C. Wang, W. Yan, J. Rong, Y. Song, Z. Xiao, A. Cai, S. H. Liang and W. Liu, ACS Catal., 2023, 13, 2761–2770 CrossRef CAS PubMed.
- (a) C. T. Yang, Z. Q. Zhang, Y. C. Liu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 3904–3907 CrossRef CAS PubMed; (b) Y.-Y. Sun, J. Yi, X. Lu, Z.-Q. Zhang, B. Xiao and Y. Fu, Chem. Commun., 2014, 50, 11060–11062 RSC; (c) E. B. McLean, V. Gauchot, S. Brunen, D. J. Burns and A.-L. Lee, Chem. Commun., 2019, 55, 4238–4241 RSC.
- (a) Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed; (b) Z. Zhang, M. J. Tilby and D. Leonori, Nat. Synth., 2024, 3, 1221–1230 CrossRef CAS.
- (a) Y. Zhou, L. Qiu, J. Li and W. Xie, J. Am. Chem. Soc., 2023, 145, 28146–28155 CrossRef CAS PubMed; (b) J. Xu, X. Zhang, Y. Cao, X. Jian, L. Song and W. Xie, Cell Rep. Phys. Sci., 2024, 5, 102050 CrossRef CAS.
- (a) J.-Y. Lee and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 5616–5617 CrossRef CAS PubMed; (b) T. Komiyama, Y. Minami and T. Hiyama, Chem. Lett., 2018, 47, 1048–1050 CrossRef CAS; (c) D. A. Powell and G. C. Fu, J. Am. Chem. Soc., 2004, 126, 7788–7789 CrossRef CAS PubMed; (d) N. A. Strotman, S. Sommer and G. C. Fu, Angew. Chem., Int. Ed., 2007, 46, 3556–3558 CrossRef CAS PubMed.
- (a) M. Stradiotto, R. J. Lundgren, Ligand Design in Metal Chemistry: Reactivity and Catalysis, Wiley, 2016, Hoboken, pp. 1–400 CrossRef; (b) D. J. Durand and N. Fey, Chem. Rev., 2019, 119, 6561–6594 CrossRef CAS PubMed.
- (a) U. B. Kim, D. J. Jung, H. J. Jeon, K. Rathwell and S.-g. Lee, Chem. Rev., 2020, 120, 13382–133433 CrossRef CAS PubMed; (b) L. K. G. Ackerman-Biegasiewicz, S. K. Kariofillis and D. J. Weix, J. Am. Chem. Soc., 2023, 145, 6596–6614 CrossRef CAS PubMed; (c) S. K. Dorn and M. K. Brown, ACS Catal., 2022, 12, 2058–2063 CrossRef CAS PubMed; (d) Y. Wu, X. Huo and W. Zhang, Chemistry, 2020, 26, 4895–4916 CrossRef CAS PubMed.
- (a) Y. Wang, Y. He and S. Zhu, Acc. Chem. Res., 2023, 56, 3475–3491 CrossRef CAS PubMed; (b) Y. Sun, B. Wang and Z. Lu, Org. Chem. Front., 2023, 10, 4146–4160 RSC.
- (a) B. P. Fors and S. L. Buchwald, J. Am. Chem. Soc., 2010, 132, 15914–15917 CrossRef CAS PubMed; (b) G. Y. Wang, Z. Ge, K. Ding and X. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307973 CrossRef CAS PubMed; (c) T. B. Hamby, M. J. LaLama and C. S. Sevov, Science, 2022, 376, 410–416 CrossRef CAS PubMed; (d) S.-H. Kim-Lee, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, Chem, 2021, 7, 2212–2226 CrossRef CAS; (e) J. Corpas, M. Gomez-Mendoza, J. Ramírez-Cárdenas, V. A. de la Peña O'Shea, P. Mauleón, R. Gómez Arrayás and J. C. Carretero, J. Am. Chem. Soc., 2022, 144, 13006–13017 CrossRef CAS PubMed.
- For the selected review on NHC–copper systems, see: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) Science of Synthesis: N-Heterocyclic Carbenes in Catalytic Organic Synthesis, ed. S. P. Nolan and C. S. J. Cazin, Thieme, Stuttgart, 2016, vol. 1–2 Search PubMed; (c) S. Díez-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- For the representative references on the formation of NHC–copper aryl complexes, see: (a) J. R. Herron and Z. T. Ball, J. Am. Chem. Soc., 2008, 130, 16486–16487 CrossRef CAS PubMed; (b) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792–5795 CrossRef CAS PubMed.
- For the generation of phenanthroline ligand supported copper ethoxide species, see: M. Ohashi, T. Adachi, N. Ishida, K. Kikushima and S. Ogoshi, Angew. Chem., Int. Ed., 2017, 56, 11911–11915 CrossRef CAS PubMed.
- Deposition number 2361575 (for Cu-1) contains the supplementary crystallographic data for this paper. The data is provided free of charge by the Cambridge Crystallographic Data Centre.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC for Cu-1, 2361575. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc07441f |
| This journal is © The Royal Society of Chemistry 2025 |