
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarmonizing proton sponge and proton reservoir in conjugated microporous polymers for enhanced photocatalytic hydrogen peroxide production†
Shiyuan
Zhou
 a,
Wenwen
Chen
a,
Xiaobo
Luo
a,
Wenxiu
Guo
a,
Jingwen
Dong
a,
Yuxi
Liu
a,
Yuzhe
Zhang
a,
Danfeng
Wang
*a,
Zhongyu
Li
*ab and
Peiyang
Gu
*ac
a,
Wenwen
Chen
a,
Xiaobo
Luo
a,
Wenxiu
Guo
a,
Jingwen
Dong
a,
Yuxi
Liu
a,
Yuzhe
Zhang
a,
Danfeng
Wang
*a,
Zhongyu
Li
*ab and
Peiyang
Gu
*ac
aJiangsu Key Laboratory of Advanced Catalytic Materials and Technology, School of Petrochemical Engineering, Changzhou University, Changzhou, 213164, P. R. China. E-mail: wangdanfeng@cczu.edu.cn; gupeiyang0714@cczu.edu.cn
bSchool of Environmental Science and Engineering, Changzhou University, Changzhou, 213164, P. R. China. E-mail: zhongyuli@mail.tsinghua.edu.cn
cEngineering Laboratory of Functional Nano- and Microstructured Materials in Petroleum and Chemical Industry, School of Petrochemical Engineering, Changzhou University, Changzhou, 213164, P. R. China
First published on 15th April 2025
Abstract
Photocatalytic technology is highly sought-after for H2O2 production; however, the low selectivity between the oxygen reduction reaction (ORR) and water oxidation reaction (WOR) pathways is the primary factor limiting photocatalytic performance. Herein, a strategy that simulates a proton sponge by integrating aliphatic tertiary amines into conjugated microporous polymers (CMPs) to synthesize PPDI-N is reported. This method uses a carboxylic acid contaminant (2,4-dichlorophenoxyacetic acid, 2,4-D) as the proton reservoir to synergistically expedite the photosynthesis of H2O2via a selective one-step 2e− ORR. Importantly, with the aid of 2,4-D (acting as both a proton supplier and hole scavenger), the H2O2 production rate of PPDI-N is 4.4-fold higher than that in pure water, reaching 8.15 mmol g−1 within 4 h irradiation time, which is 58.2 times greater at the same pH value. By mimicking a photo Fenton-like process with the assistance of Fe2+, PPDI-N exhibits an unprecedented removal efficiency (>99%) for 300 ppm of 2,4-D within 60 min. As revealed by Kelvin probe force microscopy and electric surface potential calculations, an enhanced built-in electric field was established in PPDI-N. This work provides valuable guidance for advancing CMP photocatalysts and establishes an ideal scenario for enabling simultaneous photocatalytic mineralization of organic contaminants and H2O2 production.
Introduction
Hydrogen peroxide (H2O2), an environment-friendly oxidant and clean energy source, has found promising and critical applications in modern society.1 Its current industrial-scale production primarily relies on the traditional multistep anthraquinone method or direct synthesis from O2 and H2, both of which have various drawbacks, including high cost, high energy consumption, and numerous by-products. Therefore, the development of a safe, efficient, and cost-effective H2O2 production technology with environmental sustainability is of paramount importance.2 Solar-driven photocatalytic H2O2 production in the aqueous phase is considered a cost-effective and efficient alternative with great potential to realize light-to-chemical energy conversion. In light of this, the exploration of high-performance photocatalysts for H2O2 production is of key interest.3Amongst various heterogeneous photocatalysts, the emerging class of conjugated microporous polymers (CMPs) has garnered significant attention due to their considerable advantages in light absorption and photocatalytic applications, stemming from their variable and extended π-conjugation structures and the ease of tuning their optical and electronic properties by tailoring their molecular structures.4 The challenges in designing highly efficient CMP photocatalysts have limited current H2O2 photosynthesis at the micromolar level, far below the industrial utilization requirements. Therefore, it is essential to develop versatile approaches and elucidate structure–property relationships in order to enhance the photosynthesis of H2O2 with CMPs.
Mechanistically, the oxygen reduction reaction (ORR) has faster kinetics and occurs on a shorter timescale than the water oxidation reaction (WOR). Therefore, improving the selectivity of the ORR process is key to enhancing photocatalytic H2O2 production.5 Specifically, there are three ORR pathways: direct 2e− (2H+ + 2e− + O2 → H2O2), indirect 2e− (O2 + e− → ˙O2−; ˙O2− + e− + 2H+ → H2O2), and the undesirable 4e− process (O2 + 4H+ → 2H2O). All involve proton activity, a key factor that occurs in the proton-coupled electron transfer (PCET) process, which dictates the rate-determining step and governs ORR selectivity.6,7 It has been demonstrated that H2O2 formation requires a fast proton transfer rate and otherwise leads to the production of superoxide radicals (˙O2−). To improve proton activity and achieve optimal rates between proton transfer and O–O cleavage,8 proton transfer dynamics must be enhanced.7,9 Unlike previous approaches involving the protonation of the photocatalyst by protonic acid, which risks altering catalyst activity due to the loss of surface protons,7,10 we drew inspiration from the concept of a proton sponge, which is capable of capturing protons from the reaction medium.11 We envisioned that incorporating dimethylamine groups into CMPs could induce a proton sponge effect to accelerate the PCET process. In addition to the above advantage, the introduction of amino groups offers several advantages. (1) Improved dispersibility in aqueous solution due to increased hydrophilicity, enhancing the efficiency of surface reactions. (2) A more positive zeta potential, strengthening interactions with anionic organic pollutants. (3) Enhanced O2 adsorption ability, further improving O2 utilization rate. (4) Altered intrinsic photoelectrical properties, e.g., increased photocurrent and conductivity, collectively boosting overall photocatalytic performance.
Herein, we synthesized perylene diimide (PDI)-containing CMP photocatalysts with (PPDI-N)/without (PPDI-C) dimethylamine groups, respectively, with the aim of investigating the sponge effect on enhancing photocatalytic H2O2 production (Scheme 1). As a result, PPDI-N exhibited an H2O2 production rate of 1.65 mmol g−1 h−1, 21 times higher than PPDI-C (0.08 mmol g−1 h−1). Remarkably, the addition of the model pollutant 2,4-dichlorophenoxyacetic acid (2,4-D) afforded 10.24 mmol per g H2O2 production within 4 h under irradiation, 4.4 times higher than without additive, while achieving >99% removal efficiency within 75 min. Simulating a Fenton-like reaction for in situ utilization of low-concentration H2O2 (ref. 12) further enhanced 2,4-D removal through increased hydroxyl radical (˙OH) formation, which was confirmed by both quenching experiments and electron spin resonance (ESR) spectroscopy. Mechanistic investigations via radical quenching experiments and ESR revealed the positive role of the aliphatic amino group in generating reactive oxygen species (ROS), including singlet oxygen (1O2) and ˙O2−, and promoting the selective 2e− ORR process. Electro-optical studies highlighted the importance of the terminal tertiary amine group in improving the transient photocurrent response and reducing the electric impedance, thereby enhancing the photocatalytic performance. Density functional theory calculations were employed to further elucidate the reaction pathway. This work provides a versatile strategy for achieving a selective 2e− ORR process to boost H2O2 photosynthesis, offering an ideal platform for wastewater treatment through simultaneous production and utilization of H2O2.
