
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphite mediated molecular editing via switch to meta-C–H alkylation of isoquinolines: emergence of a distinct photochemical [1,3] N to C rearrangement†
Soniya
Rani‡
 ab,
Anuj Kumar
Ray‡
c,
Devendra Kumar
Dewangan‡
ab,
Nita Aruna Ramchandra
Patil
a,
M
Aarthika
ab,
Ankan
Paul
*c and
Pradip
Maity
*ab
ab,
Anuj Kumar
Ray‡
c,
Devendra Kumar
Dewangan‡
ab,
Nita Aruna Ramchandra
Patil
a,
M
Aarthika
ab,
Ankan
Paul
*c and
Pradip
Maity
*ab
aOrganic Chemistry Division, CSIR-National Chemical Laboratory (CSIR-NCL), Pune 411 008, India. E-mail: p.maity@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
cSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, India. E-mail: rcap@iacs.res.in
First published on 12th December 2024
Abstract
The isoquinoline core is present in one of the largest subsets of bioactive natural products. The multifunctional isoquinoline core exerts diverse bioactivity, resulting in the development of numerous isoquinoline-based drugs and molecules that are currently under clinical trials. We developed a new approach for phosphite-mediated [1,2] alkyl migration for an overall ortho-C–H alkylation via N-alkylation of isoquinoline. Tuning the phosphite-mediated protocol to switch the site selectivity would expedite direct and diverse multi-C–H bond functionalization. We report a new approach starting with a simple N-alkylation of isoquinoline with sterically and electronically diverse alkyl bromides for their phosphite-mediated photochemical [1,3] N to C rearrangement followed by a rearomatization sequence that leads to meta-C–H (C4) alkylation. Combined experimental and computational studies unveiled the emergence of an unprecedented C–N bond cleavage pathway from the singlet excited state of the enamine-type intermediate. Our radical bond-cleavage pathway favors substituted alkyl group migration that complements the recently successful meta-alkylation methods with smaller and more reactive electrophiles. This switch in site selectivity via tuning the phosphite-mediated protocol resulted in sequential C–H difunctionalization of isoquinoline including regiodivergent ortho, meta-dialkylations of isoquinolines.
Introduction
Isoquinoline is an important structural core in a diverse array of natural products (over 2500) and drugs currently in use or under clinical trials.1 The synthesis of multi-functionalized isoquinoline gained significant focus due to their prevalence in pharmaceuticals,2a agrochemicals,2b chiral ligands,2c and materials.2d Direct molecular editing of isoquinoline C–H bonds through consecutive selective C–H functionalization can provide access to diverse analogs valuable in pharmaceuticals.3 Substitution at each carbon of the isoquinoline core is prevalent, including meta-functionalized bioactive compounds with substituted alkyl groups (Fig. 1A).4 Among different C–H bonds, ortho- and para-alkylations were achieved successfully with a broad range of electronically and sterically diverse substituents.5 However, meta-C–H alkylation remained a formidable challenge due to its inertness towards both electrophilic and nucleophilic reagents. As a result, most of the isoquinoline natural products containing meta-alkyl substitution were synthesized from their alkyl substituted acyclic precursors.4,6 | ||
| Fig. 1 (A) Natural products and biologically active compounds with meta-alkylated isoquinoline core. (B–D) Reported strategies for meta-C–H alkylations of isoquinolines. (E) N-Functionalization of isoquinoline and phosphite-mediated regioselective N to C migration. | ||
In a more direct approach, Minter and Re in 1988 showed a one-pot multi-step operation for effective meta-C–H alkylation of isoquinolines with aldehydes (Fig. 1B).7a This strategy is based on reductive hydroboration, followed by electrophilic functionalization of the resulting enamine, and finally, dehydration–tautomerization for the overall meta-alkylation. The reaction was shown to work only with aldehydes, limiting it to the installation of primary alkyl groups. In 2017, Du, Zhang, and co-workers reported a similar strategy via initial hydrogenation rather than hydroboration.7b Catalytic hydrogenation by formic acid resulted in less waste-generation, but it remained limited to primary alkylation.
In 2022, the Wang group developed an improved Lewis acid catalyzed reductive hydroboration to enamine and its electrophilic functionalization at meta-C–H. The catalyst activation led to successful reaction with activated ketones and imines, along with aldehydes. Additionally, a second equivalent of electrophiles oxidized the C3 functionalized enamine intermediate for an overall meta-C–H amino and hydroxyl alkylation (Fig. 1C).7c In 2023, the Wang group followed up with an asymmetric meta-allylation with reactive chiral iridium and palladium π-allyl electrophiles.7d,e In 2022, the Kuninobu group utilized a similar approach with silane as the reductant for meta-selective trifluoromethylation.7f In these redox approaches, both pre-reduction and post-oxidation steps require extra catalysts and stoichiometric reagents, leading to waste generation. Importantly, activated electrophiles were required for successful meta-functionalization, and no reaction was reported with simple alkyl electrophiles such as alkyl halides.
In 2022, Studer and co-workers reported a meta-selective radical fluoroalkylation of azaarenes via a redox neutral reaction sequence.7g,h In 2023, the Donohoe group developed an excess benzoic acid-mediated one-pot meta-alkylation of isoquinolines with unsubstituted vinyl ketones, leading to primary alkyl group installation.7i These elegant methods avoid the reduction and oxidation of azaarenes for their meta-C–H alkylation. However, they are limited to strong alkylating electrophiles only, owing to a similarly reactive enamine intermediate developed via reduction.7a–f The requirement of stoichiometric reagents to form dearomatized adducts and their removal after alkylation led to waste generation (Fig. 1D). A direct and waste-free meta-alkylation with unactivated and sterically demanding alkyl halides remains elusive.
