
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiacetylene-bridged covalent organic framework as crystalline graphdiyne analogue for photocatalytic hydrogen evolution†
Zhiqing
Lin
a,
Songyao
Dai
a,
Shan
Yao
b,
Qia-Chun
Lin
a,
Mengying
Fu
a,
Lai-Hon
Chung
 *a,
Bin
Han
*b and
Jun
He
*a
*a,
Bin
Han
*b and
Jun
He
*a
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou 510006, China. E-mail: junhe@gdut.edu.cn; laihonchung@gdut.edu.cn
bGuangdong Basic Research Center of Excellence for Ecological Security and Green Development, Key Laboratory for City Cluster Environmental Safety and Green Development of the Ministry of Education, School of Ecology, Environment and Resources, Guangdong University of Technology, Guangzhou, 510006, P. R. China. E-mail: hanbin@gdut.edu.cn
First published on 16th December 2024
Abstract
Graphdiyne (GDY) alone as a photocatalyst is unsatisfactory because of its low crystallinity, limited regulation of the band gap, weak photogenerated charge separation, etc., and heterojunctioning with other materials is necessary to activate the photocatalytic activity of GDY. Through elaborate design, a diacetylene-rich linker (S2) was prepared and employed to construct a crystalline and structurally well-defined GDY-like covalent organic framework (COF, namely S2-TP COF) which merges the merits of both COF and GDY to boost the photocatalytic hydrogen evolution reaction (HER). By theoretical prediction on the donor–acceptor (D–A) pair, two other monoacetylene-bridged COFs (S1-TP COF and S3-TP COF) were prepared for comparison. Exhibiting enhanced separation and suppressed recombination of photogenerated excitons, Pt-photodeposited S2-TP COF showed a higher HER rate (10.16 mmol g−1 h−1) than the other two non-GDY-like COFs (3.71 and 1.13 mmol g−1 h−1). A joint experimental–theoretical study suggests that the appropriate D–A structure for photogenerated charge separation and diacetylene motif as the adsorption site are the key reasons for the boosted HER. This work opens a new avenue for the rational design of COFs as GDY mimics for photocatalytic application.
Introduction
Graphdiyne (GDY), a 2D carbon-based network composed of sp and sp2 hybridized carbon, was only a fantasy on the theoretical level until the successful preparation of GDY film on copper foil by Li and co-workers.1 Since this achievement, GDY has been considered a promising platform for various solar-driven applications including the hydrogen evolution reaction (HER), CO2 reduction reaction (CO2RR), nitrogen reduction reaction (NRR), light-assisted pollutant degradation, etc. because its large porosity, alkyne-rich functionalities and high degree of π-conjugation favor substrate mass transfer, adsorption of metal ions for building active sites, light harvesting and separation/transport of charge carriers.2,3 Promising as it seems, GDY has an Achilles' heel of a narrow band gap (0.46–1.22 eV) and mostly resorts to forming a binary/ternary heterojunction with other semiconductors (e.g., CdS, g-C3N4, TiO2, etc.) for optimal band structures to actualize photocatalysis.4–7 Even though a few GDY-based single-component photocatalysts have been achieved recently by introducing defects into the GDY net through editing the structure of graphene precursors (i.e., tuning the number of alkyne groups on the aromatic core or heteroatom/functional group substitution),8–11 the amorphous nature of the as-prepared GDYs is unfavorable for structure–function correlation (e.g., the function of acetylene bridges and how active sites work during photocatalysis). Another challenge comes with the instability of multialkyne monomers (e.g. hexaalkynyl benzene) which contributes to the amorphous nature and causes structural uncertainty of the resultant GDY by side reactions.12,13 So, to keep the strengths and rectify the weaknesses of GDY, it is necessary to develop novel crystalline platforms featuring a continuum of diacetylene functionalities.Covalent organic frameworks (COFs) are ideal platforms for constructing crystalline porous polymers with rich functionality customization, well-defined structure, high stability, and tunable pores.14–18 Intriguingly, the high degree of freedom and precision in structural design is especially useful for the integration of diverse functional units. Specifically, building donor–acceptor (D–A) type COFs has emerged as a convenient and effective strategy for high-performance photocatalysts because this approach allows easy band structure tuning and skips external photosensitizers as well as heterojunction formation.19–21 In this regard, we propose the incorporation of GDY moieties into COFs to be a two-fold approach to keep the strengths of GDY (e.g., permanent porosity, abundant alkynyl active sites, intensive π-conjugation) on one hand and rectify the weaknesses of GDY (e.g., lack of bandgap tuning, photosensitizing power) on the other hand.
Since the synthesis of diacetylene-bridged COFs is not easy, only a few examples based on 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDA), 4,4′-(buta-1,3-diyne-1,4-diyl)dibenzaldehyde (BD) or dehydrobenzoannulene (DBA) monomers have been reported so far.22–27 These works indicate that the diacetylene units not only facilitate the separation and transfer of charge carriers, but also suppress reverse charge recombination to improve the D-to-A migration lifetime of charge carriers, thereby enhancing photocatalytic activity. However, the role of diyne units in COFs is still unclear from the mechanistic viewpoint, especially the comparison with monoacetylene analogues. Therefore, it is crucial to design GDY-like COFs with well-defined structures, tunable band gaps, and abundant diacetylene units to unveil the structure–property correlation and develop next-generation GDY-like catalysts.
Herein, we adopted Glaser–Hay coupling to construct a diacetylene-linked triangular triamine linker (S2, Scheme S1†) which was allowed to undergo Schiff-base condensation with classical C3-symmetric 1,3,5-triformylphloroglucinol (TP) to build a GDY-like COF. As a comparison, triamine linkers with monoacetylene of different skeletal lengths were employed to construct analogous networks for follow-up investigation on how diacetylene-based networks beat monoacetylene counterparts using the photocatalytic HER as a model reaction. Joint experimental and theoretical studies suggest that the GDY-like COF works better than monoacetylene-based analogues, which is attributed to the following advantages: (1) construction of optimal D–A junction to enhance light-harvesting ability; (2) the introduction of −C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C−CC− linkages regulates the planarity of the COF for smooth e− transport, establishes spatial distance between D–A units for extensive separation of electron–hole (e−–h+) pairs and minimizes exciton binding energy; (3) the GDY-like COF generates good reactant affinity, improving the loading and dispersion of the Pt cocatalysts, thereby achieving excellent photocatalytic performance. This work presents an unprecedented way to merge the merits of GDY into the structurally robust, diversified crystalline framework for the rational development of functional materials.
