
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolded–twisted mechanisms control dynamic redox properties, photophysics and electron transfer of anthanthrene-quinodimethanes†
Abel
Cárdenas Valdivia‡
a,
Frédéric
Lirette‡
b,
José M.
Marín-Beloqui
 *a,
Abel
Carreras
*c,
David
Casanova
de,
Joël
Boismenu-Lavoie
b,
Jean-François
Morin
*b and
Juan
Casado
*a
*a,
Abel
Carreras
*c,
David
Casanova
de,
Joël
Boismenu-Lavoie
b,
Jean-François
Morin
*b and
Juan
Casado
*a
aDepartment of Physical Chemistry, University of Málaga, Andalucia-Tech, Campus de Teatinos s/n, 29071 Málaga, Spain. E-mail: casado@uma.es; jm.marinbeloqui@uma.es
bDépartement de Chimie, Université Laval, 1045 Ave de la Médecine, Québec, G1V 0A6, Canada. E-mail: Jean-Francois.Morin@chm.ulaval.ca
cMultiverse Computing, 20014 Donostia, Euskadi, Spain. E-mail: abelcarreras83@gmail.com
dDonostia International Physics Center (DIPC), 20018 Donostia, Euskadi, Spain
eIKERBASQUE – Basque Foundation for Science, 48009 Bilbao, Euskadi, Spain
First published on 16th December 2024
Abstract
The synthesis, electrochemical, spectroelectrochemical, photophysical and light induced electron transfer reactions in two new anthanthrene quinodimethanes have been studied and analyzed in the context of dynamic electrochemistry. Their properties are dependent on the interconversion between folded and twisted forms, which are separated by a relatively small energy range, thus allowing to explore their interconversion by variable temperature measurements. The photophysics of these molecules is mediated by a diradical excited state with a twisted structure that habilitates rapid intersystem crossing. Moreover, when codissolved with an electron acceptor compound such as PCBM, direct anthanthrene to PCBM electron transfer and back-electron transfer are also observed, resulting in the formation of charged states and, then, a raise in the population of the excited state triplet manifold upon charge recombination. Some of these properties have been interpreted with the help of quantum chemical calculations. The existence of an equilibrium between folded and twisted states upon electrochemical and photoexcitation stimuli represents a novelty in comparison with existing literature on compounds also showing dynamic electrochemical properties.
1 Introduction
In the conventional reversible electrochemistry of π-conjugated molecules, the molecule that is oxidized smoothly changes its molecular structure upon electron extraction in such a way that its associated reduction back to the neutral compound occurs in a slightly altered conformation implying small structural changes (Scheme 1a).1–3 This mechanism displays separated anodic and cathodic electrochemical waves with potentials slightly differing in between. | ||
| Scheme 1 (a) Typical reversible behavior of the electrochemical process of π-conjugated organic molecules; (b) dynamic redox electrochemistry mechanism (F = folded, T = twisted). | ||
Dynamic redox electrochemistry4,5 introduces a change in this mechanism consisting in an equilibrium between distinctive conformations of the neutral and of charged species (for instance, the processes seen in Scheme 1b between AF and AT). This equilibrium causes a difference in the oxidation and reduction waves of the same redox process depending on the temperature. In the literature, most of the studied cases of polycyclic dynamic redox electrochemistry behaviour are focused in anthraquinodimethane derivatives6–9 and in their dimers10–12 (see Scheme 2). The mechanism that explains this dynamic redox electrochemistry in this family of compounds is the coupling of the electron transfer process in the electrode with an intramolecular conformational reorganization in the organic redox active molecule. This means that the most stable conformation of the neutral compound (a folded structure) is not the most stable conformation upon oxidation (typically a twisted structure). This overall results in rather different voltage peaks with redox gaps (difference between the oxidation and reduction peaks for the same electron transfer process) as high as ≈1 V between the forward oxidation and the backward reduction.
