
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond Fang's fury: a computational study of the enzyme–membrane interaction and catalytic pathway of the snake venom phospholipase A2 toxin†
Juliana
Castro-Amorim
 a,
Alexandre V.
Pinto
a,
Ashis K.
Mukherjee
b,
Maria J.
Ramos
a and
Pedro A.
Fernandes
*a
a,
Alexandre V.
Pinto
a,
Ashis K.
Mukherjee
b,
Maria J.
Ramos
a and
Pedro A.
Fernandes
*a
aLAQV/Requimte, Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Rua do Campo Alegre, s/n, 4169-007 Porto, Portugal. E-mail: pafernan@fc.up.pt
bInstitute of Advanced Study in Science and Technology, Vigyan Path Garchuk, Paschim Boragaon, Guwahati-781035, Assam, India
First published on 2nd January 2025
Abstract
Snake venom-secreted phospholipases A2 (svPLA2s) are critical, highly toxic enzymes present in almost all snake venoms. Upon snakebite envenomation, svPLA2s hydrolyze cell membrane phospholipids and induce pathological effects such as paralysis, myonecrosis, inflammation, or pain. Despite its central importance in envenomation, the chemical mechanism of svPLA2s is poorly understood, with detrimental consequences for the design of small-molecule snakebite antidotes, which is highly undesirable given the gravity of the epidemiological data that ranks snakebite as the deadliest neglected tropical disease. We study a member of the svPLA2 family, the Myotoxin-I, which is part of the venom of the Central American pit viper terciopelo (Bothrops asper), a ubiquitous but highly aggressive and dangerous species responsible for the most problematic snakebites in its habitat. Furthermore, PLA2 enzymes are a paradigm of interfacial enzymology, as the complex membrane–enzyme interaction is as important as is crucial for its catalytic process. Here, we explore the detailed interaction between svPLA2 and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 POPC/POPS membrane, and how enzyme binding affects membrane structure and dynamics. We further investigated the two most widely accepted reaction mechanisms for svPLA2s: the ‘single-water mechanism’ and the ‘assisted-water mechanism’, using umbrella sampling simulations at the PBE/MM level of theory. We demonstrate that both pathways are catalytically viable. While both pathways occur in two steps, the single-water mechanism yielded a lower activation free energy barrier (20.14 kcal mol−1) for POPC hydrolysis, consistent with experimental and computational values obtained for human PLA2. The reaction mechanisms are similar, albeit not identical, and can be generalized to svPLA2 from most viper species. Furthermore, our findings demonstrate that the sole small molecule inhibitor currently undergoing clinical trials for snakebite is a perfect transition state analog. Thus, understanding snake venom sPLA2 chemistry will help find new, effective small molecule inhibitors with anti-snake venom efficacy.
:1 POPC/POPS membrane, and how enzyme binding affects membrane structure and dynamics. We further investigated the two most widely accepted reaction mechanisms for svPLA2s: the ‘single-water mechanism’ and the ‘assisted-water mechanism’, using umbrella sampling simulations at the PBE/MM level of theory. We demonstrate that both pathways are catalytically viable. While both pathways occur in two steps, the single-water mechanism yielded a lower activation free energy barrier (20.14 kcal mol−1) for POPC hydrolysis, consistent with experimental and computational values obtained for human PLA2. The reaction mechanisms are similar, albeit not identical, and can be generalized to svPLA2 from most viper species. Furthermore, our findings demonstrate that the sole small molecule inhibitor currently undergoing clinical trials for snakebite is a perfect transition state analog. Thus, understanding snake venom sPLA2 chemistry will help find new, effective small molecule inhibitors with anti-snake venom efficacy.
Introduction
Epidemiological background
Snakebite envenoming is currently regarded as the most lethal neglected tropical disease (NTD) by the World Health Organization (WHO). With 81–138 thousand fatalities each year and 400 thousand amputations and other permanent disabilities, snakebite is responsible for more deaths than all other NTDs combined.1,2 Most snakebite victims are from low- to middle-income regions where medical resources are scarce and health systems are ineffective, mainly in southeast Asia, sub-Saharan Africa, Latin America, and parts of Oceania.3,4Antibody-based antivenom is the only available but centenary therapy that can effectively prevent or reverse the toxic effects of viper venom.4–8 However, it is quite expensive and needs to be transported and stored within a cold chain, which can be challenging. Therefore, this therapy is barely accessible to envenomed patients, particularly those from the rural areas of developing countries.6–9 Moreover, a significant number of victims (20–82%) experience adverse reactions to these antivenoms, which prompts them to stick with the traditional medicines, postponing the administration of targeted treatment.10,11 As a result, the WHO released a work plan in May 2019 to halve envenoming-related mortality and morbidity by 2030.8,12,13 This strategic initiative involves various objectives, including researching an effective and accessible treatment based on small-molecule inhibitors that can be administered outside hospital settings and stored in local communities.12–14 However, the poor atomic-level understanding of the molecular mechanisms of snake venom toxins limits the development of small-molecule toxin inhibitors.
The fearsome terciopelo (Bothrops asper) and its venomous cocktail
The medically relevant snakes belong almost exclusively to two snake families, the elapids and the viperids.15 The latter family is responsible for a substantial portion of snakebite fatalities. Proteomic analysis has revealed the presence of four prominent families of proteins in most viper venoms: Zn2+-dependent metalloprotease (41–44%), secreted phospholipase A2 (29–45%), serine protease (4–18%) and L-amino acid oxidase (5–9%)16–18 (Fig. S1,† Section I).The venom of each species comprises several svPLA2 enzyme isoforms, subdivided into acidic and basic. Due to its superior toxicity in vivo, the basic isoforms have been the focus of many studies.16,19 Basic PLA2 isoforms induce various toxic effects, whereas acidic isoforms have higher catalytic activity but do not produce obvious toxic consequences. The most common pathological effect of viper svPLA2 is myotoxicity. Other possible toxic effects include paralysis, anticoagulant action, blistering, edema-inducing activity, inflammation, and pain,17,20–22 thus playing a vital role in prey immobilization brought on by envenoming.
sPLA2s of elapids and viperids share a significant sequence identity of ≈60% with structural similarity ranging from 60% to 90%. They possess a highly conserved active site, and, expectedly, a similar reaction mechanism.23–25 Still, they differ in cell type affinities, with the viper venom attacking mostly myocytes and the elapid venom targeting motor neurons, even though exceptions are abundant.15,26 Accordingly, svPLA2s are categorized into two main groups: group IA, typically found in the venom of elapids, and group IIA, predominantly found in viperids. While both groups exhibit an overall conserved architecture, group IIA enzymes feature a more elongated C-terminal loop with 6 to 7 additional amino acids compared to group IA, influencing substrate specificity and interactions with phospholipid bilayers.25
The specific svPLA2 EC 3.1.1.4 (ref. 21, 27 and 28) studied here is found in the venom of a Latin American large pit viper, the B. asper (Viperidae family), commonly called “Terciopelo”,4,28–30 which is responsible for 50–80% of ophidic envenomations within its habitat.3,4,19,21,30 Some well-known researchers, like Picado31 and Bolaños32 have commented on its reputation as a fearsome, aggressive, agile, and unpredictable species.33 Bites from this viper may also result in death, disabilities, abortion, and other permanent sequelae due to the venom myotoxic and hemotoxic effects. In this and other vipers, the integrity of the plasma membrane of skeletal muscle fibers (sarcolemma) may also be disrupted.20,21,34 The Myotoxin-I (Mt-I) isoform, a basic Asp48 enzyme (svPLA2 numbering system), is a general representative of the large svPLA2 family of enzymes, and the focus of this paper.
Snake venom-secreted phospholipase A2 (svPLA2) Mt-I
svPLA2 myotoxins are small (14–15 kDa) globular enzymes that belong to a large family of enzymes catalyzing the hydrolysis of membrane phospholipids at the sn-2 ester bond (Fig. S2†) in a Ca2+-dependent fashion.27,35 This process yields lysophospholipids and fatty acids, precursors of several signaling molecules involved in biological processes. In addition, this leads to an increase in Ca2+ in the muscle cells, followed by the release of K+ and ATP.21,36 As a result, a cascade of events is initiated, including the loss of mitochondrial function, the efflux of muscle-derived cytosolic components, widespread proteolysis, focal hypercontraction of myofilaments, and other degenerative processes that are yet to be identified.20,21,26,37,38B. asper Mt-I (BaMt-I) consists of 122 amino acid residues with seven disulfide bridges.16,39 This enzyme forms homodimers that are not covalently bonded in their native state nor glycosylated.16,20 Additionally, it adopts the classical group IIA sPLA2 fold with (i) a N-terminal α-helix 1 (α1) followed by a short helix, (ii) a Ca2+-binding loop, (iii) two antiparallel disulfide-linked α-helixes 3 and 4 (α3 and α4), where the active site cleft is deeply buried, (iv) two-stranded antiparallel β-sheet (β-wing) and (v) a flexible C-terminal loop (Fig. 1, left).40–42
 | ||
| Fig. 1 Cartoon representation of the svPLA2 tertiary structure. The active site residues (His47, Asp48, Tyr51, Tyr64, and Asp89) are shown as grey sticks, and the Ca2+ ion as a brown sphere. The disulfide bonds are shown in yellow. A close-up view of crucial residues of the catalytic network and Ca2+-binding loop is shown on the right. | ||
Its catalytic network includes the most structurally conserved feature among sPLA2 enzymes, the His47/Asp89 dyad, which may interact through a low-barrier hydrogen bond involving the His47 nitrogen atom (Nε2) and the Asp89 carboxyl oxygen atom (Oδ1).43–45 It also contains an Asp48, a crucial element for the binding of the Ca2+ cofactor, while Asp89, Tyr51, and Tyr64 aid in maintaining the proper His47 position for the reaction via hydrogen bonding.46 The Ca2+-binding loop is formed by the Tyr27, Gly29, and Gly31 backbone carbonyl groups and by the Asp48 β-carboxyl group (Fig. 1, right).20,27,41
Membrane binding as a key to PLA2 catalysis
Despite being water-soluble, all PLA2s exhibit minimal activity towards non-aggregated substrates available in the bulk solvent. However, this behavior changes dramatically upon binding cell membranes, triggering a critical event in the PLA2s reaction cycle, known as “interfacial activation”.43,47 Hypothetically, it may facilitate the diffusion of the substrate along the hydrophobic channel for optimal catalytic activity (“substrate model”), lead to a change in the PLA2's conformation making its active site more accessible for substrate interaction (“enzyme model”),25,47 or both. The result is a dramatic increase (up to 10000-fold) in the reaction rate. Despite decades of research, the molecular basis for this rate enhancement is yet to be understood.