C−CC− linkages regulates the planarity of the COF for smooth e− transport, establishes spatial distance between D–A units for extensive separation of electron–hole (e−–h+) pairs and minimizes exciton binding energy; (3) the GDY-like COF generates good reactant affinity, improving the loading and dispersion of the Pt cocatalysts, thereby achieving excellent photocatalytic performance. This work presents an unprecedented way to merge the merits of GDY into the structurally robust, diversified crystalline framework for the rational development of functional materials.
Results and discussion
Construction of D–A pairs
Establishing clear D–A junctions in COFs promotes oriented intramolecular charge separation and introducing π-conjugated structures such as alkynyl groups suppresses charge recombination which are key prerequisites for developing a competent photocatalyst. In our plot, a classical linker with C3 symmetry (TP as an electrophilic acceptor) is allowed to form imine links with three alkyne-rich amine-terminal monomers S1, S2, and S3 to give TP-COFs. To evaluate the possibility of a D–A junction in the selected COFs, the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of all building blocks were calculated (Fig. 1). To attain the D–A junction in a system, the HOMO should be dominated by the donor while the LUMO should be mainly contributed by the acceptor.28–30 A clear D–A junction in TP-COFs is established with the alkyne-rich building blocks and TP unit taking up the role of donor and acceptor respectively. Noteworthily, an extension of the alkynyl π system (i.e., from −CC− in S1 to −CC−CC− in S2) narrows the HOMO–LUMO gap, indicating higher conjugation in diacetylene than in the monoacetylene bridge. Therefore, TP-COFs were used as platforms to study how alkynyl π-bridges on donors modulate photocatalytic properties.
 | ||
| Fig. 1 HOMO and LUMO energy level diagram of building blocks for constructing the D–A junction deduced from density functional theory (DFT) using Gaussian 16A software with B3LYP/6-31G (d) as the functional and basis sets (red and blue denote positive and negative regions, isosurface values = 0.02 a.u.). | ||
Synthesis and characterization of TP-COFs
TP-centered trialdehyde is commercially available while alkyne-bridged triamines were synthesized through Sonogashira coupling and Glaser–Hay coupling (for synthetic details, see ESI, Schemes S1, S2 and Fig. S1–S8†). Noteworthily, another common synthetic strategy to prepare linker S2 by direct coupling between 4-ethynylaniline and 1,3,5-triethynylbenzene was found to give low selectivity and yield because of the homocoupling of 4-ethynylaniline. β-Ketoenamine-linked TP-COFs as a honeycomb net were synthesized by condensation between amines and TP in the mixture of mesitylene/1,4-dioxane/6 M acetic acid (Fig. 2a). TP-COFs were prepared smoothly by the optimized protocols notwithstanding the extension of the alkynyl π system or change of electronic conjugation in triamines. Also, S1-TP COF was reported in our previous work.31 | ||
| Fig. 2 (a) Molecular building blocks and structural models for TP-COFs. (b) Experimental, simulated, and Pawley refined PXRD of S2-TP COF. (c) FT-IR spectra of S2-TP COF. (d) N2 sorption isotherms of TP-COFs. (e) HR-TEM image and (f) magnified HR-TEM image of S2-TP COF (orange inset shows lattice stripes corresponding to the (200) crystal plane). (g) Band structures of TP-COFs. | ||
Powder X-ray diffraction (PXRD) patterns show high crystallinity of TP-COFs (Fig. 2b and S9–S11†). Reasonable Rwp and Rp values (Rwp = 3.24–3.60% and Rp = 2.52–2.78%) based on the Pawley refinement indicate that these COFs have a 2D hexagonal network with space group of P6 symmetry and AA stacking mode (S1-TP COF: a = 23.18 Å and c = 3.52 Å; S2-TP COF: a = 25.10 Å and c = 3.34 Å; S3-TP COF: a = 27.75 Å and c = 3.29 Å, with c referring to the interlayer spacing, Table S1†). Note that the narrower interlayer spacing from S1-TP COF (3.52 Å) to S2-TP COF (3.34 Å) probably originates from stronger interlayer π–π interaction by extending the acetylene bridge from −CC− to −CC−CC−. Considering the Fourier-transform infrared (FT-IR) spectra of TP-COFs, the emerging C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, CC stretching, and C−N vibrational bands at ∼1623, 1565, and 1287 cm−1 together with the disappearing N−H and CO (∼1644 cm−1) signals of monomers (Fig. 2c, S12 and S13†) support the formation of the β-ketoenamine linkage via enol–keto tautomerization. Notably, in TP-COFs, −CC− stretching signals at 2207 cm−1 prove the chemical tolerance of dialkynyl bridges throughout the COF assembly. Besides, the β-ketoenamine linkages of S2-TP COF and S3-TP COF were verified by solid-state 13C NMR spectroscopy (∼147.1, 107.7, and 184.5 ppm for the enamine carbon C−N, α-enamine carbon CC, and keto carbon CO, respectively, Fig. S14 and S15†) and X-ray photoelectron spectroscopy, XPS (C−N, ∼400 eV in N 1s spectra, Fig. S16 and S17†), as was probed in previously reported S1-TP COF.31 Moreover, the Raman spectra of the samples show strong signals at ∼1182 cm−1 and ∼2217 cm−1, attributable to the in-phase stretching vibration of sp2 hybridized carbons (C−N) in aromatic rings and internal alkynes (Fig. S18†), respectively.
O stretching, CC stretching, and C−N vibrational bands at ∼1623, 1565, and 1287 cm−1 together with the disappearing N−H and CO (∼1644 cm−1) signals of monomers (Fig. 2c, S12 and S13†) support the formation of the β-ketoenamine linkage via enol–keto tautomerization. Notably, in TP-COFs, −CC− stretching signals at 2207 cm−1 prove the chemical tolerance of dialkynyl bridges throughout the COF assembly. Besides, the β-ketoenamine linkages of S2-TP COF and S3-TP COF were verified by solid-state 13C NMR spectroscopy (∼147.1, 107.7, and 184.5 ppm for the enamine carbon C−N, α-enamine carbon CC, and keto carbon CO, respectively, Fig. S14 and S15†) and X-ray photoelectron spectroscopy, XPS (C−N, ∼400 eV in N 1s spectra, Fig. S16 and S17†), as was probed in previously reported S1-TP COF.31 Moreover, the Raman spectra of the samples show strong signals at ∼1182 cm−1 and ∼2217 cm−1, attributable to the in-phase stretching vibration of sp2 hybridized carbons (C−N) in aromatic rings and internal alkynes (Fig. S18†), respectively.
The N2 sorption isotherms of these COFs (Fig. 2d and S19–S21†) were measured at 77 K and all of them demonstrate remarkable Brunauer–Emmett–Teller (BET) surface areas and clear pores (BET surface areas and pore sizes, obtained from non-local density functional theory (NLDFT)—S1-TP COF: 1054 m2 g−1, 1.73 nm; S2-TP COF: 1162 m2 g−1, 1.73 nm; S3-TP COF: 1274 m2 g−1, 2.43 nm). Note that S1-TP COF and S2-TP COF exhibit the typical type-I isotherm, indicating a microporous structure while S3-TP COF shows a typical type-IV isotherm with mesoporous characteristics. As shown by scanning electron microscopy (SEM), TP-COFs were clusters of submicron flakes (Fig. S22†). As shown in transmission electron microscopy (TEM) images, a lattice fringe (0.94 nm) corresponding to the (200) phase of S2-TP COF is visible, highlighting good crystallinity of the COF and consolidating the appropriateness of the simulated structure (Fig. 2e and f). Notably, thermogravimetric analysis (TGA) under an N2 atmosphere reveals high thermal stability of the TP-COFs with most of the weight (weight loss <5%) retained up to 400 °C of TP-COFs (Fig. S23†). Besides, consistent PXRD patterns of COFs before and after immersion in various solutions for 3 days indicate their satisfactory chemical stability, which is beneficial to photocatalysis (Fig. S24†).
To evaluate the feasibility of the COFs as HER photocatalysts, the optical absorption profiles and electronic band structures of the COFs were characterized using UV-visible diffuse reflectance spectroscopy (UV-Vis DRS) and Mott–Schottky (M–S) measurement. The D–A interaction leads to a red-shift in the absorption band of the D–A type COFs, and the red-shift is more obvious with stronger D–A interaction.32 All the TP-COFs absorb strongly in the visible spectral region with obvious redshifts when compared with monomers, S1, S2, and S3, indicative of the successful construction of the D–A junction (Fig. S25†). Among these COFs, the GDY-like COF (COF based on S2) demonstrates even red-shifted absorption tailing until around 1000 nm, featuring a wider light-harvesting spectrum than COFs built from S1 and S3 (Fig. S25†). By Kubelka–Munk conversion, the band gaps of S1-TP COF, S2-TP COF, and S3-TP COF were found to be 2.28, 2.25 and 2.38 eV respectively (Fig. S26†). M–S plots reveal all the COFs to be n-type semiconductors which bear a conduction band (CB) edge lying higher than the flat-band potential by 0.1 V.33–35 Therefore, the CB edges relative to a normal hydrogen electrode (NHE, pH = 7) of S1-TP COF, S2-TP COF, and S3-TP COF were calculated to be −1.28, −0.90 and −1.04 V, which are more negative than the redox potential required to drive H+-to-H2 reduction (−0.41 V vs. NHE, pH = 7) (Fig. 2g and S27†), suggesting these COFs to be suitable for the HER from the thermodynamic point of view.
Photocatalytic performance of COFs
Note that TP-COFs bearing polar keto and protic amine groups show good hydrophilicity as revealed by their small water contact angles (S1-TP COF: 71°; S2-TP COF: 54°; S3-TP COF: 77°, Fig. S30†) improved the hydrophobic defects of the S2 monomer itself (Fig. S29, similar to GDY). Thanks to the hydrophilic nature, TP-COFs are suitable for systematic comparison to rationalize the function of the diacetylene bridge on photocatalysis, suppressing the HER by good H2O flow in COF pores which enhances the accessibility of active sites. After optimization of reaction conditions, TP-COFs were used for the photocatalytic HER study under visible-light irradiation (λ > 420 nm) in a DMF/ascorbic acid (AA) mixture with Pt as a co-catalyst (Fig. 3a, S33–S40 and Tables S3–S9†). S2-TP COF showed a monotonic increase in H2-evolution performance (Fig. 3b) with a higher HER rate (10.16 mmol g−1 h−1) than S1-TP COF (3.71 mmol g−1 h−1) as well as S3-TP COF (1.13 mmol g−1 h−1) and this indicates that the –CC–CC– skeleton in S2-TP COF may exert some functions to promote the HER. Notably, S2-TP COF gave an unsatisfactory HER rate of 3.26 mmol g−1 h−1 in AA aqueous solution, with the amount of H2 evolving 3-fold lower than that in the mixed solvent system. As shown in Fig. S32,† TP-COFs were well dispersed (beyond 30 minutes) in the DMF/AA mixture but rapidly precipitated in the AA solution, suggesting less aggregation in the mixed system. The downsizing of TP-COFs in the DMF/AA mixture exhibited significantly enhanced H2 evolution performance because of their increased active site exposure and light-harvesting properties.36
 | ||
| Fig. 3 (a) Photocatalytic H2 evolution rates of TP-COFs with 3 wt% Pt feeding content. (b) Time-dependent H2 evolution curves of TP-COFs (5 mg) with 3 wt% Pt feeding content upon visible-light illumination (λ > 420 nm). (c) Cycling stability tests of H2 evolution (4 hours for each cycle) and (d) comparative plot between wavelength-dependent AQY and absorption profile of S2-TP COF. | ||
Besides, the TP-COFs demonstrate operational stability (say S2-TP COF maintained steady HER rates of around 10 mmol g−1 h−1 over 4 photocatalytic cycles, Fig. 3c). Moreover, consistent PXRD patterns (retained crystallinity), FT-IR spectra (unchanged chemical functionalities), and morphologies of TP-COFs before and after photocatalysis consolidate their photo- and operational stability (Fig. S41 and S42†). The best catalytic dosage was found to be 2 mg as reflected by the peak HER rate of 14.35 mmol g−1 h−1 with S2-TP COF (Fig. S37†); even though a higher input of the catalyst lowers the rate probably stemming from weaker light absorption by aggregation of catalysts, to minimize errors, a higher dosage of catalysts (i.e., 5 mg) was used for comparing the HER performance of the COFs studied in this work. To evaluate the light utilization efficiency of the COF, the best-performing S2-TP COF was adopted to study the apparent quantum yield (AQY) for the HER using different band-pass filters (365–760 nm, Fig. 3d, S43 and S44†). S2-TP COF bearing 3 wt% Pt showed the top AQY of 10.62% upon 485-nm irradiation. The consistency between the AQY trend and absorption profile in S2-TP COF revealed the reliance of HER performance on photocatalysts and incident light.37–39 It is worth noting that the AQY of S2-TP COF significantly falls regardless of the selected wavelength indicating the enhancement of light utilization by Pt. For comparison, 1,3,5-triethynylbenzene was adopted to construct the GDY derivative (HGDY) by Glaser coupling (Scheme S4†).40 Despite the appropriate band gap (Eg = 2.25 eV, Fig. S28†), HGDY only gave negligible photocatalytic HER performance (3.93 μmol g−1 h−1) under the same conditions when compared with S2-TP COF (10.16 mmol g−1 h−1). Low crystallinity of HGDY based on random coupling, lack of D–A junction, and hence weak charge separation in HGDY probably accounts for its far lower HER rate.
Mechanism insights on photocatalytic HER
To rationalize the divergence in photocatalytic activities of TP-COFs, a series of photophysical and electrochemical measurements were conducted. Electrochemical impedance spectroscopy (EIS) showed a far smaller arc radius of S2-TP COF than those of S1-TP COF and S3-TP COF (Fig. S45†), indicating the smallest charge transfer resistance in S2-TP COF. On the other hand, all TP-COFs exhibited stronger electron paramagnetic resonance (EPR) signals upon photoirradiation (Fig. 4a), revealing the formation of more unpaired electrons in S2-TP COF under illumination. Also, the strongest transient photocurrent response (photocurrent intensity = 0.21 μA cm−2, Fig. S46†), weakest photoluminescence (λex = 370 nm, Fig. 4b) and longest photoluminescent lifetime (Fig. 4c) of S2-TP COF highlight the strongest generation, the best separation/worst e−–h+ pair recombination and the longest lifespan of photogenerated carriers of S2-TP COF amongst all TP-COFs.41–43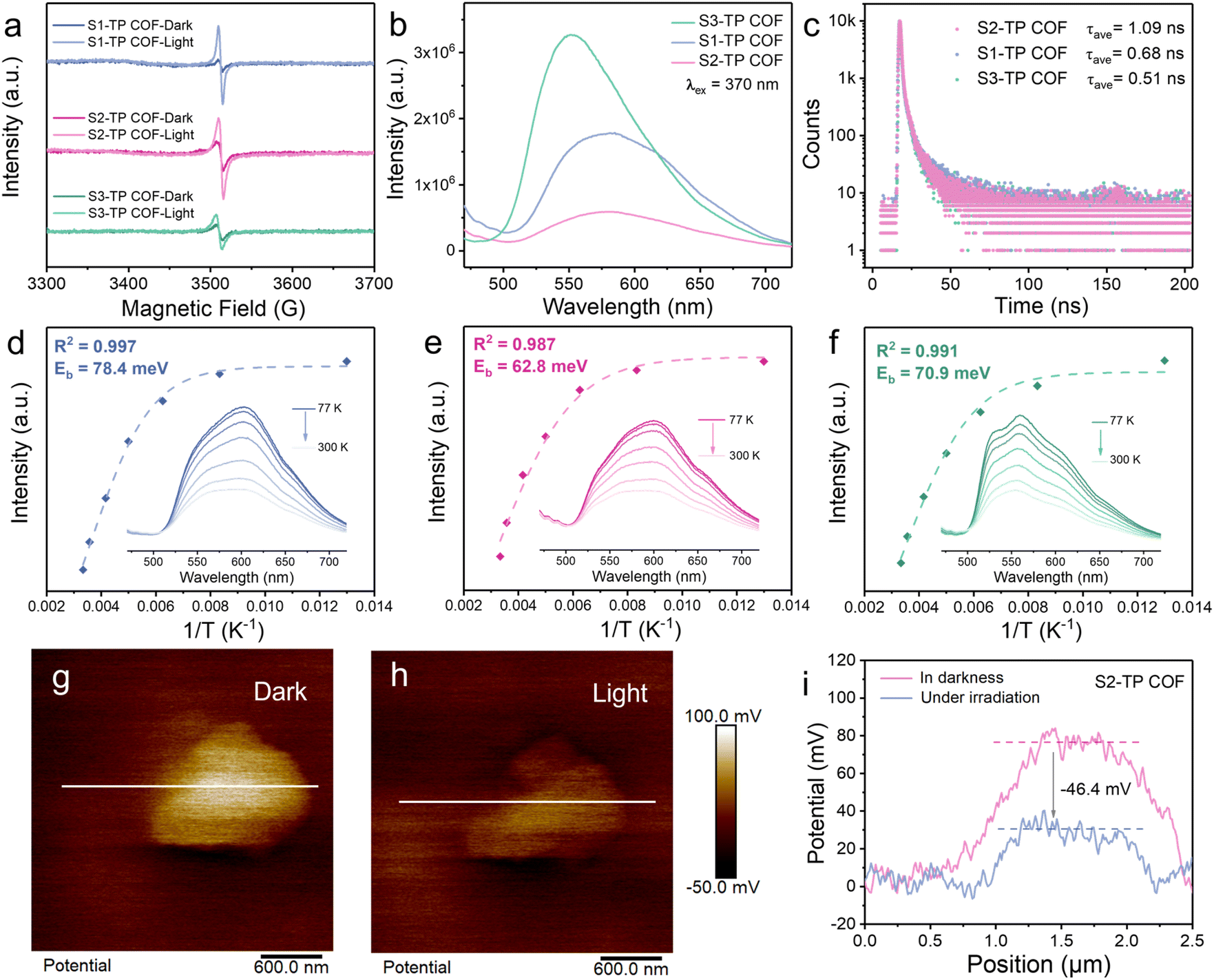 | ||
| Fig. 4 (a) EPR spectra (in the dark and upon photoirradiation, measurement after 5-minute illumination), (b) room-temperature steady-state photoluminescence spectra, and (c) time-resolved fluorescence spectra (TRFS) of TP-COFs. Integrated PL intensity as a function of temperature and the fitted exciton binding energy of S1-TP COF (d, blue), S2-TP COF (e, pink), and S3-TP COF (f, green) (inset: temperature-dependent PL spectra from 77 to 300 K under the excitation wavelength of 370 nm). Surface potential distribution images of S2-TP COF (g) in darkness and (h) under UV-light irradiation, as well as (i) the corresponding surface potential difference of the cross-section of S2-TP COF before and after photoirradiation. | ||
Given that photoinduced e−–h+ dissociation is critical to photocatalytic efficiency, exciton binding energies (Eb) of TP-COFs were used to evaluate their charge recombination tendency.44–46 By temperature-dependent fluorescence spectroscopy (77–300 K, Eb obtained by fitting emission signals of largest intensity using the Arrhenius equation, see the ESI† for fitting details), Eb values of S1-TP COF and S3-TP COF were calculated to be 78.4 and 70.9 meV respectively while that of S2-TP COF is the lowest at 62.8 meV (Fig. 4d–f and S47†), indicative of the most difficult e−–h+ recombination/effective e−–h+ dissociation in S2-TP COF. To visualize the transport of photogenerated carriers, in situ kelvin probe force microscopy (KPFM) was employed to observe the changes in surface potential of TP-COFs upon photoirradiation (Fig. 4g–i and S48†).47–49 Upon illumination, S2-TP COF darkened the most (Fig. 4i) and gave the most negative shift of surface potential by 46.4 mV (negative shifts of surface potential: S1-TP COF = 22.8 mV; S3-TP COF = 21.4 mV, Fig. S48d and S48l†), suggesting greater photoinduced aggregation of electrons on the surface of S2-TP COF than the other two analogues and indicating the strongest e−–h+ dissociation in S2-TP COF. These findings jointly highlight the best e−–h+ dissociation/separation in S2-TP COF and probably relate e−–h+ recombination suppression to the π-extended system, faster charge transport, and the enhanced flatness of the conjugated system through installing −CC−CC− motifs into the framework.
As Pt is a key player for the HER, detailed characterization was carried out on COFs after photocatalysis. Specifically, the energy-dispersive spectroscopy (EDS) mapping revealed a uniform distribution of C, O, N, and Pt elements (Fig. S49†), confirming the presence of Pt element in the sample of S2-TP COF after photocatalysis. As shown in the TEM images (Fig. S50†), numerous Pt single atoms are dispersed over the whole framework showing a corresponding lattice stripe spacing of about 0.11 nm, attributed to the (222) facet of Pt single atoms. Meanwhile, no aggregation of Pt clusters was found, demonstrating excellent dispersibility. The statistical histogram of randomly selected Pt single atoms (>100 populations) shows an average particle diameter of about 2.33 nm and a narrow size distribution consolidating the nanoscale of the Pt single atoms (Fig. S51†). To move further, XPS was adopted to probe the chemical environment of Pt, and the XPS Pt 4f spectra of TP-COFs (Fig. S52–S54†) all give signals with respect to Pt0 (4f7/2: ∼71.8 eV; 4f5/2: ∼75.3 eV) and Pt2+ (4f7/2: ∼73.5 eV; 4f5/2: ∼77.0 eV) and importantly the C 1s spectrum shows a signal at 290.3 eV, indicative of π–π* interaction between Pt atoms and S2-TP COF (Fig. 5a), and probably π-back bonding from Pt d orbitals to the π* orbital of the diacetylene bridge.40,50 Compared with the original S2-TP COF, the C−N peak at 399.82 eV in the S2-TP COF after photocatalysis was slightly blue-shifted by 0.21 eV (Fig. 5b), indicating lower electron density of N atoms originating from the interaction between ketoenamine and Pt2+.50,51 Notably, compared with S1-TP COF (0.42 wt%, monoacetylene site), lower Pt loadings of S2-TP COF (0.29 wt%, diacetylene site) estimated by inductively coupled plasma optical emission spectroscopy (ICP-OES) (Table S10†) demonstrate that the acetylene-rich active sites in the GDY-based framework promote the dispersion of the Pt atom, which is beneficial for the transfer of photogenerated carriers and hence HER performance.
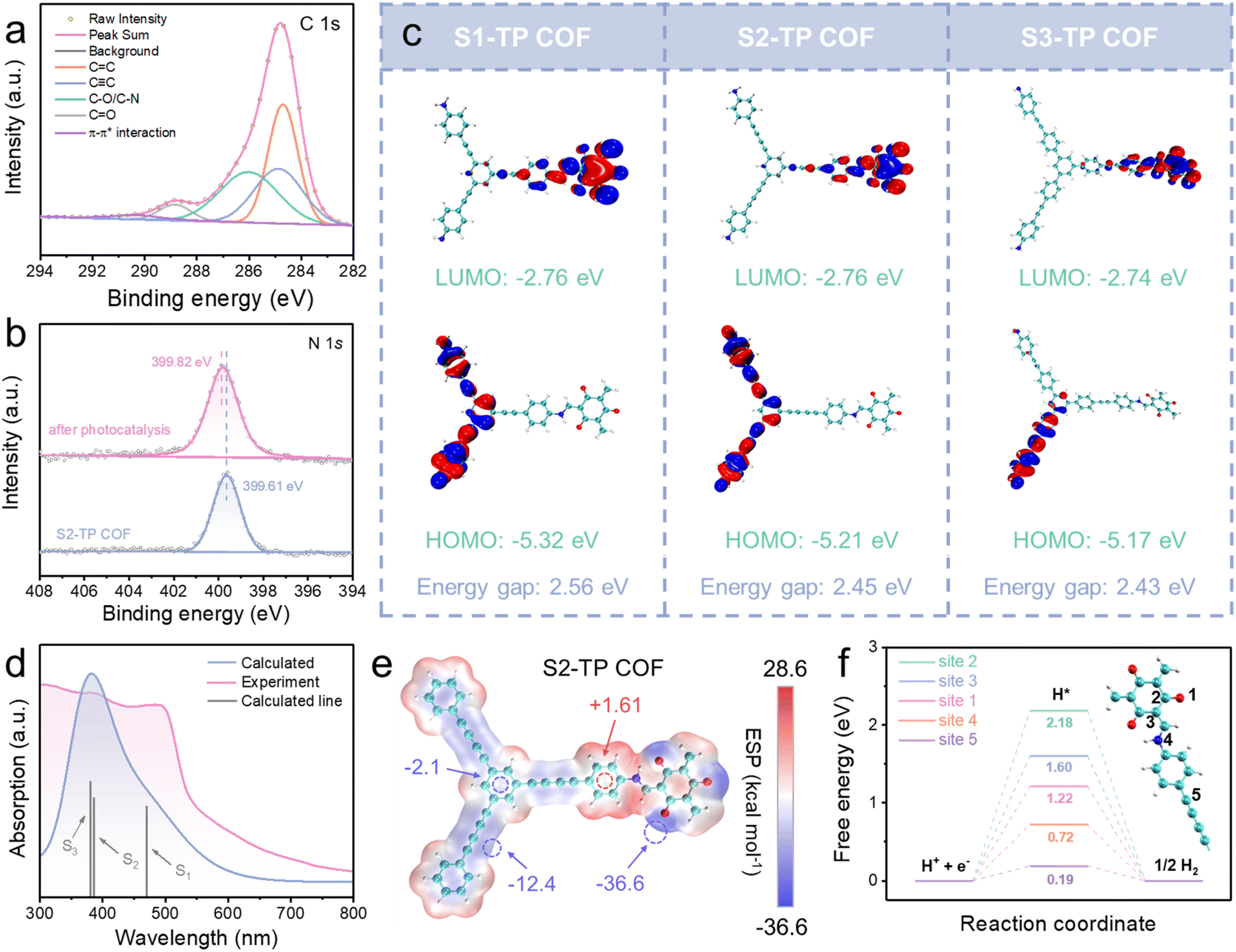 | ||
| Fig. 5 (a) C 1s and (b) N 1s XPS spectra of S2-TP COF powder after photocatalysis. (c) HOMOs and LUMOs of the theoretical models of TP-COFs (red and blue regions denote positive and negative respectively, isosurface values of 0.02 a.u.). (d) Calculated (blue) and experimental (pink) UV-Vis absorption spectra and main vertical transitions (grey) of S2-TP COF. (e) Electrostatic potential maps of the S2-TP COF model (negative and positive regions indicated in blue and red respectively). (f) Free energy plots for selected active sites of the S2-TP COF model (blue, cyan, red, and white spheres represent N, C, O, and H atoms). | ||
DFT calculations
To gain more insights into charge separation and redox active sites, density functional theory (DFT) calculation at the B3LYP/6-31G(d) level was conducted on theoretical models of TP-COFs (for details of Computational procedures, see the ESI†). The Multiwfn 3.8 and VMD 1.9.3 programs were adopted to analyze orbital composition and visualization, respectively.52,53 All atoms in the optimized models of S1-TP COF and S2-TP COF are coplanar featuring a high degree of planarity as well as electronic conjugation to exert strong interlayer π–π stacking for slower recombination and extended lifetime of excitons.54,55 As shown in Fig. 5c, HOMOs mainly lie in S1, S2, and S3 motifs while LUMOs centralize on the β-ketoenamine section, in agreement with the prediction of the D–A junction (Fig. 1). Non-overlapping between HOMOs and LUMOs in TP-COFs indicates good separation of photoinduced e−–h+ pairs which is beneficial to suppress e−–h+ recombination and hence improve HER efficiency. To correlate optical absorption and HER performance of TP-COFs, time-dependent density functional theory (TD-DFT) calculation was conducted using the best-performing S2-TP COF to scrutinize the nature of electronic transitions. As shown in Fig. 5d, the calculated absorption profile agrees well with the experimental one,56,57 and the absorption is mainly composed of dipole-allowed S0 → S1, S0 → S2 and S0 → S3 vertical transitions. Excited states S1, S2, and S3 are dominantly contributed by wavefunctions of HOMO−2 → LUMO, HOMO−1 → LUMO and the linear combination of HOMO−2 → LUMO+1/HOMO → LUMO+3 respectively. Fig. S55† reveals the isosurfaces of frontier orbitals contributing to vertical transitions (HOMO−2, HOMO−1, HOMO, LUMO, LUMO+1, and LUMO+3). Noteworthily, the main contributor, HOMO−2 → LUMO, to the most allowed lowest-energy vertical transition S0 → S1 (λabs = 470 nm) features intramolecular charge transfer (ICT) from one hand of the triazine linker to the TP core, leading to extension of light-harvesting to the visible spectral region by establishment of the D–A junction.To elucidate the HER mechanism, potential active sites were studied by a joint experimental–theoretical approach. The best-performing S2-TP COF was used for in situ IR spectroscopic measurement to monitor any change in functionalities. In pure H2O with Pt salt, significant weakening of −CC− stretching at 2215 cm−1 (Fig. S56†) was observed along with illumination, indicating that −CC−CC− motifs probably engage in Pt2+/Pt0 adsorption as well as formation of adsorbed hydrogen (H*) during the HER. Electrostatic potentials (ESPs) of TP-COFs of the optimized fragment structures show that electron density mainly distributes over acetylenic bridges and CO terminals (Fig. 5e and S57†), consolidating these sites to be potential adsorption sites for Pt2+/Pt0 and H2O.58,59 Noteworthily, rather more even distribution of ESPs in S3-TP COF probably leads to less apparent charge separation (Fig. 5e) and ineffective adsorption of Pt2+/Pt0 and H2O and these may account for the inferior HER rates.
Concerning photocatalytic HER, the Gibbs free energy change for hydrogen sorption (ΔGH*) represents a key indicator to evaluate the most energetically favorable HER active sites.60,61 The whole profile is composed of three states: initial state (H+ + e−), intermediate state (adsorbed hydrogen, H*), and final state (1/2H2) (Fig. S58†). Using S2-TP COF as a model, five potential active sites were locked on and found to give distinguishable ΔGH*. Importantly, the acetylene bridge (site 5) features the smallest ΔGH* (0.19 eV) suggesting site 5 to be the most kinetically favorable for the HER (Fig. 5f). Also worth noting are the imine linkage (site 4) and the keto-group (site 1) because these two sites possess smaller ΔGH* (0.72 and 1.22 eV) than the rest of the carbon-based sites (sites 2 and 3: 2.18 and 1.60 eV) (Fig. 5f), highlighting that sites 1 and 4 should not be excluded from energetically feasible HER sites. Note that considering the abovementioned ESPs (acetylene bridge and keto-group as plausible adsorption sites), both acetylene bridges and keto-groups probably serve as active sites for the HER. Experimental and theoretical evidence jointly reveals that introducing a GDY-like motif into the COF enhances HER performance by facilitating charge separation and lowering activation barriers.
Conclusions
Glaser–Hay coupling was adopted to prepare a diacetylene-bridged monomer, S2, which was used to construct a crystalline COF featuring a GDY-like network. By comparison with COFs built from linkers bearing a monoacetylene bridge (S1 & S3), the GDY-like COF (S2-TP COF) indeed exhibits better charge separation, faster charge transport, and suppressed charge recombination, as exemplified by its 2.7- and 9-fold enhancement in photocatalytic HER relative to S1-TP COF and S3-TP COF respectively. This work opens an avenue to crystalline GDY mimics merging the state-of-the-art design of COFs and excellent electronic properties as well as rich easy post-synthetic modification of GDY to actualize the rational design of a high-tier sustainable photocatalyst.Author contributions
L.-H. C., B. H., and J. H. conceived and designed the research. Z. L., S. D., and S. Y. performed the experiments and collected the data. Z. L., Q. L., and M. F. conducted the DFT calculations and analysed the data. Z. L., L.-H. C., B. H., and J. H. co-wrote the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Acknowledgements
Support from the National Natural Science Foundation of China (22371054, 22301045), the Foundation of Basic and Applied Basic Research of Guangdong Province (2020B1515120024, 2024A1515012801), and Science and Technology Planning Project of Guangdong Province (2021A0505030066, 2023A0505050164) is gratefully acknowledged. We also thank the Instrumental Analysis Centre of Guangdong University of Technology for SEM images, solid-state 13C NMR, EPR, and temperature-dependent and steady-state photoluminescence data collection.Notes and references
- G. Li, Y. Li, H. Liu, Y. Guo, Y. Li and D. Zhu, Chem. Commun., 2010, 46, 3256–3258 RSC.
- J. Li, L. Zhu, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2023, 62, e202301384 CrossRef CAS PubMed.
- Y. Fang, Y. Liu, L. Qi, Y. Xue and Y. Li, Chem. Soc. Rev., 2022, 51, 2681–2709 RSC.
- M. Wang, J. Pu, Y. Hu, Y. Zi, Z.-G. Wu and W. Huang, Adv. Funct. Mater., 2024, 34, 2308601 CrossRef CAS.
- J. Li, A. Slassi, X. Han, D. Cornil, M.-H. Ha-Thi, T. Pino, D. P. Debecker, C. Colbeau-Justin, J. Arbiol, J. Cornil and M. N. Ghazzal, Adv. Funct. Mater., 2021, 31, 2100994 CrossRef CAS.
- R. Wang, M. Shi, F. Xu, Y. Qiu, P. Zhang, K. Shen, Q. Zhao, J. Yu and Y. Zhang, Nat. Commun., 2020, 11, 4465 CrossRef CAS PubMed.
- F. Xu, K. Meng, B. Zhu, H. Liu, J. Xu and J. Yu, Adv. Funct. Mater., 2019, 29, 1904256 CrossRef CAS.
- L. Wang, Y. Wan, Y. Ding, S. Wu, Y. Zhang, X. Zhang, G. Zhang, Y. Xiong, X. Wu, J. Yang and H. Xu, Adv. Mater., 2017, 1702428 CrossRef PubMed.
- Z. Zhang, C. Wu, Q. Pan, F. Shao, Q. Sun, S. Chen, Z. Li and Y. Zhao, Chem. Commun., 2020, 56, 3210–3213 RSC.
- Z. Zuo, D. Wang, J. Zhang, F. Lu and Y. Li, Adv. Mater., 2019, 31, 1803762 CrossRef PubMed.
- W. Zhou, H. Shen, C. Wu, Z. Tu, F. He, Y. Gu, Y. Xue, Y. Zhao, Y. Yi, Y. Li and Y. Li, J. Am. Chem. Soc., 2019, 141, 48–52 CrossRef CAS PubMed.
- C. Bie, B. Cheng, W. Ho, Y. Li, W. Macyk, J. B. Ghasemif and J. Yu, Green Chem., 2022, 24, 5739–5754 RSC.
- X. Liu, Y. Zhao, J. Du and D. Wang, Sci. China Mater., 2024, 67, 729–751 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- Z. Yong and T. Ma, Angew. Chem., Int. Ed., 2023, 62, e202308980 CrossRef CAS PubMed.
- Z. Weng, Y. Lin, S. Guo, X. Zhang, Q. Guo, Y. Luo, X. Ou, J. Ma, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202310934 CrossRef CAS PubMed.
- N.-Y. Huang, Y.-T. Zheng, D. Chen, Z.-Y. Chen, C.-Z. Huang and Q. Xu, Chem. Soc. Rev., 2023, 52, 7949–8004 RSC.
- Z. Chen, J. Wang, M. Hao, Y. Xie, X. Liu, H. Yang, G. I. N. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2023, 14, 1106 CrossRef CAS PubMed.
- R. Luo, H. Lv, Q. Liao, N. Wang, J. Yang, Y. Li, K. Xi, X. Wu, H. Ju and J. Lei, Nat. Commun., 2021, 12, 6808 CrossRef CAS PubMed.
- J. Li, S.-Y. Gao, J. Liu, S. Ye, Y. Feng, D.-H. Si and R. Cao, Adv. Funct. Mater., 2023, 33, 2305735 CrossRef CAS.
- J.-N. Chang, J.-W. Shi, Q. Li, S. Li, Y.-R. Wang, Y. Chen, F. Yu, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e20230360 Search PubMed.
- P. Pachfule, A. Acharjya, J. Roeser, T. Langenhahn, M. Schwarze, R. Schomacker, A. Thomas and J. Schmidt, J. Am. Chem. Soc., 2018, 140, 1423–1427 CrossRef CAS PubMed.
- C.-R. Zhang, W.-R. Cui, R.-H. Xu, X.-R. Chen, W. Jiang, Y.-D. Wu, R.-H. Yan, R.-P. Liang and J.-D. Qiu, CCS Chem., 2021, 3, 168–179 CrossRef CAS.
- X.-R. Chen, W.-R. Cui, R.-P. Liang, C.-R. Zhang, R.-H. Xu, W. Jiang and J.-D. Qiu, ACS Appl. Bio Mater., 2021, 4, 6502–6511 CrossRef CAS PubMed.
- A. Khojastegi, A. Khosropour, S. Amirjalayer, I. Mosleh and A. Abbaspourrad, Adv. Funct. Mater., 2023, 33, 2309367 Search PubMed.
- L. A. Baldwin, J. W. Crowe, M. D. Shannon, C. P. Jaroniec and P. L. McGrier, Chem. Mater., 2015, 27, 6169–6172 CrossRef CAS.
- J. W. Crowe, L. A. Baldwin and P. L. McGrier, J. Am. Chem. Soc., 2016, 138, 10120–10123 CrossRef CAS PubMed.
- Y. Qian, Y. Han, X. Zhang, G. Yang, G. Zhang and H.-L. Jiang, Nat. Commun., 2023, 14, 3083 CrossRef CAS PubMed.
- J. Kou, G. Wang, H. Guo, L. Li, J. Fang, J. Ma and Z. Dong, Appl. Catal., B, 2024, 352, 124020 CrossRef CAS.
- B. C. Deibel, T. Strobel and V. Dyakonov, Adv. Mater., 2010, 22, 4097–4111 CrossRef PubMed.
- Z. Lin, Y.-H. Zhong, L. Zhong, X. Ye, L.-H. Chung, X. Hu, Z. Xu, L. Yu and J. He, JACS Au, 2023, 3, 1711–1722 CrossRef CAS PubMed.
- Y. Xia, W. Zhang, S. Yang, L. Wang and G. Yu, Adv. Mater., 2023, 35, 2301190 CrossRef CAS PubMed.
- C. Han, P. Dong, H. Tang, P. Zheng, C. Zhang, F. Wang, F. Huang and J.-X. Jiang, Chem. Sci., 2021, 12, 1796–1802 RSC.
- Z. Li, T. Deng, S. Ma, Z. Zhang, G. Wu, J. Wang, Q. Li, H. Xia, S.-W. Yang and X. Liu, J. Am. Chem. Soc., 2023, 145, 8364–8374 CrossRef CAS PubMed.
- G.-Q. Lai, Z. Jiang, H. Zhong, L.-H. Chung, N. Li and J. He, Chin. J. Struct. Chem., 2023, 42, 100090 CrossRef.
- W. Zhao, L. Luo, M. Cong, X. Liu, Z. Zhang, M. Bahri, B. Li, J. Yang, M. Yu, L. Liu, Y. Xia, N. D. Browning, W.-H. Zhu, W. Zhang and A. I. Cooper, Nat. Commun., 2024, 15, 6482 CrossRef CAS PubMed.
- K. Wang, Y. Zhong, W. Dong, Y. Xiao, S. Ren and L. Li, Angew. Chem., Int. Ed., 2023, 62, e202304611 CrossRef CAS PubMed.
- Y. Wang, Z. Hu, W. Wang, H. He, L. Deng, Y. Zhang, J. Huang, N. Zhao, G. Yu and Y.-N. Liu, Chem. Sci., 2021, 12, 16065–16073 RSC.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 62, e202305355 CrossRef CAS PubMed.
- P. Zhao, H. Jiang, H. Shen, S. Yang, R. Gao, Y. Guo, Q. Zhang and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202314121 CrossRef CAS PubMed.
- Y. Luo, C. Liu, J. Liu, X. Liu, Y. Zhou, X. Ou, B. Weng, J. Jiang and B. Han, Chem. Eng. J., 2024, 481, 148494 CrossRef CAS.
- M. Lu, S.-B. Zhang, M.-Y. Yang, Y.-F. Liu, J.-P. Liao, P. Huang, M. Zhang, S.-L. Li, Z.-M. Su and Y.-Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202307632 CrossRef PubMed.
- P.-J. Tian, X.-H. Han, Q.-Y. Qi and X. Zhao, Chem. Sci., 2024, 15, 9669–9675 RSC.
- G. Fu, D. Yang, S. Xu, S. Li, Y. Zhao, H. Yang, D. Wu, P. S. Petkov, Z.-A. Lan, X. Wang and T. Zhang, J. Am. Chem. Soc., 2024, 146, 1318–1325 CrossRef CAS PubMed.
- H. Liang, X. Bi, H. Chen, T. He, Y. Lin, Y. Zhang, K. Ma, W. Feng, Z. Ma, G. Long, C. Li, B. Kan, H. Zhang, O. A. Rakitin, X. Wan, Z. Yao and Y. Chen, Nat. Commun., 2023, 14, 4707 CrossRef CAS PubMed.
- W. Wang, H. Wang, X. Tang, J. Huo, Y. Su, C. Lu, Y. Zhang and C. Gu, Chem. Sci., 2022, 13, 8679–8685 RSC.
- M.-L. Xu, M. Lu, G.-Y. Qin, X.-M. Wu, T. Yu, L.-N. Zhang, K. Li, X. Cheng and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210700 CrossRef CAS PubMed.
- Y. You, S. Chen, J. Zhao, J. Lin, D. Wen, P. Sha, L. Li, D. Bu and S. Huang, Adv. Mater., 2024, 36, 2307962 CrossRef CAS PubMed.
- R. Chen, F. Fan, T. Dittrich and C. Li, Chem. Soc. Rev., 2018, 47, 8238–8262 RSC.
- P. Dong, Y. Wang, A. Zhang, T. Cheng, X. Xi and J. Zhang, ACS Catal., 2021, 11, 13266–13279 CrossRef CAS.
- W. Weng and J. Guo, Nat. Commun., 2022, 13, 5768 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- K. Xiong, Y. Wang, F. Zhang, X. Li and X. Lang, Appl. Catal., B, 2023, 322, 122135 CrossRef CAS.
- M. Wang, Z. Wang, M. Shan, J. Wang, Z. Qiu, J. Song and Z. Li, Chem. Mater., 2023, 35, 5368–5377 CrossRef CAS.
- Q. Zhou, Y. Guo and Y. Zhu, Nat. Catal., 2023, 6, 574–584 CrossRef CAS.
- Y. Yang, X. Chu, H.-Y. Zhang, R. Zhang, Y.-H. Liu, F.-M. Zhang, M. Lu, Z.-D. Yang and Y.-Q. Lan, Nat. Commun., 2023, 14, 593 CrossRef CAS PubMed.
- H. T. B. Pham, J. Y. Choi, S. Huang, X. Wang, A. Claman, M. Stodolka, S. Yazdi, S. Sharma, W. Zhang and J. Park, J. Am. Chem. Soc., 2022, 144, 10615–10621 CrossRef CAS PubMed.
- H. Yan, Y. Peng, Y. Huang, M. Shen, X. Wei, W. Zou, Q. Tong, N. Zhou, J. Xu, Y. Zhang, Y.-X. Ye and G. Ouyang, Adv. Mater., 2024, 36, 2311535 CrossRef CAS PubMed.
- J. Li, J. Zhou, X.-H. Wang, C. Guo, R.-H. Li, H. Zhuang, W. Feng, Y. Hua and Y.-Q. Lan, Angew. Chem., Int. Ed., 2024, 63, e202411721 CrossRef CAS PubMed.
- W. Chen, L. Wang, D. Mo, F. He, Z. Wen, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2020, 59, 16902–16909 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06633b |
| This journal is © The Royal Society of Chemistry 2025 |