 | ||
| Scheme 2 Chemical structures of anthraquinodimethane derivatives (a and b) displaying dynamic electrochemical redox behaviors as well as the anthanthrone diradical reported by us (c). Chemical structures of the anthantrone A(m-OMe) and A(p-F) derivatives in this work (d). | ||
The conformational requirements for the existence of dynamic redox electrochemistry in anthraquinodimethane derivatives are imposed by the aromatic groups attached to the exocyclic double bonds and the steric hindrance with the central anthracene-like moiety, resulting in the twisted–folded equilibrium. The anthraquinodimethane represents the smallest moiety able to have a central quinodimethane ring at the origin of the folded–twisted conformational equilibrium. Therefore, the almost exclusive use of the anthracene as the π-bridge limits the understanding of structure–properties relationships needed to tune the electrochemical responses and, hence, other central quinodimethane units are welcome to broaden the space of chemical compounds with these dynamic redox properties.
We recently reported a new quinodimethane unit based on the anthanthrene quinodimethane13–15 moiety, where the twisted–folded equilibrium caused the formation of a diradical in the neutral state upon application of external pressure. This behavior motivated us to explore this polycyclic core, the anthanthrenequinodimethane group, to search for new electrophores with dynamic redox properties. With these studies, we hope to find new structure–function connections that could help to understand the intricated mechanisms relating conformational equilibria and redox processes. In this article, the anthanthrene polycyclic aromatic core is doubly flanked by bis(phenyl substituted)methylene groups (Scheme 2d), which impart the required steric congestion to promote the folded–twisted conformational effect. Furthermore, the outer phenyl rings are substituted with methoxy groups in the meta position (i.e., A(m-OMe)) and fluorine in the para position (i.e., A(p-F)). A complete electrochemical and UV-vis-NIR spectroelectrochemical study as a function of the temperature, as well as femtosecond, picosecond and microsecond transient absorption spectroscopic analysis in conjunction with quantum chemical calculations are conducted.
Interestingly, the folded(neutral)–twisted(dication) redox process occurs in a rather unusually small redox gap in A(m-OMe), which permits to tune such equilibrium by thermal energy. Moreover, the folded–twisted interconversion also plays an important role in the photophysical process through the appearance of a diradicaloid excited state able to promote efficient intersystem crossing. The convergence of these phenomena not only showcases the novelty of this research, but also paves the way for new molecules capable of dynamic redox electrochemistry.
2 Results and discussion
2.1. Synthesis
The synthesis of A(m-OMe) and A(p-F) is shown in Scheme 3, for further details and NMR spectra, see ESI.† Starting from compound 1,14 a four-fold Suzuki coupling with either 3-methoxyphenylboronic acid or 4-fluorophenylboronic acid provided A(m-OMe) and A(p-F) in 81% and 85% yield, respectively. | ||
| Scheme 3 Synthetic route for A(m-OMe) and A(p-F). | ||
2.2. Electrochemical study
The cyclic voltammograms of the two compounds in Fig. 1, A(m-OMe) and A(p-F), are significantly different: both compounds show very similar CV shapes in the oxidation process. In both cases a single oxidation band is present, indicating the presence of a single oxidation step. As seen in similar molecules in literature,13–15 this oxidation process corresponds to the two-electron oxidation to form the A(m-OMe)2+ and A(p-F)2+ dications. The origin for this two-electron oxidation is the large distortion (folded) of the neutral molecule which originates a decoupling between the two donor moieties. Therefore, the oxidation of a terminal bis(phenyl substituted)methylene moiety occurs independently of the other unit. However, their corresponding reduction processes are completely different, with A(p-F) disclosing the typical aspect of a redox reversible step. Conversely, A(m-OMe) displays the features related with the existence of dynamic redox electrochemistry, such as the appearance of two peaks (instead of one) separated by a 0.17 V redox gap. The similar oxidation process for both molecules is in line with the calculated HOMO energy level (see Fig. S1–S3 and Table S1†) for both molecules.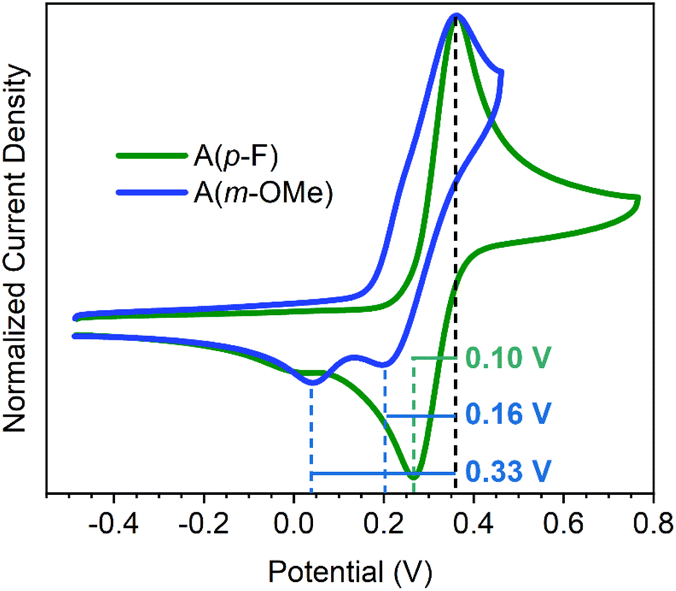 | ||