One of our research programs focuses on the facile synthesis of N-functionalized pyridinium salts for their phosphite-catalyzed migration to the ring carbons (Fig. 1E).8 The allyl and alkyl groups on pyridine nitrogen successfully migrated to the C2 position for overall ortho-allylation and alkylation.8a,b A switch in site selectivity to regiodivergent C–H alkylation from the same N-alkylpyridinium salts under tunable reaction conditions would be highly advantageous for diversity-oriented synthesis. Regiodivergent switch between ortho- and para-positions has been achieved due to their similar electrophilic reactivity pattern. The switch is usually accomplished by taking advantage of the nitrogen coordination to either direct the nucleophile to the ortho-position, or sterically block the ortho-position with bulky nitrogen-coordinating additives.9 However, switching the same functional group between ortho-/para- and meta-C–H bonds is very challenging due to their differential reactivity.
Transition metal catalyzed ortho-/para- and meta-C–H activation followed by borylation, arylation, and alkenylations of azaarenes was established recently. The switch in selectivity was achieved via different catalysts with precisely designed ligands.3,10 A more attractive electrochemical carboxylation was recently reported by Yu, Lin, and co-workers, where the switch from meta- to para-carboxylation was achieved simply by using either divided or undivided cells.11 In 2023, the Studer group reported an elegant switch to para-C–H alkylation from their previous meta-C–H fluoroalkylation via a simple change in the pH of the reaction. However, para-alkylation needs a nucleophilic alkyl radical coupling partner, while meta-fluoroalkylation works with electronically opposite electrophilic radicals.7h,12 To the best of our knowledge, a switch between ortho- and meta-positions by electronically similar alkyl groups has not been reported.
Herein, we report that direct photochemical irradiation under a 365 nm LED to the phosphite adduct (2) of N-alkyl isoquinolinium salt (1) resulted in meta-C–H alkylation. The unique feature of our approach is the dual role of the functional group on nitrogen to act as a nitrogen activator to facilitate phosphite adduct (2) formation, followed by its migration from nitrogen to ring carbon (Fig. 1E). The migration from nitrogen deactivates it, which triggers the in situ phosphite elimination for an overall C–H functionalization. The recovery and reuse of phosphite after work-up make the overall transformation net-zero in carbon waste. The one-pot method starts with either bench-stable N-alkyl isoquinolinium salt 1 or directly from isoquinoline and alkyl halides. Experimental and computational evidence was presented for a plausible mechanism for this unprecedented meta-C–H alkylation with primary, secondary, and tertiary alkyl groups. Furthermore, consecutive functionalization on isoquinoline nitrogen and its migration to the ring carbons under the same phosphite-mediated tunable reaction conditions allow for selective and regioisomeric ortho, meta-di-C–H functionalization with chemically equivalent alkyl halides. The utility of phosphite-mediated methods via N-functionalization was further demonstrated for the derivatization of meta-alkylated products.
Results and discussion
N-Alkylation of isoquinoline
First, we screened conditions for a direct alkylation on isoquinoline nitrogen. A simple solvent-free mixing of isoquinoline with primary, secondary, and tertiary alkyl halides at ambient to moderate temperature led to the corresponding N-alkyl isoquinolinium halide salt formation with good to excellent yields.13 Some of the halide salts are hygroscopic in nature and form color impurities when stored for months. Literature-modified ion exchange methods were employed to the corresponding tetrafluoroborate salts that are not hygroscopic and are bench stable for years without degradation. A direct tetrafluoroborate salt formation was also achieved via stirring isoquinoline and corresponding alkyl halide in acetone with six equivalents of sodium tetrafluoroborate as an additive.14Development of phosphite mediated photochemical meta-alkylation
We envisioned that the phosphite-adduct 2 of N-isoquinolinium salt could be perceived as an N-alkylated aryl-conjugated enamine or amine-conjugated styrene derivative. The [1,3] O-to-C rearrangement of an O-alkylated aryl-conjugated enol is well established via both ionic and radical pathways.15 The corresponding aza-version with N-to-C migration is rare, with only one thermal migration known to occur from l,4-dialkyl-l,4-dihydropyrazines via stereoinversion.16 We plan to explore a similar N-to-C [1,3] alkyl shift, owing to our previous success with other phosphite-mediated aza-migrations around azaarenes.8a,b Thermally heating the phosphite adduct 2 did not result in any migration product.Next, we explored the possibility of photochemically exciting the amine-conjugated styrene part of the adduct 2 for a possible photochemical aza-[1,3] migration. Our photophysical studies of adduct 2 showed that it could be excited with 365 nm light. A fluorescence emission study with excitations at different wavelengths established better absorption at 365 nm with the highest emission (see ESI†). Therefore, we explored a one-pot phosphite addition, followed by its direct excitation for aza-[1,3] shift and subsequent base-mediated phosphite elimination for the meta-alkylation of isoquinoline (Table 1). Delightfully, the one-pot reaction sequence with the stoichiometric phosphite additive in p-xylene with four equivalents of potassium carbonate as a base led to the meta-benzyl isoquinoline in 60% yield with complete regioselectivity (entry 1). Starting with the N-benzylation of isoquinoline and its one-pot migration also yielded the product with comparable efficiency (entry 2). Catalytic amounts of phosphite resulted in lower yield with no trace of unreacted starting material or intermediate (2a) in the crude reaction mass (entry 3). On the other hand, phosphite was regenerated quantitatively. Therefore, we chose to proceed with stoichiometric phosphite to complete 2a formation under darkness, followed by its photochemical meta-alkylation. Diethylphosphite can be separated from the product and other impurities via a simple aqueous work-up protocol (see ESI†). Reaction in other solvents also formed the product, but the p-xylene remains the best (entry 4, see ESI for details†). Changing base equivalents or other bases led to lower yields (entries 5–7). As anticipated from our photophysical studies, different light sources with higher and lower frequencies led to a drop in reaction efficiencies (entries 8–10). The light intensity is important, with low power light (18 W) leads to lower yield (entry 11). No meta-benzyl isoquinoline formation was observed without light, and a trace amount of product formation was observed without base (entries 12 and 13).
