
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMOSAEC-DB: a comprehensive database of experimental metal–organic frameworks with verified chemical accuracy suitable for molecular simulations†
Marco
Gibaldi
 a,
Anna
Kapeliukha
ab,
Andrew
White
a,
Jun
Luo
a,
Robert Alex
Mayo
a,
Jake
Burner
a and
Tom K.
Woo
*a
a,
Anna
Kapeliukha
ab,
Andrew
White
a,
Jun
Luo
a,
Robert Alex
Mayo
a,
Jake
Burner
a and
Tom K.
Woo
*a
aDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, 10 Marie Curie Private, Ottawa K1N 6N5, Canada. E-mail: twoo@uottawa.ca
bEducational and Scientific Institute of High Technologies, Taras Shevchenko National University of Kyiv, 4-g Hlushkova Avenue, Kyiv 03022, Ukraine
First published on 31st January 2025
Abstract
Ongoing developments in computational databases seek to improve the accessibility and breadth of high-throughput screening and materials discovery efforts. Their reliance on experimental crystal structures necessitates significant processing prior to computation in order to resolve any crystallographic disorder or partial occupancies and remove any residual solvent molecules in the case of activated porous materials. Contemporary investigations revealed that deficiencies in the experimental characterization and computational preprocessing methods generated considerable occurrence of structural errors in metal–organic framework (MOF) databases. The MOSAEC MOF database (MOSAEC-DB) tackles these structural reliability concerns through utilization of innovative preprocessing and error analysis protocols applying the concepts of oxidation state and formal charge to exclude erroneous crystal structures. Comprising more than 124k crystal structures, this work maintains the largest and most accurate dataset of experimental MOFs ready for immediate deployment in molecular simulations. The databases' comparative diversity is demonstrated through its enhanced coverage of the periodic table, expansive quantity of structures, and balance of chemical properties relative to existing MOF databases. Chemical and geometric descriptors, as well as DFT electrostatic potential-fitted charges, are included to facilitate subsequent atomistic simulation and machine-learning (ML) studies. Curated subsets—sampled according to their chemical properties and structural uniqueness—are also provided to further enable ML studies in recognition of the strict demand for duplicate structure elimination and dataset diversity in such applications.
Introduction
Material design and property evaluation uphold vital functions across numerous research disciplines, from drug discovery to catalysis to energy conversion and storage. Classical images of these processes involve long hours of labouring to obtain hard-fought experimental data, yet purely experimental approaches are limited in material scope proportionate to the available pool of researcher time and productivity. While experimental works remain crucial towards practical realization of these applications, interdisciplinary efforts recruiting insight from advanced computational techniques have gained favour as means to widen the design space and accelerate materials discovery. Computer-accelerated materials design naturally involves some mixture of rapid performance evaluations employing simulation and machine learning (ML) techniques, and may even include generative workflows to produce wholly new candidate structures.1 Deterministic and stochastic atomistic modelling methods, such as molecular dynamics (MD) and Monte Carlo (MC), provide opportunities to directly simulate materials properties like gas adsorption2–4 and diffusion5 in a high-throughput manner. Sophisticated neutral networks and machine learning architectures trained on experimental and simulated data allow for the classification and prediction of numerous chemically relevant properties, including adsorption energies,6 band gaps,7 intermolecular interaction energies,8 gas uptakes,9 and material stability.10 Generative algorithms evolved towards the identification of various hypothetical candidate structures11–15 with specific material properties and performance in the targeted application. With surging demand for accelerated materials discovery pipelines to solve ongoing environmental crises, these data-driven approaches continue to enrich investigations of diverse classes of materials such as drugs, polymers, porous materials, and so on.Availability of high-quality data and data curation protocols endures as an essential exercise in all computational materials screenings. When adapting experimental information (e.g., crystallographic data, isotherms, spectra, articles, etc.), it becomes necessary to standardize the data to guarantee accuracy in the subsequent computations. Reacting to this need for standardization, several databases focusing on materials and their properties emerged, covering a broad range of applications. Billions of commercially accessible compounds are assembled into the ZINC16 database, which provides a free source of small molecules for virtual screenings such as molecular docking. The ChEMBL database extracted information from medicinal chemistry journals and pharmaceutical research to generate an open-access database of over 2.4 million bioactive molecules amenable to drug discovery studies.17 A massive global collaboration aiming to compile experimental structural data of biologically relevant macromolecules –namely proteins– developed into the Protein Data Bank,18 which now boasts more than 200k experimental structures and over 1 M computed structure models. Large-scale aggregation efforts, such as those undertaken by PubChem, combine various physical and chemical data from hundreds of sources (i.e. scientific literature, patents, governmental reports, other databases, etc.) to establish searchable collections comprising millions of compounds, bioactivity measurements, safety and toxicity data, and so forth.19 Similarly, since the 1960s, the Cambridge Crystallographic Data Centre has maintained a repository of experimental organic and inorganic crystal structures known as the Cambridge Structural Database (CSD).20 The CSD has recently grown to comprise over 1.2 M experimentally characterized structures,21 and accordingly it serves as a vital starting point in the development of many computational materials databases. Particularly in the study of periodic materials such as metal–organic frameworks (MOFs), the vast majority of computational studies are propelled by a limited selection of popular databases derived from CSD deposited crystal structures undergoing additional processing stages to prepare them for computation. The most prevalent such database in the MOF research space being the “computation-ready” (CoRE) database22,23 which boasts over 19k experimental MOF crystal structures advertised as immediately viable for use in simulations. Since its creation, this catalogue of experimental MOFs has observed widespread adoption across adsorption studies and ML model training.24–27 Another commonly employed database is the newer QMOF database,28,29 comprising over 20k crystal structures possessing atomic positions relaxed by density functional theory (DFT) alongside several calculated electronic properties. Analogously, QMOF spawned many successive studies taking advantage of its practicality for the development of novel ML applications and high-throughput (HT) property screenings.2,30–32 The Cambridge Structural Database also contributed two of its own MOF-related datasets: (i) CSD MOF subset,33 presently containing over 125k experimental MOF crystal structures along with the scripts necessary to prepare them for computation, and (ii) CSD MOF Collection,34 containing over 10k 3D “computation-ready” crystal structures. Again, these databases provide sources of standardized MOF structures that require minimal further processing by the end-user, thereby representing a considerable benefit to the accessibility and consistency of published results.
The precise state of the “computation-ready” MOF structures contained in these databases has come under increasing scrutiny due to recent discoveries of prevalent structural errors in many databases.35 The generally stated goal of structure processing for computation is to resolve experimental artefacts and mimic the materials' activation such that ensuing property calculations yield conditions as close to experiment as possible. The exact structure preparation process differs on a database-by-database basis; however, it generally involves processing of the experimental crystallographic data to exclude residual solvent and other artefacts, such as disorder. For instance, authors of the CoRE MOF database22,23 outlined protocols to eliminate all disordered atom sites, retain charged counterions based on CSD charge information, and to perform residual solvent removal according to a revised graph-labelling solvent removal method. Similarly, the preprocessing methods presented with the CSD MOF subset33 and MOF collection34 capitalized upon the wealth of CSD structural data to identify molecules requiring removal. Additional structural filtering is oft employed to discard structures which do not meet specified property thresholds, such as minimum porosity or cell dimension metrics. Through previous manual inspections of thousands of MOF structures contained in these two databases, it was discovered that the accuracy of preprocessing was heavily influenced by two principal factors: (i) resolution issues in the deposited experimental crystal structure (i.e. omitted hydrogen atoms, failure to model charge-balancing counterions, etc.), and (ii) dysfunction in one or more preprocessing steps (i.e. improper solvent removal, flawed disorder handling).35–37 These issues bring about a high incidence of structural errors in many computational MOF databases,35 and ultimately mars the quality of data generated by property calculations depending on accurate chemical and physical representations of the materials in molecular simulation and ML applications. Modern database approaches employing stricter material selection and filtering criteria, such as QMOF, experienced lesser impact of these structural errors in our previous accounts. In recognizing the experimental structure accuracy problem, one may simply choose to regard any ambiguity regarding the correctness of the deposited structure –for example, presence of disorder, detection of charge-balancing species, etc.– as cause for its automatic rejection as a structure for further processing and computation. While this preprocessing philosophy proved highly effective at curtailing structural errors, the size and diversity of the structure inventory diminished due to the harshness of the imposed constraints. Ideally, database creators would prefer to employ looser criteria to retain as many candidate structures as possible, but no comprehensive methods of dealing with structural issues currently exist. Our recent accounts of chemistry-minded approaches to solvent removal37 and structural error analysis35 demonstrated a high degree of success in managing structural errors on an individual basis which obviates the need for restrictive wholesale filtering. The lack of similar established preprocessing techniques tailored to contemporary knowledge of MOF structural errors limits the balance of diverse MOF chemistry and structural accuracy present within prevailing databases—both of which are forecasted to be meaningful contributors to the success of future MOF materials discovery studies.