 | ||
| Scheme 1 Schematic illustration of the strategies for enhancing photocatalytic H2O2 production. | ||
Results and discussion
PPDI-N and PPDI-C were fabricated step-wise via Suzuki coupling and oxidative coupling reactions, as depicted in Fig. 1a and Scheme S1,† and their morphology and chemical structures were comprehensively characterized. The structures of the PDI-based monomers were confirmed using 1H nuclear magnetic resonance (1H NMR) spectra, as shown in Fig. S1–S4.† Scanning electron microscopy (SEM) revealed porous irregular structures for both PPDI-N and PPDI-C (Fig. 1b and c). Contact angles showed that PPDI-N with a terminal amine group had a lower value (28°) than PPDI-C (64°), suggesting that PPDI-N is more hydrophilic (Fig. 1d). In the solid state 13C NMR (ss-13C NMR) spectra, the peak at 164 ppm indicated the presence of amide groups, while the peaks in the range of 103–148 ppm and 6–47 ppm can be attributed to aromatic carbons and alkane carbons, respectively (Fig. 1e).13 The peak at 55 ppm in PPDI-N and a faint peak at 57 ppm in PPDI-C corresponded to the CH2 group adjacent to the N atom of the amide.14 The subtle difference in the chemical shift of the CH2 group between PPDI-N and PPDI-C results from the end group of the alkane chains. The Fourier transform infrared (FT-IR) spectra of the monomers (PDI-NH2-Cz and PDI-CH3-Cz) and polymers (PPDI-N and PPDI-C) exhibited no significant differences, as polymerization/crosslinking exclusively occurred at carbazole moieties without any pronounced changes in the functional groups (Fig. 1f). The peaks in the range of 2600–3100 and 691–936 cm−1 can be indexed to the stretching and bending vibrations of –C–H bonds attributed to alkane chains and benzene rings, while the peaks at 1414–1540 and 1551–1752 cm−1 were attributed to the stretching vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the aromatic skeletons and CO groups, respectively.15 Concerning the X-ray photoelectron spectroscopy (XPS) (Fig. 1g, S5 and S6†), both PPDI-N and PPDI-C exhibited clear C 1s, N 1s, and O 1s signals. The C 1s high-resolution XPS spectra of PPDI-N primarily consist of three peaks at 284.8, 285.3, and 288.1 eV, corresponding to the aromatic C–C/CC, CO, and C–N bonds, respectively.15 Notably, the N 1s high-resolution spectrum of PPDI-C displayed a single peak at 400.0 eV attributed to amido groups, whereas, in PPDI-N, an additional peak of 402.8 eV was observed, which can be ascribed to the tertiary amine.14
C bonds in the aromatic skeletons and CO groups, respectively.15 Concerning the X-ray photoelectron spectroscopy (XPS) (Fig. 1g, S5 and S6†), both PPDI-N and PPDI-C exhibited clear C 1s, N 1s, and O 1s signals. The C 1s high-resolution XPS spectra of PPDI-N primarily consist of three peaks at 284.8, 285.3, and 288.1 eV, corresponding to the aromatic C–C/CC, CO, and C–N bonds, respectively.15 Notably, the N 1s high-resolution spectrum of PPDI-C displayed a single peak at 400.0 eV attributed to amido groups, whereas, in PPDI-N, an additional peak of 402.8 eV was observed, which can be ascribed to the tertiary amine.14
 | ||
| Fig. 1 (a) Schematic illustration of the ‘sponge effect’ of the CMP photocatalysts and the synthetic routes. SEM image of (b) PPDI-N and (C) PPDI-C. (d) Contact angles of PPDI-N and PPDI-C. (e) ss-13C NMR spectra of PPDI-N and PPDI-C. (f) FT-IR spectra of PPDI-N, PPDI-C, and their corresponding monomers. (g) The N 1s high-resolution XPS spectra of PPDI-N and PPDI-C. | ||
The N2 adsorption–desorption isotherms and pore size distributions of PPDI-N and PPDI-C are presented in Fig. S7,† and the corresponding data are summarized in Table S1.† The Brunauer–Emmett–Teller (BET) specific surface areas of PPDI-N and PPDI-C were determined to be 8.27 and 7.67 m2 g−1, respectively, while the total pore volumes are measured to be 0.027 and 0.025 cm3 g−1, respectively, suggesting their low surface areas. The pore sizes of PPDI-N and PPDI-C were primarily distributed in 1.74 and 1.76 nm, respectively, indicating the microporous properties of these CMPs. The thermal stability of both PPDI-N and PPDI-C was assessed via thermogravimetric analysis (TGA), as shown in Fig. S8a.† At the point of 10% weight loss, PPDI-N exhibited a temperature of 287 °C, while PPDI-C displayed a considerably higher temperature of 503 °C, thus confirming the excellent thermal stability of these CMPs. Moreover, X-ray diffraction analysis revealed that PPDI-N and PPDI-C exhibit broad peaks around 20° (Fig. S8b†), uncovering their amorphous nature, which is in line with previous observations in the SEM images.
After confirming the successful preparation of PPDI-N and PPDI-C by the above characterizations, the optical and electrochemical properties were investigated to gain insight into their photo-chemical behaviors. In the UV-visible diffuse absorption spectra (Fig. 2a), PPDI-N exhibited a more red-shifted absorption than PPDI-C, corresponding to the intramolecular charge transfer (ICT) induced by the electron-donating tertiary amine group. This increases the incident photon numbers, facilitating later photochemical reactions. The calculated band gaps of PPDI-N and PPDI-C using Tauc's equation (Fig. 2b) were found to be 1.62 and 1.66 eV, respectively. To obtain detailed band structures, Mott–Schottky profiles (Fig. 2c) which enabled the estimation of the conduction band (CB) positions were obtained, with PPDI-N and PPDI-C calculated to be −0.37 eV and −0.52 eV vs. the normal hydrogen electrode (NHE), respectively.16,17 The positive slopes of both polymers revealed n-type semiconducting traits.17 Thus, the detailed band structures were clarified and presented in Fig. 2d, and both PPDI-N and PPDI-C exhibited sufficient redox potential to feasibly access the electrochemical processes of O2 → ˙O2− (−0.33 V vs. NHE), O2 → H2O2 (0.28 V vs. NHE), and H2O → H2O2 (1.35 V vs. NHE). However, as yet, we were unable to realize the generation of ˙OH from the process of H2O → ˙HO (2.31 V vs. NHE). This indicates that the WOR is an unfavorable reaction, whereas the 2e− ORR is thermodynamically favorable, which aligns with the observations made in later mechanistic investigations. Furthermore, photocurrent measurements and electrochemical impedance analyses were conducted to characterize the photoelectrochemical properties of PPDI-N and PPDI-C. As shown in Fig. 2e, the photocurrent response of PPDI-N is significantly stronger than that of PPDI-C, with the photocurrent intensity of PPDI-N being 5.23 times higher than that of PPDI-C, indicating a higher amount of populated excitons acting as a driving force for photochemical processes.18 Electrochemical impedance analyses illustrated that PPDI-N possesses a pronouncedly smaller impedance radius compared to PPDI-C, indicating higher photogenerated carrier mobility (Fig. 2f). Therefore, from the above evaluations, it can be concluded that PPDI-N would exhibit better photocatalytic performance in H2O2 production due to its superior photochemical properties.