|
|
||
|---|---|---|
| Entrya | Deviation from standard conditionsa | Yieldb (%) |
| a 0.2 mmol scale. b Isolated yield. c One-pot N-alkylation and migration to the meta-position. d For the second step. n.d. – not detected. | ||
| 1 | None | 60 |
| 2c | One-pot with 1a-bromide | 58 |
| 3 | 50 mol% phosphite | 35 |
| 4 | Other solvents | 5–44 |
| 5 | 3 equiv. of K2CO3 | 54 |
| 6 | 5 equiv. of K2CO3 | 60 |
| 7 | Other bases | 10–50 |
| 8 | λ max = 320 nm | <5 |
| 9 | λ max = 380 nm | 40 |
| 10 | λ max = 395 nm | 29 |
| 11 | λ max = 365 nm, 18 W | 34 |
| 12 | No light | n.d. |
| 13d | No base | <5 |
Substrate scope
Under the optimized reaction conditions, we screened the substrate scope (Fig. 2), focusing on the migrating alkyl group first. A chloro substituent at ortho-, meta-, and para-to the phenyl ring (4b–d) worked better than the unsubstituted benzyl migrating group. These results indicate that the steric crowd is tolerant around the aryl ring. Other halides at the meta-position were equally efficient (4e–g). The compatibility of all halides at different positions of the aryl substituents is significant since a transition metal catalyzed meta-alkylation could be problematic for these substituents. Both electron-deficient groups such as cyano (4h) and ester (4i), and electron donating methyl (4j) and methoxy (4k) were well tolerated at the meta-position. The electron-deficient cyano substitution in the para-position (4l) resulted in good yields. However, the electron-donating methyl group led to a low 24% yield (4m). With this trend, we tested other non-phenyl aryl groups on migrating carbon. An electron neutral β-naphthyl (4n) and electron-deficient 3- and 4-pyridines gave products (4o,p) with good yields, while electron-rich furan (4q) and thiophene rings (4r) led to lower yields. After the success of a wide variety of substituted benzyl migration, we tested the suitability of non-benzyl groups. An N-allyl group on nitrogen (4s) was migrated successfully with good yields. The chemoselective reduction of the corresponding meta-allyl isoquinoline led to a meta-alkylated product (4t). A direct meta-alkylation via N-alkylation and migration of alkyl groups, such as methyl and ethyl, failed under our reaction protocol. An ester substitution on migrating alkyl carbon also migrated successfully (4u) in 58% yield. | ||
| Fig. 2 Substrate scope. All reactions were performed on 0.2 mmol scale. All yields are isolated. aReaction time in the light is 60 h. bReaction time in the light is 72 h. c% Conversion is 70. d1aj (0.2 mmol), diethylphosphite (0.24 mmol), Et3N (0.3 mmol) and 2.5 ml DCM, 24 h, −40 °C; K2CO3 (0.5 mmol), p-xylene (0.1 M), 30 W LED (λmax = 365 nm), 24 h, 15 °C. | ||
We next examined the scope of our method with substituted isoquinolines. Substitution at the C1 position is not reported for meta-C–H alkylation, presumably due to their reduced reactivity towards reduction and nucleophilic addition to form the enamine-type intermediate.7 Since the phosphite anion is a better nucleophile, we tested our protocol with the 1-methylisoquinoline substrate. Both benzyl and diphenylmethyl salts of 1-methylisoquinoline formed the phosphite adduct smoothly, and led to successful product formation under the photochemical reaction conditions (4v, 4w). Methyl substitution at the C3 position yielded the meta-benzylated product (4x) successfully under prolonged irradiation. On the fused benzene ring of the isoquinoline, we explored electron-rich substituents due to their prevalence in natural products.4 Methoxy or dimethoxy substitutions on the fused benzene ring of the isoquinoline successfully yielded the corresponding products with similar efficiencies (4y,z,aa). We also tested bromo-substitution on either ring of isoquinoline (C3 and C6), but the corresponding phosphite adduct remained unreactive under the optimized reaction conditions. Longer irradiation led to slow decomposition for both the substrates. We anticipated that a shorter singlet excited state lifetime via facile singlet to triplet intersystem crossing in chromophores conjugated with bromides could be the reason for their inertness. These results reinforce our mechanistic hypothesis that the [1,3] migration proceeds via a singlet excited state.17 It is noteworthy to mention that all aryl halides are tolerated when they are not conjugated with the chromophore (4b–g).
Many of the bioactive isoquinolines with meta-alkyl substitution are tertiary alkyl groups.4,18 Therefore, we tested our method for the N-alkylation and migration of secondary alkyl halides. The more substituted isoquinolinium salt (1) from a secondary alkyl halide with alkyl and aryl substitutions led to better yields than the parent benzyl group. A methyl and phenyl substitution on the migrating carbon led to a 66% yield of the tertiary alkylated isoquinoline product (4ab). Expectedly, a methyl and electron-poor aryl substitution resulted in slightly better yields (4ac,ad). An ethyl instead of methyl as the alkyl substitution is equally efficient (4ae). Interestingly, cyclopropyl as the alkyl substitution led to normal cyclopropyl product formation (4af) without any detectable ring-opening product. The success of a wide variety of substituents on the migrating carbon makes this method attractive as a general meta-C–H alkyl protocol. The better results with sterically demanding substituted alkyl groups complement the known meta-C–H alkylation methods where steric crowding led to poor yields or failed reactions.7 Migration of dibenzylic groups was studied next, which led to the highest 78% yield for diphenyl substitution (4ag). Other diarylmethyl groups also worked well to form the triarylmethane products (4ah,ai) in good yields. We also tested the alkylation on nitrogen with the α-oxy aryl lactone (phthalide) substituted alkyl group due to their prevalence in natural products.19N-alkylation with a group containing a heteroatom is tricky due to its instability.20 A modified N-alkylation and photochemical migration protocol with a milder triethylamine base at a lower temperature resulted in the successful migration of this sensitive group with 68% yield (4aj). A methyl and ester substituted alkyl also migrates successfully (4ak). Finally, we tested the migration of a tertiary alkyl group at the meta-C–H position. The dimethylphenyl substituted substrate successfully formed the product (4al) with a 40% yield. Although the yield is lower, a quaternary carbon center formation at meta-C–H with an all carbon alkyl group has not been achieved previously. The increase in steric and nucleophilicity of the tertiary alkyl group due to the extra methyl substitution might be the cause of moderate yield with substantial dimerization of the migrating group.