The non-covalent adsorption to a membrane is thought to rely on electrostatic and hydrophobic interactions between residues on the enzyme's interfacial binding surface (IBS) and cell membrane components.48 For example, at a neutral pH, both human group IIA secretory phospholipase A2 (hGIIA sPLA2) and the cotton mouth snake (A. piscivorus piscivorus) venom sPLA2 (App-D48 sPLA2) exhibit higher activity towards membranes enriched in negatively charged phospholipids than towards zwitterionic membranes.49,50 Charge reversal mutagenesis decreases the binding affinity of App-D48 sPLA2 and hGIIA sPLA2 to negatively charged surfaces. However, charge-compensation mutants, in which positively charged lysine residues were replaced with methionine residues, exhibited a modest increase in catalytic activity at the zwitterionic interface compared to the anionic one.47,49,50
The exact mechanism by which svPLA2 myotoxins exert their toxic effects has also been the subject of debate for many years. While their catalytic activity is essential for myotoxicity, some PLA2s with high catalytic activity exhibit minimal toxicity in living creatures. This observation has led to the hypothesis that the toxicity of these enzymes goes beyond simple catalysis and may involve additional, non-enzymatic, membrane-damaging effects (i.e., packing defects, density, lipid protrusions, etc.).51 These toxic effects would depend on specific molecular domains other than the catalytic site, where the aforementioned phenomenon would play a substantial role.52
Bridging the gap: exploring PLA2 putative reaction mechanisms
The PLA2 family of enzymes is known to operate using two potential catalytic mechanisms – the “single-water mechanism” and the “assisted-water mechanism”.53 These mechanisms were proposed based on the analysis of X-ray structures of several PLA2s derived from different organisms, which were complexed to inhibitors, substrate- and transition state-analogs,54–58 alongside mutagenesis studies and the knowledge of chemical mechanisms employed by related enzymes.35,59–63 The primary distinction lies in the number of water molecules involved.The single-water mechanism proposed initially by Verheij in 1980,59 involves the presence of a hepta-coordinated Ca2+ ion formed by the two carboxylate oxygens of the Asp48 sidechain, three backbone carbonyl oxygen atoms of the Ca2+-binding loop (Tyr27, Gly29, and Gly31),43,64,65 and two water molecules.64 Both water molecules are displaced upon substrate binding, specifically by the phospholipid phosphate head and sn-2 carbonyl oxygen.43,64 The His47 residue, polarized by the His47(Nε)–(Oγ)Asp89 hydrogen bond, deprotonates a catalytic water molecule, which is not coordinated by the Ca2+ ion, via the Nδ atom. Consequently, the generated hydroxide performs a nucleophilic attack at the sn-2 ester bond of the phospholipid substrate forming a tetrahedral oxyanion intermediate.43,53,66,67 The negatively charged tetrahedral intermediate is stabilized by the Ca2+ ion and by hydrogen bonding to the backbone amine of Gly29.64 Ultimately, the tetrahedral intermediate collapses upon deprotonation of His47 NδH+ by the sn-2 oxygen of the lysophospholipid leaving group (Fig. 2).43,65,66 Once the products are released, three water molecules move into the active site, from which two coordinate the Ca2+ ion, and the third replenishes the active cycle for nucleophilic attack at the subsequent turnover.43,53
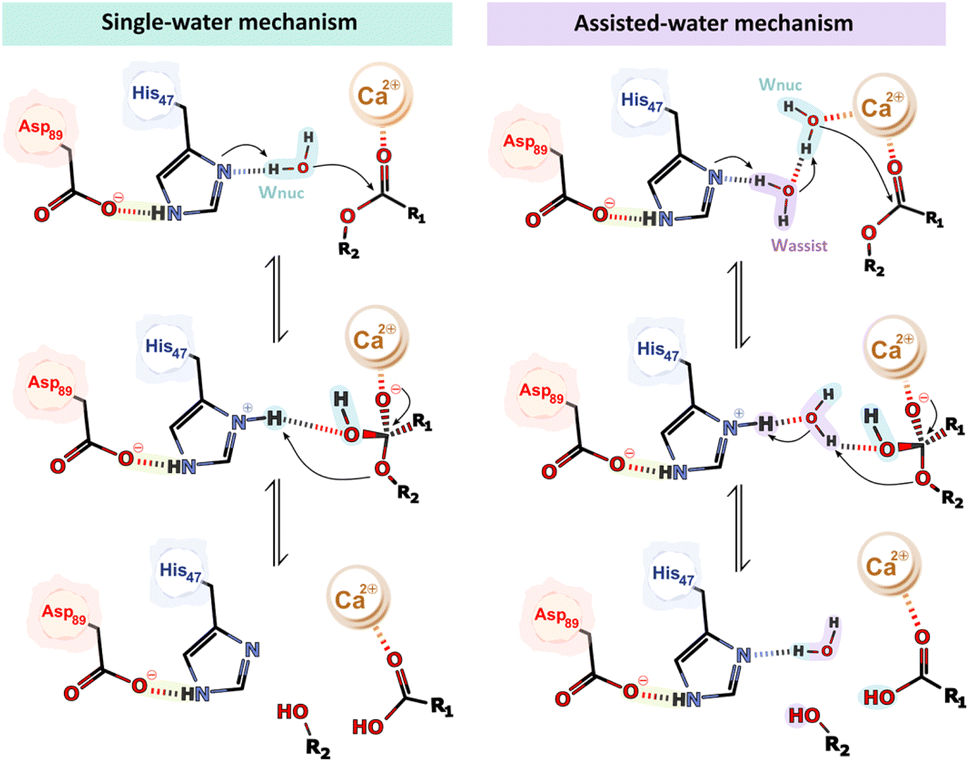 | ||
| Fig. 2 Schematic representation of the single- and the assisted-water mechanism. | ||
In 1998, Yu proposed an alternate mechanism called the “assisted-water mechanism” that involves the participation of two water molecules.68 It includes the nucleophilic water (in analogy with the first mechanism) in the Ca2+ inner part coordination sphere (Wnuc), hydrogen-bonded to an “assisting water” in the Ca2+ outer part coordination sphere (Wassist), which, in turn, is hydrogen-bonded to the His47 Nδ atom.69 According to Yu and coworkers, in the first step of the mechanism, the Ca2+-bound Wnuc deprotonates the bridging Wassist, which is in turn deprotonated by the His47 Nδ atom, thereby facilitating the reaction (Fig. 2). Subsequently, the generated Ca2+-bound hydroxide nucleophilically attacks the substrate sn-2 carbon atom. This reaction leads to the formation of a tetrahedral intermediate with a Ca2+-coordinated oxyanion. During the collapse of the tetrahedral intermediate, His47 protonates Wassist, which, in turn, protonates the departing alkoxy oxygen.68
This research aims to investigate and disclose the effects of enzyme–membrane binding in the structures of the enzyme and membrane, in addition to the dynamics and the underlying catalytic mechanisms of svPLA2 compared to human synovial sPLA2 using QM/MM MD simulations.
Both human synovial PLA2 found in arthritic fluids and BaMt-I adopt the group IIA sPLA2 fold, with the former being non-toxic and the latter highly toxic, making this difference in bioactivity poorly understood. The study is also crucial for rationally developing transition-state-analog inhibitors with antiophidic properties.
Methods
Here, we briefly summarize the methodology employed. A very detailed description of all methods employed can be found in the ESI,† Section II.We performed multiple computational methods to thoroughly investigate the catalytic mechanism of svPLA2 in a membrane model (Fig. S3, S4 and Table S1†). The process included (i) modeling of svPLA2:POPC:membrane complex in which the target protein structure with PDB code 5TFV was positioned at a weakly interacting distance (≈5 Å) over the upper leaflet of a 1:1 POPC/POPS bilayer (80 POPC:80 POPS per leaflet) (Fig. 3, left); Despite the target being homodimeric in its crystalline form, it has been demonstrated that PLA2s act as monomers when bound to lipid–water interfaces.47 Based on this evidence, we used the monomeric form to accurately reflect the enzyme's active state during interfacial activation; (ii) classical molecular dynamics (cMD) simulations using the GROMACS 2021.5 software for a total of 0.5 μs across six replicas (Table S2†); The Amber99SB-ildn force field was used for the protein, while the lipids were described by the Slipids-2020 force field;70,71 While the overall behavior among the replicas was relatively similar, the fifth replica was selected for subsequent studies due to its greater stability throughout the trajectory and more catalytically favorable distances; (iii) clustering analysis of the MD trajectories to identify structures with the active site properly preorganized to catalyze the phosphodiester hydrolysis for starting the mechanistic studies (Fig. S5 and S6†); (iv) DFT/MM MD umbrella sampling (US) simulations with the PBE functional employing the Gaussian double-ζ valence polarized (DZVP) basis set. DFT/MM calculations were carried out with the Gaussian plane wave formalism as implemented in CP2K, where an auxiliary plane-wave cutoff of 300 Ry was applied for the valence electrons, while the core electrons were treated using Goedecker–Teter–Hutter (GTH-PBE) pseudopotentials.72 The QM unit cell contained a total of 167 atoms with a neutral charge and a singlet spin (Fig. S7†). The reaction coordinates (ξ) for each mechanistic hypothesis were employed as collective variables (CVs) and were sampled at the DFT/MM level for 20 ps. For the single water (SW) mechanism, CV1 was defined as (d1 + d2) and CV2 as (d3 + d4), while for the assisted-water (AW) mechanism, CV1 was (d1 + d2 + d3) and CV2 was (d4 − d5), as depicted in Fig. 3, right. CV1 represents the protonation of the catalytic His47 and the nucleophilic attack by a water molecule on the carbonyl carbon of the substrate while CV2 represents both the cleavage of the substrate's ester bond and protonation of the departing lysophospholipid group (full details provided in the ESI†).
 | ||
| Fig. 3 The modeled BaMt-I:POPC:membrane complex in a solvated box; (Left): the protein structure depicted in rainbow cartoon and the POPC substrate in lime green sticks. A light blue surface represents the solvent environment, while green and purple spheres denote chloride (Cl−) and sodium (Na+) ions. The lipid membrane is depicted in brown sticks, emphasizing the phosphate groups as dark orange spheres. (Right): A close-up view of the QM layer and the reaction coordinates (ξ1 – CV1 and ξ2 – CV2) used for the QM/MM MD calculations. Blue and purple circles represent the reaction coordinates used for the single – (SW) and the assisted-water (AW) pathways, respectively. In both cases, CV1 is represented by the same respective colors (blue and purple), and CV2 by black color in dashed lines. | ||
Results and discussion
Mapping BaMt-I:membrane interactions and their impact on the protein and membrane structure and dynamics
All the analysis regarding the svPLA2-membrane dynamic behavior can be found on the ESI, Fig. S8–S16,† Section III.Since sPLA2 optimal activity is primarily restricted to the water–lipid interface, the presence of two binding sites, the catalytic domain and the aforementioned IBS, is crucial. The latter is located on a flat external region that surrounds the opening of the hydrophobic channel25,73 (Fig. S16†). Despite their conserved architecture and catalytic site, the sPLA2s have distinct interfacial binding properties, leading to distinct membrane affinities.73,74 Variations in the primary sequence of different PLA2s are depicted in Fig. S17.† In general, the IBS comprises a ring of basic and hydrophobic residues, in which the former interact electrostatically with the negatively charged head groups of anionic lipids25 and the latter, particularly the bulkier aromatic ones, interact and penetrate the phospholipid bilayer into the hydrophobic tail region, causing local membrane disordering. Fig. S16† reveals that the BaMt-I surface contains an IBS that is, indeed, arranged in a large patch of hydrophobic residues (Leu2, Ile3, Ala6, Leu10, Leu16, Phe18, Tyr20, Trp30, Met108, Ala109) flanked by several basic residues (Lys7, Lys58, Lys60, Arg63, Lys105) and some non-charged hydrophilic residues (Thr22, Thr23, Thr61).
The number of atomic contacts between BaMt-I and the mixed POPC/POPS bilayer along with an illustration of the residues interacting with the membrane is presented in Fig. 4A and B, respectively. On average, BaMt-I residues established a more significant number of contacts with the zwitterionic POPC lipids (3602 ± 561) than anionic POPS lipids (2887 ± 807). However, at approximately 170 ns, there was a noticeable increase in interactions between hydrophobic residues and the anionic phospholipids. In contrast to the hydrophobic residues, basic residues preferentially interacted with the headgroups rather than the tails, with which very few contacts were established (Fig. 4A).
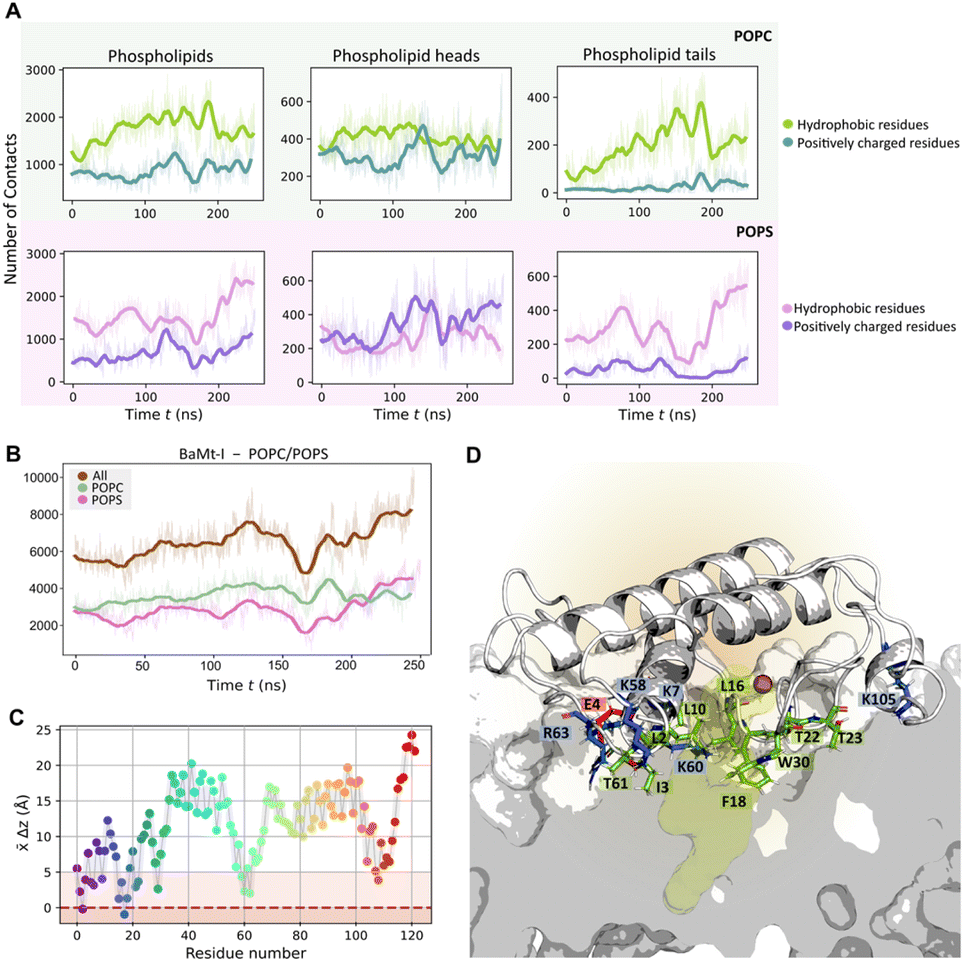 | ||
| Fig. 4 Protein–lipid interatomic contacts and a close-up view of the residues interacting with the membrane. (A) The average number of interatomic contacts with each phospholipid region over time. (B) The average number of interatomic contacts between BaMt-I and POPC (pale green) and POPS (pink) phospholipids over time; a minimum distance cutoff of 6 Å was applied to define a contact. (C) The average penetration depth of the BaMt-I residues into the lipid bilayer. The dark red dashed line represents the average membrane phosphate groups, and the light brown area represents the region occupied by the phospholipid heads (D) Snapshot illustrating the enzyme's position within the lipid bilayer and a closer view of the primary residues that penetrate it. Positively and negatively charged residues are depicted in blue and red sticks, respectively, hydrophobic anchors and non-charged hydrophilic residues in green. | ||
The evolution of the total BaMt-I:membrane contacts throughout the simulation suggested a progressive insertion of BaMt-I into the membrane core. To assess the extent of penetration of BaMt-I into the lipid bilayer, we monitored the z-coordinate of the center of mass (COM) of each residue (Fig. 4C) and averaged along the whole trajectory. A residue was considered as inserted into the membrane if the z-distance of its COM was <5 Å of the average z-coordinate of the phosphate groups.
Fig. 4D highlights those residues that interacted and got partially inserted into the membrane – hydrophobic (Leu2, Ile3, Ala6, Leu10, Leu16, Phe18 and Trp30), basic (Lys7, Lys58, Lys60, Arg63 and Lys105), and non-charged hydrophilic residues (Thr22, Thr23 and Thr61).
Based on these findings, it might be possible that Lys7, Arg63, and Lys105 are the leading promoters of protein insertion into the 1:1 POPC/POPS bilayer. These residues likely aid in pulling the protein into the anionic lipid interface through electrostatic interactions. Furthermore, hydrophobic residues like Ile3, Phe18, Leu10, and Leu16 probably contribute to the enzyme's anchoring.75,76 Most of the residues align with those suggested by Salvador et al.20 and are believed to be essential for BaMt-I binding to the membrane.
The visual inspection of the trajectory showed that BaMt-I promptly reached the membrane interface but did not penetrate extensively, embedding approximately 11 Å into the membrane, as indicated by the partial density profile (Fig. 5). In alignment with the previous results, Fig. 5 shows an evident predominance of hydrophobic residues at the IBS, compared to basic or acidic residues. The hydrophobic residues did not penetrate deeply into the membrane hydrophobic core, except for Ile3, Leu16, Leu10, and Phe18. Throughout the simulation, BaMt-I underwent rotational adjustments, characterized by increased penetration in the N-terminus region and an elevation of the C-terminal area concerning the membrane interface.