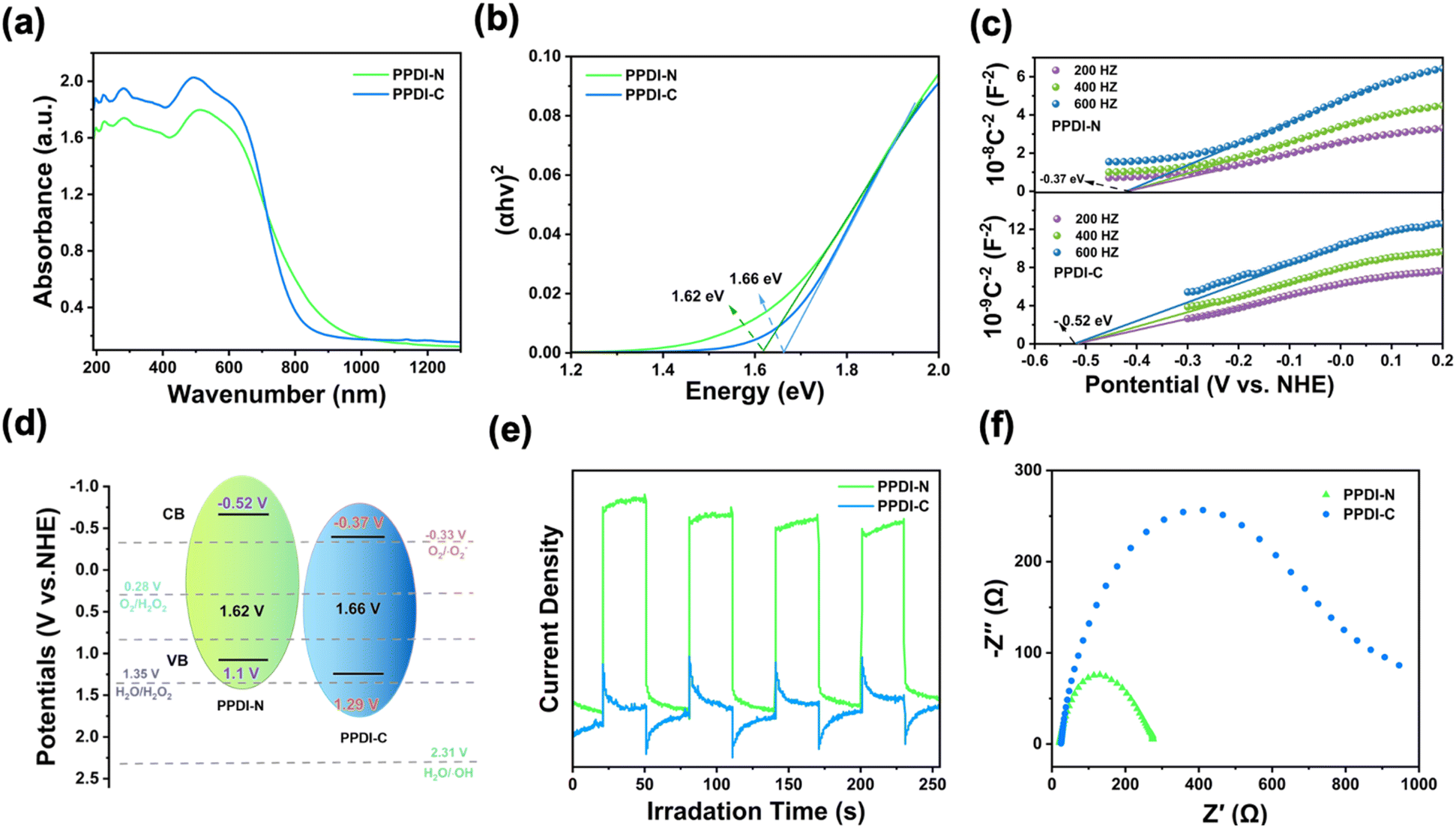 | ||
| Fig. 2 (a) The UV-vis diffuse reflectance absorption spectra at the solid state, (b) the Tauc plots, and (c) the Mott–Schottky plots of PPDI-N and PPDI-C obtained at 200, 400, and 600 Hz, respectively. (d) Detailed band structures of both PPDI-N and PPDI-C. The ORR and WOR-related potentials referred to the pH at 7. (e) Photocurrent responses and (f) electrochemical impedance of both PPDI-N and PPDI-C. | ||
After obtaining fundamental photo and chemical information about the as-prepared CMPs photocatalysts, we then focused on investigating their photocatalytic performance to further prove our concept. Initially, artificial photosynthesis of H2O2 was performed under different atmospheres of air, O2, and N2 within 1 h of irradiation (Fig. 3a and d). As anticipated, PPDI-C showed inferior photocatalytic activity to PPDI-N, which could barely produce H2O2 (0.02–0.03 mmol g−1 h−1). In contrast, PPDI-N was found to be efficient in generating H2O2 under both air (1.65 mmol g−1 h−1) and O2 (2.25 mmol g−1 h−1), but only produced trace amounts of H2O2 under N2, suggesting the absence of the 2e− WOR process and thus identifying the occurrence of the selective 2e− ORR process. The on/off-like effect on the photocatalytic production of H2O2 is likely due to the proton sponge effect, as PPDI-C without the ability to capture H+ from water resulted in the inactivation of the photocatalytic activity.