Mechanistic studies
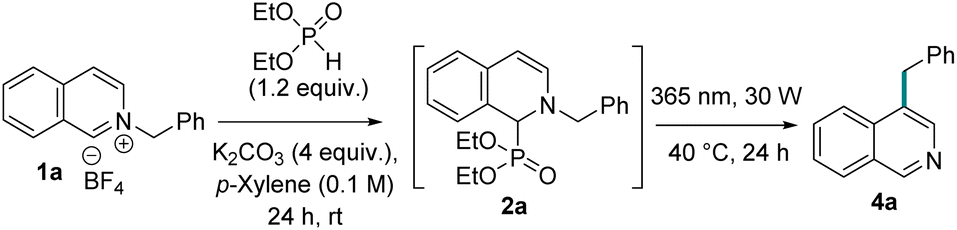
Although photochemical aza-[1,3] migration is not known, various other photochemical [1,3] migrations are reported to undergo via a concerted pericyclic pathway21 and dissociative mechanism with radical recombination22 or radical chain propagation.23 To understand our reaction path, we carefully analyzed our reactions to detect any intermediate and side-products formed in the reaction. In that effort, we found a 5–20% dimer (5) of the migrating alkyl group depending on their reactivity. Electron-rich benzyl radicals and tertiary alkyl radicals were more prone to dimerization. The degree of dimerization is inversely correlated with the yields of meta-alkylated product formation. This dimer formation suggests a homolytically dissociative mechanism for our meta-alkyl migration. We also isolated small amounts (5–10%) of ortho-phosphonate isoquinoline (6). We presume this might be formed via the single electron oxidation of the other radical partner (R1) (Fig. 3A). The classical TEMPO trapping experiment led to the benzyl-TEMPO adduct (7a), further supporting homolytic C–N dissociation as the reaction initiation step (Fig. 3B). A direct photochemical excitation of the adduct 2 excites it to the singlet state, and therefore, the dissociation from either a singlet or triplet state is feasible. To experimentally distinguish between these two possibilities, we set to attain singlet and triplet excited states selectively to study their reactivity. A triplet quencher like naphthalene diminishes the product formation by only 7%.24 However, an iridium based triplet photosensitizer with higher triplet energy than the adduct 2 (calculated to be 53.6 kcal mol−1) led to 10% product formation under 450 nm light irradiation (Fig. 3C).25 No reaction occurred with direct 450 nm light excitation without the iridium photosensitizer. These results indicate a major reaction path from the singlet excited state with a minor triplet state component.To shed light on the reaction mechanism, we conducted TDDFT and DFT computations with model dimethylphosphite adduct 2a bearing a benzyl as the migrating group. We truncated the ethyl groups present on the phosphite to methyl groups in order to reduce the computational cost. The first singlet excited state (S1) was found to be the bright state, and the S1 optimized intermediate 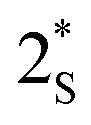 lies at 77.7 kcal mol−1 above the adduct 2a (Fig. 3E). The activation total energy
lies at 77.7 kcal mol−1 above the adduct 2a (Fig. 3E). The activation total energy  for the C–N bond dissociation at the S1 surface was estimated to be +5.7 kcal mol−1 above the Franck–Condon geometry on the S1 surface. The
for the C–N bond dissociation at the S1 surface was estimated to be +5.7 kcal mol−1 above the Franck–Condon geometry on the S1 surface. The  from the S1 equilibrium was found to be +14.8 kcal mol−1. Alternatively,
from the S1 equilibrium was found to be +14.8 kcal mol−1. Alternatively,  could undergo inter-system crossing (ISC) to its triplet state T1 forming intermediate
could undergo inter-system crossing (ISC) to its triplet state T1 forming intermediate  . To check whether ISC is feasible, we computed the spin orbit coupling matrix elements (SOCME) at the CASSCF level including 3 singlets and 2 triplets at S0, S1 and T1 geometries (see ESI for details†). Since, all the spin orbit coupling matrix elements have paltry values (<1 cm−1) the rate for ISC is expected to be significantly slow, making the dissociation at the S1 surface the favored pathway.26 This observation is expected according to El-sayed's rule27 as the photoexcitations involved in 2a are of only π–π* nature (see ESI†). In order to obtain further insights into the photoexcited bond dissociation process, we performed relaxed potential energy scans considering the singlet ground state (S0) and two most important excited states, namely, first excited singlet (S1) and triplet states (T1) using DFT/TDDFT (M062X/6-31++G(d,p)) (see ESI for details†). The density functional studies rather suggested a substantial barrier to dissociation on the S1 surface.
. To check whether ISC is feasible, we computed the spin orbit coupling matrix elements (SOCME) at the CASSCF level including 3 singlets and 2 triplets at S0, S1 and T1 geometries (see ESI for details†). Since, all the spin orbit coupling matrix elements have paltry values (<1 cm−1) the rate for ISC is expected to be significantly slow, making the dissociation at the S1 surface the favored pathway.26 This observation is expected according to El-sayed's rule27 as the photoexcitations involved in 2a are of only π–π* nature (see ESI†). In order to obtain further insights into the photoexcited bond dissociation process, we performed relaxed potential energy scans considering the singlet ground state (S0) and two most important excited states, namely, first excited singlet (S1) and triplet states (T1) using DFT/TDDFT (M062X/6-31++G(d,p)) (see ESI for details†). The density functional studies rather suggested a substantial barrier to dissociation on the S1 surface.
Since the chemical transformation involves a bond breaking scenario under photoexcitation, for obtaining more reliable estimates on energetics proper treatment of static and dynamic electron correlation is needed.28 Hence, we carried out single point calculations on relevant DFT optimized geometries with strongly contracted n-electron valence state perturbation theory (SC-NEVPT2)29 (see ESI†). According to the computed NEVPT2 energetics the barriers associated with the desired C–N bond cleavage are similar on both S1 and T1 surfaces. The NEVPT2 studies revealed that the TS lies just 4.6 kcal mol−1 above the Franck–Condon region on the S1 excited state surface (see ESI†). Hence, it can be expected that a significant fraction of photo-excited molecules will undergo C–N bond dissociation, while a larger fraction will relax to the S1 minimum leading to fluorescence (which we experimentally observe). Furthermore, we theoretically studied the congener of 2a having p-CN substitution at the migrating phenyl group (see ESI†), which provides a higher yield of the photoproduct compared to that of 2a experimentally. It was found that the TS for C–N photo-dissociation lies below the Franck–Condon geometry on the S1 surface and only 2.6 kcal mol−1 above the S1 minimum. Based on these observations and low computed SOCME values we propose this channel at S1 to be the dominant pathway for C–N photo-dissociation.