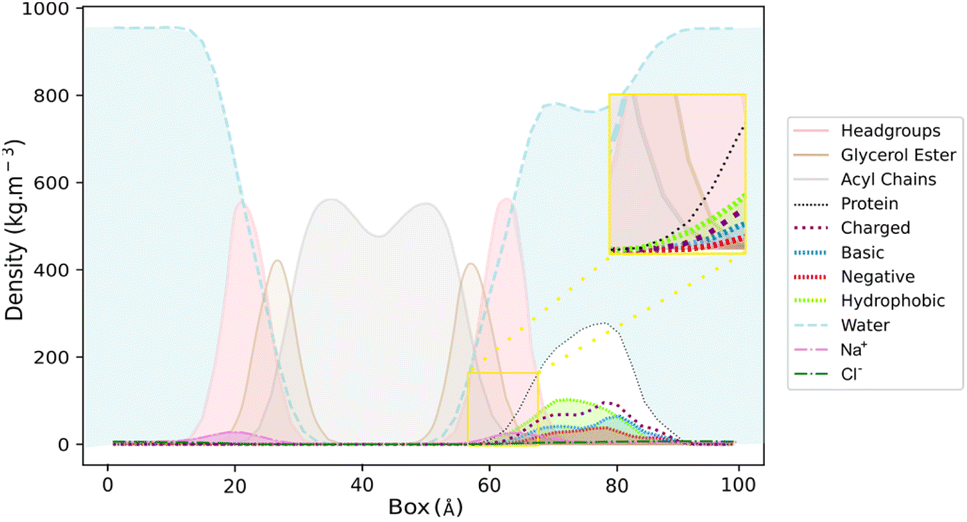 | ||
| Fig. 5 Partial density profile across the BaMt-I:membrane complex for membrane headgroups (pink), glycerol ester (tan) and acyl chains (grey), and protein (black dashed lines) with their hydrophobic (green), basic (marine blue) and negative (red) residues. The aqueous environment (blue), and counterions Na+ (magenta) and Cl− (dark green) are also represented. A close-up view of the area of the protein that penetrated both the glycerol ester, and the acyl chains of the membrane is presented in a yellow box. | ||
Finally, we evaluated also the destabilizing impact of the enzyme on the molecular organization of the lipid bilayer by assessing the order parameter profiles of the phospholipids within three distinct regions (Fig. 6). It is a measure of the degree of rigidity or fluidity, i.e., order or disorder, in lipid acyl chains within a membrane, quantifying their orientation relative to the membrane's normal axis. These regions were located at 8, 16, and 24 Å from the BaMt-I IBS. The deuterium order parameters (−SCD) for carbons of the sn-1 and sn-2 acyl chains of POPC and POPS were calculated within these regions. Results revealed that the acyl chain carbon's order parameters of both POPC and POPS in the vicinity of the enzyme were lower (lower −SCD values) than the most distant ones (16 and 24 Å). These observations indicate a more disordered packing of the lipids in the presence of the enzyme. The observed results are justified by the substantial amount of the already demonstrated hydrophobic residues on the enzyme's IBS and the interaction of basic residues with the membrane. According to Salvador et al.,20 some of the residues that penetrate the lipid bilayer belong to a “myotoxic cluster” and are believed to be membrane-docking/disrupting sites. Their presence favors dispersion interactions with the bilayer non-polar hydrocarbon tails, which might change their local properties and further impact the neighboring areas.
 | ||
| Fig. 6 Definition of regions in the membrane surrounding BaMt-I. Three regions—8 (pink), 8–16 (iceblue), and 16–24 Å (orange)—were defined, each of which denotes a distance layer from the enzyme (white cartoon). Deuterium order parameters for carbon atoms in the sn-1 (line) and sn-2 (dashed line) hydrocarbon chains of POPC (top) and POPS (bottom) lipids were calculated in the defined regions. | ||
Notably, BaMt-I IBS is characterized by the predominance of hydrophobic residues. Leu2, Ile3, Ala6, and Leu10 are part of the amphipathic N-terminal α-helix (residues 2-13), which participates in the substrate binding pocket25,77–79 and plays a crucial role in the orientation and binding mode to the membrane.50,77–82 These residues, along with Leu16, Phe18 and Trp30, stabilize the enzyme in the bilayer environment by hydrophobically anchoring it.
Studies have shown that mutations in bulky aromatic residues at the IBS significantly impact the affinity for zwitterionic membranes.75,81,83,84 This was the case for the acidic PLA2 from N. naja atra, where introducing mutations at Trp and Phe residues on the IBS led to a significant decrease in both zwitterionic membrane penetration and enzymatic activity (up to 50-fold decrease).85 In contrast, hGIIA PLA2, which lacks these bulky residues, displays low binding affinity towards zwitterionic membranes, favoring the binding to anionic membranes. However, when both Val3 and Val31 were mutated to a Trp residue, the hydrolytic activity towards zwitterionic interfaces improved.86 In the case of BaMt-I, Phe18 and Trp30 could have an essential role in the interfacial binding process in addition to substrate recognition.25,87
Unlike many other PLA2 enzymes (e.g., human and porcine pancreatic PLA2),88 BaMt-I does not possess clusters of positive residues surrounding the IBS. Lys7, Arg63, and Lys105, also common in Lys49-PLA2-like proteins, have been proposed to form hydrogen bonds and salt bridges with the carbonyl oxygens of anionic lipids.89 Lys60 is thought to contribute to interfacial adsorption and electrostatic interactions with the substrate phosphate. Lys7 is highly conserved among PLA2s, and its mutation to glutamate in both the basic App-D48 sPLA2 myotoxin and in the hGIIA sPLA2, significantly decreases the adsorption and catalysis on anionic lipid bilayers.50,81,89 This residue and a patch of additional lysine residues at the end of the N-terminal segment—which are absent from BaMt-I—are thought to be responsible for PLA2's anionic membrane selectivity.73,78 Moreover, Lys105Ala mutants in BthTX-I, a PLA2-like enzyme from the pit viper B. jararacussu, exhibited decreased membrane damaging activity yet did not eradicate it. These enzymes, along with Myotoxin II (B. asper) and crotoxin (C. durissus terrificus),90 preferentially bind to anionic phospholipids, emphasizing the role of electrostatic interactions in their interfacial binding affinity. Experimental data suggested that ca. 20% negatively charged phospholipids are needed for these enzymes to bind the membrane with high affinity due to charge–charge attractions.83,91
Therefore, the hydrophobic/basic nature of BatMt-I IBS may result in an enhanced ability to interact with both zwitterionic and anionic phospholipids, relative to the enzymes mentioned above, i.e. a higher affinity towards membranes.
Structural insights into the pre-reactive state of svPLA2
The clustering of the whole molecular dynamics trajectory, based on reactive distances, revealed three main clusters of conformations primarily dictated by the distances between His47 Nδ–Cpopc, and Tyr51 OH–Asp89 Oδ1.These distances exhibited considerable variations during the simulations (Fig. S9†). As illustrated in Fig. 7, only a minor portion of the trajectory contained distance-suitable conformations that agreed with the expected reactive-prone conformations proposed in the literature. This was caused by the distance between Tyr51 OH and Asp89 Oδ1, which was large for most of the simulation (Fig. S9†). Notably, this dynamic interplay had a significant impact on the availability of productive conformations within the trajectory, resulting in only approximately 20% of the entire trajectory, represented in purple as shown in Fig. 7.
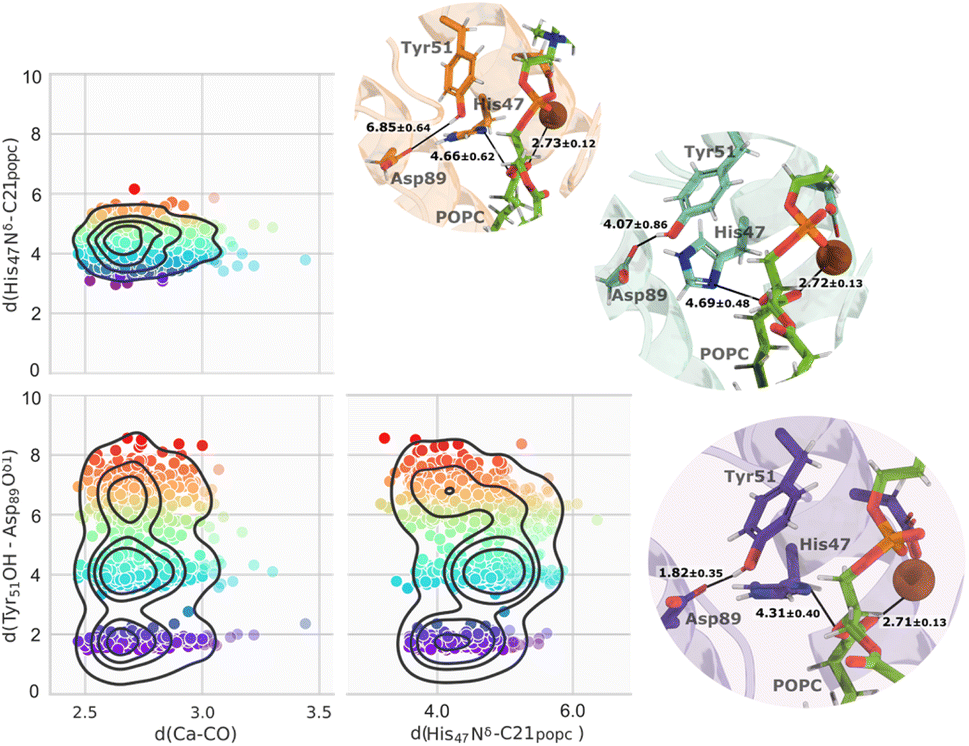 | ||
| Fig. 7 The three main clusters identified after filtering the Ca2+–COpopc, His47 Nδ–Cpopc, and Tyr51 OH–Asp89 Oδ1 distances, represented by a color gradient. As the color changes from purple to red, it indicates that the structure becomes less ideal. Close-up views of representative structures from the three main clusters and respective distances are also shown. The tonality of each representative structure aligns with its corresponding cluster. | ||
We re-clustered the conformations retrieved from the initial clustering process. A dominant cluster encompassed almost 75% of the retrieved microstates from the first clustering process with an average RMSd of 0.97 Å to the crystallographic structure, while the second cluster contained about 13% of the structures.
Upon examining the structures of each cluster representative, it was evident that the structural elements can be almost perfectly superimposed with minor variations around catalytic residues (Fig. S6†). The chosen structure, which contained two water molecules in the active site interacting with the Ca2+ ion and the His47 Nδ atom, was used for both mechanistic hypotheses.
Three different solvation spheres are evident in the radial distribution function (RDF) of water molecules around the midpoint of the Ca2+–His47 Nδ–C21popc plane (Fig. 8A). The first and second solvation spheres, spanning approximately from 1.65 Å to 2.50 Å and 2.50 Å to 3.10 Å, exhibit well-defined peaks and accommodate an average of 0.8 and 2.1 water molecules, respectively. The third solvation sphere (≈3.10 Å to 4.20 Å), displays a broader distribution and contains an average of 6.0 water molecules. This third solvation sphere encompasses water molecules near the Asp89 Oδ1 and His47 Hε2 atoms.
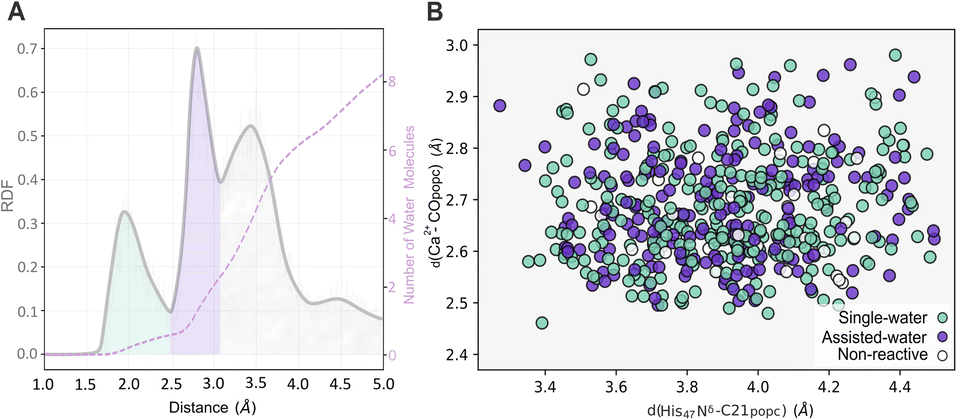 | ||
| Fig. 8 The RDF depicts the spatial distribution of water molecules relative to the midpoint of the Ca2+–His47 Nδ–C21popc plane. The dashed pink line represents the cumulative quantity of water molecules (A). Distribution of productive conformations for the single- and the assisted-water mechanisms in function of crucial distances between His47 Nδ–Cpopc and the Ca2+–C21popc (B). | ||
This study also explored the potential existence and dominance of productive conformations regarding the single- and assisted-water mechanisms. Thus, the productive conformations retrieved from the latter clustering step were further evaluated by examining the number of water molecules within the active center concerning the His47 Nδ–Cpopc and the Ca2+–COpopc distances (Fig. 8B). Among the examined frames, 54% and 40% were suitable for the single- and the assisted-water mechanisms, respectively, while 6% were disregarded as non-productive conformations. These findings suggest that the energy penalty associated with the conformational arrangements required for catalysis is similar, making both pathways accessible.
Despite undergoing a complex clustering process to ensure conformity with literature and recent mechanistic studies on the PLA2 enzyme,48 the starting structure for the mechanistic study is thought to have minimal impact on the calculated free energy profile. This is because the QM/MM MD simulations mitigate any dependence on the starting structure chosen during the clustering analysis process. However, by selecting a structure with optimized catalytic distances with a low RMSD to the crystallographic reference, it is possible to carry out the mechanistic calculations with faster convergence results.
Exploring svPLA2 possible reaction pathways
In the reactant state of the single-water mechanism (Fig. 9 – REACT), the nucleophilic water (Wnuc) was well-positioned with respect to the His47 Nδ and the Cpopc atoms, with distances of 1.72 and 4.03 Å, respectively, suggesting a favorable geometry for the first catalytic step. Notably, a hydrogen bond was formed between Wnuc and the Oδ2 atom of the Asp48 residue. It has been shown that mutations at the Asp48 to Glu or Lys compromise the enzyme's activity by affecting its binding affinity for both Ca2+ and the phospholipid substrate within the active site.60,63 Despite not being coordinated with the Ca2+ ion (dWnuc–Ca2+ ≈ 4 Å), the Wnuc, forms a short hydrogen-bond with the His47 Nδ atom and the negatively charged carboxyl group of the Asp48 Oδ base, resulting in a polarized state. | ||
| Fig. 9 The free energy profile shows the single-water mechanism. The first peak represents the first transition state (TS1), where the nucleophilic attack takes place, and the second peak represents the second transition state (TS2) with partial hydrolysis of the sn-2 ester bond. Only the residues of the QM layer directly involved in the reaction are shown, while the MM layer is omitted for clarity. All distances are given in Å. | ||
The umbrella sampling simulations along the reaction coordinates identified a stepwise pathway characterized by two distinct transition states (TS1 and TS2). At ξ = +3.00 Å, the first transition state (TS1) was formed. Here, the polarized Wnuc was deprotonated by the basic His47 residue, generating a nucleophilic hydroxide ion (HO−) hydrogen-bonded to Asp48 Oδ2, which, in turn, initiated the nucleophilic attack on the phospholipid substrate's ester bond (Cpopc). These events occurred synchronously, as evidenced by distances between H2wat–His47 Nδ (d1) and OWat–Cpopc (d2) in TS1, which decreased to 1.18 and 1.74 Å, respectively. This further indicates that the catalytic water was fully deprotonated and that the newly formed hydroxide ion species, while not fully stabilized, was already partially bonded to Cpopc (Fig. 9 – TS1).
Along the TS1 decay, the OWat–Cpopc distance decreased from 1.74 to 1.50 Å. As a result, a tetrahedral oxyanion intermediate was formed at ξ = +2.54 Å, where the oxygen on the oxyanion hole, stabilized by the Ca2+ cofactor, became negatively charged and Cpopc adopted a sp3 hybridization (Fig. 9 – INT). This first step yielded a free energy barrier of 18.97 kcal mol−1, which aligns with the experimental data for hydrolase-related reaction mechanisms.48,85,92
As the reaction progressed to the TS2 at ξ = +4.23, the active site underwent a significant hydrogen-bond network rearrangement (Fig. 9 – TS2). The initial hydrogen bond between the OH− at the sn-2 ester bond and the Asp48 Oδ2 atom is disrupted due to the entrance of a water molecule that bridges this interaction. This water molecule also forms a hydrogen bond with the carbonyl oxygen of the substrate while simultaneously coordinating with the Ca2+ ion (depicted by light gray dashed lines). These events caused the carbonyl oxygen of the substrate (COpopc) to reorient away from the Ca2+ ion, up to an average distance of 3.45 Å, which may have contributed to an increase in the barrier. The free energy barrier associated with TS2 was 20.14 kcal mol−1 concerning the initial reactant state.
Finally, the sn-2 ester bond of the substrate quickly broke, accompanied by a notable increase in the Cpopc–Opopc (d3) distance, which rose from 1.57 to 3.80 Å. Synchronously, the resulting negatively charged oxygen on the lysophospholipid group deprotonated the positive His47 NδH. The newly formed fatty acid was also observed to be transiently deprotonated by the neighboring Asp48 until the final product was obtained (Fig. 9 – PROD). These results further confirm that Asp48 plays a significant role in catalysis. Specifically, the hydrogen bonds formed between Asp48 and the catalytic water (Asp48 Oδ2–Wnuc), as well as with the fatty acid group (Asp48 Oδ1–COO−popc), prove its role in activating the nucleophile and stabilizing the products, which were stabilized by several active site residues. The phosphate group of the lysophospholipid product got stabilized through interactions with the Ca2+ ion, Lys60, and Gly31 from the Ca2+-binding loop. The free energy associated with the enzyme-bound products was 13.52 kcal mol−1, showing an endergonic character.
Representative snapshots of each stationary state and respective interatomic distances are presented in Fig. 9.
Concerning the alternative assisted-water mechanism, in the reactant state (Fig. 10 – REACT), the Wnuc H–Wassist O, Wassist H–His47 Nδ and Wnuc O–Cpopc average distances were 2.59, 1.87, and 3.73 Å, respectively, favoring both the proton transfers and the nucleophilic attack. Furthermore, consistent with earlier literature and in contrast to the previous observations in the single-water pathway, the catalytic water (Wnuc) coordinates with the Ca2+ ion.
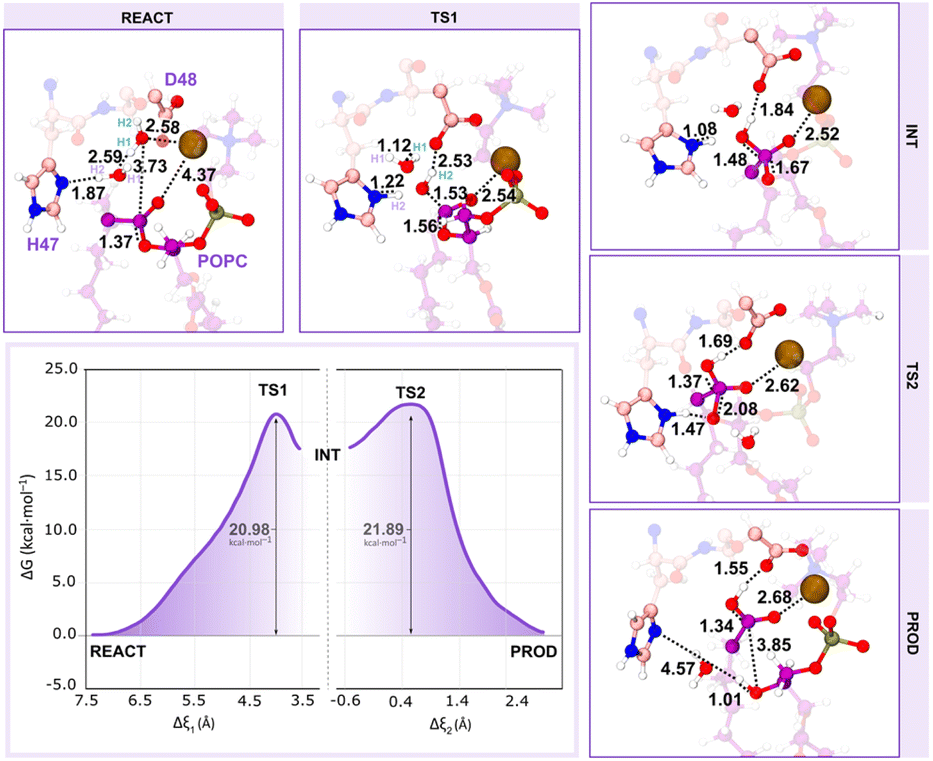 | ||
| Fig. 10 The free energy profile shows the assisted-water mechanism. The first peak represents the first transition state (TS1), where the nucleophilic attack takes place, and the second peak represents the second transition state (TS2) with partial hydrolysis of the sn-2 ester bond. Only the residues of the QM layer that are directly involved in the reaction are shown, while the MM layer is omitted for clarity. All distances are given in Å. | ||
The coordination of the Wnuc to the Ca2+ ion completes the characteristic Ca2+ coordination sphere observed in sPLA2s, which may lead to the movement of the COpopc from the cofactor to approximately 4.37 Å.
We carried out umbrella sampling simulations, and the results indicate that the assisted-water mechanism also follows a stepwise reaction, featuring two transition states (TS1 and TS2) at ξ = +4.10 Å and +0.45 Å, respectively. The reaction is initiated by the deprotonation of the Wnuc by Wassist, generating a nucleophilic HO− and a highly unstable H3O+. Simultaneously, the catalytic His47 residue deprotonates the bridging water molecule (Wassist), and the previously produced HO−, which is again hydrogen bonded to His47 Nδ and Asp48 Oδ2, attacks the substrate sn-2 bond. This resulted in the formation of the TS1 (Fig. 10 – TS1), in which the OWnuc–Cpopc shortened to 1.53 Å and the Cpopc–Opopc distance increased to 1.56 Å. Synchronously, as the COpopc becomes negatively charged after the HO− nucleophile, the distance between the COpopc and the Ca2+ ion decreased from 4.37 to 2.54 Å. The free energy barrier associated with this step was 20.98 kcal mol−1. As a result, a tetrahedral oxyanion intermediate was formed (Fig. 10 – INT), with the sp3 hybridization of the Cpopc bond and the stabilization of the COpopc negative charge by the Ca2+ ion. At TS2 (Fig. 10 – TS2), the Cpopc–Opopc bond stretched up to 2.08 Å as the His47 suffered an orientation shift to transfer its proton to the leaving lysophospholipid group (Opopc). The active role of the Wassist concluded after the deprotonation of Wnuc, resembling the second step of the single-water pathway. The free energy barrier of this step is the highest of the reaction cycle, 21.89 kcal mol−1 above the reactants, becoming the rate-limiting transition state.
Ultimately, the substrate sn-2 ester bond broke along the path from the TS2 to the products, resulting in a distance of 3.85 Å and the collapse of the tetrahedral structure. As in the single-water mechanism, the newly formed fatty acid product also interacted with the neighboring Asp48, evidencing the catalytic role of the latter (Fig. 10 – PROD). The second step exhibits a pronounced free energy decrease at PROD, reaching 0.1 kcal mol−1. Furthermore, some active site residues played a stabilizing role regarding the products. The phosphate group of the lysophospholipid product got stabilized through interactions with the Ca2+ ion, and residues Lys60, Trp30, and Gly31 from the Ca2+-binding loop. Additionally, the Ca2+ ion, Asp48, and Gly29 stabilized the fatty acid product. Except for the Gly29 and Trp30 residues, the same interactions were observed in the single-water pathway.
Fig. S18–S24† illustrate the convergence of the free energy profiles and Fig. S25 and S26† depict changes in atomic distances over time, along with the corresponding standard deviations.
The results have shown that both the single- and the assisted-water mechanisms proceeded in a stepwise fashion. The former displayed a lower activation energy of 20.14 kcal mol−1 compared to the 21.89 kcal mol−1 necessary for the latter. The obtained free energy barriers are close when considering the inherent MUE associated with computational methods (≈3 kcal mol−1). A comparison of the activation free energies associated with the breaking and formation of the same bond type leads to a significant error cancellation, driving the comparison into the area of meaningfulness. Additionally, the calculated energies correspond well to those obtained from computational studies.48 Although both reactions exhibited endergonic character (ΔG° = 13.52 kcal mol−1 and 0.1 kcal mol−1), the assisted-water pathway displayed a thermodynamically and kinetically more favorable product state.
While product formation occurs in both reaction cycles, the enzyme's complete structural and functional reconstitution is not achieved. Prior research indicates that the full regeneration of PLA2 enzymes typically requires the influx of three water molecules into the active site following the release of products,25,43 likely equalizing the free energy at the end of the cycle. Among these, two coordinate the Ca2+ ion while the third replenishes the active cycle for subsequent nucleophilic attack. In this case, the absence of such a water exchange process could contribute to the observed endergonic character. Furthermore, slight conformational changes within the active site, coupled with the aforementioned observation, likely hinder the precise realignment of the active site for subsequent turnover.
Overall, the interplay between the lower activation energy obtained for the single-water pathway and the less endergonic nature of the assisted-water pathway compensate for each other's thermodynamic limitations.
Using the QM/MM transition states to design potent svPLA2 transition-state-analog inhibitors
Understanding how PLA2 enzymes behave at the atomic level is critical to designing next-generation drugs that can effectively block the enzyme's activity, complementing or even outperforming antibody-based therapies. By designing transition state analogs, molecules that mimic the short-lived, high-energy transition state of the reaction, it is possible to develop highly potent and specific svPLA2 inhibitors with minimal off-target effects. Varespladib, a PLA2s inhibitor and the only small-molecule antidote reaching clinical trials against snakebite has proven effective against multiple catalytic PLA2s from snake venom and non-catalytic PLA2-like toxins from several medically important snakes.93–95In vitro and in vivo studies have also been carried out on BaMt-I, showing that Varespladib neutralizes its cytotoxic and myotoxic effects.95 Studies suggest that Varespladib can interfere with the interfacial activation process by interfering with or reducing the ability of the enzyme to disrupt the integrity of the plasma membrane in muscle cells.95By superimposing the obtained rate-limiting transition state geometries (TS2 from both catalytic pathways) with Varespladib from the co-crystallized X-ray structure of Mt-II isolated from Bothrops moojeni (PDB code: 6PWH)96 (Fig. 11), we observed that the inhibitor mimics the crucial interactions found at the transition states reported here. Particularly, the carboxylate group of the drug mimics the negatively charged oxygen of the substrate phosphate group and the carboxamide group emulates the hydroxide ion (TS2, Fig. 9 and 10). In addition, the amine moiety of the carboxamide forms hydrogen bonds with the catalytic His47 and Asp48, and the Gly29 of the Ca2+-binding loop, mimicking the catalytic water molecule. We also observed π-stacking interactions between Trp30 and the benzyl group.
 | ||
| Fig. 11 (Left) Superimposition of the assisted-water rate-limiting transition state structure obtained through the QM/MM MD calculations reported here (purple sticks) and the structure of the hGIIA PLA2 inhibitor, Varespladib (pink sticks) from the structure with the PDB ID 6PWH. (Right) A close-up view of the reaction region from another angle with the hydrogen bonds evidenced by dashed black lines demonstrates a high degree of similarity between the two ligands. Given the remarkable similarity in the position and structure of the single-water transition state, it was not included for clarity. | ||
The fact that varespladib so accurately mimics the obtained rate-limiting transition states demonstrates that it is an effective transition-state analog. This supports the approach of guiding drug discovery on the accurate structures of rate-limiting transition states identified by QM/MM calculations. Therefore, the results presented here can be employed to enhance existing drugs' efficacy and selectivity or develop entirely new ones.
Conclusions
In this work, we performed molecular dynamics simulations of the svPLA2 toxin, BaMt-I, bound to a POPC substrate in a 1:1 POPC/POPS mixture membrane bilayer. Analysis of the bound complex showed an abundance of hydrophobic residues in its interfacial binding surface, which distinguishes it from other PLA2 enzymes that are characterized by a stronger cationic nature (e.g. hGIIA PLA2, porcine PLA2, AppD48). The latter selectively bind anionic interfaces. Additionally, simulation results have confirmed that BaMt-I mainly interacted peripherally with the membrane, with low penetration depth. Following the existing literature, the key residues responsible for facilitating penetration were identified as Phe18, Ile3, and Trp30. On the other hand, basic residues, such as Lys7 and Lys105, were found to play a significant role in forming electrostatic and hydrogen bonds with the membrane. These results further suggest that BaMt-I may interact with zwitterionic and anionic interfaces. This hypothesis is corroborated by the surface charge distribution on its interfacial binding surface, the observed binding interactions, and the contacts established between BaMt-I and both types of phospholipids during simulation.
Furthermore, BaMt-I disrupted the lipid packing within the membrane, while the membrane environment, in turn, prompted conformational changes in the protein. Thus, the direct hydrolysis of phospholipids seems to be, indeed, a prominent mechanism driving toxicity, although membrane destabilization may occur as a secondary effect of enzyme activity through its interaction and light penetration. Moreover, the combined action of multiple svPLA2 enzymes may also play a role in destabilizing the phospholipid bilayer. After binding to the lipid bilayer, these enzymes accumulate at the interface, releasing hydrophobic reaction products, i.e., lysophospholipids and fatty acids. These products can activate additional PLA2 enzymes and alter the physical properties of the membrane, destabilizing it.
We have investigated two distinct reaction mechanisms: the single-water pathway, which proceeded in two steps with a nucleophilic water molecule uncoordinated to the Ca2+ cofactor, and the assisted-water pathway, which also followed two steps, however, with a metal-coordinating nucleophilic water molecule. Although both pathways were mechanistically viable, with a small barrier gap (≈2 kcal mol−1), and catalytically-competent, it seems that the single-water pathway is more likely to occur, as evidenced by the analysis of reactive conformations and the lower activation-free energy barrier. However, the assisted-water pathway is thermodynamically more favorable. While the single water yielded a more endergonic product than the assisted-water, the final free energy will likely be equivalent after the reaction cycle is completed. Nonetheless, while it is suggested that the reaction mechanism may be conserved across species due to the significant structural similarity among PLA2 enzymes, further investigation is necessary to validate this generalization, particularly concerning PLA2s from elapids. Additionally, the subtle differences between the two proposed mechanistic pathways suggest that these mechanisms could potentially converge in other snake species.
Furthermore, mimicking rate-limiting transition states has emerged as an attractive approach for the design of next-generation inhibitory drugs. The evident similarity that emerged from this study between the coordination mode of the clinical trial candidate, Varespladib, and the QM/MM rate-limiting transition states demonstrated and reinforced the potential of using this powerful strategy in drug discovery.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
J. C. A.: conceptualization, methodology, software, validation, formal analysis, investigation, visualization, writing – original draft, writing – review & editing. A. V. P.: methodology, software, writing – review & editing. A. K. M.: writing – review & editing. M. J. R.: conceptualization, writing – review & editing, supervision, funding acquisition. P. A. F.: project administration, conceptualization, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from FCT/MCTES—the Portuguese Fundação para a Ciência e Tecnologia, through project PTDC/QUI-OUT/1401/2020 and from the Laboratório Associado para a Química Verde (LAQV) (reference REQUIMTE 2021-46), which is financed by FCT/MCTES within the scope of project UIDB/50006/2020. AVP thanks FCT for financing his PhD project through the grant 2021.05918.BD. We further thank the HPC RIVR consortium and EuroHPC JU for providing computing resources of the HPC system Vega supercomputer at the scope of the EURO-HPC project through the grant EHPC-REG-2022R03-205.Notes and references
- J. P. Chippaux, J. Venomous Anim. Toxins Incl. Trop. Dis., 2017, 23, 38 CrossRef PubMed.
- A. Kasturiratne, A. R. Wickremasinghe, N. de Silva, N. K. Gunawardena, A. Pathmeswaran, R. Premaratna, L. Savioli, D. G. Lalloo and H. J. de Silva, PLoS Med., 2008, 5, e218 CrossRef PubMed.
- J. M. Gutierrez, J. Venomous Anim. Toxins Incl. Trop. Dis., 2014, 20, 7 CrossRef PubMed.
- R. Otero-Patino, Toxicon, 2009, 54, 998–1011 CrossRef CAS PubMed.
- C. Xie, L. O. Albulescu, M. A. Bittenbinder, G. W. Somsen, F. J. Vonk, N. R. Casewell and J. Kool, Biomedicines, 2020, 8, 297 CrossRef CAS PubMed.
- A. G. da Costa Neves-Ferreira, R. H. Valente, J. Perales and G. B. Domont, in Handbook of Venoms and Toxins of Reptiles, ed. S. P. Mackessy, Taylor & Francis/CRC Press, 2009, pp. 259–284 Search PubMed.
- B. Kalita, S. P. Mackessy and A. K. Mukherjee, Expert Rev. Proteomics, 2018, 15, 837–849 CrossRef CAS PubMed.
- L. O. Albulescu, C. Xie, S. Ainsworth, J. Alsolaiss, E. Crittenden, C. A. Dawson, R. Softley, K. E. Bartlett, R. A. Harrison, J. Kool and N. R. Casewell, Nat. Commun., 2020, 11, 6094 CrossRef CAS.
- C. C. Liu, C. J. Wu, Y. C. Hsiao, Y. H. Yang, K. L. Liu, G. J. Huang, C. H. Hsieh, C. K. Chen and G. W. Liaw, J. Proteomics, 2021, 234, 104084 CrossRef CAS.
- R. Otero-Patino, A. Segura, M. Herrera, Y. Angulo, G. Leon, J. M. Gutierrez, J. Barona, S. Estrada, A. Pereanez, J. C. Quintana, L. J. Vargas, J. P. Gomez, A. Diaz, A. M. Suarez, J. Fernandez, P. Ramirez, P. Fabra, M. Perea, D. Fernandez, Y. Arroyo, D. Betancur, L. Pupo, E. A. Cordoba, C. E. Ramirez, A. B. Arrieta, A. Rivero, D. C. Mosquera, N. L. Conrado and R. Ortiz, Toxicon, 2012, 59, 344–355 CrossRef CAS PubMed.
- U. Puzari, P. A. Fernandes and A. K. Mukherjee, J. Ethnopharmacol., 2022, 292, 115208 CrossRef CAS PubMed.
- D. J. Williams, M. A. Faiz, B. Abela-Ridder, S. Ainsworth, T. C. Bulfone, A. D. Nickerson, A. G. Habib, T. Junghanss, H. W. Fan, M. Turner, R. A. Harrison and D. A. Warrell, PLoS Neglected Trop. Dis., 2019, 13, e0007059 CrossRef.
- L. M. Preciado, J. A. Pereañez and J. Comer, Toxins, 2020, 12, 8 CrossRef CAS.
- U. Puzari, P. A. Fernandes and A. K. Mukherjee, J. Med. Chem., 2021, 64, 13938–13979 CrossRef CAS PubMed.
- A. L. Oliveira, M. F. Viegas, S. L. da Silva, A. M. Soares, M. J. Ramos and P. A. Fernandes, Nat. Rev. Chem, 2022, 6, 451–469 CrossRef PubMed.
- Y. Angulo and B. Lomonte, Toxicon, 2009, 54, 949–957 CrossRef CAS PubMed.
- A. Alape-Giron, L. Sanz, J. Escolano, M. Flores-Diaz, M. Madrigal, M. Sasa and J. J. Calvete, J. Proteome Res., 2008, 7, 3556–3571 CrossRef CAS.
- J. Fernandez, J. M. Gutierrez, Y. Angulo, L. Sanz, P. Juarez, J. J. Calvete and B. Lomonte, Biochimie, 2010, 92, 273–283 CrossRef CAS PubMed.
- J. M. Gutierrez and B. Lomonte, Toxicon, 1995, 33, 1405–1424 CrossRef CAS PubMed.
- G. H. Salvador, J. I. Dos Santos, B. Lomonte and M. R. Fontes, Biochimie, 2017, 133, 95–102 CrossRef CAS.
- J. Fernandez, P. Caccin, G. Koster, B. Lomonte, J. M. Gutierrez, C. Montecucco and A. D. Postle, FEBS J., 2013, 280, 3878–3886 CrossRef CAS PubMed.
- A. K. Mukherjee, Biochimie, 2014, 99, 153–161 CrossRef CAS PubMed.
- C. A. Fernandes, R. J. Borges, B. Lomonte and M. R. Fontes, Biochim. Biophys. Acta, 2014, 1844, 2265–2276 CrossRef CAS PubMed.
- A. L. N. de Oliveira, M. T. Lacerda, M. J. Ramos and P. A. Fernandes, Toxins, 2024, 16, 71 CrossRef CAS PubMed.
- J. Castro-Amorim, A. Novo de Oliveira, S. L. Da Silva, A. M. Soares, A. K. Mukherjee, M. J. Ramos and P. A. Fernandes, J. Med. Chem., 2023, 66, 5364–5376 CrossRef CAS.
- J. M. Gutierrez and C. L. Ownby, Toxicon, 2003, 42, 915–931 CrossRef CAS.
- L. Bitar, D. Jundi, M. Rima, J. Al Alam, J.-M. Sabatier and Z. Fajloun, Venom. Toxins, 2021, 1, 1–12 CrossRef.
- E. Jimenez-Charris, A. Montoya-Gomez, J. K. Torres, M. Gomez-Diaz and W. Bolivar-Garcia, Biochimie, 2022, 194, 19–27 CrossRef CAS PubMed.
- M. Sasa, D. K. Wasko and W. W. Lamar, Toxicon, 2009, 54, 904–922 CrossRef CAS PubMed.
- D. Mora-Obando, D. Salazar-Valenzuela, D. Pla, B. Lomonte, J. A. Guerrero-Vargas, S. Ayerbe, H. L. Gibbs and J. J. Calvete, J. Proteomics, 2020, 229, 103945 CrossRef CAS PubMed.
- C. Picado Twight, Serpientes venenosas de Costa Rica: sus venenos seroterapia anti-ofídica, CR Alsina, 1931 Search PubMed.
- R. Bolaños, Editorial Universidad de Costa Rica, 1984 Search PubMed.
- N. Scott, Costa Rican Natural History, The University of Chicago Press, Chicago and London, 1983, pp. 383–384 Search PubMed.
- D. Mora-Obando, J. Fernandez, C. Montecucco, J. M. Gutierrez and B. Lomonte, PLoS One, 2014, 9, e109846 CrossRef.
- K. Sekar, B. Z. Yu, J. Rogers, J. Lutton, X. Liu, X. Chen, M. D. Tsai, M. K. Jain and M. Sundaralingam, Biochemistry, 1997, 36, 3104–3114 CrossRef CAS PubMed.
- P. Caccin, P. Pellegatti, J. Fernandez, M. Vono, M. Cintra-Francischinelli, B. Lomonte, J. M. Gutierrez, F. Di Virgilio and C. Montecucco, Biochem. Biophys. Res. Commun., 2013, 430, 1289–1293 CrossRef CAS.
- R. Doley, G. F. King and A. K. Mukherjee, Arch. Biochem. Biophys., 2004, 425, 1–13 CrossRef CAS.
- D. Saikia, N. K. Bordoloi, P. Chattopadhyay, S. Choklingam, S. S. Ghosh and A. K. Mukherjee, Biochim. Biophys. Acta, 2012, 1818, 3149–3157 CrossRef CAS.
- J. E. Burke and E. A. Dennis, J. Lipid Res., 2009, 50(Suppl), S237–S242 CrossRef.
- C. Peggion and F. Tonello, Toxins, 2021, 13, 290 CrossRef CAS.
- R. M. Kini, Toxicon, 2003, 42, 827–840 CrossRef CAS.
- J. E. Burke and E. A. Dennis, Cardiovasc. Drugs Ther., 2009, 23, 49–59 CrossRef CAS PubMed.
- D. L. Scott, S. P. White, Z. Otwinowski, W. Yuan, M. H. Gelb and P. B. Sigler, Science, 1990, 250, 1541–1546 CrossRef CAS PubMed.
- R. R. Kim, Z. Chen, T. J. Mann, K. Bastard, F. S. K and W. B. Church, Molecules, 2020, 25, 4459 CrossRef CAS PubMed.
- T. S. Kang, D. Georgieva, N. Genov, M. T. Murakami, M. Sinha, R. P. Kumar, P. Kaur, S. Kumar, S. Dey, S. Sharma, A. Vrielink, C. Betzel, S. Takeda, R. K. Arni, T. P. Singh and R. M. Kini, FEBS J., 2011, 278, 4544–4576 CrossRef CAS.
- R. J. Borges, G. H. M. Salvador, H. B. Campanelli, D. C. Pimenta, M. de Oliveira Neto, I. Uson and M. R. M. Fontes, Int. J. Biol. Macromol., 2021, 191, 255–266 CrossRef CAS.
- O. G. Berg, M. H. Gelb, M.-D. Tsai and M. K. Jain, Chem. Rev., 2001, 101, 2613–2654 CrossRef CAS.
- A. V. Pinto, P. Ferreira, A. V. Cunha, R. W. A. Havenith, A. L. Magalhaes, M. J. Ramos and P. A. Fernandes, Chem. Sci., 2024, 15, 9793–9805 RSC.
- Y. Snitko, R. S. Koduri, S. K. Han, R. Othman, S. F. Baker, B. J. Molini, D. C. Wilton, M. H. Gelb and W. Cho, Biochemistry, 1997, 36, 14325–14333 CrossRef CAS PubMed.
- S. K. Han, E. T. Yoon, D. L. Scott, P. B. Sigler and W. Cho, J. Biol. Chem., 1997, 272, 3573–3582 CrossRef CAS.
- D. Saikia, R. Thakur and A. K. Mukherjee, Toxicon, 2011, 57, 841–850 CrossRef CAS.
- D. Mora-Obando, C. Diaz, Y. Angulo, J. M. Gutierrez and B. Lomonte, PeerJ, 2014, 2, e569 CrossRef PubMed.
- V. O. Zambelli, G. Picolo, C. A. H. Fernandes, M. R. M. Fontes and Y. Cury, Toxins, 2017, 9, 406 CrossRef.
- M. M. Thunnissen, E. Ab, K. H. Kalk, J. Drenth, B. W. Dijkstra, O. P. Kuipers, R. Dijkman, G. H. de Haas and H. M. Verheij, Nature, 1990, 347, 689–691 CrossRef CAS PubMed.
- K. Sekar, S. Eswaramoorthy, M. K. Jain and M. Sundaralingam, Biochemistry, 1997, 36, 14186–14191 CrossRef CAS PubMed.
- S. P. White, D. L. Scott, Z. Otwinowski, M. H. Gelb and P. B. Sigler, Science, 1990, 250, 1560–1563 CrossRef CAS PubMed.
- D. L. Scott, S. P. White, J. L. Browning, J. J. Rosa, M. H. Gelb and P. B. Sigler, Science, 1991, 254, 1007–1010 CrossRef CAS.
- D. L. Scott, Z. Otwinowski, M. H. Gelb and P. B. Sigler, Science, 1990, 250, 1563–1566 CrossRef CAS.
- H. M. Verheij, J. J. Volwerk, E. H. Jansen, W. C. Puyk, B. W. Dijkstra, J. Drenth and G. H. de Haas, Biochemistry, 1980, 19, 743–750 CrossRef CAS.
- C. J. Van Den Bergh, A. J. Slotboom, H. M. Verheij and G. H. De Haas, Eur. J. Biochem., 1988, 176, 353–357 CrossRef CAS PubMed.
- J. M. Maraganore, G. Merutka, W. Cho, W. Welches, F. J. Kezdy and R. L. Heinrikson, J. Biol. Chem., 1984, 259, 13839–13843 CrossRef CAS.
- C. Diaz-Oreiro and J. M. Gutierrez, Toxicon, 1997, 35, 241–252 CrossRef CAS.
- Y. Li, B. Z. Yu, H. Zhu, M. K. Jain and M. D. Tsai, Biochemistry, 1994, 33, 14714–14722 CrossRef CAS PubMed.
- C. Betzel, T. P. Singh, D. Georgieva and N. Genov, in Handbook of Metalloproteins, ed. A. Messerschmidt, R. Huber, T. Poulas, K. Wieghardt, M. Cygler and W. Bode, Wiley, 2004 Search PubMed.
- H. Verheij, A. Slotboom and G. De Haas, Rev. Physiol. Biochem., 1981, 91, 91–203 CAS.
- A. R. Peters, N. Dekker, L. van den Berg, R. Boelens, R. Kaptein, A. J. Slotboom and G. H. de Haas, Biochemistry, 1992, 31, 10024–10030 CrossRef CAS PubMed.
- E. A. Dennis, J. Cao, Y. H. Hsu, V. Magrioti and G. Kokotos, Chem. Rev., 2011, 111, 6130–6185 CrossRef CAS.
- B.-Z. Yu, J. Rogers, G. R. Nicol, K. H. Theopold, K. Seshadri, S. Vishweshwara and M. K. Jain, Biochemistry, 1998, 37, 12576–12587 CrossRef CAS.
- T. M. Epstein, B. Z. Yu, Y. H. Pan, S. P. Tutton, B. P. Maliwal, M. K. Jain and B. J. Bahnson, Biochemistry, 2001, 40, 11411–11422 CrossRef CAS PubMed.
- J. P. Jambeck and A. P. Lyubartsev, J. Chem. Theory Comput., 2013, 9, 774–784 CrossRef PubMed.
- J. P. Jambeck and A. P. Lyubartsev, J. Phys. Chem. B, 2012, 116, 3164–3179 CrossRef PubMed.
- M. Krack, Theor. Chem. Acc., 2005, 114, 145–152 Search PubMed.
- B. I. Lee, R. Dua and W. Cho, Biochemistry, 1999, 38, 7811–7818 CrossRef CAS PubMed.
- M. Gudmand, S. Rocha, N. S. Hatzakis, K. Peneva, K. Müllen, D. Stamou, H. Uji, J. Hofkens, T. Bjørnholm and T. Heimburg, Biophys. J., 2010, 98, 1873–1882 CrossRef CAS PubMed.
- A. L. Lomize, I. D. Pogozheva, M. A. Lomize and H. I. Mosberg, BMC Struct. Biol., 2007, 7, 44 CrossRef.
- T. V. Pogorelov, J. V. Vermaas, M. J. Arcario and E. Tajkhorshid, J. Phys. Chem. B, 2014, 118, 1481–1492 CrossRef CAS PubMed.
- S. Qin, A. H. Pande, K. N. Nemec and S. A. Tatulian, J. Mol. Biol., 2004, 344, 71–89 CrossRef CAS.
- B. Z. Yu, J. Rogers, M. D. Tsai, C. Pidgeon and M. K. Jain, Biochemistry, 1999, 38, 4875–4884 CrossRef CAS.
- C. C. Yang and L. S. Chang, Toxicon, 1988, 26, 721–731 CrossRef CAS.
- S. Qin, A. H. Pande, K. N. Nemec, X. He and S. A. Tatulian, J. Biol. Chem., 2005, 280, 36773–36783 CrossRef CAS PubMed.
- R. V. Stahelin and W. Cho, Biochemistry, 2001, 40, 4672–4678 CrossRef CAS.
- X. Liu, H. Zhu, B. Huang, J. Rogers, B. Z. Yu, A. Kumar, M. K. Jain, M. Sundaralingam and M. D. Tsai, Biochemistry, 1995, 34, 7322–7334 CrossRef CAS PubMed.
- S. Bezzine, J. G. Bollinger, A. G. Singer, S. L. Veatch, S. L. Keller and M. H. Gelb, J. Biol. Chem., 2002, 277, 48523–48534 CrossRef CAS PubMed.
- S. F. Baker, R. Othman and D. C. Wilton, Biochemistry, 1998, 37, 13203–13211 CrossRef CAS PubMed.
- S. F. Sousa, A. R. Calixto, P. Ferreira, M. J. Ramos, C. Lim and P. A. Fernandes, ACS Catal., 2020, 10, 8444–8453 CrossRef CAS.
- S. A. Beers, A. G. Buckland, N. Giles, M. H. Gelb and D. C. Wilton, Biochemistry, 2003, 42, 7326–7338 CrossRef CAS PubMed.
- M. Sumandea, S. Das, C. Sumandea and W. Cho, Biochemistry, 1999, 38, 16290–16297 CrossRef CAS.
- K. Diraviyam and D. Murray, Biochemistry, 2006, 45, 2584–2598 CrossRef CAS.
- Y. Snitko, S. K. Han, B. I. Lee and W. Cho, Biochemistry, 1999, 38, 7803–7810 CrossRef CAS PubMed.
- C. Diaz, G. Leon, A. Rucavado, N. Rojas, A. J. Schroit and J. M. Gutierrez, Arch. Biochem. Biophys., 2001, 391, 56–64 CrossRef CAS.
- A. G. Buckland and D. C. Wilton, Biochim. Biophys. Acta, 2000, 1483, 199–216 CrossRef CAS PubMed.
- K. K. Lucas and E. A. Dennis, Prostaglandins Other Lipid Mediators, 2005, 77, 235–248 CrossRef CAS.
- Y. Wang, J. Zhang, D. Zhang, H. Xiao, S. Xiong and C. Huang, Molecules, 2018, 23, 391 CrossRef PubMed.
- G. H. M. Salvador, R. J. Borges, B. Lomonte, M. R. Lewin and M. R. M. Fontes, Biochim. Biophys. Acta Gen. Subj., 2021, 1865, 129913 CrossRef CAS PubMed.
- W. Bryan-Quiros, J. Fernandez, J. M. Gutierrez, M. R. Lewin and B. Lomonte, Toxicon, 2019, 157, 1–7 CrossRef CAS PubMed.
- G. H. M. Salvador, A. A. S. Gomes, W. Bryan-Quiros, J. Fernandez, M. R. Lewin, J. M. Gutierrez, B. Lomonte and M. R. M. Fontes, Sci. Rep., 2019, 9, 17203 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06511e |
| This journal is © The Royal Society of Chemistry 2025 |