 | ||
| Fig. 3 (a) Time course and (d) comparisons at 60 min time scale for H2O2 production performance of PPDI-N and PPDI-C under air, N2, and O2, respectively. (b) The effect of pH and (e) the effect of concentration of 2,4-D on photocatalytic H2O2 production performance using PPDI-N under air conditions within 4 h. (c) Simulation of the Fenton reaction and (f) comparisons on 60 min time scale for 300 ppm of 2,4-D photodegradation and the assessment of Fe2+ concentrations. | ||
As previously stated, proton activity plays a key role in the PCET process, determining the selectivity of the 2e− ORR. It can therefore be postulated that the proton source may significantly affect the photocatalytic behavior. Thus, the influence of pH was investigated to evaluate the availability of readily dissociated free H+. However, beyond free H+, proton sources stored in organic molecules can also be utilized. Thus, we conceived the strategic approach of simultaneous photosynthesis of H2O2 and photodegradation of carboxylic acids. As such, this combination offers two key advantages. Firstly, carboxylic acids serve as H+ donors and h+ scavengers, enhancing the photosynthesis rate of H2O2. Secondly, they facilitate the utilization of the in situ-formed H2O2 in low concentration via a Fenton-like process. As shown in Fig. 3b, under air atmosphere, H2O2 production at different pH values, including pH = 7, 9, and 11 showed no significant difference (2.32–2.45 mmol g−1) over 4 h. However, a more acidic environment (pH = 3 and 5) significantly suppressed H2O2 production. The suppression likely results from excess free H+ binding with tertiary amines to form ammonium salts, thereby eliminating their proton sponge effect. More importantly, excess H+ readily consumes energetic photoelectrons, competing with H2O2 production. Interestingly, a 300 ppm solution of 2,4-D exhibited a pH value of 3.07 but resulted in a markedly enhanced H2O2 production rate of 8.15 mmol g−1 within 4 h irradiation time, which is 58.2 times higher than the previous result at pH = 3. Such a huge discrepancy may originate from the difference in the dissociation constants of HCl and 2,4-D (Kd (HCl) ≫ Kd (2,4-D)), with 2,4-D acting as a proton buffer.19 Further, to probe the intermolecular interaction between the photocatalyst PPDI-N and 2,4-D at various pH values, the zeta potentials were measured, giving 25.7, 10.3, −3.2, −8.42, and −25.7 mV at pH 3, 5, 7, 9, and 11, respectively (Fig. S9†). A more positive zeta potential indicates stronger electrostatic interactions with anionic 2,4-D. This stronger interaction is believed to promote surface reactions, particularly the PCET process, due to the increase of mass transfer efficiency, which aligns with the photocatalytic performance including H2O2 photosynthesis and the later photodegradation of 2,4-D. Therefore, the significantly enhanced proton activity in PCET results in vastly improved photocatalytic H2O2 production, indicating the synergistic effect between the tertiary amine as a proton sponge and 2,4-D as a proton buffer. The 2,4-D concentration profile depicted in Fig. 3e indicates that a higher concentration can lead to a higher H2O2 production rate, peaking at 10.24 mmol g−1 within 4 h irradiation in the concentration range of 500–900 ppm, which is a 4.4-fold increase compared to the water/air conditions (2.35 mmol g−1). Moreover, the reusability of PPDI-N was assessed by evaluating its photocatalytic H2O2 production performance over five cycles, with each cycle subjected to 4 h of irradiation (Fig. S10†). The H2O2 production amount exhibited a minimal decrease from 2.35 to 2.28 mmol g−1 after five cycles, demonstrating the excellent reusability of PPDI-N. Additionally, the FT-IR spectra and SEM images of PPDI-N before and after the cycles revealed that the chemical structure and porous skeleton were maintained, as evidenced by the similar porous irregular morphology and characteristic group peaks, further underscoring the outstanding structural stability of PPDI-N.
Given the reality that in situ generated H2O2 is present in low concentration and thus challenging to collect, the Fenton reaction offers a potential solution to bridge this gap via the process of H2O2 + Fe2+ → ˙HO + OH− + Fe3+.20 Prior to this, photocatalytic degradation experiments of PPDI-N toward methyl orange (MO), bisphenol A (BPA) and 2,4-D were performed, as shown in Fig. S11–S17,† which demonstrates the excellent photodegradation performance of PPDI-N for anionic organic pollutants. Subsequently, the simulation of Fenton reaction by adding Fe2+ showed a markedly accelerated degradation rate of 300 ppm of 2,4-D in 40 min with over 98% removal (Fig. 3c). Variations in the concentration of Fe2+ (30, 50, 70, and 100 ppm) showed subtle differences in the kinetics and the residual rate was further decreased from 11% to 2% (Fig. 3f), highlighting the effectiveness of the Fenton process in removing contaminants. To the best of our knowledge, this photodegradation performance has surpassed currently reported photocatalysts, either via the Fenton process or not, and the comparison is listed in Table S2† to further underscore the superior performance of PPDI-N. Moreover, unlike the conventional Fenton reaction with Fe3+ → Fe2+ as the rate-determining step, the active e− generated from the photo-self-Fenton reaction can effectively reduce Fe3+ to Fe2+ to expedite the overall photocatalytic process to fulfill the catalytic cycle. To prove this, we experimented with adding Fe3+ (Fig. S18†), and the results were similar to those of Fe2+. These results further substantiated the effectiveness and efficiency of the photo-self-Fenton reaction in utilizing the low concentration of H2O2 to boost the rate of photodegradation of organic contaminants.
To gain a better understanding of the role of different active species during the photocatalytic H2O2 production, and the origin of the selectivity of different ORR pathways, radical quenching experiments were performed. Scavengers including AgNO3, MeOH, tert-butylalcohol (tBA), p-benzoquinone (pBQ), and β-carotene were employed to capture e−, h+, ˙OH, ˙O2− and 1O2, respectively. As depicted in Fig. 4a, PPDI-C was not further studied due to its ineffectiveness in producing H2O2. As for PPDI-N, the H2O2 production was significantly inhibited with the addition of AgNO3, indicating that e− are essential to the production of H2O2, and combined with the fact that pBQ exerts no influence at all on the overall production rate, the selective direct 2e− ORR process can therefore be identified. The other scavengers of tBA and MeOH both showed small contributions to the H2O2 production, hence h+ and ˙OH were not the principal reactive species, i.e., the WOR pathway can be regarded as a minor reaction pathway. In addition, PPDI-N led to significant decomposition of H2O2 under the N2 atmosphere, and because of the escape of O2 and the lack of O2 participating, the selective 2e− ORR process can no longer proceed (Fig. 4b and S19†). This result further confirmed that the primary process for the PPDI-N photocatalyst is the 2e− selective ORR. To further track the oxygen pathway during the process of photocatalytic H2O2 generation, we conducted an isotope labelling experiment using H218O and 18O2.21 As depicted in Fig. S20,† in the GC-MS of the headspace after irradiation for 1 h using H218O as the solution, peaks at m/z = 34 (16O18O) and 36 (18O2) were observed for PPDI-N, whereas virtually no peaks at these masses were detected for PPDI-C. This indicates that PPDI-N facilitates a 4e− WOR pathway for the formation of 18O2. Therefore, by detecting m/z = 36 in the headspace after adding MnO2 to the aqueous phase of PPDI-N suspension, it can be concluded that H218O2 was produced by the stored 18O2. However, the produced H2O2 rapidly decomposed for PPDI-N (Fig. 4b), and this is the primary reason for the distinct behaviour of peaks at m/z = 34 and 36 observed in the isotope labelling experiment for the two photocatalysts, despite their similar photocatalytic H2O2 production performance under an N2 atmosphere. Moreover, we also conducted the isotopic labelling experiment using 18O2 as the reaction atmosphere. As shown in Fig. S21,† for PPDI-N, an obvious peak at m/z = 36 attributed to 18O2 was observed in the mixed gases that came from the decomposition of formed H2O2, whereas no peak was detected for PPDI-C. The result was consistent with the photocatalytic performance of the two photocatalysts. The peak intensity at m/z = 36 was much stronger than that achieved in the experiment using H218O as the reaction solution, this indicates that the reduction of 18O2 was the dominant pathway for the photosynthesis of H218O2.