Next, we tried to understand the C–C bond formation mechanism from the bis-radical intermediacy (R1 & R2). Most photochemical [1,3] rearrangements were proposed to form C–X bonds via a radical-chain propagation mechanism.22 However, photoexcited aryl enamines with bicyclic N–O substituted compounds are reported to undergo a [1,3] shift via intramolecular radical recombination.22 For our [1,3] alkyl shift, either the benzyl–benzyl (R1 & R2) radical recombination to intermediate 3, or a benzyl radical (R2) addition to the electron-rich enamine (2) for chain propagation could be kinetically challenging (Fig. 3D). Both mechanistic pathways explain poor yields with electron-rich migrating alkyl radicals while better results with electron-deficient ones. We performed a crossover experiment with 1h and 1z to gain experimental evidence. The recombination mechanism should predominantly produce only normal products (4h and 4z), provided the concentration of the radicals is low at any point. On the other hand, the chain mechanism would generate all four products, including 4a and 4aa (Fig. 3F).23 The fact that we obtained major normal products with significant cross-products indicates the possibility of both pathways operating at variable degrees. The fact that the radical trapping experiment with TEMPO (Fig. 3B) did not completely shut down the product formation also suggests partial radical recombination via solvent-trapped intermediates (R1 & R2).15b,30 The feasibility of the radical chain propagation path was calculated next. The electrophilic migrating radical R2 generated via C–N bond dissociation can attack another electron-rich adduct 2a at the meta-position to form intermediate 2-R2. The activation barrier for such addition is 16.0 kcal mol−1 with the benzyl radical. Subsequent N–benzyl bond dissociation would form the aza-[1,3] migrated product (3) with another benzyl radical (R2) for chain propagation (Fig. 3G). The activation barrier for the C–N bond dissociation from 2-R2 was calculated to be 27.7 kcal mol−1, making it feasible at a reaction temperature of 40 °C.
 | ||
| Fig. 3 Mechanistic investigation. All reactions were performed on 0.2 mmol scale. Isolated yields are given. aMore than 95% intermediate remaining. b15% intermediate remaining. n.d. – not detected. | ||
The final meta-alkylation product formation from the aza-[1,3] alkyl shifted intermediate 3 is proposed to undergo via a base-mediated 1,4-phosphite elimination. We exposed adduct 2a to 365 nm light without any base to trap intermediate 3a, but it resulted in very little product formation with no detectable intermediate. Quenching the reaction at different times and the analysis of the crude reaction mixture also failed to detect intermediate 3avia mass or NMR. However, we could isolate around 9–13% of 8a, which might form via aerobic oxidative aromatization of the dihydroisoquinoline intermediate 3a. To validate this hypothesis, we ran the reaction in the presence of air, and that led to a higher amount of 8a formation (20%) along with 4a (Fig. 3H).
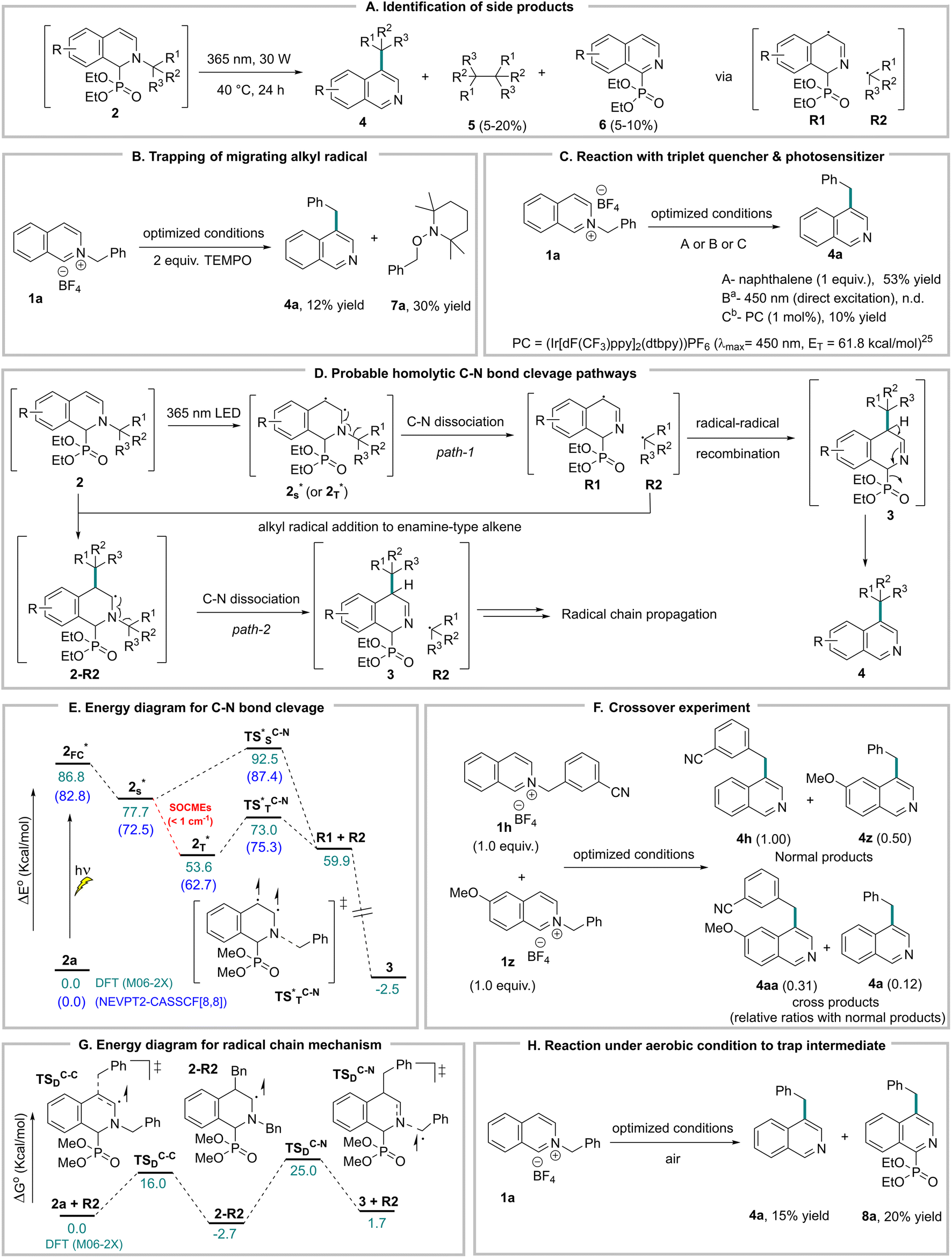
Molecular editing via phosphite mediated sequential N-functionalization and regio-selective C–H functionalization
Sequential and regioselective C–H bond functionalization of isoquinolines is immensely attractive for the derivatization of the bioactive isoquinolines to improve and expand their potential drug candidacy. For example, ortho, meta-dialkyl isoquinolines are frequently screened for SAR studies in bioactivity assessment.4c,d,31 The synthesis of these regioselective double alkylated isoquinolines requires multiple steps with different alkylating reagents and catalysts. With our previous ortho-alkylation method from the same starting material 1 in hand,8b we tested the feasibility of sequential N-alkylation and phosphite mediated regioselective rearrangements for multiple C–H alkylations with different alkyl groups regioselectively. In our first sequence, we alkylated the isoquinoline nitrogen with the meta-fluoro benzyl group (1g) for its photochemical migration to the meta-position (4g). Subsequently, 