This work aims to augment the accessible inventory of experimental MOF crystal structures ready for molecular simulations and other computational studies through development of a novel computational MOF database. Through utilization of recently published crystal structure error analysis35 and solvent removal37 methods which employ metal oxidation states and ligand formal charges as diagnostic tools, probable structural errors were expelled thereby achieving a higher fidelity database preprocessing protocol. This database—dubbed MOSAEC-DB due to the integral role of the eponymous algorithm in its construction—constitutes the largest contemporary collection of processed MOF crystal structures, comprising over 124k structures possessing varying degrees of activation and porosity. Importantly, framework charge accounting demonstrated in the SAMOSA solvent removal method37 was applied to generate a dataset of charge-labelled ionic MOF frameworks, including more than 17k distinct charged framework entries. This represents the first collection of its kind, permitting new avenues towards high-throughout investigation of a diverse range of cationic and anionic MOF chemistry which was previously cumbersome without automated framework charge accounting and validation procedures. Analysis of computed geometric and chemical descriptors followed database construction to characterize the pore environments and chemical substructure diversity available in MOSAEC-DB. Additionally, such geometric and chemical properties were applied in the development of numerous diverse subsets aiming to encompass a broad range of experimental MOF chemistry while minimizing the possibility of crystal structure duplication. While the existence of duplicate crystal structures carries minor consequences in HT screenings, they represent significant data leakage risks to any ensuing ML studies. Evaluations concerning the influence of previously reported database curation protocols and the degree of activation simultaneously provided insight regarding how preprocessing choices and structural errors impact the final chemical space present in these databases. We conclude by discussing this database's position with respect to the current state-of-the-art and how its inclusive construction approach allows future screening efforts to investigate a deeper set of materials with lessened fear of structural inaccuracies.
Methodology
Source of experimental MOF crystal structures
The foundation of the MOSAEC database was built upon the processing of experimental MOF crystal structures available in the CSD MOF subset,38 currently amounting to more than 125k structures (CSD version 5.4.5). This dataset contains a combination of 1D, 2D, and 3D experimentally synthesized MOFs, as well as a mixture of disordered and non-disordered crystal structures. Additional MOF and coordination polymer structures not present in this subset were retrieved through an enhanced chemical substructure search of periodic materials within the CSD, as well as an examination of the materials present in other popular MOF databases, such as the CoRE22,23 and QMOF28,29 databases. The supplemental search process utilized the CCDC Conquest39 program's query functions with a broader definition of chemical motifs relevant to MOF chemistry. A summary of this additional search process and the underlying chemical motifs is reported in the ESI (Fig. S1).† It is important to note that these criteria intentionally allowed for the admittance of periodic structures—such as coordination polymers, sulfates, phosphates, etc.—which do not strictly coincide with the definition of a MOF (i.e., those lacking any hydrogen or carbon atoms), thus it is recommended that users filter out any crystal structures that do not fit their chemical composition expectations. Experimental crystallographic information files (.cif) were retrieved directly from the CSD, then subjected to the preprocessing protocol described in the following sections.Crystallographic data processing
Subsequent processing and analysis protocols require that the symmetry of each structure be converted to P1, which was achieved using an in-house written Python script that utilizes Pymatgen40 symmetry operation modules. Disordered solvent proved to be highly consequential in our previous reports on solvent removal37 accuracy, thus removal of any guest, solvent, or framework atom sites labelled as disordered or possessing partial occupancies was implemented to handle the high degree of crystallographic disorder in many CSD MOF structures. This indiscriminate approach to disorder handling was deemed acceptable as any instances where it produced unreasonable chemistry by eliminating essential framework atoms would be nonetheless discarded at the later error analysis stages.Solvent removal
The recently published solvent removal method, called SAMOSA, informed by ligand formal charges37 was applied to produce high quality structures cleaned for molecular simulation. Validation of the SAMOSA method against past preprocessing efforts suggested that more reliable removal of solvent molecules and retention of charged ligand species could be achieved through the consideration of ligand charge and materials-specific rules. In accordance with experimental observations regarding the difficulties associated with reaching complete pore activation, this method incorporates both full (i.e. free and bound solvent) and partial (i.e. free solvent) solvent removal options to better approximate real crystal structure activation outcomes. All prospective experimental MOF crystal structures underwent both full and partial solvent removal, however, instances spawning identical activated structures were removed to minimize structure duplication. MOSAEC-DB crystal structure filenames contain either “_full” or “_partial” labels to differentiate between these degrees of activation completeness. An illustrative example demonstrating the differences in activation state is displayed in Fig. S2.†Further, the novel charge accounting protocol established by SAMOSA allows for discrimination between neutral and charged frameworks that is not attainable in prior studies. Calculation of these charge labels grants the opportunity to include a collection of charged MOF structures alongside the more traditionally employed neutral databases. An instance wherein a cationic framework is produced following structure processing is outlined in the ESI (Fig. S3).† MOSAEC-DB structures ending in “_p(x)” or “_n(y)” labels possess net framework charges corresponding to either positive |x| or negative |y|, respectively. The computed framework charge labels were verified through a modified version of the structure error analysis protocol described in the upcoming section; therefore, only structures possessing reasonable metal oxidation states upon consideration of their charge label were included. While these charged frameworks may not be appropriate for all situations, inclusion of credible, charge-labelled frameworks allows researchers to study a diverse set of ionic frameworks which were previously neglected in MOF database curation.
Structural error analysis
Following structure cleaning and preprocessing, previously described methods of identifying crystal structures with structural errors were employed to discard materials with questionable structural fidelity. The MOSAEC algorithm35 leverages the concept of ligand formal charges and metal oxidation states to judge whether a given crystal structure possesses structural errors. Instances where all metals contain valid oxidation states suggest that the underlying crystal structure does not contain common structural errors with a high degree of confidence. Conversely, when even one metal site possesses an impossible (i.e. exceeding the number of valence electrons available) or improbable (i.e. zero valent, scarcely observed experimentally, etc.) oxidation state, then MOSAEC flags that crystal structure due to the high likelihood that it contains one or more structural error. Analysis of a manually validated test set of ∼16k crystal structures showed that MOSAEC is 95% accurate when flagging a structure as containing an error and approximately 85% accurate when determining that a structure lacks any error. The higher incidence of incorrect classification of structures as error-free is often the result of metals possessing multiple plausible oxidation states (e.g., Mn, Fe, Co, Cu, etc.) wherein MOSAEC will correctly assess that the implied oxidation states are both possible and probable but manual validation indicated that the crystal structure differed from the original experimental report; for example, the MOSAEC-computed oxidation state determines Cu(I) but comparison to the true crystallographic data from the publication suggested the absence of anionic counterions resulting in a true oxidation state of Cu(II). All studied MOF structures were subjected to crystal structure error analysis and those receiving a classification indicative of errors were excluded from the final database. A concise summary of the MOSAEC algorithm's operation in the database construction process is provided in the ESI.†Database construction
A detailed schematic of the full database construction workflow is presented in Fig. 1. Various structure filtration steps were applied to discard problematic structures and to classify the MOFs based on several essential properties, such as porosity, framework charge, solvation status, and so on. Atom-pair distances were computed to eliminate structures with potentially overlapping atoms—herein defined as distances less than 70% of the sum of covalent bond radii41 of the atom pair in question. Further, the connectivity of each MOF structure graph was analyzed to recognize hypervalent atoms (e.g., hydrogen atoms possessing more than 1 bond, carbon atoms with more than 4 bonds, etc.) in the organic linkers and remove any such overbonded structures. Any structure passing all stages of the structure filtration and MOSAEC error analysis was regarded as much less likely to contain significant structural errors, and thus could be reliably added. The attached database contains all structures which have been modified by the above construction workflow. Cases requiring no solvent removal or any other alterations relative to their CSD entry structure are not included in this attached set of MOSAEC-DB crystallographic files; however, any unmodified CSD refcode which passed all stages of error analysis—amounting to approximately 45k crystal structures—are provided with the attached files such that CSD-licensed users may regenerate the structures independently using the provided structure conversion codes. The public MOSAEC-DB structure quantity will thus differ from the total quantities reported hereafter until these unmodified structures are properly regenerated. | ||
| Fig. 1 Concise summary of the workflow employed to construct a database of simulation ready MOFs from raw experimental crystal structures. Abridged illustrations of each of the primary stages are included on the periphery. | ||
Geometric and chemical descriptor calculations
The geometric properties –accessible surface area (ASA), largest cavity diameter (LCD), pore-limiting diameter (PLD), void fraction, etc.– of each crystal structure were determined using the Zeo++ pore analysis package (version 0.3.0).42 Comparison of these properties allows for determination of structural variations between MOFs prepared by various computation solvent removal and preprocessing protocols, such as those established by the CSD MOF,33,34 CoRE 2019,23 and QMOF28,29 databases. The recommended number of MC steps and high accuracy settings were employed for all geometric calculations, along with probe radii of 1.45 Å corresponding to the kinetic diameter of H2. Framework dimensionality was assessed using the method described in previous characterizations of the CSD MOF subset.43 Simply put, this script identifies a given structures' smallest repeating unit and then expands this unit to varying degrees to determine how the dimensions of the minimum bounding box evolve. The ratios of the minimum bounding box dimension between the larger and smaller expanded periodic unit are utilized to evaluate how many dimensions changed significantly when the polymeric unit expanded.Bond connectivity for each crystal structure was generated using the nearest-neighbour algorithm designed by Isayev et al.44 in order to complete several analyses. This algorithm employs both a Voronoi tessellation-based and a bond distance-based criterion to determine when a pair of atomic sites is considered bonded. This bond connectivity information is then converted to fragments containing small groups of atoms for comparison with the unique definitions of various common substructures (e.g., aromatics, amines, carboxylates, etc.). Also, the bonding connectivity tables are supplied to the CrystalNets45 package when determining the network topology of each framework. Settings identical to those applied in our previous analysis of the ARC-MOF46 database were employed as they were found to properly reflect the topological information of hypothetical MOFs (hMOF) with a high degree of accuracy. Further, we performed an analysis of the bonding connectivity of the metal atoms within MOSAEC-DB, which was facilitated by an open metal site (OMS) detection algorithm first reported with the CoRE 2019 database.23
Descriptors characterizing the local chemical environment near metal and linker atom sites were computed through the previously described revised autocorrelation (RAC) method.47–49 This protocol provides a description of various distinct subdomains within the crystal structure according to common roles in the chemical structure graph. The RAC descriptors are then calculated as the sum over a product or difference of the atomic properties (e.g., nuclear charge, electronegativity, topology, identity, and atomic radius) between each atom belonging to the subdomain and their neighbouring atoms at specified through-bond walk distances in the chemical structure graph. All RAC subdomains were considered in this analysis including the metal-centered, ligand-centered, linker-centered, functional group-centered, and SBU-centered descriptors up to a maximum through-bond depth of 3. The SBU-centered features encompass the inorganic nodes, including the constituent metals and their coordination sphere, while the metal features only perform the sum over the metal atoms in these identified subgraphs. The ligand and linker atom subdomains are differentiated according to the number of SBUs they bridge in the graph, i.e. a single SBU in the case of ligands and multiple SBUs for linkers. The functional group RACs focus on heteroatoms in the linker structure that are not bound to metal atoms, while the linker-connecting features focus on the atoms connecting the linker and SBU substructures. Visualization of the chemical environments found in various databases as characterized in the RACs was achieved by dimensionality reduction via the t-stochastic neighbor embedding (t-SNE) method.50 Analysis of the resulting clusters in the reduced t-SNE descriptor space allows for exploration of the chemical motif differences in the inorganic node and organic linkers of the contrasted databases. This analysis was repeated on additional categories of descriptor employed to represent MOF crystal structures, such as the above-described geometric properties, to assess the structural diversity of MOSAEC-DB in relation to commonly screened databases. Additional categories of MOF descriptors, such as the atomic-property weighted radial distribution functions (AP-RDF) and atom-specific persistent homology,51,52 were computed wherever possible to provide additional descriptors characterizing the local atomic and pore environments, and enable their immediate use in future ML studies. Beyond database diversity comparison, these descriptors were utilized to generate various subsets of MOSAEC-DB through a farthest point sampling approach wherein the full vectors were analyzed to select crystal structures with the maximal diversity possible in the considered feature space. Details regarding the composition of these diverse subsets are outlined in the results section.
Duplicate crystal structure identification
Several quintessential MOF crystal structures and families of structures—for example, MOF-5, UiO-66, MOF-74, MIL-120, CuBTC, and so on—observe enormous quantities of repetition within experimental repositories such as the CSD. These duplicated crystal structures may be collected from a variety of X-ray crystallographic instruments, synthetic conditions, and even research groups with broad interests and intended applications, yet many instances resolve a nearly identical unit cell of relative atomic positions. This phenomenon is mostly inconsequential for HT screenings employing atomistic simulations beyond the wasted computational resources associated with performing calculations of duplicate systems, whereas the effect is more insidious for ML studies where duplication of data across the training and test datasets leads to false reporting of model efficacy. Previous investigations into machine learning on MOFs have reported methods of combating such structural duplication, such as through the comparison of their bond connectivity graphs.52 Other methods exist that apply geometry-based descriptors, which are generally rapidly computed, to compare the real space relationships between atomic positions in the unit cell and identify closely related structure. This database opts to apply one such method known as the pointwise distance distribution53,54 (PDD) which analyzes the distances between the nearest neighbours for each atom in a given unit cell and compares that matrix to the analogous one calculated for all possible duplicate crystal structures. The overall duplicate identification workflow involved first comparing each MOF structure's empirical formula. Then performing PDD computations on all structure pairs sharing a common formula. Any pair possessing a PDD score below a defined threshold value were classified as duplicate structures, and only one instance of each group of duplicates was retained in the unique dataset of MOSAEC-DB MOFs. A more extensive description of the implementation of this duplicate crystal structure identification protocol is provided in the ESI (Fig. S4).†Results & discussion
Database summary
Table 1 summarizes the contents of the MOSAEC database, including the different structural categories and the quantity of structures present within each subset. Recall, only one form (i.e. the fully activated structure) was retained when identical structures were produced by full and partial activation, culminating in the significantly greater number of those structures. A total of 143.9k candidate crystal structures were considered upon combining the CSD MOF subset with the previously discussed additions to the chemical substructure search criteria. Following the oxidation state-based and physical (i.e. overlapping and hypervalent atoms) validation steps exercised to remove erroneous structures, 124.4k crystal structures remained belonging to 91.7k unique CSD entries. A striking proportion of MOSAEC-DB possesses minimal porosity—amounting to only 15.3% and 3.9% of the neutral fully and partially activated structures possessing void fractions (ϕ) greater than 0.10, respectively—despite employing an H2 probe molecule during textural analyses. The charged framework subsets observe sizable enhancements (2–4×) to their porosity compared to their neutral counterparts, but this effect is overestimated due to the absence of the non-coordinating counterions from the pore network. The imperative restoration of these charge-balancing species to the charged crystal structures before simulation would likely erase much of these gains in pore volume. Notably, shifting the porosity definition to include all structures possessing a non-zero void fraction boosts the fraction of porous, neutral MOFs to 31.4% (full) and 14.6% (partial); however, this observation maintains that the bulk (> approximately 80%) of the considered experimental MOF crystal structures are effectively non-porous. While these non-porous entities may not be optimal in the gas adsorption and separation roles typically associated with MOFs, they may possess unique electronic, chemical, or other properties which render them relevant to other applications. A similar design philosophy was adopted with respect to the diverse framework dimensionalities present in MOSAEC-DB. The database is approximately equally composed of one-dimensional (36.76%), two-dimensional (27.54%), and three-dimensional (29.13%) crystal frameworks according to analyses of the increasing dimension(s) of polymeric unit expansion. Depending on the outlined function, one may have certain bias and uncertainty regarding lower-dimensional frameworks' synthesis and reproducibility; however, these structures may retain unique properties that could prove to be useful in future screening and data-mining investigations. In summation, this database elected for an inclusive approach with the goal of maximizing the total structure space, thus users are encouraged to refine the screening pool to best fit their target applications' specifications using the available dimensionality and geometric data.| Degree of activation | Charge state | Structure quantity | Porous (ϕ > 0.10) | Framework dimensionality | ||
|---|---|---|---|---|---|---|
| 1D | 2D | 3D | ||||
| Full | Neutral | 78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 176 176 |
15.32% | 34.24% | 27.92% | 31.39% |
| Charged | 13302 |
30.39% | 44.02% | 22.37% | 26.76% | |
| Partial | Neutral | 28373 |
3.93% | 37.64% | 29.49% | 26.28% |
| Charged | 4618 | 16.70% | 53.10% | 24.01% | 15.31% | |
| Total |
124![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) 469 469
|
14.39% | 36.76% | 27.54% | 29.13% | |
Chemical composition and substructure determination
A diverse selection of chemical compositions and moieties exist within the MOSAEC database as a consequence of its large structure quantity and expanded search criteria. A demonstration of its far-reaching chemical composition space is shown in Fig. 2 which highlights the total structure count breakdown by element. The composition distribution is clearly strongly influenced by the specific material and substructure definitions selected when identifying MOFs in the CSD. Creation of the CSD MOF subset leaned heavily on chemical moiety descriptions involving nitrogen, carbon, oxygen, hydrogen, and generic metal atoms,33 thereby explaining those atoms' high incidence rates. The additional search criteria (Fig. S1†) employed in this work allowed for a greater representation of other elements such as boron, sulfur, selenium, and phosphorus compared to existing databases. The decision to establish looser substructure search definitions allowed for the inclusion of isostructural variants through substitutions by elements with common chemical properties and reactivity, for instance those observed in the relationship between carboxylates and thiocarboxylates. Moreover, several metal elements such as technetium, tantalum, osmium, and iridium which are rarely or never observed in the ARC-MOF, CoRE, or QMOF databases—as demonstrated by their respective periodic table visualizations (Fig. S5†)—may be accessed. While studies seeking only such metals in particular may be uncommon, this further evidenced the novel chemical diversity supplied by this database relative to existing computational MOF databases. A comprehensive comparison to all structures contained in the CSD (Fig. S6†), beyond solely the MOFs or coordination polymers targeted by MOSAEC-DB, indicates that the primary element omissions from this database involve other relatively rare elements including He, Ne, Ra, Pa, Bk, and Cf. Furthermore, compared to the entire CSD, the relative representation of many elements—such as B, F, Si, P, Cl and several catalytically relevant metals like Ru, Rh, and Pd—in MOSAEC-DB is clearly lowered. This is somewhat unsurprising due to these elements' underrepresentation as linkers in periodic crystal structures and common occurrence in drug and other organic molecules, and transition metal complexes. In many cases, the wide net cast by this database construction approach led to the admittance of hundreds or thousands of additional crystal structures for even the rarest metal species, such as the lanthanides and precious metals, and linkers relevant to the chemistry of MOFs and other coordination polymers.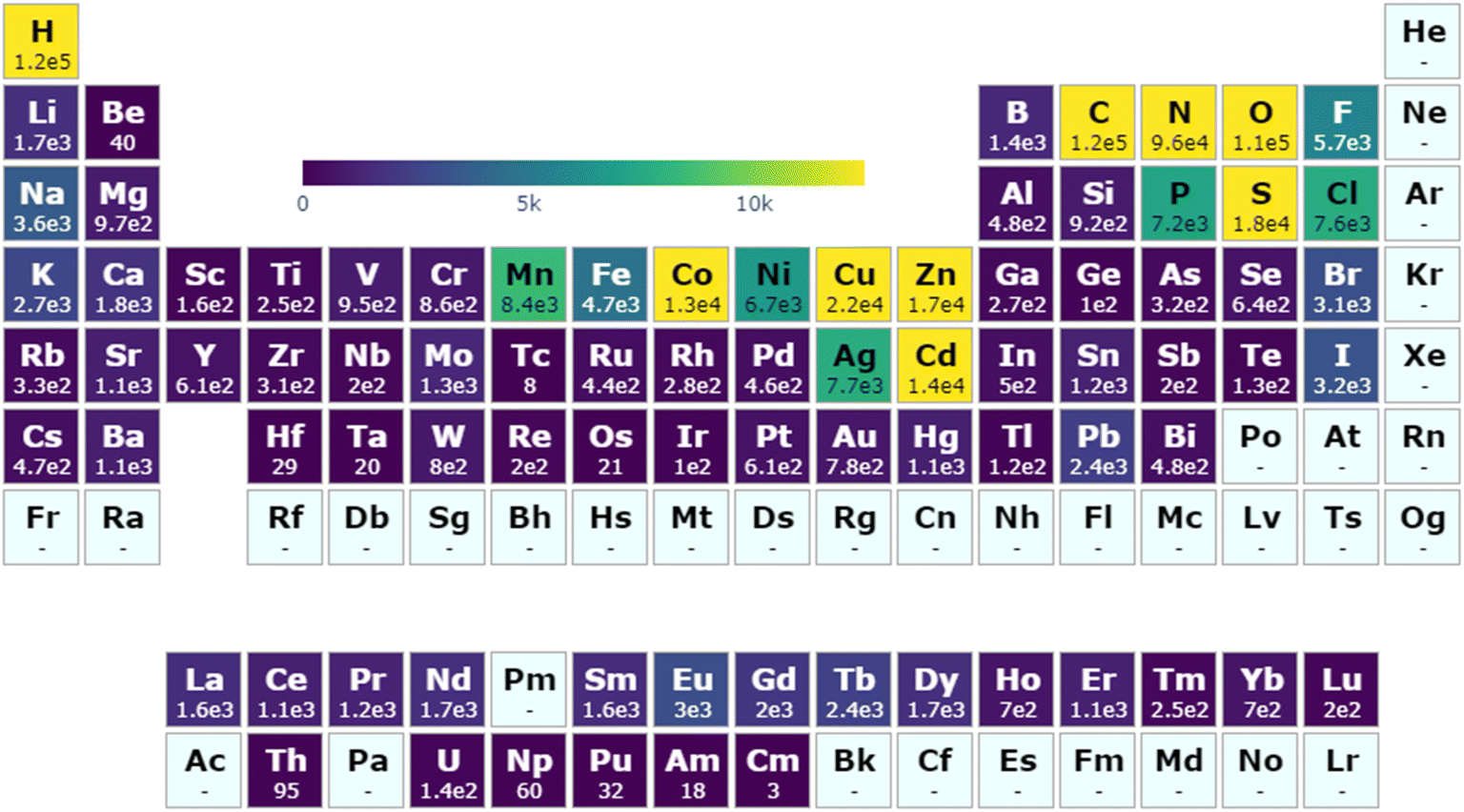 | ||
| Fig. 2 Account of MOSAEC-DB crystal structures containing given elements across the periodic table. A colour gradient maximum equal to 10% of the total structure count is enforced to facilitate visualization (generated via pymatviz55). | ||
To further appraise the accessible chemistry, the presence of various characteristic chemical moieties was determined through analysis of each structures' bond connectivity. Fig. 3 compiles the principal findings concerning the predominant chemical substructures and linker molecules found in this work. The most frequently occurring chemical substructures consisted of aromatics (77.1%), carboxylates (40.9%), amines (24.7%), halogens (14.6%), and alkenes (7.5%), which is roughly coincident with similar investigations performed on ARC-MOF,46 a database principally composed of hMOFs and a smaller proportion of experimental structures. Notably, far fewer alkenes and alkynes were observed in MOSAEC-DB relative to ARC-MOF which could be predicted from the regularity with which these substructures appear in the organic structural building unit (SBU) libraries applied during the construction of hMOF crystal structures, particularly those with the objective of producing isoreticular hMOF series. Further, the extended search criteria (Fig. S1†) implemented during candidate structure retrieval intensified the incidence of the substructures which were overlooked by the original CSD MOF subset definitions, such as sulfates (1.3%); however, in general, many of the added substructures (e.g., thiolates, dithioates, selenocyanates, borates, and so on) captured by this broadened search criteria were fairly rare, causing their influence on the overall database substructure statistics (Fig. 3a) to be relatively minor. While infrequent, the combined impact of these moieties' inclusion remains a beneficial expansion to the chemical diversity available in simulation ready MOFs.
 | ||
| Fig. 3 Analysis of the (a) frequency of chemical substructures and (b) quantity of crystal structures containing common linker molecules within the MOSAEC database. Analogous chemical substructures are grouped together when calculating their frequency, for example the amine category represents all (primary, secondary and tertiary) amines and the aromatic category represent all subcategories of heterocyclic aromatic compounds. Skeletal representations of only one of the several possible constitutional isomers (e.g., 1,4-benzene dicarboxylate, 1,3-benzene dicarboxylate in the case of the C8H4O4 linkers) are depicted for simplicity. | ||
A comprehensive investigation of the constituent building blocks forming each crystal structure was also performed as depicted in Fig. 3b. The observed top 10 organic linker SBUs according to structure count were consistent with the chemical substructure data (Fig. 3a) and chemical intuition with respect to popular compounds employed in MOF syntheses. As anticipated, carboxylates and aromatic molecules—including N-heterocyclic aromatics—pervade the frequently occurring linkers and historically significant molecules such as benzene dicarboxylic acid (BDC), formic acid, and bipyridine appear often. Full details regarding each structures' component SBUs are provided in the MOSAEC-DB Zenodo repository described below; thus, one may target specific building blocks in future screening or ML studies when preferable.
Geometric characteristics
The geometry properties of MOSAEC-DB crystal structures were then juxtaposed with those found within other leading MOF databases—namely, ARC-MOF, CoRE 2019, QMOF, and the CSD MOF collection—to assess its coverage of the diverse pore geometries available to MOFs. Probability distributions of the volume fractions, gravimetric surface area, largest cavity diameter (LCD), and pore limiting diameter (PLD) are depicted in Fig. 4, while comparisons of additional geometric properties are provided in the ESI (Table S3).† ARC-MOF provides the largest overall range of coverage across all compared properties and total quantity of structures in the high porosity domain. This is to be expected owing to its heavy leaning (ca. 97%) towards hMOFs which allow for the establishment of larger pore structures than typically observed in experimental crystal structures. MOSAEC-DB observes similar distributions to other databases consisting principally of experimental MOF crystal structures (i.e. CoRE 2019, CSD MOF, and QMOF), which generally bias towards higher density and lower pore sizes. The apparent differences in the CSD MOF Collection distribution at volume fractions nearing 0.30 arises due to the considerable duplication of MOF-74(Cu) crystal structures—for example, the hundreds of crystal structures reported under the CSD refcodes MOJPOT, MOLLAD, and MOKYAP—which leads to a high relative representation compared to other entries. The similarities in the experimental MOF database distributions is further emphasized by Table S3† wherein comparable means, standard deviations, and ranges of values are observed across all considered geometric properties when comparing the experimental databases. The sole exception being QMOF which possesses a greater fraction of structures with high volume fraction and surface area, likely due to its decision to include a small subset of hMOF structures. Notably, MOSAEC-DB possesses a significant fraction of non-porous crystal structures which would not be present in these related databases due to the authors' decision to eliminate MOFs outside of a given PLD threshold, historically set at 2.4 Å in CoRE and QMOF databases. Implementing an analogous PLD cut-off threshold in MOSAEC-DB reduces the database size to ca. 47k (37.95%), which still far surpasses what can currently be accessed in these existing experimental datasets. Further analyses related to the internal disparity between the various categories of MOFs outlined in this database (e.g., neutral vs. charged, full activation vs. partial activation, etc.) are presented in the ESI (Fig. S7).† As expected from the previously discussed discrepancies in porosity within these structural subsets, a great difference in pore diameters, surface areas, and void fractions are observed between the neutral and charged structure categories. The mean LCD and PLD values are approximately 1.5 Å larger in the charged MOF frameworks due to elimination of the non-coordinating counterions. As previously stated, this dissimilarity will likely be cancelled once the counterions are restored before simulation. The discrepancy resulting from the degree of activation is comparatively lesser, observing more modest reductions in the mean pore cavity diameters (ca. 0.5–1 Å) and void fraction (ca. 0.04) in the partially activated subcategories. | ||
| Fig. 4 Distributions of geometric properties within the MOSAEC database contrasted against other commonly used MOF databases, such as ARC-MOF, CSD MOF Collection, CoRE 2019, and QMOF databases. Distributions of (a) accessible volume fraction, (b) gravimetric surface area, (c) largest cavity diameter (LCD), and (d) pore limiting diameter (PLD) were calculated by Zeo++ using a probe radius corresponding to the kinetic diameter of an H2 molecule (1.45 Å). Zero values are excluded from the analysis for clarity. | ||
Topology classification
Reporting of net topology data is inconsistent throughout the sourced MOFs' literature, thus we relied on the CrystalNets45 package to compute these values across all MOSAEC-DB crystal structures rather than any potential text mining workflows. This technique appears to be limited in its capacity to handle lower-dimensional (i.e. 1D) frameworks, resulting in approximately 63% of the database possessing incomplete topological information. Roughly half of the 3D and 2D frameworks were successfully assigned topological nets, while topological analysis could not be completed for any 1D frameworks. An analysis of the most frequently occurring, underlying net topologies discovered in the remainder of MOSAEC-DB and their relative frequency is presented in Table 2. In total, approximately 400 unique topologies are identified in the final MOSAEC-DB. Structure possessing sql (8.99%), hcb (4.34%), dia (1.65%), pcu (1.29%), and fes (1.11%) net topologies constitute the five most common found within the database. This distribution of topologies represents a significant departure from the observations of our previous analysis on ARC-MOF which determined that pcu (38.0%), fsc (17.2%), nbo (16.3%), pts (14.1%), and sra (4.0%) topologies dominated this largely hMOF-containing databases. Furthermore, 69 distinct topologies were found to possess over 50 structural entries which eclipses the 46 such topologies identified in ARC-MOF. This information is perhaps unsurprisingly as the “top-down” crystal construction approaches used to generate hypothetical structures are limited to only producing topological nets for which they possess a compatible set of SBUs, and prior hMOF databases generally put forth minimal effort to exhaustively explore the SBU-topology space due to the sheer number of possible combinations available. The same restriction does not exist for experimental MOF structures which are only bounded by the thermodynamic accessibility of the requisite metal-linker coordination geometries required to form any given topology. Therefore, access to a wider array of topologies underscores a key advantage of MOSAEC-DB compared to its predecessors towards various state-of-the-art screening and ML implementations.| Topology | MOSAEC-DB representation | |
|---|---|---|
| Structure count | Frequency (%) | |
| sql | 11195 |
8.99% |
| hcb | 5404 | 4.34% |
| dia | 2048 | 1.65% |
| pcu | 1609 | 1.29% |
| fes | 1390 | 1.11% |
| bey | 635 | 0.51% |
| pts | 572 | 0.46% |
| ths | 584 | 0.45% |
| cds | 544 | 0.44% |
| bex | 531 | 0.43% |
Solvent removal and open metal sites
The primary innovation contributed by this database—beyond its sheer quantity—stems from the accurate solvent removal protocol (SAMOSA) which generates high-quality, activated crystal structures without introducing structural errors seen in prior entries. The true experimental feasibility of the activation assumed by the solvent removal routine will vary by material according to its physical properties, and no generalized tools currently exist to predict the likelihood or stability of a given activated structure. In particular, the removal of bound solvent in the full activation scheme producing an open metal site (OMS) is somewhat contested as it may not be energetically accessible in strongly binding metal-solvent pairs. For such reasons, QMOF databases limit themselves to the removal of unbound solvent (i.e. partial activation).28 Additionally, the presence of OMSs may not be compatible in the desired application conditions if environmental contaminants are apt to irreversibly bind with the metal. In recognition of this relatively common reluctance to treat materials containing OMSs, their presence has been catalogued in MOSAEC-DB and duly provided in the ESI.† An overview of the representation of OMSs and various solvent binding relationships is provided in Fig. 5. Naturally, the full activation procedure yields considerably higher incidence of structures possessing at least one OMS than partial activation (Fig. 5a), amounting to a more than 2-fold increase between the two degrees of activation. Analysis of the metal identity at these sites revealed the copper, zinc, and silver were the three most frequently existing as OMS, corresponding to 13.56%, 7.52%, and 5.77% of all database structures, respectively. The top OMS metal species identified in Fig. 5b roughly follow the trends in overall representation of these metals within the database displayed in Fig. 2, thereby not suggesting any strong correlation between metal identity and propensity to form OMSs within this dataset. This information does, however, provide a substantial pool of crystal structures ready for use in screening studies seeking to utilize specific metal OMSs as catalytic sites. Conversely, one may wish to exclude these OMS-containing structures when performing molecular simulations that do not accurately model the metal sites' interactions within the system in question; for instance, applying Grand-Canonical Monte Carlo (GCMC) simulations of polar adsorbates like H2O which will probably interact more strongly with OMSs than classical force fields would generally predict. Nevertheless, this once again highlights how this databases' inclusive approach aims to provide users with the requisite information to prepare subsets that best meet their particular demands. | ||
| Fig. 5 Survey of the open metal site (OMS) and solvent removal statistics observed in the processed MOSAEC-DB MOFs, including: (a) frequency of OMS across the activation state and framework charge subcategories, (b) most common OMS species by metal identity, and (c) most common free solvent, bound solvent, and counterions removed during solvent processing. | ||
Further, investigation into the solvent removal statistics provides more insight into OMSs and the activation of the enclosed crystal structures. Concisely, the SAMOSA protocol transformed the activation state in 63.82% of the 124.4k MOSAEC-DB structures while the remainder of the structures were already sufficiently activated prior to processing. The most commonly removed free solvent, bound solvent, and non-coordinating counterion species encountered during database processing are summarized in Fig. 5c. Various representations of water (i.e. H2O and O2− with unresolved protons) dominate both the free and bound solvent lists, followed by other usual MOF synthesis solvents—for example, dimethyl formamide, methanol, and acetonitrile—at much more moderate representations. Nothing particularly surprisingly arises from these quantities with respect to previous intuition gained during the development of the solvent removal protocol; however, the reported solvent identity may be employed to further refine the dataset according to OMS occurrence pending knowledge of the solvent's binding energy or other factors contributing to the porous materials' feasibility of activation. For example, as data-mining efforts and predictive ML models of thermal and activation stability56 continue to develop, insights into the important features influencing their stability predictions could be utilized to inform the computational processing and solvent removal workflow.
Diversity analysis
A detailed analysis of the structural and chemical diversity was initiated to estimate how well this novel database covers the MOF design space in relation to existing databases. The selected approach builds upon prior works49 wherein dimensionality reduction techniques were applied to high-dimensional vectors containing chemical and geometric descriptors in order to yield an interpretable, lower dimensional (i.e. 2D or 3D) visualization of entire databases. Akin to these past efforts, RAC descriptors were selected to characterize each structures' chemical environment alongside the t-SNE algorithm to produce the mappings of the combined MOF design space. Fig. 6 demonstrates the reduced, 2D descriptor space of the MOSAEC-DB contrasted with the ARC-MOF, CoRE 2019, and QMOF databases in consideration of four distinct RAC descriptor categories. Broadly speaking, the distributions representing MOSAEC-DB crystal structures span the entirety of the aggregate chemical design space for all four descriptors. ARC-MOF and QMOF possess several areas of coverage in the metal (Fig. 6a) and SBU (Fig. 6d) chemistry plots that are distinct to those in MOSAEC-DB, but the admittance of hMOF structures in said databases likely accounts for these discrepancies. Inspection of Fig. S8,† which combines the hypothetical crystal structures into a separate entity, confirms this assertion as the bulk of the chemical descriptor domains lacking MOSAEC-DB representation correspond to hMOFs. Relative to the other largely experimental database (CoRE-2019), the database outlined in this work almost fully envelops its footprint in the aggregated descriptor space illustrated in Fig. 6. The outlying crystal structures found in only CoRE 2019 may be related to this database's high incidence of structural errors which were herein omitted due to the rigorous structural validation protocol discussed in this work. This suggestion is reasoned given similar findings in past studies of structural diversity and errors in experimental MOFs36 which determined that much of the chemical space not covered by a corrected SBU database corresponded to erroneous crystal structures, such as those with missing atoms, crystallographic disorder or imbalanced framework charges. Moreover, an analysis of reduced space of geometric descriptors (i.e. density, void fraction, surface areas, PLD, LCD, etc.) was undertaken to understand the extent that each database encompasses the pore geometry space. The outcomes of this analysis are summarized in Fig. S9,† clearly demonstrating that ARC-MOF provides the most significant and diverse collection of geometric properties. This finding is consistent with the geometric property probability distributions discussed earlier in the context of which the principally experimental databases—namely MOSAEC-DB, CoRE-2019, and QMOF—showed lesser ranges of common descriptors of pore geometry. In the subclass of experimental databases, we again noticed that the enclosed database possessed the next highest diversity in the t-SNE-reduced geometric space after ARC-MOF. Overall, this database encompasses a significant degree of the diverse MOF chemistry and pore geometry reported in other state-of-the-art computation-ready databases of both hypothetical and experimental MOF. MOSAEC-DB serves as an excellent resource for those wishing to sample a comprehensive array of experimental MOF chemistries and pore structures in future materials discovery efforts. | ||
| Fig. 6 Comparison of the two-dimensional projections of revised autocorrelation (RACs) descriptor space computed within the MOSAEC, ARC-MOF, CoRE 2019, and QMOF databases. Probability density distributions across each of the reduced t-SNE axes are depicted to provide additional contrast between the datasets. The (a) metal, (b) function group, (c) ligand, and (d) SBU RAC descriptors subcategories are distinctly characterized to display the relative coverage of each database in various domains of chemical space. Dimensionality reduction is performed using the t-SNE algorithm on the combined descriptor space of all relevant structures. | ||
Available descriptor data and subsets
As in our previous database curation efforts, a primary objective of MOSAEC-DB is to provide a standard dataset for atomistic simulations and ML studies. For this reason, a number of common global features used to describe MOF crystal structures were computed for as many structures as possible to facilitate the ML featurization process. This list of precomputed descriptors includes geometric, RAC, AP-RDF, and atom-specific persistent homology features. Further, inspired by the diversity analysis, several subsets of the database were prepared by selecting MOFs from the pool of unique crystal structures according to the diversity of their descriptor vectors. The goal of these datasets is to improve the efficiency of future screenings and limit the opportunity for data leakage in ML model training and evaluation. Finally, partial atomic charges were calculated for a substantial portion (>70%) of the neutral MOSAEC-DB structures to accelerate their application in atomistic simulations such as GCMC and MD. Both DFT-derived REPEAT57 charges and charges predicted by the recently published MEPO-ML58 graph attention network model are provided in distinct datasets. A summary of the contents of the provided neutral subsets and the availability of partial atomic charges within each dataset is outlined in Table 3.| MOSAEC-DB subset | Neutral structure count | Charged structure count | Partial atomic charges (structure count) | Sampled descriptor | |
|---|---|---|---|---|---|
| REPEAT | MEPO-ML | ||||
| Uniq-neutral-porous-vf | 11014 |
— | 7614 | 11014 |
Void fraction > 0.10 |
| Uniq-neutral-porous-pld | 30105 |
— | 21123 |
30105 |
PLD > 2.4 Å |
| Diverse-neutral-func | 20000 |
— | 14726 |
20000 |
Functional group RAC |
| Diverse-neutral-ligand | 20000 |
— | 14510 |
20000 |
Ligand RAC |
| Diverse-neutral-linker | 20000 |
— | 14977 |
20000 |
Linker-connecting atom RAC |
| Diverse-neutral-metal | 20000 |
— | 14228 |
20000 |
Metal RAC |
| Diverse-neutral-sbu | 20000 |
— | 13766 |
20000 |
SBU RAC |
| Diverse-neutral-geom | 20000 |
— | 13058 |
20000 |
Geometric (PLD, LCD, etc.) |
| Diverse-neutral-phom | 20000 |
— | 13462 |
20000 |
Atom-specific persistent homology |
| Overall | 106549 |
17920 |
78161 |
106549 |
— |
Conclusions
In summary, we described the construction and validation of the largest accessible database of computation-ready experimental MOF crystal structures, totaling over 124k distinct entries including the first instance of charge-labelled frameworks. Insights gained through prior manual examinations of crystal structures contained in related databases were applied to establish a novel holistic approach mindful of common MOF structural errors at all stages of structure processing, from solvent removal to error analysis and filtering. Further, structure comparisons of MOSAEC-DB to its predecessors determined that the incorporation of these chemistry-minded protocols significantly reduced occurrences of systematic errors introduced in prominent MOF databases. Appropriate chemical and geometric properties—including chemical composition and moieties, surface area and other pore structure features, topologies, and so forth—are made available to facilitate applications in HT screening and ML applications. Related property distributions were computed for the competing, state-of-the-art databases (e.g., CoRE 2019, QMOF, CSD MOF subset, etc.) to assess key differences brought about by disparate choices in database construction. Contributions towards the diversification of experimental MOF databases were gauged through comparison of chemical and geometric descriptor spaces using t-SNE dimensionality reduction. This database encompasses a broader scope of the total MOF chemical space relative to the earlier works processing experimental MOFs along with the elimination of several clusters corresponding to erroneous structural motifs discussed here and in our previous reports. Deviations in geometric properties followed expectations associated with degree of activation employed during solvent removal, namely sweeping increases in nearly all properties upon full activation. It is important to note that the reported fully activated structure may not be feasible or stable under real experimental conditions, thus caution must be exercised regarding the materials' activation uncertainty when simulating properties possessing high correlation with these geometric properties, for example gas adsorption. Nonetheless, MOSAEC-DB attained analogous properties and structural fidelity to the strictest database preparation methods such as QMOF, while maintaining a great quantity of entries due to its more inclusive approach to candidate structure searching and processing.We forecast that this rich source of chemically diverse and predominantly error-free experimental MOF structures will afford researchers with ample opportunities to develop novel ML models and HT screenings with improved confidence in the constituent structures' viability. Furthermore, integration of MOSAEC-DB into such previously published works may provide a path towards more reliable model performance and the identification of additional top-performing materials from candidates which were omitted by the narrower structural definitions utilized in past databases. While MOSAEC-DB captures the current state of experimental MOF crystallography deposited in the CSD, this database will be continually updated as the supply of experimentally characterized MOFs grows and as increasingly sophisticated crystal structure error analysis and repair tools advance towards reclaiming the discarded erroneous structures. Furthermore, this work establishes several diverse subsets of the MOF crystal structure space sampled according to their metal and linker chemistries, as well as geometric and pore chemistry, which will serve as efficient standard datasets that reduce the risk of duplicated and closely related structures in any subsequent computations. Generally, computational MOF researchers would benefit from the adoption of such standard databases and data handling practices to enhance the reliability of any chemical understanding (e.g., structure–property relationships) inferred from their HT screenings and ML models, and to improve their ability to directly benchmark competing methods' performance. We believe that the workflow shown in this study, in conjunction with our previous accounts concerning structural error detection and solvent removal, will greatly contribute to this standardization effort and form a strong basis for numerous future materials discovery applications.
Data availability
The ESI† is made available free of charge at DOI: https://doi.org/10.1039/d4sc07438f. The MOSAEC database (CIF files) and its computed properties (CSV files) are freely available to download on Zenodo (https://doi.org/10.5281/zenodo.14025238). A summary of the relevant codes utilized at various stages of database creation and analysis can be found on the uOttawa Woo Lab GitHub (https://github.com/uowoolab/MOSAEC-DB). Additional information regarding the database construction process (i.e. candidate search, solvent removal, etc.) and structural properties (i.e. chemical composition, geometric distribution, diversity analysis, etc.) of the enclosed MOSAEC-DB crystal structures are provided in the attached documents (PDF).Author contributions
M. G. and T. K. W. conceptualized the project. A. K., A. W., and M. G. developed the core methodology and software. M. G., J. L., R. A. M., and J. B. contributed to data curation. M. G. performed the experimental investigation, visualization, and manuscript writing – original draft. All authors participated in writing – reviewing & editing. T. K. W. provided project supervision and funding acquisition.Conflicts of interest
Separate patent applications have been filed for the method of assigning metal oxidation states and method of processing chemical compounds for computation employed in this work. There are no other conflicts of interest to declare.Acknowledgements
Financial support from the Natural Sciences and Engineering Research Council of Canada (DISCOVERY Grant), the University of Ottawa, MITACS (ACCELERATE), and TotalEnergies is greatly appreciated, as well as the computing resources provided by the Canada Foundation for Innovation, TotalEnergies and the Digital Research Alliance of Canada.References
- R. Pollice, G. dos Passos Gomes, M. Aldeghi, R. J. Hickman, M. Krenn, C. Lavigne, M. Lindner-D'Addario, A. Nigam, C. T. Ser, Z. Yao and A. Aspuru-Guzik, Data-Driven Strategies for Accelerated Materials Design, Acc. Chem. Res., 2021, 54(4), 849–860, DOI:10.1021/acs.accounts.0c00785.
- G. Ercakir, G. O. Aksu, C. Altintas and S. Keskin, Hierarchical Computational Screening of Quantum Metal–Organic Framework Database to Identify Metal–Organic Frameworks for Volatile Organic-Compound Capture from Air, ACS Eng. Au, 2023, 3(6), 488–497, DOI:10.1021/acsengineeringau.3c00039.
- S. Lee, B. Kim, H. Cho, H. Lee, S. Y. Lee, E. S. Cho and J. Kim, Computational Screening of Trillions of Metal–Organic Frameworks for High-Performance Methane Storage, ACS Appl. Mater. Interfaces, 2021, 13(20), 23647–23654, DOI:10.1021/acsami.1c02471.
- P. G. Boyd, A. Chidambaram, E. García-Díez, C. P. Ireland, T. D. Daff, R. Bounds, A. Gładysiak, P. Schouwink, S. M. Moosavi, M. M. Maroto-Valer, J. A. Reimer, J. A. R. Navarro, T. K. Woo, S. Garcia, K. C. Stylianou and B. Smit, Data-Driven Design of Metal–Organic Frameworks for Wet Flue Gas CO2 Capture, Nature, 2019, 576(7786), 253–256, DOI:10.1038/s41586-019-1798-7.
- T. Watanabe and D. S. Sholl, Accelerating Applications of Metal–Organic Frameworks for Gas Adsorption and Separation by Computational Screening of Materials, Langmuir, 2012, 28(40), 14114–14128, DOI:10.1021/la301915s.
- S. Pablo-García, S. Morandi, R. A. Vargas-Hernández, K. Jorner, Ž. Ivković, N. López and A. Aspuru-Guzik, Fast Evaluation of the Adsorption Energy of Organic Molecules on Metals via Graph Neural Networks, Nat. Comput. Sci., 2023, 3(5), 433–442, DOI:10.1038/s43588-023-00437-y.
- A. Jose, E. Devijver, N. Jakse and R. Poloni, Informative Training Data for Efficient Property Prediction in Metal–Organic Frameworks by Active Learning, J. Am. Chem. Soc., 2024, 146(9), 6134–6144, DOI:10.1021/jacs.3c13687.
- R. Goeminne, L. Vanduyfhuys, V. Van Speybroeck and T. Verstraelen, DFT-Quality Adsorption Simulations in Metal–Organic Frameworks Enabled by Machine Learning Potentials, J. Chem. Theory Comput., 2023, 19(18), 6313–6325, DOI:10.1021/acs.jctc.3c00495.
- K. Choudhary, T. Yildirim, D. W. Siderius, A. G. Kusne, A. McDannald and D. L. Ortiz-Montalvo, Graph Neural Network Predictions of Metal Organic Framework CO2 Adsorption Properties, Comput. Mater. Sci., 2022, 210, 111388, DOI:10.1016/J.COMMATSCI.2022.111388.
- A. Nandy, S. Yue, C. Oh, C. Duan, G. G. Terrones, Y. G. Chung and H. J. Kulik, A Database of Ultrastable MOFs Reassembled from Stable Fragments with Machine Learning Models, Matter, 2023, 6(5), 1585–1603, DOI:10.1016/j.matt.2023.03.009.
- Z. Yao, B. Sánchez-Lengeling, N. S. Bobbitt, B. J. Bucior, S. G. H. Kumar, S. P. Collins, T. Burns, T. K. Woo, O. K. Farha, R. Q. Snurr, A. Aspuru-Guzik, B. Sanchez-Lengeling, N. S. Bobbitt, B. J. Bucior, S. G. H. Kumar, S. P. Collins, T. Burns, T. K. Woo, O. K. Farha and R. Q. Snurr, others. Inverse Design of Nanoporous Crystalline Reticular Materials with Deep Generative Models, Nat. Mach. Intell., 2021, 3(1), 76–86, DOI:10.1038/s42256-020-00271-1.
- H. Park, S. Majumdar, X. Zhang, J. Kim and B. Smit, Inverse Design of Metal–Organic Frameworks for Direct Air Capture of CO2 via Deep Reinforcement Learning, Digital Discovery, 2024, 3(4), 728–741, 10.1039/D4DD00010B.
- J. Park, Y. Lee and J. Kim, Multi-Modal Conditioning for Metal-Organic Frameworks Generation Using 3D Modeling Techniques, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-w8fps.
- X. Fu, T. Xie, A. S. Rosen, T. Jaakkola, and J. Smith, MOFDiff: Coarse-Grained Diffusion for Metal-Organic Framework Design, arXiv, 2023, preprint, arXiv:2310.10732, DOI:10.48550/arXiv.2310.10732.
- F. Cipcigan, J. Booth, R. N. Barros Ferreira, C. Ribeiro dos Santos and M. Steiner, Discovery of Novel Reticular Materials for Carbon Dioxide Capture Using GFlowNets, Digital Discovery, 2024, 3(3), 449–455, 10.1039/D4DD00020J.
- J. J. Irwin, K. G. Tang, J. Young, C. Dandarchuluun, B. R. Wong, M. Khurelbaatar, Y. S. Moroz, J. Mayfield and R. A. Sayle, ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery, J. Chem. Inf. Model., 2020, 60(12), 6065–6073, DOI:10.1021/acs.jcim.0c00675.
- D. Mendez, A. Gaulton, A. P. Bento, J. Chambers, M. De Veij, E. Félix, M. P. Magariños, J. F. Mosquera, P. Mutowo, M. Nowotka, M. Gordillo-Marañón, F. Hunter, L. Junco, G. Mugumbate, M. Rodriguez-Lopez, F. Atkinson, N. Bosc, C. J. Radoux, A. Segura-Cabrera, A. Hersey and A. R. Leach, ChEMBL: Towards Direct Deposition of Bioassay Data, Nucleic Acids Res., 2019, 47(D1), D930–D940, DOI:10.1093/nar/gky1075.
- H. M. Berman, The Protein Data Bank, Nucleic Acids Res., 2000, 28(1), 235–242, DOI:10.1093/nar/28.1.235.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, PubChem 2023 Update, Nucleic Acids Res., 2023, 51(D1), D1373–D1380, DOI:10.1093/nar/gkac956.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72(2), 171–179, DOI:10.1107/S2052520616003954.
- R. Taylor and P. A. A. Wood, Million Crystal Structures: The Whole Is Greater than the Sum of Its Parts, Chem. Rev., 2019, 119(16), 9427–9477, DOI:10.1021/acs.chemrev.9b00155.
- Y. G. Chung, J. Camp, M. Haranczyk, B. J. Sikora, W. Bury, V. Krungleviciute, T. Yildirim, O. K. Farha, D. S. Sholl and R. Q. Snurr, Computation-Ready, Experimental Metal–Organic Frameworks: A Tool To Enable High-Throughput Screening of Nanoporous Crystals, Chem. Mater., 2014, 26(21), 6185–6192, DOI:10.1021/cm502594j.
- Y. G. Chung, E. Haldoupis, B. J. Bucior, M. Haranczyk, S. Lee, H. Zhang, K. D. Vogiatzis, M. Milisavljevic, S. Ling, J. S. Camp, B. Slater, J. I. Siepmann, D. S. Sholl and R. Q. Snurr, Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019, J. Chem. Eng. Data, 2019, 64(12), 5985–5998, DOI:10.1021/acs.jced.9b00835.
- S. Kancharlapalli, A. Gopalan, M. Haranczyk and R. Q. Snurr, Fast and Accurate Machine Learning Strategy for Calculating Partial Atomic Charges in Metal-Organic Frameworks, J. Chem. Theory Comput., 2021, 17(5), 3052–3064, DOI:10.1021/acs.jctc.0c01229.
- L. Li, Z. Shi, H. Liang, J. Liu and Z. Qiao, Machine Learning-Assisted Computational Screening of Metal-Organic Frameworks for Atmospheric Water Harvesting, Nanomaterials, 2022, 12(1), 1–14, DOI:10.3390/nano12010159.
- S. Kancharlapalli and R. Q. Snurr, High-Throughput Screening of the CoRE-MOF-2019 Database for CO 2 Capture from Wet Flue Gas: A Multi-Scale Modeling Strategy, ACS Appl. Mater. Interfaces, 2023, 15(23), 28084–28092, DOI:10.1021/acsami.3c04079.
- S.-Y. Kim, S.-I. Kim and Y.-S. Bae, Machine-Learning-Based Prediction of Methane Adsorption Isotherms at Varied Temperatures for Experimental Adsorbents, J. Phys. Chem. C, 2020, 124(36), 19538–19547, DOI:10.1021/acs.jpcc.0c01757.
- A. S. Rosen, S. M. Iyer, D. Ray, Z. Yao, A. Aspuru-Guzik, L. Gagliardi, J. M. Notestein and R. Q. Snurr, Machine Learning the Quantum-Chemical Properties of Metal–Organic Frameworks for Accelerated Materials Discovery, Matter, 2021, 4(5), 1578–1597, DOI:10.1016/j.matt.2021.02.015.
- A. S. Rosen, V. Fung, P. Huck, C. T. O'Donnell, M. K. Horton, D. G. Truhlar, K. A. Persson, J. M. Notestein and R. Q. Snurr, High-Throughput Predictions of Metal–Organic Framework Electronic Properties: Theoretical Challenges, Graph Neural Networks, and Data Exploration, npj Comput. Mater., 2022, 8(1), 112, DOI:10.1038/s41524-022-00796-6.
- H. Xiao, R. Li, X. Shi, Y. Chen, L. Zhu, X. Chen and L. Wang, An Invertible, Invariant Crystal Representation for Inverse Design of Solid-State Materials Using Generative Deep Learning, Nat. Commun., 2023, 14(1), 7027, DOI:10.1038/s41467-023-42870-7.
- H. Park, Y. Kang and J. Kim, Enhancing Structure–Property Relationships in Porous Materials through Transfer Learning and Cross-Material Few-Shot Learning, ACS Appl. Mater. Interfaces, 2023, 15(48), 56375–56385, DOI:10.1021/acsami.3c10323.
- V. Fung, J. Zhang, E. Juarez and B. G. Sumpter, Benchmarking Graph Neural Networks for Materials Chemistry, npj Comput. Mater., 2021, 7(1), 84, DOI:10.1038/s41524-021-00554-0.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Development of a Cambridge Structural Database Subset: A Collection of Metal–Organic Frameworks for Past, Present, and Future, Chem. Mater., 2017, 29(7), 2618–2625, DOI:10.1021/acs.chemmater.7b00441.
- A. Li, R. B. Perez, S. Wiggin, S. C. Ward, P. A. Wood and D. Fairen-Jimenez, The Launch of a Freely Accessible MOF CIF Collection from the CSD, Matter, 2021, 4(4), 1105–1106, DOI:10.1016/j.matt.2021.03.006.
- A. White, M. Gibaldi, J. Burner, and T. K. Woo, Alarming Structural Error Rates in MOF Databases Used in Computational Screening Identified via a Novel Metal Oxidation State-Based Method, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-ftsv3.
- M. Gibaldi, O. Kwon, A. White, J. Burner and T. K. Woo, The HEALED SBU Library of Chemically Realistic Building Blocks for Construction of Hypothetical Metal–Organic Frameworks, ACS Appl. Mater. Interfaces, 2022, 14(38), 43372–43386, DOI:10.1021/acsami.2c13100.
- M. Gibaldi, A. Kapeliukha, A. White, and T. Woo, Incorporation of Ligand Charge and Metal Oxidation State Considerations into the Computational Solvent Removal and Activation of Experimental Crystal Structures Preceding Molecular Simulation, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-7vq41.
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Development of a Cambridge Structural Database Subset: A Collection of Metal-Organic Frameworks for Past, Present, and Future, Chem. Mater., 2017, 29(7), 2618–2625, DOI:10.1021/acs.chemmater.7b00441.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, New Software for Searching the Cambridge Structural Database and Visualizing Crystal Structures, Acta Crystallogr., Sect. B:Struct. Sci., 2002, 58(3), 389–397, DOI:10.1107/S0108768102003324.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Python Materials Genomics (Pymatgen): A Robust, Open-Source Python Library for Materials Analysis, Comput. Mater. Sci., 2013, 68, 314–319, DOI:10.1016/j.commatsci.2012.10.028.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Covalent Radii Revisited, J. Chem. Soc., Dalton Trans., 2008,(21), 2832–2838, 10.1039/b801115j.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Algorithms and Tools for High-Throughput Geometry-Based Analysis of Crystalline Porous Materials, Microporous Mesoporous Mater., 2012, 149(1), 134–141, DOI:10.1016/j.micromeso.2011.08.020.
- P. Z. Moghadam, A. Li, X. W. Liu, R. Bueno-Perez, S. D. Wang, S. B. Wiggin, P. A. Wood and D. Fairen-Jimenez, Targeted Classification of Metal-Organic Frameworks in the Cambridge Structural Database (CSD), Chem. Sci., 2020, 11(32), 8373–8387, 10.1039/d0sc01297a.
- O. Isayev, C. Oses, C. Toher, E. Gossett, S. Curtarolo and A. Tropsha, Universal Fragment Descriptors for Predicting Properties of Inorganic Crystals, Nat. Commun., 2017, 8, 1–12, DOI:10.1038/ncomms15679.
- L. Zoubritzky and F.-X. Coudert, CrystalNets.Jl: Identification of Crystal Topologies, SciPost Chem., 2022, 1(2), 005, DOI:10.21468/SCIPOSTCHEM.1.2.005.
- J. Burner, J. Luo, A. White, A. Mirmiran, O. Kwon, P. G. Boyd, S. Maley, M. Gibaldi, S. Simrod, V. Ogden and T. K. Woo, ARC–MOF: A Diverse Database of Metal-Organic Frameworks with DFT-Derived Partial Atomic Charges and Descriptors for Machine Learning, Chem. Mater., 2023, 35(3), 900–916, DOI:10.1021/acs.chemmater.2c02485.
- J. P. Janet and H. J. Kulik, Resolving Transition Metal Chemical Space: Feature Selection for Machine Learning and Structure–Property Relationships, J. Phys. Chem. A, 2017, 121(46), 8939–8954, DOI:10.1021/acs.jpca.7b08750.
- E. I. Ioannidis, T. Z. H. Gani and H. J. Kulik, MolSimplify: A Toolkit for Automating Discovery in Inorganic Chemistry, J. Comput. Chem., 2016, 37(22), 2106–2117, DOI:10.1002/jcc.24437.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Understanding the Diversity of the Metal-Organic Framework Ecosystem, Nat. Commun., 2020, 11(1), 4068, DOI:10.1038/s41467-020-17755-8.
- L. Van Der Maaten and G. Hinton, Visualizing Data Using T-SNE, J. Mach. Learn. Res., 2008, 9, 2579–2625 Search PubMed.
- Y. Jiang, D. Chen, X. Chen, T. Li, G.-W. W. Wei and F. Pan, Topological Representations of Crystalline Compounds for the Machine-Learning Prediction of Materials Properties, npj Comput. Mater., 2021, 7(1), 1–8, DOI:10.1038/s41524-021-00493-w.
- K. M. Jablonka, A. S. Rosen, A. S. Krishnapriyan and B. Smit, An Ecosystem for Digital Reticular Chemistry, ACS Cent. Sci., 2023, 9(4), 563–581, DOI:10.1021/acscentsci.2c01177.
- D. Widdowson, M. M. Mosca, A. Pulido, A. I. Cooper and V. Kurlin, Average Minimum Distances of Periodic Point Sets – Foundational Invariants for Mapping Periodic Crystals, MATCH Commun. Math. Comput. Chem., 2022, 87(3), 529–559, DOI:10.46793/match.87-3.529W.
- D. E. Widdowson and V. A. Kurlin, Resolving the Data Ambiguity for Periodic Crystals, Adv. Neural Inf. Process. Syst., 2022, 35, 1–14 Search PubMed.
- J. Riebesell, H. Yang, R. Goodall, and S. G. Baird, Pymatviz: Visualization Toolkit for Materials Informatics, 2022, DOI:10.5281/zenodo.7486816.
- A. Nandy, C. Duan and H. J. Kulik, Using Machine Learning and Data Mining to Leverage Community Knowledge for the Engineering of Stable Metal–Organic Frameworks, J. Am. Chem. Soc., 2021, 143(42), 17535–17547, DOI:10.1021/jacs.1c07217.
- C. Campañá, B. Mussard and T. K. Woo, Electrostatic Potential Derived Atomic Charges for Periodic Systems Using a Modified Error Functional, J. Chem. Theory Comput., 2009, 5(10), 2866–2878, DOI:10.1021/ct9003405.
- J. Luo, O. B. Said, P. Xie, M. Gibaldi, J. Burner, C. Pereira and T. K. Woo, MEPO-ML: A Robust Graph Attention Network Model for Rapid Generation of Partial Atomic Charges in Metal-Organic Frameworks, npj Comput. Mater., 2024, 10(1), 224, DOI:10.1038/s41524-024-01413-4.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07438f |
| This journal is © The Royal Society of Chemistry 2025 |