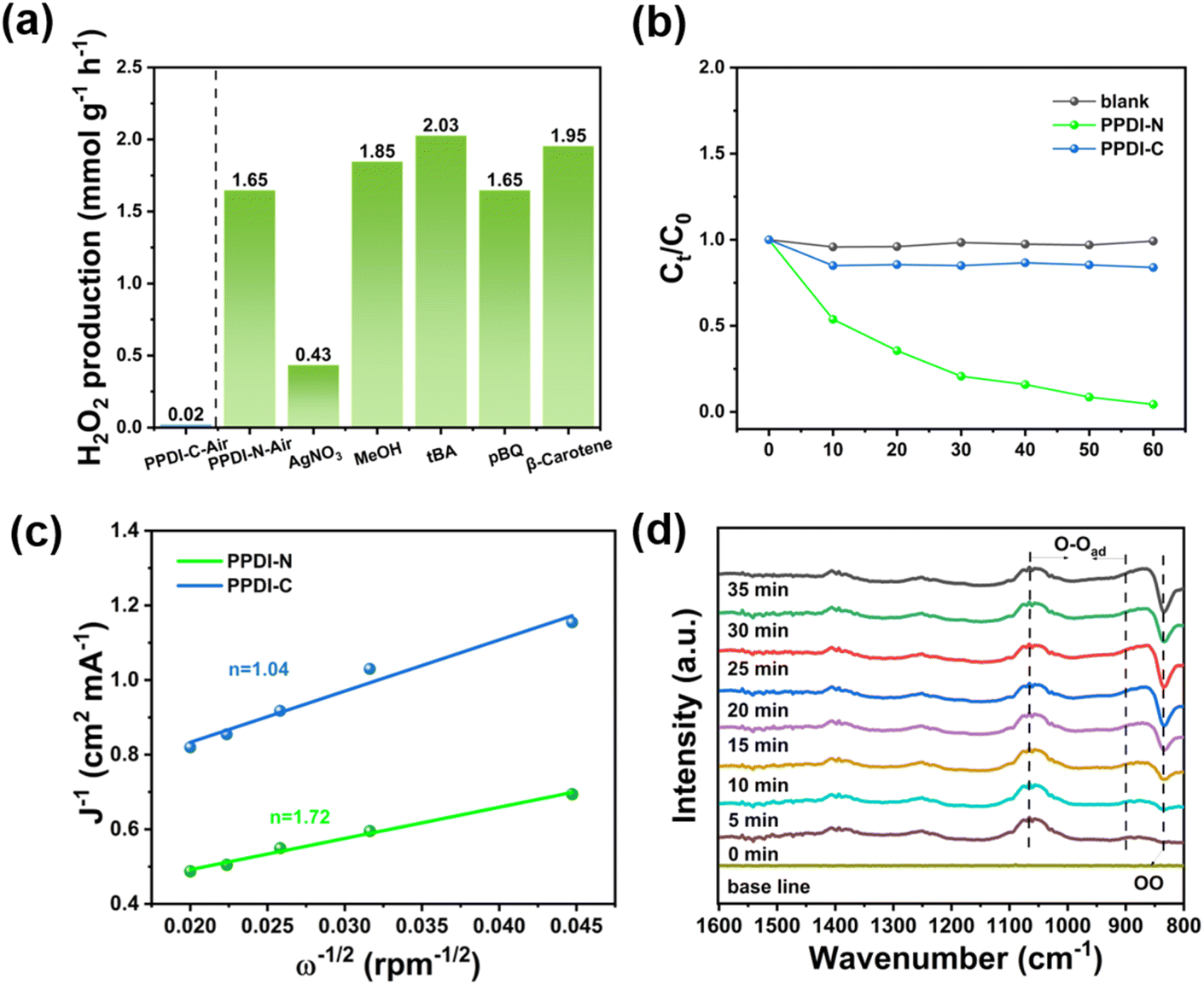 | ||
| Fig. 4 (a) The H2O2 production rate of PPDI-N and PPDI-C in the presence of different scavengers. (b) Photocatalytic decomposition of 10 mM H2O2 with/without photocatalysts under N2 atmosphere. (c) The Koutecky–Levich plots of PPDI-N and PPDI-C obtained from rotating disk electrode experiments. (d) Time-dependent in situ DRIFTS spectra for the photocatalytic system of PPDI-N. | ||
To clarify the average electron transfer during the photocatalytic H2O2 production, we conducted rotating disk electrode experiments (Fig. 4c and S22†). The average electron transfer number (n) was found to be 1.72 and 1.04 for PPDI-N and PPDI-C, respectively. This observation validated the exceptional selectivity of PPDI for the 2e− ORR process over PPDI-C, highlighting the significance of the tertiary amine group. Moreover, to further provide intuitive and accurate evidence of the selective 2e− process, time-dependent in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were performed. As shown in Fig. 4d, in the dark reaction phase, PPDI-N exhibited a weak vibration peak, and along with the increase of irradiation time, gradually augmented vibrational peaks were observed in the range of 897–1064 cm−1, corresponding to the vibration of –O–O–, which indicates that the initial adsorption of O2 and the *O2 is mainly attributed to the Yeager-type.22,23 Such O2 adsorption conformation is conceived to be the direct 2e− ORR process, as the other Pauling-type conformation of *O2 leads to the population of *OOH, a species that is highly active in breaking the bonding between the photocatalyst and O atom, facilitating the formation of ˙O2− to undergo an unfavorable inefficient one-step 2e− ORR pathway. In addition, another set of distinct peaks gradually appeared at 833 cm−1, which could be attributed to the formation of 1,4-endoperoxide species,23,24 further confirming the Yeager-type conformation in the O2 adsorption. Importantly, to ensure that high selectivity effectively enhances the efficiency of H2O2 formation, an equilibrium between the population of Yeager-type *O2 species and sufficiently active protons from the PCET process is essential. Accordingly, a 2,4-D-assisted PPDI-N photocatalytic system was presented as a proof-of-concept, showcasing a synergistic effect between a proton sponge and a proton reservoir.