4g nitrogen was alkylated with the meta-chloro benzyl group for its ortho-alkyl migration method, successfully generating the ortho, meta-dialkylated product 10gc with 45% yield. It is significant to note that the benzyl group on nitrogen can selectively migrate to either the ortho- or meta-position via the same phosphite adduct 2, only by varying basic vs. photochemical reaction conditions. To further demonstrate the utility of this consecutive regioselective approach, we reverse the sequence of N-alkylation with meta-fluoro and meta-chlorobenzyl groups. As a result, the regiodivergent dialkyl isoquinoline 10cg formed successfully with complete regioselectivity. This unprecedented regiodivergency via a common phosphite adduct of N-alkyl isoquinoline gives us great control in functionalizing multiple C–H bonds to access densely functionalized isoquinolines (Fig. 4A). | ||
| Fig. 4 Molecular editing of isoquinoline C–H bonds via sequential N-functionalization and phosphite-mediated migration. | ||
To further demonstrate the synthetic utility of the meta-alkylated product 4, we attempted an N-acylation and unreported phosphite-mediated migration of an acyl (benzoyl) group to the ortho-position.32 To our delight, the corresponding phosphite adduct underwent base-mediated benzoyl migration to the ortho-position smoothly to form ortho-acyl-meta-alkyl isoquinoline 11aa with 68% yield. Next, we did N-benzylation of 4a, and the intermediate was oxidized in air via another phosphite-mediated method to form N-benzylated isoquinolone 12aa in good yield (Fig. 4B).14b
Conclusions
In summary, we have developed a new photochemical method for the migration of an N-alkyl group on isoquinoline to its meta-position. This meta-C–H alkylation of isoquinoline works with all primary, secondary, and tertiary alkyl migrating groups to form sterically demanding secondary, tertiary, and all-carbon quaternary substitutions. A variety of functional groups on both migrating carbon and isoquinoline were tolerated. Sequential N-alkylation, followed by phosphite-mediated method manipulations, led to double C–H alkylation to regiodivergent ortho, meta-dialkylation of isoquinoline. Other phosphite-mediated methods were also developed and utilized for further C–H functionalization of meta-C–H alkylated products. Both experimental and computational mechanistic studies were conducted to establish a probable reaction mechanism. Currently, we are studying the synthesis of N-functionalized azaarenium salts with other functional groups for their meta-migration as a general waste-free strategy for meta-C–H functionalization of azaarenes.Data availability
The complete data supporting this article have been uploaded as part of the ESI material.†Author contributions
S. R. and D. K. D. conducted the experiments and analysed the data for the optimization study, substrate scope synthesis and experimental mechanistic study under the supervision of P. M. M. A. performed the experiments for sequential functionalization of isoquinoline. A. K. R. designed and performed the computational study under the supervision of A. P. N. A. R. P. conducted the NMR study and analysed the data. P. M. and A. P. directed the project and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the SERB (CRG/2019/001065 and SCP/2022/000465), Govt. of India. S. R. and A. K. R. thank CSIR, India for their research fellowship. D. K. D. and M. A. thank DST, India for INSPIRE fellowship. We thank Dr Nilofar Baral for her experimental help. The support from the central analytical facility, NCL is greatly acknowledged.Notes and references
- (a) C. Luo, M. Ampomah-Wireko, H. Wang, C. Wu, Q. Wang, H. Zhang and Y. Cao, Anti-Cancer Agents Med. Chem., 2021, 21, 811–824 CrossRef CAS PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 1992, 9, 365–391 RSC; (c) M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341–3370 CrossRef CAS PubMed; (d) M. Chrzanowska, A. Grajewska and M. D. Rozwadowska, Chem. Rev., 2016, 116, 12369–12465 CrossRef CAS; (e) A. N. Kim, A. Ngamnithiporn, E. Du and B. M. Stoltz, Chem. Rev., 2023, 123, 9447–9496 CrossRef CAS.