| Fig. 1 Cyclic voltammograms of (A(m-OMe), blue line) and (A(p-F), green line) at 298 K at a scan rate of 100 mV s−1 in 0.1 M TBAPF6 in CH2Cl2. Potentials have been corrected using ferrocene as reference. | ||
For A(p-F), the decoupling of the oxidation and reduction peak of 0.10 V involving the same species is characteristic of reversible processes in organic molecules.1–3 On the other hand, the CV of A(m-OMe) displays two peaks in the reduction process, spaced by 0.16 and 0.33 V from the voltage of the oxidation peak. In the reported cases of dynamic redox electrochemistry involving anthracene derivatives,6–10 only a reduction peak is observed, with redox gaps of 0.60–0.90 V from the oxidation peak. Hence, the features of A(m-OMe) reveal an interesting distinctive case in which: (i) two peaks appear, one likely due to the reversible process spaced by 0.16 V similar to 0.10 V in the reversible case of A(p-F), and another separated by 0.33 V, which we tentatively associate to the dynamic redox electrochemical behavior; and (ii) this latter peak appears at significantly smaller redox gaps, or voltages differences, compared to those typically described in anthracene derivatives. These differences in the behavior from anthracene to anthanthrene quinodimethanes highlight the important role of the polycyclic aromatic group.
Oxidation of the A(m-OMe) and A(p-F) compounds in the folded structure involves two electrons extraction leading to the formation of the dicationic species (see below for the spectroscopic characterization of the divalent species). The presence of single peak two-electron process indicates a decoupling of the external methylene carbons in the neutral state as a result of the folded conformation, where the positive charges are going to be localized upon oxidation.
2.3. Spectroelectrochemical analysis
In situ UV-vis-NIR spectroelectrochemical experiments have been carried out for both compounds at room temperature (Fig. S4†). Oxidation of A(m-OMe) to A(m-OMe)2+ in CH2Cl2 solution at 300 K gives way to the appearance of only one distinctive electronic absorption spectrum with NIR absorption bands at 1110 nm (shoulder) and 1365 nm accompanied by a weaker and narrow band at 703 nm. The position of the A(m-OMe)2+ absorption band is in line with other anthanthrene derivative dications previously published, confirming this assignment.16 Cooling this solution to 200 K produces the regression of the 1365 nm bands in favor of the 1110 nm (owing to the 1110 nm shoulder seen at 298 K) through a well-defined isosbestic point (see Fig. S2†). This isosbestic point is in line with the 1110/1365 nm pair of bands merging from two species in thermal equilibrium.Similarly, oxidation of A(p-F) at 300 K exhibits behavior akin to A(m-OMe). The oxidation of A(p-F) produces a spectrum featuring a near-infrared absorption band at 952 nm, comparable to the 1110 nm band of A(m-OMe) and likewise associated with the dication species. Cooling the solution of the dication of A(p-F) does not produce any significant changes (the band at 952 nm shifts to 934 nm, see Fig. S4†), highlighting the existence of only one form of the dication. In addition, the inversion of the voltage polarity after formation of the dications of A(m-OMe) and A(p-F) completely recovers the total absorbance and spectrum of the initial neutral species, in agreement with the absence of irreversible processes in the two cases.
Quantum chemical calculations within the density functional theory (DFT), the B3LYP exchange–correlation functional17,18 and the 6-31G(d,p) basis set (see ESI for further details†) for the dication state of A(m-OMe) have been carried out for the twisted and folded structures (Fig. 2b). In the twisted conformation, time-dependent DFT (TD-DFT)19 calculations predict two transitions with similar oscillator strength (i.e., intensity) at 700 and at 1200 nm. In the folded dication of A(m-OMe), TD-DFT excited state calculations estimate a single main transition band at 1000 nm with larger oscillator strength than the two bands of the twisted form. These three calculated excitations in the NIR region of the two dication conformations of A(m-OMe) can be correlated with the experimental absorption bands at 703 and 1365 (twisted dication), and 1110 nm (folded dication), respectively. Therefore, the theoretical spectra help to rationalize that at 300 K we have a mixture of folded and twisted dications, whereas the folded conformation is the dominant one at 200 K.
 | ||
| Fig. 2 (a) UV-vis-NIR absorption spectroelectrochemistry in 0.1 M TBAF CH2Cl2 of the neutral A(m-OMe) and A(p-F) together with absorption spectra of the chemically oxidized A(m-OMe) and A(p-F) at 300 K and at 200 K. (b) Simulated absorption spectrum for the folded (blue) and twisted (orange) forms of A(m-OMe) dication obtained at the B3LYP/6-31G(d,p) level. | ||
A van't Hoff plot (Fig. S5†) assuming an intramolecular conformational reaction (see Scheme 1 for the folded–twisted interconversion equilibrium of the dication, AT2+ ↔ AF2+) has been done from the spectra taken at different temperatures. From the slope of the van't Hoff plot, we estimated a total variation of the standard enthalpy of −8.0 kcal mol−1, that is, the enthalpy required to transform the dication from the twisted structure to the folded one. From the cyclic voltammetry at room temperature (Fig. 1), we measure a voltage difference between the oxidation peak and the two peaks of the reduction (due to the two conformations of the dication) of 0.16 V and 0.33 V, which correspond to a free energy variation20 of −7.4/−15.2 kcal mol−1, within the range of the thermodynamic data deduced from the spectroscopic results, and in agreement with the computed folded–twisted energy difference (−10.1 kcal mol−1).
The influence of temperature on the absorption spectra, driven by the conformational equilibrium, leads us to test how the electrochemical process is affected by temperature as well. For that, we design the CV experiment in low temperature conditions by cooling overnight the whole electrochemical setup (except the reference electrode) and then carrying out CV experiments until recovery of the room temperature. This CV experiment at low temperature was carried out for A(m-OMe) and A(p-F) (Fig. 3). We observe that by lowering the temperature, the CV of A(m-OMe) recovers almost full reversibility shape: the two peaks of the reduction backward process coalesce into one peak at similar voltage of the first reduction. This behavior evidences the dynamic redox electrochemical properties of A(m-OMe) and corroborates the conversion in the UV-vis-NIR spectra between the two conformations of the same dication. At low temperatures, the most stable folded conformer is increasingly populated at the expenses of the twisted form. Conversely for A(p-F), the lowering of the temperature does not produce any significant change in the recorded CVs. In fact, both CVs of A(m-OMe) and A(p-F) disclose very similar profiles at low temperature.
 | ||
| Fig. 3 Cyclic voltammetries of A(m-OMe) and A(p-F) in 0.1 M TBAPF6 in CH2Cl2 at room (300 K, darker lines) and at low (190 K, lighter lines) temperatures. Potentials have been corrected using ferrocene as internal reference. | ||
2.4. Transient absorption measurements
Pump-probe time resolved femtosecond excited state absorption measurements have been carried out for the two A(m-OMe) and A(p-F) compounds in an attempt to correlate the excited state neutral species with the folded and twisted conformations of the dications. In our previous publication of an anthantrene derivative15 (Fig. 1), the transition from a closed-shell neutral ground state to an open-shell state drives the formation of the twisted structure upon application of pressure.To this end, picosecond transient absorption spectra (ps-TAS) of A(m-OMe) in methyl cyclohexane solution at 298 K were obtained, as shown in Fig. 4a. Additionally, a tentative Jablonski photophysical scheme is proposed in Fig. 5 to explain the formation of the transient species. Results of A(p-F) were almost identical and are shown in Fig. S6,† indicating a very similar behaviour for both molecules upon photoexcitation. In addition, global analysis has been performed to help with the TAS signal assignment (Fig. S7†). At early times in the ps-TAS experiment of A(m-OMe), the spectrum presents two positive TAS bands, at 730 and 1115 nm. This profile with two different bands is very similar to the absorption spectra of the twisted dication with bands centered at ca. 700 and 1350 nm. Therefore, we assign the initially formed species to the relaxed S1 state of A(m-OMe) with a twisted structure. The planarization of the central anthanthrene core in the twisted form after excited state relaxation causes the common NIR absorption profile of the photo-generated species and the twisted dication A(m-OMe) species. Moreover, we argue that the S1 state must hold a large diradical character in order to stabilize the twisted conformation, [S1(twisted)] in Fig. 5. This description is in agreement to the diradical state formed upon pressure in the anthanthrene analogue15 in Fig. 1. After 5 ps, the spectrum evolves to a narrower shape with wavelengths changes and two additional positive TAS bands appearing at 1311 and 580 nm.
 | ||
| Fig. 4 Femtosecond transient absorption measurements of pristine A(m-OMe) in methylcyclohexane after pumping at 480 nm left) and of the a A(m-OMe):PCBM solution excited at 532 nm (middle) and at 470 nm (right). Excitation densities were maintained at 0.25 mW. | ||
 | ||
| Fig. 5 Photophysical Jablonski diagram for the excitation/de-excitation of A(m-OMe) in the presence and absence of PCBM acceptor. In the box, the redox processes are described. | ||
Electronic structure calculations at the twisted geometry of A(m-OMe) predicts near degeneracy of the S1 state with a triplet state (T3) with almost identical spatial distribution of the two unpaired electrons (Table S2 and Fig. S8†). The small singlet–triplet gap (amounting to 0.3 kcal mol−1), and the non-negligible spin–orbit coupling (Table S1†) suggests the existence of fast S1 → T3 intersystem crossing, in agreement with the quick appearance (5 ps) of the two-bands feature. Subsequently, the two-bands pattern converts to a single band at 715 nm which we associate as resulting from optical excitations from the lowest energy triplet excited state after T3 → T1 internal conversion. Finally, after 80 ps, the TA spectrum displays a single band at 580 nm. This single band spectrum of the T1 state is similar to the single band ground folded electronic state of A(m-OMe), which suggests that the triplet created upon photoexcitation might adopt a folded or pseudo-folded conformation. This triplet species in folded conformation is in line with the folded conformation of the naphthalene dimer published elsewhere.21
To further enrich the discussion, a solution of A(m-OMe) with PCBM acceptor was also analyzed upon photo-excitation. Fig. 5 shows the paths to account for the experiments in binary solutions, which were carried out in two conditions:
(i) by exciting the PCBM at 532 nm, where A(m-OMe) lacks of absorbance (Fig. 4b), a clear band at 1030 nm due to the PCBM radical anion [PCBM˙−]22 arises at early times accompanied by another band at 710 nm and a shoulder at 1100 nm that resemble very much the 715/1115 nm pair of bands of the A(m-OMe) recorded in the absence of the acceptor. This species might consist of a positively charged species of A(m-OMe), which keeps the twisted structure of its precursor S1 (relaxed) and that is in agreement with the similar spectral pattern of twisted structures in the dication of A(m-OMe) observed in the spectroelectrochemical experiments. The formation of the dication species in solution is likely due to the PCBM ability to accommodate up to six negative charges and also to the excess of PCBM in the experiment allowing the twisted dication state to be compensated by two radical anions.23 This 710/1150 nm band of the charged state formed by electron transfer decays very slowly, in the order of nanoseconds, until it is transformed to a band at 700 nm at 5 ns, which corresponds with the absorption of the PCBM triplet, T1(PCBM).24 After prompt formation of the PCBM:A(m-OMe) charged state, this radical pair might undergo back-electron transfer by charge recombination that largely populates the PCBM triplet manifold responsible of the 700 nm band surviving in nanoseconds.
(ii) Furthermore, μs-TAS experiments were carried out in the PCBM:A(m-OMe) solution (Fig. S9†) with excitation at 532 nm with the objective to form PCBM triplets able to be transferred to A(m-OMe). At early μs-timescales, a band at 700 nm is formed that matches with the PCBM triplets, which posteriorly decays in favor of a new band at 600 nm. This 600 nm band surviving in μs-timescales was reversibly sensitive to oxygen and was therefore assigned to anthanthrene triplet excited state species. This is in agreement with the assignment of the 580 nm band in the picosecond experiments also to the triplet states of A(m-OMe).
(iii) By performing ps-TAS experiments exciting on the A(m-OMe) absorption band at 475 nm (Fig. 4c) of the binary solution, the same band at 710 nm PCBM triplet at early timescales was detected as well as the double band feature at 560 and 680 nm also assigned to the triplet species of A(m-OMe). These triplet bands finally transform into the band at 600 nm formed upon 100 ps that can be related with the presence of pseudo-folded triplet excited states of A(m-OMe), such as observed in ps-TAS experiments of pristine A(m-OMe).
The change on conformation of A(m-OMe) upon photoexcitation was further confirmed by the use of steady-state emission spectra (Fig. S10†). A(m-OMe) emission results show mirror-like spectra relative to the 0–0 absorption transition with a considerable Stokes shift and vibronic progressions with significant spacing. These Stokes shift and vibronic progressions are in line with the large change of conformation upon photoexcitation, between the absorbing (folded) and the fluorescent emitting (twisted) states. In addition, the similarity between the PL results of A(m-OMe) and A(p-F) further verifies the resemblance between photoexcitation dynamics of A(m-OMe) and A(p-F).
3 Conclusions
In this article, anthraquinodimethane, the general example of molecules displaying dynamic electrochemical redox properties has been extended with an anthanthrene core with a quinodimethane naphthalene moiety. This singular molecule is able to show dynamic electrochemical redox properties with redox gaps between the oxidation and reduction much smaller than the typical ones found in anthracene and bisanthracene derivatives. The conversion between the folded (neutral) and the twisted (dications) forms thus discloses suitable small redox gaps that allow to explore their interconversion by temperature variable measurements further permitting to obtain the thermodynamics properties of the reaction interconversion. The photophysics of the given compounds has been studied by transient absorption spectroscopy from picoseconds to microsecond time-resolved spectra. The main photophysical properties can be interpreted in the folded–twisted interconversion leading to a diradical excited state that promotes intersystem crossing. These excited states can also act as electron-donor in the presence of PCBM fullerene acceptor, allowing the formation of photo-produced radical cations likely stabilized in a twisted structure. This study highlights the crucial role of anthanthrene derivatives in triplet formation, particularly due to their recent use as singlet fission sensitizers. Our findings unequivocally demonstrate these derivatives remarkable ability to produce triplets.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
F. L. and J. B.-L. did the synthesis. A. C. V. carried out the electrochemical and spectroscopic work assisted by J. M. M. B., A. C. and D. C. did most calculations. J. F. M. and J. C. conceived the work. All authors contribute to write and discuss the scientific contents.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support by the MCIN/AEI of Spain (projects PID2021-127127NB-I00, PID2022-136231NB-I00 and RED2022-134939-T – Red de Fotovoltaica), and the Junta de Andalucía (PROYEXCEL-0328) are acknowledged. We also thank the Research Central Services (SCAI) and Supercomputing and Bioinnovation Center (SCBI) of the University of Málaga. J. M. M. B. wants to acknowledge the Spanish University Ministry and the European Union for his Maria Zambrano fellowship with NextGen-EU funding.References
- W. Ten Hoeve, H. Wynberg, E. E. Havinga and E. W. Meijer, J. Am. Chem. Soc., 1991, 113, 5887–5889 CrossRef CAS.
- P. Bäuerle, U. Segelbacher, K. Gaudl, D. Huttenlocher and M. Mehring, Angew Chem. Int. Ed. Engl., 1993, 32, 76–78 CrossRef.
- P. Bäuerle, T. Fischer, B. Bidlingmeier, J. P. Rabe and A. Stabel, Angew Chem. Int. Ed. Engl., 1995, 34, 303–307 CrossRef.
- T. Suzuki, M. Kondo, T. Nakamura, T. Fukushima and T. Miyashi, Chem. Commun., 1997, 2325–2326 RSC.
- T. Suzuki, J. Nishida and T. Tsuji, Angew Chem. Int. Ed. Engl., 1997, 36, 1329–1331 CrossRef CAS.
- Y. Ishigaki, T. Hashimoto, K. Sugawara, S. Suzuki and T. Suzuki, Angew. Chem., Int. Ed., 2020, 59, 6581–6584 CrossRef CAS.
- Y. Ishigaki, K. Sugawara, M. Yoshida, M. Kato and T. Suzuki, Bull. Chem. Soc. Jpn., 2019, 92, 1211–1217 CrossRef CAS.
- Y. Ishigaki, Y. Hayashi and T. Suzuki, J. Am. Chem. Soc., 2019, 141, 18293–18300 CrossRef CAS PubMed.
- K. Sugawara, T. Ono, Y. Yano, T. Suzuki and Y. Ishigaki, Mater. Chem. Front., 2023, 7, 1591–1598 RSC.
- Y. Hamamoto, Y. Hirao and T. Kubo, J. Phys. Chem. Lett., 2021, 12, 4729–4734 CrossRef CAS.
- Y. Hayashi, S. Suzuki, T. Suzuki and Y. Ishigaki, J. Am. Chem. Soc., 2023, 145, 2596–2608 CrossRef CAS PubMed.
- T. Nishiuchi, S. Aibara, H. Sato and T. Kubo, J. Am. Chem. Soc., 2022, 144, 7479–7488 CrossRef CAS PubMed.
- J. Giguère, Q. Verolet and J. Morin, Chem.–Eur. J., 2013, 19, 372–381 CrossRef PubMed.
- J.-B. Giguère, J. Boismenu-Lavoie and J.-F. Morin, J. Org. Chem., 2014, 79, 2404–2418 CrossRef PubMed.
- M. Desroches, P. Mayorga Burrezo, J. Boismenu-Lavoie, M. Peña Álvarez, C. J. Gómez-García, J. M. Matxain, D. Casanova, J. Morin and J. Casado, Angew. Chem., 2017, 129, 16430–16435 CrossRef.
- M. Desroches and J. Morin, Macromol. Rapid Commun., 2018, 13, 39 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- J. R. Runo and D. G. Peters, J. Chem. Educ., 1993, 70, 708 CrossRef CAS.
- X. Wang, W. G. Kofron, S. Kong, C. S. Rajesh, D. A. Modarelli and E. C. Lim, J. Phys. Chem. A, 2000, 104, 1461–1465 CrossRef CAS.
- S. Yamamoto, J. Guo, H. Ohkita and S. Ito, Adv. Funct. Mater., 2008, 18, 2555–2562 CrossRef CAS.
- M. Bühl and A. Hirsch, Chem. Rev., 2001, 101, 1153–1184 CrossRef.
- S. Cook, H. Ohkita, J. R. Durrant, Y. Kim, J. J. Benson-Smith, J. Nelson and D. D. C. Bradley, Appl. Phys. Lett., 2006, 89, 101128 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis details; Analytical data; Results of electrochemistry, Spectroscopic characterization, Transient absorption spectroscopy and Theoretical calculations. See DOI: https://doi.org/10.1039/d4sc06442a |
| ‡ These authors contribute equally to the article. |
| This journal is © The Royal Society of Chemistry 2025 |