Further investigations into the ROSs involved in this reaction were gained through ESR experiments conducted on PPDI-N and PPDI-C suspensions in the presence of DMPO (for ˙O2−) and TEMP (for 1O2), respectively. As illustrated in Fig. 5a and b, no signal was detected under dark conditions. Under irradiation, both PPDI-N and PPDI-C exhibited enhanced intensity with characteristic triplet signals (intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, AH = 1.61 mT), attributed to the TEMP-1O2 adduct.25 Additionally, the photocatalysts also showed the typical six peaks (AH = 1.04 mT, AN = 1.46 mT) belonging to DMPO-˙O2− with prolonged irradiation time.26 The electron paramagnetic resonance (EPR) spectra of PPDI-N and PPDI-C at the solid state were used to further differentiate the abilities of the photocatalysts to generate radicals. PPDI-N exhibited enhanced intensity and two transitions with central peaks at 335.1 mT (g value of 2.004) and 335.8 mT (g value of 2.001), respectively, originating from the spin density on the nitrogen atoms of the tertiary amines (Fig. 5c). In contrast, the EPR spectrum of PPDI-C remained nearly unchanged both in the dark and under visible light irradiation. The intensity enhancement under visible light irradiation indicated the presence of radical cations on the tertiary amines, which enhances the absorption of O2.27 Therefore, both PPDI-N and PPDI-C are photoactive and were capable of generating reactive oxygen species such as ˙O2− and 1O2. This seems paradoxical to the previously assumed direct 2e− mechanism. To further quantify their abilities in producing ˙O2− and 1O2, nitro blue tetrazolium (NBT) and 1,3-diphenylisobenzofuran (DPBF) were employed in order to exclusively trap ˙O2− and 1O2, respectively (Fig. S23 and S24†). The results indicated that PPDI-N produced a higher concentration of ˙O2− (760.0 μM g−1 h−1) than PPDI-C (140.8 μM g−1 h−1), and a slightly higher concentration of 1O2 (332.6 mM g−1 h−1) than PPDI-C (320.9 mM g−1 h−1), following the trend of the photosynthesis of H2O2 and the photodegradation of contaminants. As demonstrated before, the direct 2e− ORR is the principal process, and the formation of ˙O2− and 1O2 is disadvantageous as they contribute to the paradoxical stepwise 2e− ORR process. Thus this may be the reason for the limited H2O2 production rate. However, thanks to the enhanced PCET process due to the synergistic effect of the proton sponge and proton reservoir, the direct 2e− ORR is much preferred. This is evident in Fig. 3e, where the presence of 2,4-D as the additive leads to a steep increase in H2O2 production, in contrast to the flat slope observed under standard conditions in the time course of H2O2 production. This can be ascribed to the more significant roles of ˙O2− and 1O2 in photodegradation, rather than favoring H2O2 photosynthesis.
:1:1, AH = 1.61 mT), attributed to the TEMP-1O2 adduct.25 Additionally, the photocatalysts also showed the typical six peaks (AH = 1.04 mT, AN = 1.46 mT) belonging to DMPO-˙O2− with prolonged irradiation time.26 The electron paramagnetic resonance (EPR) spectra of PPDI-N and PPDI-C at the solid state were used to further differentiate the abilities of the photocatalysts to generate radicals. PPDI-N exhibited enhanced intensity and two transitions with central peaks at 335.1 mT (g value of 2.004) and 335.8 mT (g value of 2.001), respectively, originating from the spin density on the nitrogen atoms of the tertiary amines (Fig. 5c). In contrast, the EPR spectrum of PPDI-C remained nearly unchanged both in the dark and under visible light irradiation. The intensity enhancement under visible light irradiation indicated the presence of radical cations on the tertiary amines, which enhances the absorption of O2.27 Therefore, both PPDI-N and PPDI-C are photoactive and were capable of generating reactive oxygen species such as ˙O2− and 1O2. This seems paradoxical to the previously assumed direct 2e− mechanism. To further quantify their abilities in producing ˙O2− and 1O2, nitro blue tetrazolium (NBT) and 1,3-diphenylisobenzofuran (DPBF) were employed in order to exclusively trap ˙O2− and 1O2, respectively (Fig. S23 and S24†). The results indicated that PPDI-N produced a higher concentration of ˙O2− (760.0 μM g−1 h−1) than PPDI-C (140.8 μM g−1 h−1), and a slightly higher concentration of 1O2 (332.6 mM g−1 h−1) than PPDI-C (320.9 mM g−1 h−1), following the trend of the photosynthesis of H2O2 and the photodegradation of contaminants. As demonstrated before, the direct 2e− ORR is the principal process, and the formation of ˙O2− and 1O2 is disadvantageous as they contribute to the paradoxical stepwise 2e− ORR process. Thus this may be the reason for the limited H2O2 production rate. However, thanks to the enhanced PCET process due to the synergistic effect of the proton sponge and proton reservoir, the direct 2e− ORR is much preferred. This is evident in Fig. 3e, where the presence of 2,4-D as the additive leads to a steep increase in H2O2 production, in contrast to the flat slope observed under standard conditions in the time course of H2O2 production. This can be ascribed to the more significant roles of ˙O2− and 1O2 in photodegradation, rather than favoring H2O2 photosynthesis.
 | ||
| Fig. 5 The ESR signals of (a) TEMP-1O2 and (b) DMPO-˙O2− of PPDI-N and PPDI-C in solution under simulated sunlight irradiation. (c) The EPR spectra of PPDI-N and PPDI-C in the solid state. The surface potential images and corresponding CPD profiles under dark and light conditions for (d–f) PPDI-N and (g–i) PPDI-C, respectively. | ||
In addition to understanding the internal photochemical process of the photocatalysts, mechanistic investigations were conducted to probe the surface processes that are essential to the overall photocatalytic process. Kelvin probe force microscopy (KPFM) was employed to measure the distribution of surface potential and charges of the photocatalysts at the nanometer scale. As shown in Fig. 5d–i, potential variations were found in both PPDI-N and PPDI-C within the scanning range, and the potential of PPDI-N under dark conditions was greater than PPDI-C, suggesting the more intense intramolecular charge transfer of PPDI-N than PPDI-C. Upon irradiation, evident potential changes of PPDI-N were observed, while only minor potential changes in PPDI-C were found. Such a potential increase is caused by photocarrier separation induced changes in the surface potential in n-type semiconductors. Additionally, the contact potential difference (CPD) measured by the potential difference between the tip and the photocatalyst showed that PPDI-N possesses greater ΔCPD (0.023 V) than PPDI-C (0.011 V). In principle, a higher ΔCPD indicates a more effective separation of photo-induced electrons and holes, which is believed to originate from a built-in electric field.28 Hence, it can be concluded that a stronger built-in electric field was successfully constructed in PPDI-N bearing tertiary amine groups compared to PPDI-C with only aliphatic chains.
To further elucidate the reaction mechanism of the selective direct 2e− ORR process for photocatalytic H2O2 production, density functional theory (DFT) calculations were carried out. Given the nucleophilic nature of O2 molecules, the optimal adsorption site is generally located at the electron-deficient center of the photocatalyst. Therefore, the highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) of PPDI-N were calculated to simulate the excited state charge transfer. In addition, the electrostatic surface potential (ESP) distribution was calculated to precisely determine the active site for O2 adsorption. As depicted in Fig. 6a–d, for both PPDI-N and PPDI-C, the HOMOs were mainly distributed on the carbazole segment but partially distributed on the PDI motif, while the LUMOs were primarily delocalized on the PDI part, suggesting that the electrons are transferred from the carbazole to the PDI. The HOMO and LUMO values of both PPDI-N and PPDI-C were quite close. However, in the ESP results, the dipole moments of PPDI-N and PPDI-C exhibited pronounced differences, showing that PPDI-N has a markedly larger dipole moment (2.08 D) than PPDI-C (Fig. 6e and f). This enhancement refers to the improved built-in electric field, aligning well with the conclusions drawn from KPFM (Fig. 5d–i). A stronger built-in electric field is thought to enhance photocatalytic performance.29 Furthermore, DFT calculations were employed to investigate the free energy of different transition states in the ORR process (Fig. 6g). Initially, the Gibbs free energy (ΔG) value of *O2 was calculated to be 0.12 eV, this step with an uphill energy barrier can be identified as the rate-determining step of the entire direct 2e− ORR process (* + O2 → *O2 → *HOOH → H2O2). *HOOH is an important species that can be spontaneously produced by simultaneously accepting two protons from the 2e− ORR process (O2 + 2H+ + 2e− → *HOOH) and its free energy was estimated to be −1.36 eV.
 | ||
| Fig. 6 The (a and b) LUMOs, (c and d) HOMOs, and (e and f) ESP of PPDI-N and PPDI-C, respectively. (g) Free-energy diagrams for the reduction of O2 to H2O2 on PPDI-N. (h) Primary steps of photocatalytic H2O2 production for PPDI-N. | ||
Finally, the desorption of H2O2 could be readily realized due to its low value (−1.52 eV). Based on the above results of characterizations and theoretical calculations, a feasible pathway was proposed (Fig. 6h). The two C atoms belonging to the carbonyl groups of the PDI moiety serve as the O2 adsorption sites and form a Yeager-type conformation. Then, this conformation can simultaneously afford two protons (partly from the protonated tertiary amine groups) and two electrons to produce *HOOH and further generate H2O2. Therefore, the efficient reduction from O2 to H2O2 catalyzed by PPDI-N is illustrated in detail.
Conclusions
We demonstrate that selective and efficient photocatalytic H2O2 production via 2e− ORR can be achieved by leveraging the proton sponge and sponge reservoir effects introduced through aliphatic terminal amines in CMP frameworks and carbonyl acid-based organic contaminants (e.g. 2,4-D). In addition to enhancing the PCET process, the incorporation of amine groups immensely improves photoinduced charge separation and transport and promotes surface reactions due to improved dispersibility. All these advantages contribute to an overall enhancement in photocatalytic performance. Accordingly, in the presence of 2,4-D, there is a 4.4-fold enhancement in photocatalytic H2O2 production compared to standard conditions. Furthermore, a Fenton-like process bridges low concentrations of the in situ produced H2O2 and enhances photodegradation efficiency, facilitating remarkable efficiency for removing 300 ppm of 2,4-D (>99%) within 60 min. Importantly, this work highlights a promising strategy for the simultaneous photosynthesis of H2O2 and mineralization of organic contaminants, providing new insights for the advancement of wastewater treatment.Data availability
All data associated with this study are available in the article and ESI.†Author contributions
S. Zhou: data curation, investigation, writing – original draft. W. Chen: data curation, formal analysis. X. Luo, W. Guo: data curation. J. Dong, Y. Liu: supervision, validation. Y. Zhang: supervision. D. Wang: conceptualization, data curation, writing – review & editing. Z. Li: resources, visualization. P. Gu: funding acquisition, methodology, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Major Basic Research Project of the Natural Science Foundation of the Jiangsu Higher Education Institutions (24KJA150001), Applied Basic Research Programs of Science and Technology Commission Foundation of Jiangsu Province (No. BK20231340), Changzhou Introduction Program of Innovative Leading Talents (No. CQ20220111) and the National Natural Science Foundation of China (51803143). The authors also want to thank the postgraduate research & practice innovation program of Jiangsu province. We also thank the Analysis and Testing Center, NERC Biomass of Changzhou University for the assistance with NMR analysis.Notes and references
- (a) A. G. Fink, R. S. Delima, A. R. Rousseau, C. Hunt, N. E. LeSage, A. Huang, M. Stolar and C. P. Berlinguette, Nat. Commun., 2024, 15, 766 CrossRef CAS PubMed; (b) J. Qiu, D. Dai and J. Yao, Coordin. Chem. Rev., 2024, 501, 215597 CrossRef CAS; (c) J. Du, S. Jiang, R. Zhang, P. Wang, C. Ma, R. Zhao, C. Cui, Y. Zhang and Y. Kang, ACS Catal., 2023, 13, 6887–6892 CrossRef CAS; (d) C. Ye, Y. Zhou, H. Li and Y. Shen, Green Chem., 2023, 25, 3931–3939 RSC.
- (a) D. Dai, J. Qiu, G. Xia, Y. Tang, Q. Liu, Y. Li, B. Fang and J. Yao, Small, 2024, 2403268 CrossRef CAS; (b) W. Wei, L. Zou, J. Li, F. Hou, Z. Sheng, Y. Li, Z. Guo and A. Wei, J. Colloid. Inter. Sci., 2023, 636, 537–548 CrossRef CAS PubMed; (c) Y. Yang, C. Wang, Y. Li, K. Liu, H. Ju, J. Wang and R. Tao, J. Mater. Sci. Technol., 2024, 200, 185–214 CrossRef CAS.
- (a) Y. Zhi, Z. Wang, H. Zhang and Q. Zhang, Small, 2020, 2001070 CrossRef CAS; (b) X. Xia, J. Feng, Z. Zhong, X. Yang, N. Li, D. Chen, Y. Li, Q. Xu and J. Lu, Adv. Funct. Mater., 2024, 34, 2311987 CrossRef CAS; (c) Y. Mao, H. Fan, H. Yao and C. Wang, Sci. Total Environ., 2024, 917, 170357 CrossRef CAS PubMed; (d) S. Xu and Q. Zhang, Mater. Today. Energy, 2021, 20, 100635 CrossRef CAS.
- (a) J. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120(4), 2171–2214 CrossRef CAS PubMed; (b) Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC; (c) D. Zhou, K. Zhang, S. Zou, X. Li and H. Ma, J. Mater. Chem. A, 2024, 12, 17021 RSC; (d) Q. Dong, T. Naren, L. Zhang, W. Jiang, M. Xue, X. Wang, L. Chen, C. S. Lee and Q. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202405426 CrossRef CAS PubMed.
- (a) D. Liu, X. Yang, P. Chen, X. Zhang, G. Chen, Q. Guo, H. Hou and Y. Li, Adv. Mater., 2023, 35, 2300655 CrossRef CAS PubMed; (b) W. Kang, R. Wei, H. Yin, D. Li, Z. Chen, Q. Huang, P. Zhang, H. Jing, X. Wang and C. Li, J. Am. Chem. Soc., 2023, 145, 3470–3477 CrossRef CAS; (c) J. Tang, J. R. Durrant and D. R. Klug, J. Am. Chem. Soc., 2008, 13, 13885–13891 CrossRef.
- (a) B. Lei, W. Cui, P. Chen, L. Chen, J. Li and F. Dong, ACS Catal., 2022, 12, 9670–9678 CrossRef CAS; (b) A. Rana, Y.-M. Lee, X. Li, R. Cao, S. Fukuzumi and W. Nam, ACS Catal., 2021, 11, 3073–3083 CrossRef CAS.
- Q. Zhu, L. Shi, Z. Li, G. Li and X. Xu, Angew. Chem., Int. Ed., 2024, 63, e202408041 CrossRef CAS PubMed.
- E. C. Tse, C. J. Barile, N. A. Kirchschlager, Y. Li, J. P. Gewargis, S. C. Zimmerman, A. Hosseini and A. A. Gewirth, Nat. Mater., 2016, 15, 754–759 CrossRef CAS PubMed.
- P. Zhang, J. Zhang, D. Wang, F. Zhang, Y. Zhao, M. Yan, C. Zheng, Q. Wang, M. Long and C. Chen, Appl. Catal. B: Environ., 2022, 318, 121749 CrossRef CAS.
- S. Wang, Z. Xie, D. Zhu, S. Fu, Y. Wu, H. Yu, C. Lu, P. Zhou, M. Bonn and H. I. Wang, Nat. Commun., 2023, 14, 6891 CrossRef CAS PubMed.
- (a) P. Pradhan, S. Moktan, A. Biswas, A. Das, R. Lenka and P. K. Kancharla, Org. Lett., 2024, 26, 3563–3568 CrossRef CAS; (b) L. Fan, C.-Y. Liu, P. Zhu, C. Xia, X. Zhang, Z.-Y. Wu, Y. Lu, T. P. Senftle and H. Wang, Joule, 2022, 6, 205–220 CrossRef CAS; (c) R. V. Benjaminsen, M. A. Mattebjerg, J. R. Henriksen, S. M. Moghimi and T. L. Andresen, Mol. Ther., 2013, 21, 149–157 CrossRef CAS PubMed; (d) L. M. Vermeulen, S. C. De Smedt, K. Remaut and K. Braeckmans, Eur. J. Pharm. Biopharm., 2018, 129, 184–190 CrossRef CAS PubMed.
- (a) X. Cheng, L. Liang, J. Ye, N. Li, B. Yan and G. Chen, Sci. Total Environ., 2023, 888, 164086 CrossRef CAS PubMed; (b) C. Zhang, N. Ding, Y. Pan, L. Fu and Y. Zhang, Chinese Chem. Lett., 2024, 109579 CrossRef CAS; (c) H. H. Vigil-Castillo, E. J. Ruiz-Ruiz, K. López-Velázquez, L. Hinojosa-Reyes, O. Gaspar-Ramírez and J. L. Guzmán-Mar, Chemosphere, 2023, 338, 139585 CrossRef CAS PubMed; (d) Y. Ahmed, J. Zhong, Z. Yuan and J. Guo, J. Hazard. Mater., 2022, 430, 128408 CrossRef CAS PubMed.
- S. Huang, H. Zhang, W. Li, L. Liu, J. Xu, M. Chong, J. Li and Y. Zhu, Appl. Catal. B-Environ. Energy., 2024, 347, 123790 CrossRef CAS.
- Y. Zhang, Y. Liu, Y. Zhang, X. Hu, K. Jiang, P. Gu, S. Zhou and Z. Li, Sep. Purif. Technol., 2024, 347, 127639 CrossRef CAS.
- S. H. Goudar, D. S. Ingle, R. Sahu, S. Kotha, S. K. Reddy, D. J. Babu and V. R. Kotagiri, ACS Appl. Polym. Mater., 2023, 5, 2097–2104 CrossRef CAS.
- L. Jing, D. Wang, M. He, Y. Xu, M. Xie, Y. Song, H. Xu and H. Li, J. Hazard. Mater., 2021, 401, 123309 CrossRef CAS PubMed.
- M. Xu, D. Li, K. Sun, L. Jiao, C. Xie, C. Ding and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 16372–16376 CrossRef CAS PubMed.
- Y.-Y. Tang, X. Luo, R.-Q. Xia, J. Luo, S.-K. Peng, Z.-N. Liu, Q. Gao, M. Xie, R.-J. Wei and G.-H. Ning, Angew. Chem., Int. Ed., 2024, 63, e202408186 CrossRef CAS PubMed.
- G. Pandey, S. Koley, R. Talukdar and P. K. Sahani, Org. Lett., 2018, 20, 5861–5865 CrossRef CAS PubMed.
- (a) J. Liu, B. Wang, Z. Li, Z. Wu, K. Zhu, J. Zhuang, Q. Xi, Y. Hou, J. Chen and M. Cong, J. Alloy Compd., 2019, 771, 398–405 CrossRef CAS; (b) M. Jing, H. Zhao, L. Jian, C. Pan, Y. Dong and Y. J. Zhu, Hazard. Mater., 2023, 449, 131017 CrossRef CAS PubMed; (c) T. Lu, H. Zhao, L. Jian, R. Ji, C. Pan, G. Wang, Y. Dong and Y. Zhu, Environ. Res., 2023, 222, 115361 CrossRef CAS PubMed.
- (a) J. Cheng, W. Wang, J. Zhang, S. Wan, B. Cheng, J. Yu and S. Cao, Angew. Chem., Int. Ed., 2024, 63, e202406310 CrossRef CAS PubMed; (b) L. Zhang, C. Wang, Q. Jiang, P. Lyu and Y. Xu, J. Am. Chem. Soc., 2024, 146, 29943–29954 CrossRef CAS PubMed.
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata and C. Su, Nat. Catal., 2021, 4, 374–384 CrossRef CAS.
- Y. Luo, B. Zhang, C. Liu, D. Xia, X. Ou, Y. Cai, Y. Zhou, J. Jiang and B. Han, Angew. Chem., Int. Ed., 2023, 6, e202305355 Search PubMed.
- R. Schulte-Ladbeck, A. Edelmann, G. Quintas, B. Lendl and U. Karst, Anal. Chem., 2006, 78, 8150–8155 CrossRef CAS PubMed.
- K. Wang, J. Dai, G. Zhan, L. Zhao, R. Wang, X. Zou, J. Wang, Q. Zheng, B. Zhou, R. Zhao, Y. Zhang, W. Lian, Y. Yao and L. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202412209 CrossRef CAS PubMed.
- M. Sun, M. Murugananthan, Z. Zhou, Y. Shen, Y. Zhang and X. Wang, Chem. Eng. J., 2024, 486, 150245 CrossRef CAS.
- Y. Liu, Z. A. Page, D. Zhou, V. V. Duzhko, K. R. Kittilstved, T. Emrick and T. P. Russell, ACS Cent. Sci., 2018, 4, 216–222 CrossRef CAS PubMed.
- (a) P. Zhang, Y. Tong, Y. Liu, J. J. M. Vequizo, H. Sun, C. Yang, A. Yamakata, F. Fan, W. Lin and X. Wang, et al. , Angew. Chem., Int. Ed., 2020, 59, 16209–16217 Search PubMed; (b) Y. Zhang, Q. Cao, A. Meng, X. Wu, Y. Xiao, C. Su and Q. Zhang, Adv. Mater., 2023, 3, 2306831 CrossRef PubMed.
- L. Jian, Y. Dong, H. Zhao, C. Pan, G. Wang and Y. Zhu, Appl. Catal. B-Environ. Energy, 2024, 342, 123340 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06603k |
| This journal is © The Royal Society of Chemistry 2025 |