- (a) C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed; (b) E. A. Bakhite, I. S. Marae, M. A. Gad, S. K. Mohamed, J. T. Mague and S. Abuelhassan, J. Agric. Food Chem., 2022, 70, 9637–9644 CrossRef CAS PubMed; (c) N. W. Alcock, J. M. Brown and D. I. Hulmes, Tetrahedron: Asymmetry, 1993, 4, 743–756 CrossRef CAS; (d) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS.
- (a) Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk and J.-Q. Yu, Nature, 2022, 610, 87–93 CrossRef CAS PubMed; (b) C. M. Josephitis, H. M. H. Nguyen and A. McNally, Chem. Rev., 2023, 123, 7655–7691 CrossRef CAS; (c) S. Maity, A. Bera, A. Bhattacharjya and P. Maity, Org. Biomol. Chem., 2023, 21, 5671–5690 RSC.
- (a) V. Palani, C. L. Hugelshofer, I. Kevlishvili, P. Liu and R. Sarpon, J. Am. Chem. Soc., 2019, 141, 2652–2660 CrossRef CAS PubMed; (b) D. Bhowmik, F. Buzzetti, G. Fiorillo, F. Orzi, T. M. Syeda, P. Lombardi and G. S. Kumar, Med. Chem. Commun., 2014, 5, 226–231 RSC; (c) Q. Zhang, G. Tu, Y. Zhaoa and T. Cheng, Tetrahedron, 2002, 58, 6795–6798 CrossRef CAS; (d) R. Nishikawa-Shimono, Y. Sekiguchi, T. Koami, M. Kawamura, D. Wakasugi, K. Watanabe, S. Wakahara, K. Matsumoto and T. Takayama, Bioorg. Med. Chem. Lett., 2012, 22, 3305–3310 CrossRef CAS PubMed; (e) J. W. Hernandez, J. Pospech, U. Klöckner, T. W. Bingham and D. Sarlah, J. Am. Chem. Soc., 2017, 139, 15656–15659 CrossRef.
- (a) F. Minisci, E. Vismara, F. Fontana, G. Morini, M. Serravalle and C. Giordano, J. Org. Chem., 1986, 51, 4411–4416 CrossRef CAS; (b) J. C. Lewis, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332–5333 CrossRef CAS; (c) T. Andou, Y. Saga, H. Komai, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2013, 52, 3213–3216 CrossRef CAS PubMed.
- (a) H. M. S. Haley, S. E. Payer, S. M. Papidocha, S. Clemens, J. Nyehuis and R. Sarpong, J. Am. Chem. Soc., 2021, 143, 4732–4740 CrossRef CAS; (b) G. Laudadio, P. Neigenfind, A. Peter, C. Z. Rubel, M. A. Emmanuel, M. S. Oderinde, T. E.-H. Ewing, M. D. Palkowitz, J. L. Sloane, K. W. Gillman, D. Ridge, M. D. Mandler, P. N. Bolduc, M. C. Nicastri, B. Zhang, S. Clementson, N. N. Petersen, P. Martin-Gago, P. Mykhailiuk, K. M. Engle and P. S. Baran, Angew. Chem., Int. Ed., 2024, e202314617 CAS.
- (a) D. E. Minter and M. A. Re, J. Org. Chem., 1988, 53, 2653–2655 CrossRef CAS; (b) F. Xie, R. Xie, J.-X. Zhang, H.-F. Jiang, L. Du and M. Zhang, ACS Catal., 2017, 7, 4780–4785 CrossRef CAS; (c) Z. Liu, J.-H. He, M. Zhang, Z.-J. Shi, H. Tang, X.-Y. Zhou, J.-J. Tian and X.-C. Wang, J. Am. Chem. Soc., 2022, 144, 4810–4818 CrossRef CAS PubMed; (d) Z. Liu, Z.-J. Shi, L. Liu, M. Zhang, M.-C. Zhang, H.-Y. Gou and X.-C. Wang, J. Am. Chem. Soc., 2023, 145, 11789–11797 CrossRef CAS PubMed; (e) J.-J. Tian, R.-R. Li, G.-X. Tian and X.-C. Wang, Angew. Chem., Int. Ed., 2023, 62, e202307697 CrossRef CAS PubMed; (f) R. Muta, T. Torigoe and Y. Kuninobu, Org. Lett., 2022, 24, 8218–8222 CrossRef CAS; (g) H. Cao, Q. Cheng and A. Studer, Science, 2022, 378, 779–785 CrossRef CAS; (h) P. Xu, Z. Wang, S.-M. Guo and A. Studer, Nat. Commun., 2024, 15, 4121 CrossRef CAS; (i) A. J. Day, T. C. Jenkins, M. Kischkewitz, K. E. Christensen, D. L. Poole and T. J. Donohoe, Org. Lett., 2023, 25, 614–618 CrossRef CAS.
- (a) A. Motaleb, S. Rani, T. Das, R. G. Gonnade and P. Maity, Angew. Chem., Int. Ed., 2019, 58, 14104–14109 CrossRef CAS; (b) S. Rani, S. R. Dash, A. Bera, M. N. Alam, K. Vanka and P. Maity, Chem. Sci., 2021, 12, 8996–9003 RSC; (c) N. Baral, S. Rani, P. Saikia and P. Maity, Eur. J. Org Chem., 2023, 26, e202201238 CrossRef CAS.
- (a) Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS; (b) S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370–11375 CrossRef CAS PubMed; (c) W. Lee, S. Jung, M. Kim and S. Hong, J. Am. Chem. Soc., 2021, 143, 3003–3012 CrossRef CAS PubMed; (d) J. Choi, G. Laudadio, E. Godineau and P. S. Baran, J. Am. Chem. Soc., 2021, 143, 11927–11933 CrossRef CAS PubMed; (e) S. Jung, H. Lee, Y. Moon, H.-Y. Jung and S. Hong, ACS Catal., 2019, 9, 9891–9896 CrossRef CAS; (f) Y. Gu, Y. Shen, C. Zarate and R. Martin, J. Am. Chem. Soc., 2019, 141, 127–132 CrossRef CAS PubMed; (g) T. Nishida, H. Ida, Y. Kuninobu and M. Kanai, Nat. Commun., 2014, 5, 3387–3392 CrossRef PubMed; (h) M. Jaric, B. A. Haag, A. Unsinn, K. Karaghiosoff and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 5451–5455 CrossRef CAS.
- K. Murakami, S. Yamada, T. Kaneda and K. Itami, Chem. Rev., 2017, 117, 9302–9332 CrossRef CAS PubMed.
- G.-Q. Sun, P. Yu, W. Zhang, W. Zhang, Y. Wang, L.-L. Liao, Z. Zhang, L. Li, Z. Lu, D.-G. Yu and S. Lin, Nature, 2023, 615, 67–72 CrossRef CAS PubMed.
- H. Cao, D. Bhattacharya, Q. Cheng and A. Studer, J. Am. Chem. Soc., 2023, 145, 15581–15588 CrossRef CAS.
- S. Alwarsh, Y. Xu, S. Y. Qian and M. C. McIntosh, Angew. Chem., Int. Ed., 2016, 55, 355–358 CrossRef CAS.
- (a) V. K. Aggarwal, I. Bae, H.-Y. Lee and D. T. Williams, Angew. Chem., Int. Ed., 2003, 42, 3274–3278 CrossRef CAS; (b) A. Motaleb, A. Bera and P. Maity, Org. Biomol. Chem., 2018, 16, 5081–5085 RSC.
- (a) C. G. Nasveschuk and T. Rovis, Org. Biomol. Chem., 2008, 6, 240–254 RSC; (b) M. N. Alam, S. R. Dash, A. Mukherjee, S. Pandole, U. K. Marelli, K. Vanka and P. Maity, Org. Lett., 2021, 23, 890–895 CrossRef CAS.
- J. W. Lown, M. H. Akhtar and R. S. McDaniel, J. Org. Chem., 1974, 39, 1988–2006 CrossRef.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed; (b) J. C. Koziar and D. O. Cowa, Acc. Chem. Res., 1978, 11, 334–341 CrossRef CAS; (c) Z. He, W. Zhao, J. W. Y. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed.
- T. Fan, Y. Cheng, W. Wei, Q. Zeng, X. Guo, Z. Guo, Y. Li, L. Zhao, Y. Shi, X. Zhang, J. Jiang, Y. Wang, W. Kong and D. Song, J. Med. Chem., 2022, 65, 7399–7413 CrossRef CAS PubMed.
- (a) M. D. P. C. Soriano, N. Shankaraiah and L. S. Santos, Tetrahedron Lett., 2010, 51, 1770–1773 CrossRef; (b) Y. Li and C. D. Smolke, Nat. Commun., 2016, 7, 12137 CrossRef CAS PubMed.
- J. Zhu and A. J. Bennet, J. Am. Chem. Soc., 1998, 120, 3887–3893 CrossRef CAS.
- (a) J. Slutsky and H. Kwart, J. Am. Chem. Soc., 1973, 95, 8678–8685 CrossRef CAS; (b) T. Yamabe, K. Nakamura, Y. Shiota, K. Yoshizawa, S. Kawauchi and M. Ishikawa, J. Am. Chem. Soc., 1997, 119, 807–815 CrossRef CAS; (c) N. Hammer, M. L. Christensen, Y. Chen, D. Naharro, F. Liu, K. A. Jørgensen and K. N. Houk, J. Am. Chem. Soc., 2020, 142, 6030–6035 CrossRef CAS PubMed.
- (a) E. R. Wearing, D. E. Blackmun, M. R. Becker and C. S. Schindler, J. Am. Chem. Soc., 2021, 143, 16235–16242 CrossRef CAS; (b) M. R. Gatazka, E. C. McFee, C. H. Ng, E. R. Wearing and C. S. Schindler, Org. Biomol. Chem., 2022, 20, 9052–9068 RSC.
- (a) X. Su, H. Huang, Y. Yuan and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 1338–1341 CrossRef CAS; (b) L. Xie, X. Zhen, S. Huang, X. Su, M. Lin and Y. Li, Green Chem., 2017, 19, 3530–3534 RSC; (c) H. Wang, P. Bellotti, X. Zhang, T. O. Paulisch and F. Glorius, Chem, 2021, 7, 3412–3424 CrossRef CAS; (d) F. C. S. Silva, K. Doktor and Q. Michaudel, Org. Lett., 2021, 23, 5271–5276 CrossRef.
- B. Giese, P. Wettstein, C. Stähelin, F. Barbosa, M. Neuburger, M. Zehnder and P. Wessig, Angew. Chem., Int. Ed., 1999, 38, 2586–2587 CrossRef CAS PubMed.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- A. Mavroskoufis, M. Lohani, M. Weber, M. N. Hopkinson and J. P. Gotze, Chem. Sci., 2023, 14, 4027–4037 RSC.
- M. Baba, J. Phys. Chem. A, 2011, 115, 9514–9519 CrossRef CAS.
- J. Vijayasundar, V. Subramanian and B. Rajakumar, Phys. Chem. Chem. Phys., 2019, 21, 438–447 RSC.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef CAS.
- (a) V. W. Bowry, J. Lusztyk and K. U. Ingold, J. Am. Chem. Soc., 1991, 113, 5687–5698 CrossRef CAS; (b) A. A. Martin-Esker, C. C. Johnson, J. H. Horner and M. Newcomb, J. Am. Chem. Soc., 1994, 116, 9174–9181 CrossRef CAS.
- (a) B. M. Trost, C.-I. J. Hung and Z. Jiao, J. Am. Chem. Soc., 2019, 141, 16085–16092 CrossRef CAS PubMed; (b) C. Gaoa, Y. Dua, X. Wanga, H. Caoa, B. Linb, Y. Liua and X. Di, Bioorg. Med. Chem. Lett., 2018, 28, 2265–2269 CrossRef PubMed.
- (a) H. W. Gibson, K. K. Brumfield, R. A. Grisle and C. K. F. Hermann, J. Polym. Sci., Part A:Polym. Chem., 2010, 48, 3856–3867 CrossRef CAS; (b) V. Boekelheide and J. Weinstock, J. Am. Chem. Soc., 1952, 74, 660–663 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07127a |
| ‡ S. R., A. K. R. and D. K. D. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |