
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–reactivity relationships in CO2 hydrogenation to C2+ chemicals on Fe-based catalysts
Jie
Zhu
 ,
Shamil
Shaikhutdinov
* and
Beatriz Roldan
Cuenya
,
Shamil
Shaikhutdinov
* and
Beatriz Roldan
Cuenya
Department of Interface Science, Fritz Haber Institute of the Max Plank Society, Faradayweg 4-6, 14195 Berlin, Germany. E-mail: shaikhutdinov@fhi-berlin.mpg.de
First published on 16th December 2024
Abstract
Catalytic conversion of carbon dioxide (CO2) to value-added products represents an important avenue towards achieving carbon neutrality. In this respect, iron (Fe)-based catalysts were recognized as the most promising for the production of C2+ chemicals via the CO2 hydrogenation reaction. However, the complex structural evolution of the Fe catalysts, especially during the reaction, presents significant challenges for establishing the structure–reactivity relationships. In this review, we provide critical analysis of recent in situ and operando studies on the transformation of Fe-based catalysts in the hydrogenation of CO2 to hydrocarbons and alcohols. In particular, the effects of composition, promoters, support, and particle size on reactivity; the role of the catalyst's activation procedure; and the catalyst's evolution under reaction conditions will be addressed.
1. Introduction
The continuously increasing emission of carbon dioxide (CO2) into the Earth's atmosphere and related climate changes have given rise to enormous interest in the chemical conversion of CO2 as a renewable carbon source into value-added chemicals through catalytic reactions. Using “green” hydrogen, CO2 hydrogenation is considered to be a promising strategy to achieve a CO2-neutral economy.1–3 While considerable progress has been made in converting CO2 into C1 products such as CO,4,5 CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 6,7 and methanol,8–10 the production of C2+ chemicals (hydrocarbons and alcohols) remains highly desirable due to their broader industrial applications.11,12 To some extent, this latter process resembles the famous Fischer–Tropsch Synthesis (FTS) that uses syngas (CO + H2) as a feedstock. Moreover, the CO2 hydrogenation reaction to C2+ is often referred to as CO2-FTS. For the classical FTS process, the catalysts based on iron (Fe), cobalt (Co) and ruthenium (Ru) are the most efficient for carbon chain growth.13 However, for the hydrogenation of CO2, Ru- and Co-based catalysts were found to largely produce methane (CH4), with only limited C2+ production.14–16 On the other hand, Fe-based catalysts showed great potential for producing long-chain hydrocarbons, ranging from C2–C4 olefins to diesel-range hydrocarbons,17–20 and also for producing C2+ alcohols.21 A combination of the Fe catalysts with zeolite catalysts can further upgrade the product distribution through oligomerization, isomerization, and aromatization reactions.22 Due to the superior chain growth ability and also their low cost, Fe-based catalysts are currently considered as the most promising candidates for the production of C2+ chemicals via CO2 hydrogenation on an industrial scale.18
6,7 and methanol,8–10 the production of C2+ chemicals (hydrocarbons and alcohols) remains highly desirable due to their broader industrial applications.11,12 To some extent, this latter process resembles the famous Fischer–Tropsch Synthesis (FTS) that uses syngas (CO + H2) as a feedstock. Moreover, the CO2 hydrogenation reaction to C2+ is often referred to as CO2-FTS. For the classical FTS process, the catalysts based on iron (Fe), cobalt (Co) and ruthenium (Ru) are the most efficient for carbon chain growth.13 However, for the hydrogenation of CO2, Ru- and Co-based catalysts were found to largely produce methane (CH4), with only limited C2+ production.14–16 On the other hand, Fe-based catalysts showed great potential for producing long-chain hydrocarbons, ranging from C2–C4 olefins to diesel-range hydrocarbons,17–20 and also for producing C2+ alcohols.21 A combination of the Fe catalysts with zeolite catalysts can further upgrade the product distribution through oligomerization, isomerization, and aromatization reactions.22 Due to the superior chain growth ability and also their low cost, Fe-based catalysts are currently considered as the most promising candidates for the production of C2+ chemicals via CO2 hydrogenation on an industrial scale.18
Structural and chemical changes, observed for Fe-based catalysts during the synthesis and the reaction itself, along with the complex reaction network, all present significant challenges for in-depth understanding of the structure–reactivity relationships for these catalysts. Typically, the catalyst synthesis starts with iron oxide as a precursor which undergoes reduction, carburization, and re-oxidation during its initial activation and reaction,17,23,24 often resulting in the simultaneous presence of multiple iron phases, including metallic Fe(0), and Fe(II) and Fe(III) oxides (FeO, Fe3O4, Fe2O3) and also carbides (Fe3C, Fe5C2).25–28 The structural dynamics of the Fe catalysts has been intensively studied in the closely related FTS process, which revealed the compositional and morphological changes, both in the bulk and at the surface.29–33 However, unlike FTS, where both CO and H2 behave as reducing agents, CO2 may additionally cause considerable oxidation of Fe. Surface reactions including oxygen removal, carbon deposition, carburization, oxidation, and hydrogenation become more complex. Obviously, there is a dynamic interplay between the reaction microenvironment and the surface structure of the catalyst that in turn alters surface reactions.34 In addition, metallic iron and iron carbide phases are sensitive to air exposure, which introduces some uncertainty in their identification. In this respect, ex situ studies which link the reactivity and the structural properties of a catalyst either prior to or after the catalytic tests need to be taken with certain precautions and critically analyzed, since in most cases the active sites are formed during activation or in the course of the chemical reaction. Therefore, studies on the dynamics of catalysts during the reaction become crucial for identifying the active phases/sites and for gaining a deeper understanding of the reaction mechanisms, which are pre-requisites for the rational design of more efficient and durable catalysts.35
In the past decade, several comprehensive reviews on CO2 hydrogenation to C2+ products have been published in the literature, focusing on catalyst structures, reaction mechanisms, and even on reactor design for various metal catalysts.36–39 Also, there are excellent review/perspective papers highlighting the dynamic evolution of heterogeneous catalysts in a broad range of reactions.35,40–42 Most recently, Ding et al. published an excellent review on the dynamic structure of Fe-based catalysts in COx hydrogenation, but mainly of CO.33 Thus, we are here exclusively focusing on CO2 hydrogenation to C2+ hydrocarbons and alcohols, discussing the most recent studies on the structural and chemical evolution of Fe-based catalysts. In particular, we focus on the effects of composition, promoters, support, and particle size on reactivity (Fig. 1). We also highlight the importance of in situ and operando characterization using advanced techniques described in detail in several prior reviews, including those from our own group.43,44 In the concluding section, we discuss the challenges and opportunities for future studies of this industrially important reaction.
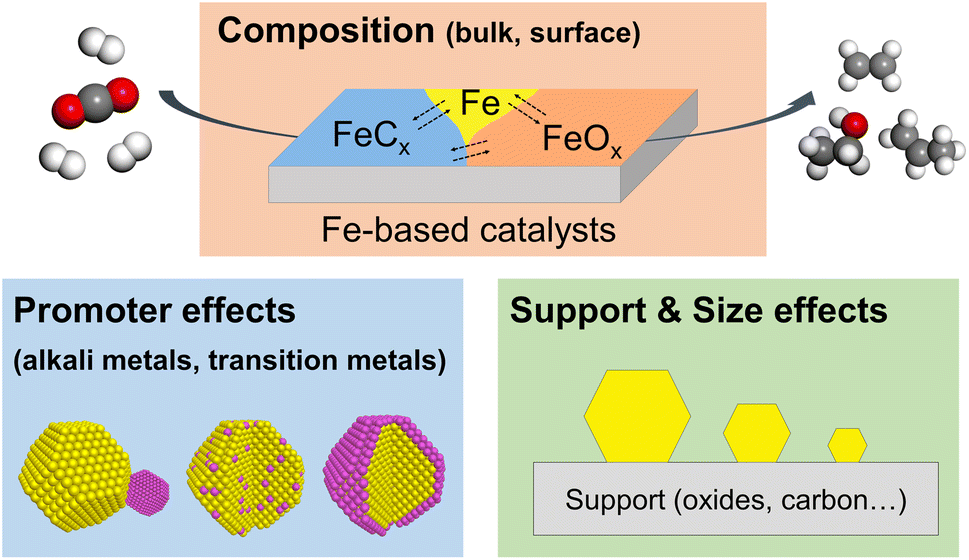 | ||
| Fig. 1 Schematic representation of several factors affecting the reactivity of Fe-based catalysts in the CO2 hydrogenation reaction. | ||
2. Phase transformations and surface composition
Preparation of the iron catalysts usually starts with iron oxides such as Fe2O3 and Fe3O4, which are stable under ambient conditions. Pristine Fe-oxides in the CO2 hydrogenation reaction primarily yield CO and water via the reverse water gas shift (RWGS) reaction.45,46 Depending on the reaction conditions, the oxides transform into metallic Fe and iron carbide phases (FeCx) during the reaction.47,48 The latter shifts the product distribution towards C2+ hydrocarbons,46 suggesting that, to make the catalyst active, the oxides must be first reduced or “activated”. In situ X-ray diffraction (XRD) measurements revealed sequential reduction of Fe2O3 to Fe3O4 and then to Fe during heating to 400 °C in H2 as shown in Fig. 2a.49 Subsequent introduction of the reaction mixture of CO2 and H2 (1:3 molar ratio) at 320 °C showed fingerprints of FeCx carbide formation within the first 20 min. After 6 h of time on stream (TOS), the Fe phase fully transformed into a mixture of Fe5C2, Fe3C, and Fe3O4. The spatial distribution of oxide and carbide phases was obtained by scanning transmission electron microscopy (STEM) combined with electron energy loss spectroscopy (EELS) in so-called quasi in situ measurements. (Henceforth, the term “quasi in situ” stands for the measurements on samples transferred from the reactor to the corresponding analytical tool without exposure to the ambient atmosphere.) The results showed that oxygen migrates from the surface inwards into the particle, while carbon remains at the surface (Fig. 2b).34 Based on additional high-resolution transmission electron microscopy (HRTEM) images, it was concluded that the initially metallic Fe particles transformed into a core–shell like structure, with the core primarily composed of Fe3O4, while the surface contained both Fe3O4 and Fe5C2, after 10 h of TOS (320 °C; 30 bar). The results also indicated that structural transformations at the surface are quite different from those in the bulk. While (bulk-sensitive) quasi in situ Mössbauer spectra showed a mixture of oxide and carbide phases reaching the steady state at ca. 3 h of TOS, the surface composition studied by quasi in situ X-ray photoelectron spectroscopy (XPS) showed continuous surface oxidation for more than 10 hours.34 Importantly, the transformation of the metallic surface into FeCx and FeOx is accompanied by an increase of CO2 conversion and C2+ hydrocarbon selectivity, from 18 to 39%, and from 20 to 57%, respectively (see region I in Fig. 2c). However, further surface oxidation slows down the activity (region II in Fig. 2c), indicating that excess surface FeOx leads to catalyst deactivation.34
 | ||
| Fig. 2 Structural evolution and catalytic performance of Fe catalysts during CO2 hydrogenation. (a) In situ XRD patterns showing the reduction of Fe2O3 to Fe in pure H2 (8 bar) and the phase transition during the reaction (H2/CO2 = 3; 320 °C; 8 bar). Adapted with permission from ref. 49. Copyright 2023, Elsevier. (b) Element distribution maps of spent catalysts after 1, 3 and 10 hours of reaction and (c) catalytic performance as a function of the reaction time (H2/CO2 = 3; 320 °C; 30 bar). Scale bars, 10 nm. Adapted with permission from ref. 34. Copyright 2022, The Authors, published by AAAS. (d) Quasi in situ Fe 2p and C 1s XPS spectra of an Fe2O3 catalyst measured after different treatments in a high-pressure cell, as indicated. Reaction conditions: H2/CO2 = 3; 300 °C; 1 bar. Adapted with permission from ref. 50. Copyright 2023, American Chemical Society. | ||
In a similar study performed at ambient pressure (1 bar), Kondratenko and co-workers50 using quasi in situ XPS showed that the surface consists of Fe(0) with small amounts of FeOx after activation in H2. During CO2 hydrogenation, metallic Fe transformed into an FeCx phase, which was concluded based on the small shift of the Fe 2p3/2 XPS peak from 706.6 to 707.0 eV and appearance of “carbidic” carbon (at 283.4 eV) in the C 1s region (Fig. 2d). In situ XRD showed rapid formation of Fe5C2 and Fe3C phases. As the reaction proceeded, the catalyst lost its activity and selectivity to hydrocarbons in favor of CO, although the surface and bulk did not undergo considerable oxidation during this period. However, in situ Raman in combination with C 1s XPS data indicated coke formation. By correlating the structural information with temporal analysis of H2 and CO2 activation and steady-state isotopic transient kinetic analysis (SSITKA) results, the authors came to the conclusion that coke inhibits the adsorption and activation of both CO2 and H2, and suppresses the C–C coupling reaction.50
Compared to the commonly studied hematite (α-Fe2O3) precursor, maghemite (γ-Fe2O3) behaves differently.51 During the reduction in H2, these two oxide phases transformed into α-Fe and γ-Fe, respectively, albeit with a portion of Fe3O4 as observed by in situ XRD and Raman. Interestingly, operando XRD measurements showed the formation of χ-Fe5C2 from α-Fe, and θ-Fe3C from γ-Fe phases, respectively, during the CO2 hydrogenation reaction (H2/CO2 = 3; 25 bar; 350 °C).51
Therefore, Fe carbides, which are widely recognized as the active phases for the classical FTS process,52 appear to be also crucial for CO2 hydrogenation, since the formation of FeCx is accompanied by the increased selectivity to C2+ hydrocarbons. Such correlations have inspired researchers to directly synthesize FeCx catalysts, with treatment in a CO atmosphere (so called “activation” in CO) being the most straightforward and efficient method. In situ XRD and Raman studies showed that Fe2O3 was first reduced to Fe and then carburized to form Fe5C2 as the temperature increased to 350 °C.53 The prepared Fe5C2 catalysts exhibited 54% selectivity to C2+ hydrocarbons and only 3% selectivity to CO. However, the Fe5C2 phase was further transformed during the reaction. Operando Raman spectra revealed the gradual appearance of FeOx-related bands after 60 hours on stream, and complementary XPS, XRD and Mössbauer data confirmed a partial oxidation of Fe5C2 into Fe3O4, which is accompanied by a decrease in activity.53 It is interesting to note a quite low selectivity to CO and substantial selectivity to CH4, which were observed on the pure Fe5C2 catalysts. Also, Liu et al. reported 50% CO2 conversion, with 51% of the products being C2+ hydrocarbons, 46% CH4, and the remaining 3% CO.54 Extrapolation of the product distribution to zero conversion led to the conclusion that RWGS and methanation are the primary reactions on pure Fe5C2, and that most C2+ hydrocarbons resulted from the secondary hydrogenation reaction of CO produced through the FTS route.
In addition to Fe5C2, the Fe3C phase is also active in the CO2 hydrogenation reaction. However, its role remains poorly understood even for a much more explored FT synthesis.39,55,56 Theoretical calculations predicted both Fe3C and Fe5C2 to show a lower barrier for CO2 dissociation and hydrogenation than metallic Fe or Fe3O4.57 Experimentally, it was shown that Fe3C exhibits a high RWGS rate at atmospheric pressure,47,48 while it facilitates hydrocarbon formation at elevated pressures.58 However, the product distribution obtained for the Fe3C and Fe5C2 phases seems to critically depend on the catalyst preparation and the reaction conditions used. For example, in Zhang's work,51 a mixture of Fe3O4 and Fe3C formed in situ from the γ-Fe2O3 precursor showed much higher selectivity towards C5+ hydrocarbons than a mixture of Fe3O4 and Fe5C2 formed from α-Fe2O3 (16% vs. 3%). In contrast, Zhu et al. demonstrated that the individual Fe3C phase exhibits a similar hydrocarbon distribution to Fe5C2 (both prepared by CO pretreatment of the α-Fe2O3 precursor), but a slightly lower CO2 conversion (31% vs. 38%).34 Note also that Fe3C may be an intermediate phase in the evolution of the iron catalyst, i.e., the carburization Fe → Fe3C (carbon deficient) → Fe5C2 (carbon rich),34 and re-oxidation Fe5C2 → Fe3C → Fe3O4.53
Obviously, if not controlled, the catalyst activation in CO resulting in Fe carbide formation may additionally cause coke deposition. In the great majority of cases, there is an overlayer of carbonaceous species formed on the carbide surface which cannot be ignored when testing the catalytic performance of the “as-prepared” FeCx catalysts. For example, this carbon overlayer can block certain active sites, thus causing an inaccurate comparison of intrinsic activity when normalized to the surface area.
The carburization and re-oxidation processes are not limited to the Fe catalysts prepared from the Fe-oxide precursors. Also, iron nitride Fe2N nanoparticles (NPs), despite being encapsulated by a carbon shell, were found to undergo phase transformation to Fe5C2 under CO2 hydrogenation conditions.59 XRD data showed the carburization to occur in the CO2 + H2 mixture at temperatures as high as 175 °C. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) results revealed the formation of Fe–NCO species, which were further hydrogenated into gas-phase NH3 and carbonyl iron (Fe–CO) intermediates, the latter leading to the Fe5C2 formation. The resulting catalysts showed a selectivity of 54% for C2+ products and 31% for C2–C4 olefins at 250 °C and 10 bar.
Active phases
Certain correlations observed between the chemical composition of the Fe catalysts and their catalytic performance provided some rationale about the possible active phases (active sites) in this reaction. In a widely reported model, CO2 is first hydrogenated to CO on the Fe oxide surface via the RWGS reaction, and the produced CO further reacts with H2 on the Fe carbide to form C2+ chemicals through the FTS route.22,60 This model suggests that the Fe oxide is essential to initiate the reaction. However, the above-mentioned dynamic studies showed that a higher content of surface FeCx (usually Fe5C2) resulted in a higher CO2 conversion and yield of C2+ hydrocarbons, while the formation of excessive FeOx led to catalyst deactivation.46 These findings made researchers revisit the necessity and the role of the Fe oxide in this reaction.61 Indeed, there are results showing that the Fe carbide phase is also active in the RWGS reaction, even with a higher activity than that of Fe oxides.47,48 Moreover, both theoretical and experimental studies indicated that CO2 and H2 activation proceed more easily on the Fe carbide than oxide surfaces.57,62,63 Therefore, it is plausible that the sequential RWGS-FTS tandem route can, in principle, occur on the single Fe5C2 phase, i.e., without invoking the FeOx phase.54 Kondratenko's group also suggested the C2+ production on FeCx without the formation of CO in the gas phase.61 These studies can rationalize the superior catalytic performance of pure Fe5C2 catalysts towards C2+ hydrocarbons.It is interesting that pure FeCx catalysts formed by activation in CO prior to the reaction showed high CH4 selectivity, which is at variance with the FeCx surface formed in situ during the CO2 hydrogenation reaction over the Fe catalyst activated in H2. Note that adding Fe3O4 to Fe5C2 can reduce the CH4 selectivity.54,64 This could be indicative of a synergistic effect between Fe3O4 and FeCx, which can be influenced by their ratio and even spatial proximity (see more details below).64,65 All in all, the FeCx carbides are considered thus far as the major active phases, with FeOx suppressing CH4 production and enhancing the C2+ selectivity, whereas the excessive oxidation of FeCx leads to deactivation.
Reaction microenvironment
During the CO2 hydrogenation reaction, the chemical compositions of both the bulk and the catalyst surface evolve into a mixture of FeOx and FeCx irrespective of the initial state of the pre-catalyst. Based on classical thermodynamics, the carburization and oxidation of Fe depend on the chemical potentials of carbon (μC) and oxygen (μO) above the surface, which may be significantly influenced by reactants, intermediates, and products.66 For initially pure FeOx, the product is mainly CO and the microenvironment favors its evolution to FeCx. As the FeCx content increases and further hydrogenation of CO proceeds, a substantial amount of H2O is produced, causing an increase in μO and hence making FeOx thermodynamically more favorable.34 Overall, these two processes continuously compete with each other, altering the catalytic performance, which in turn affects the reaction microenvironment. As a result, a delicate balance between carburization and oxidation seems to exist during the reaction. Consequently, the catalyst surface may always consist of a mixture of FeOx and FeCx. Note that metallic Fe can be oxidized by either CO2 or water, and obviously more water is formed in the CO2 hydrogenation reaction than in FTS. Thus, the substantial oxidation of the catalyst surface stands out as the significant difference between these two, FTS and CO2-FTS, processes. Bulk structure evolution is further influenced by factors such as kinetics and the mobility of carbon and oxygen atoms in the surface and the bulk.The effect of water became an interesting topic that has drawn increasing attention from researchers.67 Co-feeding 5 vol% H2O significantly accelerated the surface oxidation, as found by quasi in situ XPS.34 To remove the water formed during the reaction, Chaudret et al. used a molecular sieve that adsorbs water.68 The authors observed the transformation of Fe NPs into FeCx in a CO2 hydrogenation atmosphere even at 230 °C, whereas only oxidation was found at this temperature in the absence of the molecular sieve. Such an approach was even applied in reactor designs.69 The hydrophilic/hydrophobic properties of the catalyst surfaces may affect the interaction between water and surface iron species and hence the reaction-induced surface transformation. For instance, Xu et al. coated Fe–Mn catalysts with hydrophobic silane species, which reduce water retention on the catalyst surface during FTS and thereby protect iron carbides from water-induced oxidation.70,71 In principle, this approach is applicable to the CO2 hydrogenation reaction.72 However, a too thick hydrophobic layer may have a negative effect, i.e., accelerating the oxidation of FeCx.73 Also, the hydrophobic carbon shell formed on the Fe carbide particles during the carburization step can minimize the water effect. In particular, alkali metal promoters, which usually enhance carbon deposition, suppress water-induced oxidation in both FTS74 and CO2-FTS (see more details below). This protective effect of carbon overlayers can explain the considerably slower oxidation of Fe5C2 particles initially prepared by CO activation,53 as compared to the FeCx carbide phase formed in situ during the reaction.
Finally, CO2 hydrogenation on Fe catalysts exhibits strong pressure dependence. For example, Visconti et al. found that at atmospheric pressure the CO selectivity was close to 95%, but higher reaction pressures suppressed CO selectivity to 12% at 5 bar and 10% at 10 bar, thus shifting the product distribution towards C2+ hydrocarbons.75 Note that different partial pressures of products (e.g., CO and H2O) may also lead to different degrees of carburization and oxidation at the surface. In fact, changing the reaction conditions, including temperature, pressure, feed gas composition (e.g., H2/CO2 ratio) and even space velocity, can alter the reaction microenvironment and thus the surface composition of the working Fe catalysts.34 Therefore, the different catalytic performance may result from both the reaction conditions and the dynamic surface composition. In such a highly sensitive catalytic system, some factors are difficult to decouple, and real-time monitoring of the catalyst structure is of particular importance.
3. Promoter effects
Despite many efforts, pure Fe catalysts showed low selectivity to C2+ products. To improve the catalytic performance, alkali metals were extensively investigated as promoters in this reaction that: (i) suppresses CH4 formation and shifts the product distribution towards long-chain hydrocarbons, particularly to olefins; (ii) improves long-term stability.76–83 For example, selectivity towards C2–C4 olefins increased to 2, 22 and 27% after adding, respectively, 1, 2, and 5 wt% potassium (K) to the FeOx precursor.84 As for sodium (Na)-promoted catalysts with only 0.01 wt% added, the CH4 selectivity decreased from 41 to 24%. Further increasing the Na content to 0.5% reduced CH4 selectivity to 7%, and simultaneously increased the selectivity towards total olefins from 6 to 64% (Fig. 3a).85 In this section, we discuss the effects of alkali metals on the nature of the Fe phases and elementary reaction steps such as adsorption, dissociation, C–C coupling, and hydrogenation.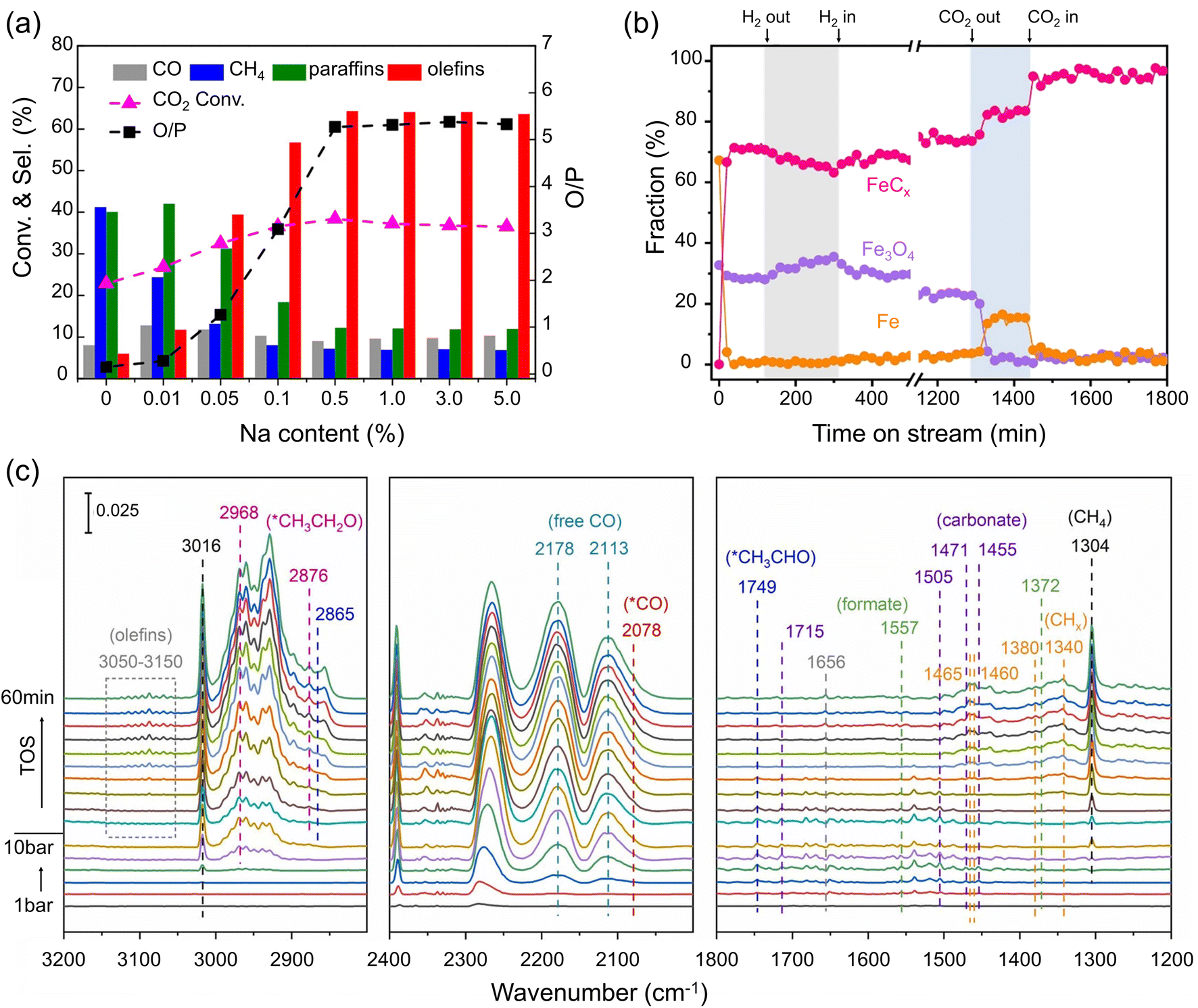 | ||
| Fig. 3 (a) Catalytic performance (conversion, selectivity, and olefin-to-paraffin ratio (O/P)) measured on Na-promoted Fe catalysts as a function of the Na content. Reaction conditions: H2/CO2 = 3; 30 bar; 320 °C. Adapted with permission from ref. 85. Copyright 2018, American Chemical Society. (b) In situ XRD-based fraction of the different Fe phases in the Na-promoted Fe catalysts (3 at%) during CO2 hydrogenation (H2/CO2 = 3; 300 °C). Arrows indicate the time when neither H2 nor CO2 was fed in the shaded area. Adapted with permission from ref. 86. Copyright 2023, Elsevier. (c) In situ DRIFTS spectra on an Fe catalyst promoted with Na and S. The spectra were collected while increasing the pressure from 1 bar to 10 bar and under reaction conditions (320 °C, 10 bar, H2/CO2 = 3) (from bottom to top). Adapted with permission from ref. 87. Copyright 2024, Elsevier. | ||
First, alkali metals promote the chemisorption of CO2 and weaken that of H2.79,88,89 Li, Na, K, Rb, and Cs were found to affect the local electronic state of Fe sites in the FeCx phase.90 Microkinetic analysis by temporal analysis of products (TAP) experiments suggested that CO2 adsorption and dissociation were enhanced by alkali metals in the order: Li < Na < K (all at 0.1 at% loading). Conversely, the ability of FeCx to activate CO and H2 was hindered, and K showed a stronger effect than Li and Na. It was further proposed that the Allen scale electronegativity is a good descriptor for both activity and product selectivity.90 Density functional theory (DFT) calculations also suggested that the presence of K lowers the energy barriers for CO2 dissociation.62 As a consequence, even microenvironments with moderate μC may promote the C–C coupling process triggering the production of C2+ hydrocarbons. Additionally, alkali metals impede olefin adsorption, thus suppressing their subsequent hydrogenation to paraffins, overall resulting in a higher olefin-to-paraffin (O/P) ratio.90,91
Alkali metals facilitate the formation of FeCx during the reaction.49,81,84,92,93 Apparently, the close proximity of Fe and the promoter results in a stronger effect.79 In addition, alkali metals inhibit the oxidation of FeCx during the reaction,49,85,86 although the fundamental reasons for this effect remain unclear. Yang et al. used in situ XRD with Rietveld analysis to investigate the effect of Na under controllably varied reaction conditions.86 At steady state, the unpromoted catalyst consisted of FeCx and Fe3O4. Removing H2 from the feed led to a decrease in FeCx and concomitant increase in Fe3O4 content due to oxidation by CO2, finally resulting in reduced catalytic activity towards C2+ hydrocarbons. Conversely, in the absence of CO2, i.e., in a pure H2 environment, both FeCx and Fe3O4 were reduced to metallic Fe. The addition of Na stabilized the catalyst composition during these “pulse” experiments, protecting catalytically active FeCx from oxidation and reduction (Fig. 3b), thereby enhancing its catalytic stability.86
The state of alkali metal species present during the reaction remains not fully understood, as they may easily interconvert during the reaction. In the “as-prepared” catalysts, K may exist as K2O, K2CO3, and KOH, but they become unstable at reaction temperatures. Gascon et al. used XPS and 39K nuclear magnetic resonance (NMR) spectroscopy to show that K2CO3 on the Fe catalysts evolved mainly into KOOCH, with small amounts of KHCO3 and K2CO3. The authors proposed that K firstly promotes the RWGS reaction: CO2 initially reacts with K2CO3 to form KHCO3, which then progressively transform into KOOCH, finally releasing CO.93 The produced CO can spill to neighboring Fe sites to carburize the surface to be ultimately hydrogenated into olefins via FTS. This mechanism explains why a carbon-containing K precursor, such as K2CO3, showed a stronger promotional effect than KCl and K2SO4.79
Compared to hydrocarbon production, the synthesis of C2+ alcohols requires not only C–C bond coupling, but also the insertion of oxygenate groups. In situ DRIFTS and theoretical calculations suggested that introducing a sulfur (S) promoter enhances the concentration and stability of the CO* intermediate on the surface.21 Given that alkali metals promote both the  “monomer” formation and carbon chain growth, a simultaneous use of alkali metals and sulfur as promoters may show cooperative effects on the C2+ alcohol production. The key is, however, to adjust the rates of C–C coupling and CO* insertion. For example, in Yao et al.'s study,87 the promotional effect of Li (0.3 wt%) on C–C coupling was rather limited, which only resulted in a slight increase in methanol selectivity when the Fe catalyst was modified with Li and S promoters. Conversely, the K promoter (3 wt%) showed a much stronger effect on C–C coupling, leading to increased selectivity towards C5+ hydrocarbons (42%) and C2+ alcohols (8%). A moderate promotional effect was observed on the Na and S-promoted catalyst at similar loadings, where the catalyst showed a CO2 conversion of 32% and 16% selectivity to C2+ alcohols.21,87In situ DRIFTS showed that in the presence of Na and S, both carbonate and formate species appeared upon exposure to the CO2 + H2 reaction mixture. Under reaction conditions, *CO, alkyl species, *CH3CHO and CH3CH2O* species appeared sequentially as the reaction proceeded (see Fig. 3c), pointing to the coupling reaction between alkyl and *CO ad-species. DFT calculations demonstrated that a delicate balance between the rates of dissociative and non-dissociative CO adsorption must have been achieved in these experiments.
“monomer” formation and carbon chain growth, a simultaneous use of alkali metals and sulfur as promoters may show cooperative effects on the C2+ alcohol production. The key is, however, to adjust the rates of C–C coupling and CO* insertion. For example, in Yao et al.'s study,87 the promotional effect of Li (0.3 wt%) on C–C coupling was rather limited, which only resulted in a slight increase in methanol selectivity when the Fe catalyst was modified with Li and S promoters. Conversely, the K promoter (3 wt%) showed a much stronger effect on C–C coupling, leading to increased selectivity towards C5+ hydrocarbons (42%) and C2+ alcohols (8%). A moderate promotional effect was observed on the Na and S-promoted catalyst at similar loadings, where the catalyst showed a CO2 conversion of 32% and 16% selectivity to C2+ alcohols.21,87In situ DRIFTS showed that in the presence of Na and S, both carbonate and formate species appeared upon exposure to the CO2 + H2 reaction mixture. Under reaction conditions, *CO, alkyl species, *CH3CHO and CH3CH2O* species appeared sequentially as the reaction proceeded (see Fig. 3c), pointing to the coupling reaction between alkyl and *CO ad-species. DFT calculations demonstrated that a delicate balance between the rates of dissociative and non-dissociative CO adsorption must have been achieved in these experiments.
In summary, alkali metals, particularly potassium and sodium, can modulate the reaction microenvironment by increasing CO2 adsorption and dissociation while weakening H2 adsorption, promote the formation of FeCx and prevent its excessive oxidation during the reaction, improving both activity and stability towards C2+ production. Thanks to their ability to tune the coupling of  species, a moderate combination with promoters like sulfur, which stabilize CO* intermediates, can achieve the production of C2+ alcohols (see more details below).
species, a moderate combination with promoters like sulfur, which stabilize CO* intermediates, can achieve the production of C2+ alcohols (see more details below).
4. Support effects
For catalytic reactions using precious or noble metals, it is common to use oxide supports to increase metal dispersion (to reduce the cost) and also to prevent thermally- or reaction-induced metal sintering. In the case of 3d-metal catalysts, in particular iron oxides, which are one of the most abundant compounds on the Earth, there is no real reason to use a support in its classical meaning, unless the oxide behaves as a structural promoter, primarily to increase the specific surface area of the active phase. In addition, a support is unavoidable for catalytic studies aimed at examining the size effect on reactivity, in particular for NPs in the sub-nanometer range, which would otherwise be impossible to stabilize against sintering at catalytically relevant temperatures.The results obtained for oxide-supported Fe-based catalysts in the CO2 hydrogenation reaction indicate that the support can considerably influence the catalytic performance.81,94–96 The supports may affect the chemical state of Fe during both activation and reaction. For example, FeO as an intermediate phase was observed during activation in CO on a catalyst supported on a monoclinic (m-) ZrO2, but not on a catalyst supported on tetragonal (t-) ZrO2.95 Moreover, less coke was formed on the former catalyst as monitored by in situ XRD and Raman spectroscopy. Consequently, m-ZrO2-supported K-promoted Fe catalysts exhibited 39% CO2 conversion and a high selectivity towards C2–C4 olefins (43% among all hydrocarbons). The morphology of the nanocrystalline support also affected the reduction of Fe-oxides. For example, CeO2 nanocubes exposing (100)-oriented facets were found to facilitate the reduction, as compared to CeO2 nanorods primarily exposing the (110) planes. Using the latter support resulted in catalysts showing a higher olefin/paraffin ratio.97
Alumina (Al2O3) is widely used as a support, and its interaction with Fe can regulate the chemical compositions of the catalyst surface. Increasing the calcination temperature of Na-promoted FeOx–Al2O3 pre-catalysts causes a stronger interaction, hindering the reduction and carburization of the Fe-oxide.98 The catalysts pre-calcined in air at 900 °C contained 25% Fe5C2 after the CO activation step, while those calcined at 350 °C showed a higher degree of carburization, resulting in 50% Fe5C2 and 13% Fe7C3. Correlation between the catalytic performance and surface composition, together with in situ DRIFTS and DFT calculations, demonstrated that a higher content of surface FeCx leads to a higher CO2 conversion, and a higher proportion of Fe5C2 in the carbide phase results in a higher chain growth possibility.
It should be noted that small Fe NPs, especially those smaller than 10 nm, behave quite differently during the reaction (see more details in Section 5). The effect of the oxide support on the surface and bulk evolution of such small NPs was investigated by Luna et al.99 FeOx NPs with a narrow size distribution around 4 nm were prepared by an inverse micelle encapsulation method. The micelles were deposited on nanocrystalline SiO2 and Al2O3 supports for in situ X-ray absorption spectroscopy (XAS) studies, and also on SiO2/Si(001) and Al2O3(0001) substrates for model studies using near ambient pressure (NAP)-XPS. The NAP-XPS spectra (Fig. 4a) showed that Fe(III) was reduced to Fe(II) and partially to Fe on a model Fe/SiO2 catalyst upon activation at 400 °C in 1 mbar H2, with Fe being re-oxidized during the CO2 hydrogenation at total 1 mbar pressure at 300 °C. In contrast, the Fe/Al2O3 model catalyst remained mainly in the Fe(III) state after both activation and reaction. Moreover, the state of Fe formed during the reaction was independent of the initial state of the pre-catalyst, i.e., Fe oxide or pure metallic Fe NPs prepared on both supports by physical vapor deposition (PVD). Quasi in situ XPS measurements performed after reduction at a catalytically relevant pressure (1 bar) revealed a higher degree of Fe reduction on the Al2O3-supported NPs as compared to SiO2 (Fig. 4b). After the CO2 hydrogenation reaction at 10 bar, the surface was found to be re-oxidized, with Fe(II) and Fe(III) species dominating the XPS spectra, independently of the oxide supports.
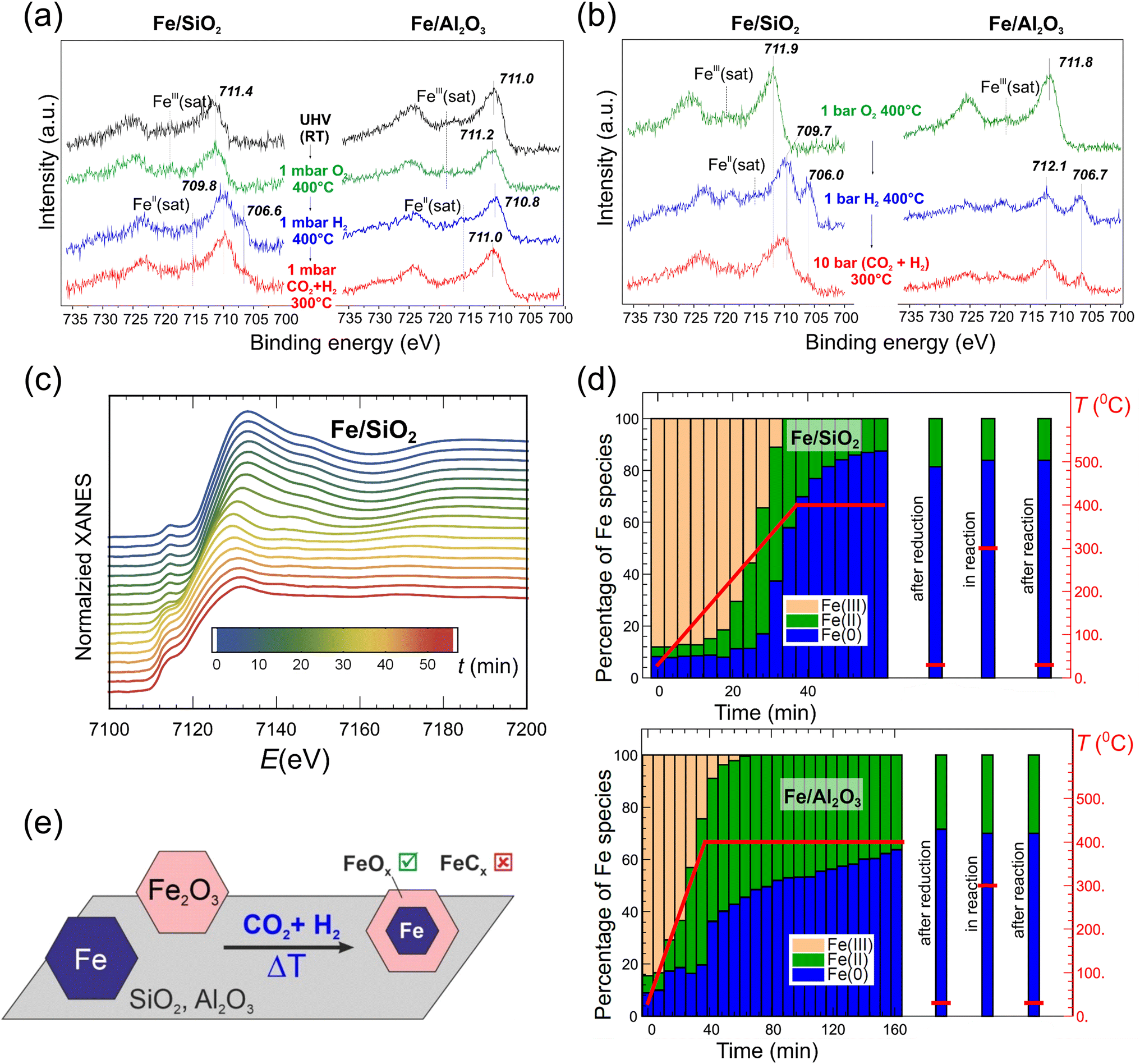 | ||
| Fig. 4 (a) Fe 2p region of the NAP-XPS spectra and (b) quasi in situ XPS spectra of model catalysts, prepared on SiO2/Si(001) and Al2O3(0001) substrates using polymer-free Fe-oxide micelles (4 nm in size). The NAP conditions (in a) and ex situ treatments (in b) are indicated. (c) In situ Fe K-edge XANES spectra of nanocrystalline (powder) SiO2-supported Fe-oxide catalysts, prepared using the same micelles as for the model catalysts (a) and (b), during heating to 400 °C in H2. (d) Fraction of different Fe species, obtained by linear combination analysis of XANES spectra (top, Fe/SiO2; bottom, Fe/Al2O3), during reduction in H2, under reaction conditions (10 bar; H2/CO2 = 3; 300 °C), and after cooling to room temperature. (e) Schematic representation of the structural evolution of the nano-sized Fe catalysts. Adapted with permission from ref. 99. Copyright 2021, American Chemical Society. | ||
A complementary in situ Fe K-edge X-ray absorption near edge structure (XANES) study showed that the fraction of metallic Fe species in the Fe/Al2O3 catalyst was significantly lower than in Fe/SiO2 (65 and 85%, respectively), indicating that FeOx NPs on Al2O3 are more resistant to reduction. Nonetheless, the state of Fe formed by the H2 activation step remained unchanged during the reaction (Fig. 4c and d), i.e., in contrast to the XPS results clearly showing surface re-oxidation in the reaction atmosphere. The main findings obtained by bulk-sensitive XAS and surface-sensitive XPS, namely, a core (metal-rich)-shell (oxide-rich) structure, are schematically depicted in Fig. 4e. Interestingly, there were no signs of Fe carbide formation during the reaction in XAS and XPS measurements on these nano-particulate catalysts, which produced light hydrocarbons, with the O/P ratio being considerably affected by the nature of the oxide support used. Still, it remains to be studied whether these findings can be assigned to pure support effects or whether they are also affected by the nano-sized nature of the active phase.
Compared to oxide supports, carbon supports were thought to exhibit a weaker interaction with Fe oxide. On the other hand, a carbon support may serve as a source of carbon for Fe carbide formation. Using in situ XANES spectroscopy, Muhler and co-workers found that SiO2-supported FeOx NPs can only be reduced to Fe(II) in H2 at 380 °C, while NPs supported on nitrogen-doped carbon nanotubes (CNTs) underwent full reduction to the metallic state.100 Consequently, the lower activity and C2+ selectivity of the Fe/SiO2 catalysts were attributed to the strong iron-silica interaction, which prevents reduction and hence carburization of Fe. In another study, Wu et al. prepared Fe/C catalysts using honeycomb-structured graphene as the support and potassium as the promoter, which showed 59% selectivity towards C2–C4 olefins, stable during 120 hours on stream.80 The long-term stability was attributed to the confinement effect of the porous structure of the support, which prevented the sintering of FeCx NPs during the reaction. Indeed, the mean size of the FeCx particles only slightly increased from 14 nm after 24 hours to 16 nm after 120 hours on stream.
In principle, the support not only influences and stabilizes the particular state of iron, but can directly participate in the reaction through the interaction with gas molecules and spillover-based mechanisms. For example, acid sites on the amorphous alumina support can promote the oligomerization of olefins first produced on the FeCx sites, as shown by in situ DRIFTS.24 Too strong acidity led to the pyrolysis of long-chain hydrocarbons, while moderate acidity in the Fe/AlOx catalysts showed a high selectivity (52%) to linear α-olefins (78% in C4+ olefins) that was stable for 450 h of TOS.24 For the case of a single-wall CNT support, those with a large curvature facilitated the dissociation of C–O bonds, thus promoting the formation of  monomers. Additionally, the confined space in CNTs can serve as a “nano-reactor”, where the residence time of light olefins can be longer, thus providing the possibility for oligomerization reactions and for achieving a high C5+ hydrocarbon selectivity, up to 40%.101
monomers. Additionally, the confined space in CNTs can serve as a “nano-reactor”, where the residence time of light olefins can be longer, thus providing the possibility for oligomerization reactions and for achieving a high C5+ hydrocarbon selectivity, up to 40%.101
Therefore, for supported Fe catalysts, not only the textural properties of a support (e.g., morphology, pore structure, specific surface area), but also their surface properties, such as acidity and hydrophilicity,73,102 play a significant role in the catalyst evolution and the surface reactions.
5. Size effects
Similar to many reactions on metal catalysts, CO2 hydrogenation is also quite sensitive to the metal particle size. In case of Ru,103 Rh,104 Ir105 and Ni106 catalysts, large particles favored CH4 formation, whereas reducing the NP size down to single atoms shifted the product distribution towards CO. The reactivity of Fe-based catalysts also showed size dependence, albeit being more complex because of a relatively large variety of products. It should be mentioned that sometimes the particle size referred to the size of Fe particles in the “as-prepared” (i.e., Fe-oxide) catalyst, or “activated” (reduced), or even spent catalyst. The latter constitutes a problem in the field, since the structure of these catalysts, including their size, likely changes during the reaction, leading to questionable size–reactivity correlations. The problem is especially drastic for single-atom pre-catalysts, where C–C coupling products might be assigned to the concomitant presence of small clusters or nanoparticles formed during operation.Based on the extended X-ray absorption fine structure (EXAFS) results of MoS2-supported Fe catalysts, Zheng et al. concluded that Fe was present primarily as single atoms even in the highly loaded catalysts, up to 10 wt%.107 The catalysts reduced in H2 showed 100% CO selectivity at 300 °C at atmospheric pressure. Increasing the pressure to 10 bar only led to the formation of small amounts of CH4 (<2%) and traces of C2 and C3 hydrocarbons, with CO dominating the product distribution (Fig. 5a). Note that close to 100% selectivity to CO remained for more than 80 hours, and no Fe–Fe bonds were found in EXAFS spectra measured on the 10 wt% Fe/MoS2 catalyst after reaction. CO2 conversion increased as the Fe loading increased from 3 to 10 wt%, presumably due to the higher density of the Fe single atoms. However, further increase of the Fe loading to 15 and 20 wt% resulted in decreased CO2 conversion due to the formation of Fe clusters, although CO was the main product.
 | ||
| Fig. 5 (a) Catalytic performance of Fe catalysts supported on MoS2 as a function of Fe loading (in wt%). Reaction conditions: 10 bar; 300 °C; H2/CO2 = 3. Adapted with permission from ref. 107. Copyright 2021, American Chemical Society. (b) Product selectivity as a function of Fe particle size on H2-activated Fe/ZrO2 catalysts. Reaction conditions: 30 bar; 320 °C, H2/CO2 = 3. (NB: The space velocity was adjusted for each catalyst to reach a similar CO2 conversion (∼13%)). Adapted with permission from ref. 45. Copyright 2020, American Chemical Society. (c) Composition of fresh, reduced, carburized, and post-reaction Fe catalysts determined by quasi in situ Mössbauer spectroscopy for two different initial particle sizes. Adapted with permission from ref. 108. Copyright 2024, Elsevier. | ||
Leybo et al. synthesized Fe phthalocyanine-derived single-atom catalysts supported on boron nitride.109 Again, the “as-prepared” catalysts exhibited 100% selectivity towards CO at 20 bar and 200–230 °C. However, as the reaction temperature increased to 320 °C, the product distribution shifted towards CH4 (15%) and C2+ hydrocarbons (10%). Interestingly, the latter products were observed even at lower reaction temperatures, if the catalyst was pre-reduced in H2 at 350 °C prior to the reaction. Based on a TEM study, the effect was explained by the formation of small Fe NPs (∼3 nm) at elevated temperatures, either during the reduction step or under reaction conditions. Therefore, the observed Fe sintering largely eliminates the initial difference in particle size.
This general trend that larger Fe NPs favor hydrocarbon production was further proven by Xie et al. who used Al2O3 supports with different pore sizes to prepare Fe2O3 particles ranging from 5 to 23 nm.110 The selectivity to C2+ and C5+ hydrocarbons showed a volcano-type relationship with respect to the initial particle size, with a maximum C2+ selectivity achieved at around 5–8 nm. Note, however, that the alumina supports were synthesized by quite distinct methods, so the results obtained may be influenced by both, size and support, effects.
Zhu et al. prepared a series of ZrO2-supported Fe catalysts with particle sizes in the reduced catalysts varying from 3 to 13 nm, as determined by a number of techniques such as CO chemisorption, XRD and TEM.45 As the particle size increased, selectivity to C2+ hydrocarbons and CH4 continuously increased from 9 to 16% and 22 to 34%, respectively, while that of CO decreased from 69 to 50% (Fig. 5b). Interestingly, the authors observed that the CO2 conversion and C2+ selectivity increase with TOS on the smallest 3 nm NPs, and attributed this behavior to the size effect via reaction-induced sintering. Kondratenko's group examined unsupported Fe2O3 NPs of larger sizes, i.e. 15–30 nm. In this study, smaller particles, possessing more defects, were found to facilitate the reduction and formation of defective Fe5C2 NPs, which showed enhanced CO2 and CO adsorption.111
One reason for the particle size effect is that small Fe NPs/clusters and single atoms often exhibit non-metallic properties. When supported, they may be harder to reduce because of their strong interaction with the underlying support.112,113 A lower degree of reduction is not conducive to the in situ formation of active FeCx.111,114 According to in situ XRD results, the reduction of ZrO2-supported FeOx particles starts at a lower temperature for 13 nm NPs, as compared to 6 nm NPs, and the formation of FeCx during CO2 hydrogenation proceeds much faster.45 In another case of carbon-supported K-promoted catalysts,108 the Fe2O3 NPs showed a similar degree of reduction to FeO in H2 at 400 °C for two samples with 7 and 9 nm initial average particle size (Fig. 5c). However, during the activation in the mixture of H2 and CO at 280 °C, these two samples showed considerably different compositions. The “7 nm” sample contained 21% of Fe2.2C, while the “9 nm” sample had 15% of Fe5C2 and 15% of Fe2.2C. More significantly, after the CO2 hydrogenation reaction (300 °C, 11 bar), the “9 nm” sample became almost fully carburized (85% Fe5C2) while the “7 nm” sample showed no changes.
The coordination of the Fe atoms at the particle surface may also play a role. Indeed, DRIFTS spectra of CO, used as a probe molecule, showed that the ratio of bridged and linear CO adsorption sites increased as the particle size increased from 3 to 13 nm, indicating a higher fraction of low-coordinated Fe sites on the smallest Fe particles.45 Since the carbon chain growth requires a close proximity of 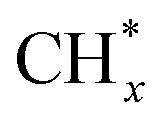 “monomers”, the C–C coupling reaction becomes more favorable on the well-ordered facets dominating on the largest particles.
“monomers”, the C–C coupling reaction becomes more favorable on the well-ordered facets dominating on the largest particles.
In summary, the particle size effects on reactivity may originate from both electronic and geometric effects, although the predominance of one versus the other is strongly linked to the nanoparticle/cluster size range considered, with electronic effects becoming most relevant for sizes in the sub-nanometer range. So far, the activity of catalysts containing single Fe atoms and small clusters in C2+ production has been very low, most likely because of: (i) the complex C–C coupling reactions requiring more than a single site; and (ii) the low degree of Fe reduction for the sub-nm particles and hence the limited formation of the FeCx carbide phase due to their strong interaction with the support. Nonetheless, a single-atom catalyst can serve as a “pre-catalyst” for preparation of catalysts with a narrow particle size distribution. The optimal particle size of Fe in the CO2-FT reaction seems to be in the range of 10–15 nm.
6. Bimetallic Fe-based catalysts
Adding a second metal (such as a 3d transition metal or noble metal) to Fe is an effective strategy to improve the selectivity and catalytic stability of the Fe catalysts.15,25,115,116 Several studies have shown that easily reducible metals, such as Pt,47 Pd117 and Cu,118 promote the reduction of the Fe-oxide through facile H2 dissociation on these metals and subsequent hydrogen spillover onto the Fe-oxide surface, thereby promoting the formation of the FeCx carbide phase under reaction conditions.For example, Cargnello's group prepared colloidal particles in order to provide a close contact between the Ru and Fe precursors, and the particles were deposited onto the γ-Al2O3 support (with a total metal loading of 1 wt%).119 After calcination at 700 °C to remove organic ligands, Ru was partially oxidized and Fe was in the form of γ-Fe2O3. Based on in situ XAS results in a H2 environment, upon the complete reduction of Ru, the Fe2O3 phase was fully reduced to metallic Fe at ∼300 °C, whereas the Ru-free, reference Fe2O3 catalyst underwent a much slower transition from Fe3O4 to FeO, with no complete reduction to Fe being observed until 500 °C (Fig. 6a). In situ Fe K-edge XANES spectra indicated that the Ru–Fe catalyst predominantly consisted of metallic Fe and FeCx during the reaction, with no observable contribution from FeOx. Interestingly, STEM images of the spent catalyst combined with energy dispersive spectroscopy (EDS) showed the formation of “core–shell” particles having a metallic Ru core and an FeOx shell about 4 nm in thickness. (In fact, the shell was composed of Fe and FeCx under reaction conditions, but was oxidized during the sample transfer through air.)
 | ||
| Fig. 6 (a) Compositional changes monitored by in situ XANES in the Fe2O3 and Ru–Fe2O3 catalysts during heating in H2. (b) Catalytic performance of Ru–Fe catalysts with different Fe contents that form 4 nm- and 1 nm-thick Fe shells during the reaction. Reaction conditions: 300 °C, 6 bar, H2/CO2 = 3. Adapted with permission from ref. 119. Copyright 2019, John Wiley and Sons. | ||
In situ EXAFS results revealed Ru–Fe bond formation at the interface between the Ru core and the Fe shell. The authors proposed that a relatively thick Fe shell in these particles obscured the electronic effect of Ru, and the difference in the catalytic performances of these two catalysts, i.e., with and without Ru, largely stems from the different degrees of reduction of the Fe phase. Indeed, when the Ru-free catalyst was reduced in H2 at 550 °C, it showed similar selectivity to the Ru-promoted catalyst, where Fe was fully reduced at 300 °C. In order to prepare the catalyst, with the surface exposing more Fe atoms in direct contact with Ru, the authors synthesized Ru particles covered by a thinner Fe shell (∼1 nm), and this catalyst showed a 4-fold increase in the hydrocarbon yield (Fig. 6b), implying the strong electronic effect of Ru on the reactivity in such hetero-structures.119
For the Pd-promoted catalysts, in situ XRD showed the formation of a Pd–Fe alloy during activation in H2.120 The catalyst underwent complete Fe carburization during the CO2 hydrogenation reaction, in contrast to the physically mixed Pd–Fe2O3 catalyst under the same reaction conditions. Since the latter does not form the Pd–Fe alloy in the reduction step, it is the alloy formation that promotes the formation of Fe5C2 in the reaction atmosphere. Based on the DRIFTS results, the alloy phase was proposed to be responsible for the RWGS reaction and CO non-dissociative activation, while Fe5C2 is responsible for the chain growth. The reaction at the PdFe/Fe5C2 interface seems to enhance the production of C2+ alcohols, achieving 27% selectivity at 300 °C and 50 bar.120
Copper (Cu) also improves the reducibility of FeOx, and hence facilitates the formation of FeCx,25 and also enhances the adsorption of CO2 and H2.115,121 Compared to the K-promoted FeCu/Al2O3 catalyst prepared by sequential impregnation, the catalyst prepared by co-impregnation of Cu and Fe precursors exhibited a strong interaction between Fe and Cu and showed a promotional effect, with selectivity to C5+ hydrocarbons increasing from 10 to 14%.122 In a similar Fe–Cu–K–Al system, Jun et al. used XRD, XPS and XAS to demonstrate that K promotes Cu incorporation into the lattice of either metallic Fe or Fe carbide phases during the reaction.123 The synergistic effect of Cu and K led to a C5+ yield of 18% compared to 13% obtained on the Cu-free, Fe–K catalyst.
Fe–Cu binary oxides have emerged as superior precursors for preparing effective catalysts. Comparative studies of CuFeO2 delafossite, CuFe2O4 spinel, and physically mixed Fe and Cu oxides showed that the fraction of C5+ in all hydrocarbons produced at 300 °C and 10 bar (with ∼30% CO selectivity) increases in the order CuO–Fe2O3 (3%) < CuFe2O4 (11%) < CuFeO2 (66%).25 Note, however, that the CuFeO2 catalyst contained traces of Na (0.03%). For a similar CuFeO2 catalyst, Li et al. reported 67% of C4+ olefins (44% of CO excluded) even at ambient pressure and 320 °C.124
Nonetheless, among the 3d transition metals, cobalt (Co) stands out as one of the most extensively studied,15,116,125–128 owing to its wide application in the conventional FTS process, where metallic Co showed a much higher chain growth factor than the Fe-based catalysts, and as such it is largely used to produce heavy hydrocarbons. However, in the CO2 hydrogenation reaction, pure Co showed high CH4 formation, with only limited C2+ production, and was therefore used primarily as the methanation catalyst. Studies on the Fe–Co catalysts showed that the spatial distance of Fe and Co significantly influences their catalytic behavior. When two phases are well separated, the CO2 hydrogenation reaction occurs independently on each component, resulting in substantial CH4 formation on the Co sites. In contrast, intimate contact or even close proximity between Fe and Co allows the CO formed on the Fe sites (via the RWGS reaction) to spill over to the Co sites, which enhances the chain growth in the FTS step and promotes heavy hydrocarbon production.
Jiang et al. addressed the role of the inter-particle distance between Fe and Co by employing different preparation methods, including co-impregnation and physical mixing.129 When Fe and Co were co-impregnated on a SiC support promoted by K, the selectivity towards C2+ hydrocarbons increased from 38 to 57%, and the CO2 conversion increased from 17 to 30%, compared to the Co-free Fe catalyst. However, the physically mixed FeK/SiC and Co/SiC catalyst, i.e., with a much larger inter-particle distance, mainly produced CH4 (79% selectivity), while it is only 3% on the Co-free Fe–K catalyst.
To controllably tune the proximity of Fe and Co phases, Tsubaki's group used graphene oxide as a “fence” to separate Fe and Co precursors (Fig. 7a).130 For Fe and Co to be in direct contact, all precursors of Fe, Co, and K were impregnated and uniformly dispersed on the exterior surface of the graphene. When the Fe precursor was first introduced for the hydrothermal treatment of graphene, Fe was found both on the graphene surface and between the graphene layers (intercalated). Finally, Co and K were impregnated onto the exterior graphene layers. Spatial distribution was analyzed using scanning electron microscopy (SEM) with EDS mapping. Using in addition in situ XRD, EXAFS and XPS, the authors showed that the catalysts consisted of Fe5C2 and metallic Co under reaction conditions. In comparison to the reference Fe catalysts, which exhibited 31% selectivity to C2–C4 olefins, the Fe–Co sites formed by direct contact revealed a higher (i.e., 50%) selectivity. Conversely, the spatially separated Fe–Co NPs produced almost no C2–C4 olefins, but achieved 44% selectivity to C3–C4 paraffins. It was proposed that the individual Co NPs enhance the secondary hydrogenation reactions of olefins produced on the Fe5C2 phase.
 | ||
| Fig. 7 (a) Scheme showing different approaches for the synthesis of spatially distributed Fe and Co on a graphene oxide (GO) support. Reproduced with permission from ref. 130. Copyright 2024, The Authors, published by Springer Nature. (b) Schematic diagram of alloying and de-alloying behaviors of Fe–Co bimetallic catalysts during activation and the CO2 hydrogenation reaction. | ||
When Fe and Co precursors form a single compound, such as CoFe2O4 and Fe–Co layered double hydroxide (LDH), then reduction in H2 results in Fe–Co alloying.131–135 However, further evolution of the alloy during CO2 hydrogenation strongly depends on the Fe/Co atomic ratio. To recall, for individual Fe and Co catalysts, metallic Fe transforms into the FeCx phase, while Co predominantly remains metallic, although there is some probability of Co-carbide formation. Accordingly, for an Fe-rich FeCo alloy, it mostly transforms into FeCx, with Co incorporated into its lattice, thus forming an “Fe–Co carbidic alloy”.
Kim et al. performed in situ XRD studies of a Na-promoted CoFe2O4 catalyst supported on CNTs.134 After reduction in H2, XRD showed diffraction patterns of an Fe–Co alloy. Due to the pressure limitation of their XRD setup, to simulate partial pressure of CO under realistic CO2 hydrogenation reaction conditions, the authors used pure CO at atmospheric pressure to treat the H2-activated catalyst. Only the FeCx phase was observed, with no signature of pure metallic Co.134 Based on theoretical considerations, the authors inferred the formation of (Fe1−xCox)5C2 carbide, where x is lower than 0.2, and even predicted its crystal structure, although direct experimental proof of the proposed structure is still missing. Nonetheless, implementation of this carbidic alloy into the fitting model showed consistency with Liu et al.‘s XRD, XAS and Mössbauer results, indirectly validating the carbidic alloy formation.132 Liu et al. also pointed out that when the Co/Fe molar ratio exceeds 0.5, the formation of the alloy carbide is suppressed, and that of the Co2C phase becomes favorable. Nonetheless, the formation of the Fe–Co carbidic alloy enhanced the production of low-carbon (C2–C4) olefins.15,132
At higher Co/Fe ratios, the alloy remains in the metallic state during the reaction. For example, a Co-rich Co7Fe3 alloy was formed after activation in H2.136 After reaction at 200 °C, both XRD and EXAFS revealed that the bulk composition remained as Co7Fe3, and quasi in situ XPS showed that both Fe and Co at the surface are in the metallic states. No carbides were observed, either in the bulk or at the surface. Theoretical calculations suggested that the Co-rich alloy is the active phase in the C–C coupling reaction between surface carbonaceous species. In contrast to the carbidic alloy that favored C2–C4 olefin production, the Co7Fe3 metallic alloy exhibited a high selectivity (63%) to jet-fuel-range (C8–C16) hydrocarbons at 10% CO2 conversion.136
De-alloying may also occur during the reaction, leading to the formation of separate phases of FeCx and Co (or CoCx).137 Chen et al. synthesized Fe–Co alloy catalysts by ball milling of a physically mixed Fe, Co3O4 and K2CO3 powder.137 After 6 hours of milling treatment, XRD patterns showed the catalysts consisting of 20% Fe–Co alloy, with the rest being CoOx and Fe. After the subsequent CO2 hydrogenation reaction, the fraction of Fe–Co alloy decreased to about 10%, with the major phases being Fe5C2 and Co2C, indicating alloy segregation. DRIFTS spectra complemented with DFT calculations suggested that CO2 is initially hydrogenated to CO on the Fe–Co alloy surface, which then reacted with surface carbon species on both iron and cobalt carbides for the C–C coupling step.
Several possible scenarios of Fe–Co catalyst evolution are depicted in Fig. 7b. They may additionally be affected by the proximity effects and by Fe/Co ratios, as well as the activation and reaction conditions. The bimetallic catalysts can form alloys, segregated phases, or a mixture of both.127 It appears that a moderate Fe–Co distance allows the rates of the RWGS reaction, methanation, 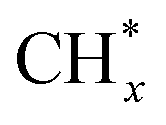 coupling and the secondary hydrogenation of olefins to be balanced, and thereby the distribution of hydrocarbon products to be tuned.
coupling and the secondary hydrogenation of olefins to be balanced, and thereby the distribution of hydrocarbon products to be tuned.
Metal oxides can also act as electronic or structural promoters.138–140 For instance, both MnOx and Na facilitated the carburization of Fe during the reaction. However, quasi in situ structural characterization showed that simultaneous modification with Na and Mn weakened the Fe–Mn interaction and decreased the content of the formed Fe5C2 as compared to the Na-free Fe–Mn catalyst, while MnOx itself was transformed into MnCO3 under reaction conditions.23 On the basis of reaction kinetics analysis, it was concluded that Na and Mn-promotion of Fe catalysts allows the reaction rates of RWGS and FTS steps to be matched, and thus results in an enhanced overall reactivity and olefin selectivity. As for the ZnO promoter, ZnFe2O4 spinel is normally used as the catalyst precursor.141,142 During activation in CO, it first separates into ZnO and FeO phases, and the latter transforms into Fe5C2.142,143 Both ZnO and Na promoters stabilize Fe5C2 against over-oxidation during the subsequent reaction, as shown by in situ XRD, Raman and NAP-XPS.143,144 The in situ formed interface between ZnO and Fe5C2 seems to be responsible for the enhanced production of light olefins.
Bimetallic Fe-based catalysts showed higher potential for C2+ alcohol production as compared to monometallic Fe catalysts. FeCx is effective for the formation and coupling of 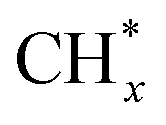 monomers. However, to produce C2+ alcohol, an additional component is needed for the formation of oxygenate intermediates such as CO* or CHO*. Too strong chain growth ability leads to the production of solely C2+ hydrocarbons, as shown in the above-mentioned studies on Fe–Cu and Fe–Zn catalysts. Thus, to improve the selectivity to C2+ alcohols, one needs to balance the rate of chain growth and oxygenate insertion, which necessitates proper modification of the catalysts and optimization of the reaction conditions. For example, an amorphous ZrO2 support facilitated non-dissociative CO adsorption on Fe–Cu–K catalysts, resulting in a C2+ alcohol selectivity of 28% and CO2 conversion of 31% at 320 °C and 50 bar.145 A carbon-supported, Na-promoted Fe–Zn catalyst evolved into a ternary ZnOx–Fe5C2–Fe3O4 compound during the reaction, as shown by in situ XRD and quasi in situ XPS.146 It was proposed that ZnO donates electrons to the Fe sites and to the carbon support, thereby strengthening the adsorption of CO. Consequently, the catalyst exhibited 19% selectivity to ethanol at a CO2 conversion of 34% at 320 °C and 50 bar, with no deactivation over more than 500 hours on stream. In situ DRIFTS confirmed the CO-insertion mechanism for ethanol production. In addition to CO*, CHO* intermediates can also be produced on the Zn-containing phase, i.e., ZnFe2O4.147 The interface between ZnFe2O4 and Fe5C2 on the optimized catalyst boosted the production of C2+ alcohols, with a proportion of 16% for all the hydrocarbon and oxygenate products at 300 °C and 50 bar. Of all alcohols produced, 98% were C2+ alcohols and more than 40% were C3+ alcohols.
monomers. However, to produce C2+ alcohol, an additional component is needed for the formation of oxygenate intermediates such as CO* or CHO*. Too strong chain growth ability leads to the production of solely C2+ hydrocarbons, as shown in the above-mentioned studies on Fe–Cu and Fe–Zn catalysts. Thus, to improve the selectivity to C2+ alcohols, one needs to balance the rate of chain growth and oxygenate insertion, which necessitates proper modification of the catalysts and optimization of the reaction conditions. For example, an amorphous ZrO2 support facilitated non-dissociative CO adsorption on Fe–Cu–K catalysts, resulting in a C2+ alcohol selectivity of 28% and CO2 conversion of 31% at 320 °C and 50 bar.145 A carbon-supported, Na-promoted Fe–Zn catalyst evolved into a ternary ZnOx–Fe5C2–Fe3O4 compound during the reaction, as shown by in situ XRD and quasi in situ XPS.146 It was proposed that ZnO donates electrons to the Fe sites and to the carbon support, thereby strengthening the adsorption of CO. Consequently, the catalyst exhibited 19% selectivity to ethanol at a CO2 conversion of 34% at 320 °C and 50 bar, with no deactivation over more than 500 hours on stream. In situ DRIFTS confirmed the CO-insertion mechanism for ethanol production. In addition to CO*, CHO* intermediates can also be produced on the Zn-containing phase, i.e., ZnFe2O4.147 The interface between ZnFe2O4 and Fe5C2 on the optimized catalyst boosted the production of C2+ alcohols, with a proportion of 16% for all the hydrocarbon and oxygenate products at 300 °C and 50 bar. Of all alcohols produced, 98% were C2+ alcohols and more than 40% were C3+ alcohols.
To sum up, the bimetallic Fe-based catalysts offer a promising approach to improve the catalytic performance for the production of both C2+ hydrocarbons and alcohols. A combination of Fe with a more easily reducible metal considerably facilitates the reduction of the Fe-oxide precursor and promotes FeCx formation. The spatial proximity and the molar ratio of the two metal precursors significantly influence alloying/de-alloying processes during the catalyst preparation, activation, and reaction. Certainly, the effects of the promoter, support, and particle size, observed for monometallic Fe catalysts, become more complex for the bimetallic systems.
7. Outlook
Over the past few decades, enormous efforts have been devoted to establishing structure-reactivity relationships for Fe-based catalysts in CO2 hydrogenation to C2+ chemicals. Given the complex and sensitive structural evolution, there remain some challenges and promising opportunities for future research.Controllable synthesis of iron carbide catalysts
Compared to χ-Fe5C2 and θ-Fe3C, other iron carbides like ε-Fe2C and Fe7C3, which showed superior performance in the FTS process,148,149 are less investigated in CO2 hydrogenation. Meanwhile, the poorly defined structure of the FeCx phases in the existing studies on CO2 hydrogenation renders determination of their intrinsic activity rather difficult. It is therefore essential to synthesize single-phase iron carbide catalysts for further fundamental studies of reactions at their surfaces. Control of the elemental composition of the FeCx phase, its crystal structure, particle size, and shape in Fe-carbide synthesis remains a significant challenge. The same applied also to their subsequent phase stabilization during the reaction. Preparation approaches like wet chemical synthesis have been developed,150 but the instability of iron carbides upon air exposure can readily cause surface restructuring during sample transfer. In this respect, vacuum based thin-film technologies can be a good option.151–153 For example, atomically defined FeCx films were recently prepared by ethylene decomposition over the Fe thin films grown on an Au(111) substrate.151,152 These films can serve not only as model catalysts for fundamental studies, but also as a prototype for monolith-type catalysts suited for industrial applications. Here, a scalable industrial technology already exists for the preparation of thin film catalysts, namely that employed for solar cells (PVD) that can be easily adapted for reproducible catalyst synthesis.The controllable synthesis of iron carbides can provide the opportunity to investigate also the shape effect in this reaction. Shape selectivity has recently been investigated for CO2 hydrogenation to methanol over ZnO-supported Cu2O nanocubes exposing solely the (001) facets.154 To date, the shape effect on the reactivity of Fe-carbides in CO2-FTS is primarily studied through theoretical simulations. For instance, Nie et al. found that the χ-Fe5C2(510) surface exhibits higher activity for the direct dissociation of CO2 into CO* and O*, while the (111) surface is more favorable for CO2 hydrogenation into the HCO* intermediate.155 Despite the different reaction pathways, both the (510) and (111) surfaces appear to be better candidates for C2+ hydrocarbon production as compared to (100), (11−1), (110) and (10−1) surfaces. Experimental efforts to control the initial shape of the Fe catalysts are currently limited to Fe-oxide precursors, but the question still remains on how to retain such shape under the reaction environment. The latter might be achieved by careful selection of the appropriate underlying support and carefully controlled treatments. Chen et al. synthesized α-Fe2O3 nanodisks of certain thickness and diameter which were enclosed with (0001) basal facets and (11−20) side facets and applied them for CO2 hydrogenation.156 However, the post-reacted catalysts displayed a quite rough surface, cracks and severe sintering, although the disk shape remained at a large scale. Very recently, Wu et al. reported synthesis of χ-Fe5C2 nanoparticles with specifically exposed surfaces using the conformal reconstruction of well-defined Fe3O4 nanocrystals during pre-reduction in H2 and activation in syngas.157 In fact, the prepared particles showed an Fe3O4 core/χ-Fe5C2 shell structure, with the χ-Fe5C2(202) surface being formed on Fe3O4 nanocubes exposing the (400)-oriented facets, while Fe3O4 octahedra primarily exposing the (111) facets favored the formation of the χ-Fe5C2(112) surface. This preparation allowed a look into the facet effects on reactivity of the Fe5C2 carbide in FTS. We believe that such an approach can also be applied to the CO2 hydrogenation reaction.
Operando characterization under catalytically relevant conditions
Given the dynamic nature of this catalytic system, the real-time analysis of the catalyst structure at different time and length scales from the atomic level, meso and microscale becomes crucial. Bulk-sensitive techniques such as XRD, Raman, and XAS are currently well-suited for operando studies. Future efforts should be focused on improving the detection sensitivity, time and spatial resolution. For example, accurate identification of the atomic structure of the above-mentioned Fe–Co alloy catalyst is not trivial due to the naturally close similarities in the structural and electronic parameters of neighboring Fe and Co in the periodic table. Moreover, spectroscopic ensemble-averaging techniques might miss key spatially separated changes in the catalyst structure and composition, such as different oxidation states or carbide phases at different locations within the same sample or even within different regions of the same large nanoparticle. This can for instance apply to nanoparticles of a different size or those located on support regions of different characteristics or specific defects within a sample with heterogeneous characteristics either in the as-prepared state, or during reaction (changes in particle size and phase composition).158 Such complexity is starting to be addressed in the catalysis community by combining multi-technique ensemble-averaging characterization approaches with locally resolved spectro-microscopy methods, including synchrotron-based transmission X-ray microscopy or low energy electron microscopy combined with X-ray photoemission electron microscopy among others.Moreover, the surface structure of the working catalyst remains poorly understood. Atomic-level understanding can, in principle, be obtained on the basis of “surface science” studies of model systems using a large variety of surface sensitive techniques. For example, Guo et al. visualized in real time the chain growth process during ethylene polymerization monitored by scanning tunneling microscopy (STM) on a carburized Fe(110) single crystal surface.159 Nevertheless, it is in many cases still unclear whether such model systems are really representative of all or at least some of the key characteristics of the real industrial catalyst, and thus, bridging the materials gap still remains a challenge. Another question to address in this community is the possible relevance of the pressure gap that most traditional surface science experiments inherently suffer from. Nilsson's group's work has recently resulted in a major leap forward in this direction by developing an advanced NAP-XPS setup enabling in situ measurements at pressures up to 500 mbar,160 whereas conventional setups mostly operate at pressures in the 1–10 mbar range. In particular, this group has investigated the surface evolution of the Fe(110) single crystal surface during CO2 hydrogenation. As the reaction temperature increases, no carbide formation was observed due to the very low CO concentration formed via the RWGS reaction on the low-surface-area single crystal catalyst. However, adding CO to the feed gas resulted in carburization of the Fe surface. Moreover, it seems possible to discriminate octahedral and trigonal prismatic carbides formed at elevated reaction temperatures.
Still, CO2 hydrogenation is known to be highly sensitive to the reaction pressure, where the product distribution shifts from over 90% CO at atmospheric pressure to hydrocarbons at higher pressures.75 Therefore, vacuum-based in situ characterization (at NAP conditions) needs to be complemented with quasi in situ measurements after high-pressure treatments to address the “pressure gap”.
Theoretical simulation of catalyst dynamics
Theoretical calculations provide fundamental insights into the reaction mechanism at the atomic level, including the determination and distinction of reaction intermediates from spectator species, both of which would be detected experimentally and at times wrongly assigned. Nevertheless, the empirically derived ideal and still mostly “static” model for the catalyst structure and reaction microenvironment may not accurately reflect the real situation under the working conditions. The complexity in phase transition, surface reconstruction, and the interplay between the catalyst surface structure and gas-phase environment are usually underestimated. Therefore, theoretical simulations on catalyst dynamics and the corresponded reaction environment are essential for rationally establishing structure–reactivity relationships,161,162 which can be achieved by combining DFT, Monte Carlo-based approaches, first-principles thermodynamics and microkinetic simulations. In this respect, machine learning could be particularly effective for such complex systems.8. Synopsis
It is generally accepted in the catalysis community that rational design of an efficient catalyst relies on precise structure–reactivity relationships which must be established for the catalyst in its working state, through comprehensive in situ/operando characterization. This situation holds true also for Fe-based catalysts in CO2 hydrogenation to C2+ chemicals, where considerable structural and chemical changes take place both in the bulk and at the surface during the reaction. This structural evolution may be affected by the reaction microenvironment formed above the catalyst surface, which also changes as the reaction proceeds. Nonetheless, fine-tuning the composition, as well as electronic and geometric structure, of the Fe-based pre-catalysts can serve to influence their subsequent transformation during the reaction. In particular, it has been shown to offer opportunities to balance multiple elementary reactions, including CO2 dissociation, chain growth, and its termination, oxygenate insertion, secondary hydrogenation and oligomerization of olefins, thereby directing the production of desired C2+ products.For synthesis of C2+ hydrocarbons from CO2 hydrogenation, FeCx carbides are believed to be the essential phases, with a certain amount of FeOx phase improving the C2+ selectivity, and excessive oxidation of FeCx leading to catalyst deactivation. Alkali metals promote FeCx formation and prevent its over-oxidation during the reaction, significantly enhancing C2+ production. Both the support and size of Fe NPs affect the reducibility of the Fe-oxide precursor to metallic Fe, which is, in turn, a pre-requisite for Fe-carbide formation. Apparently, a particle size in the range of 10–15 nm is optimal for C2+ hydrocarbon production. In bimetallic systems, the second metal primarily facilitates Fe reduction. However, the ratio and proximity of the second metal to Fe both influence the alloying/de-alloying behavior and hence the reaction pathway. For C2+ alcohol production, FeCx is responsible for 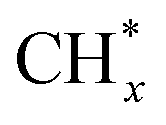 monomer formation and chain growth, cooperatively working with another active component responsible for the insertion of oxygenate groups. Promoters such as S, Cu, Pd, and ZnO can be efficient for this purpose.
monomer formation and chain growth, cooperatively working with another active component responsible for the insertion of oxygenate groups. Promoters such as S, Cu, Pd, and ZnO can be efficient for this purpose.
Given the dynamic nature and complexity of this catalytic system, it is crucial to establish the role of the promoter, the possible effects of size, support and shape (affecting the particle-support contact area) on reactivity, and formation of the “real catalyst” from the pre-catalyst upon activation, and its further evolution under reaction conditions. We hope that this review of in situ/operando studies aids in providing a better understanding of the Fe-based catalysts “at work”, and provides insights into active phase(s) of the catalysts ultimately resulting in the production of C2+ hydrocarbons and alcohols from CO2.
Data availability
This study was carried out using publicly available data from the references cited.Author contributions
J. Z. drafted the manuscript and prepared all figures. S. S. and B. R. C. conceptualized the work and edited the final manuscript. All authors approved the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the German Federal Ministry of Education and Research (BMBF) via Grant No. 03 EW0015B (CatLab) and by the Deutsche Forschungsgemeinschaft (DFG) – project no. 406944504 – SPP 2080.References
- R. Daiyan, I. MacGill and R. Amal, Opportunities and Challenges for Renewable Power-to-X, ACS Energy Lett., 2020, 5, 3843–3847 CrossRef.
- J. A. Martens, A. Bogaerts, N. De Kimpe, P. A. Jacobs, G. B. Marin, K. Rabaey, M. Saeys and S. Verhelst, The Chemical Route to a Carbon Dioxide Neutral World, ChemSusChem, 2017, 10, 1039–1055 CrossRef PubMed.
- C. F. Shih, T. Zhang, J. Li and C. Bai, Powering the Future with Liquid Sunshine, Joule, 2018, 2, 1925–1949 CrossRef.
- T. S. Galhardo, A. H. Braga, B. H. Arpini, J. Szanyi, R. V. Goncalves, B. F. Zornio, C. R. Miranda and L. M. Rossi, Optimizing Active Sites for High CO Selectivity during CO2 Hydrogenation over Supported Nickel Catalysts, J. Am. Chem. Soc., 2021, 143, 4268–4280 CrossRef.
- M. M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. V. Levchenko, R. Schlogl and E. Frei, Ni Single Atom Catalysts for CO2 Activation, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef PubMed.
- C. Vogt, M. Monai, G. J. Kramer and B. M. Weckhuysen, The renaissance of the Sabatier reaction and its applications on Earth and in space, Nat. Catal., 2019, 2, 188–197 CrossRef.
- J. Cored, A. Garcia-Ortiz, S. Iborra, M. J. Climent, L. Liu, C. H. Chuang, T. S. Chan, C. Escudero, P. Concepcion and A. Corma, Hydrothermal Synthesis of Ruthenium Nanoparticles with a Metallic Core and a Ruthenium Carbide Shell for Low-Temperature Activation of CO2 to Methane, J. Am. Chem. Soc., 2019, 141, 19304–19311 CrossRef.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts, Science, 2012, 336, 893–897 CrossRef PubMed.
- S. Kuld, M. Thorhauge, H. Falsig, C. F. Elkjær, S. Helveg, I. Chorkendorff and J. Sehested, Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis, Science, 2016, 352, 969–974 CrossRef PubMed.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts, Science, 2017, 355, 1296–1299 CrossRef.
- G. Prieto, Carbon Dioxide Hydrogenation into Higher Hydrocarbons and Oxygenates: Thermodynamic and Kinetic Bounds and Progress with Heterogeneous and Homogeneous Catalysis, ChemSusChem, 2017, 10, 1056–1070 CrossRef PubMed.
- A. Goryachev, A. Pustovarenko, G. Shterk, N. S. Alhajri, A. Jamal, M. Albuali, L. van Koppen, I. S. Khan, A. Russkikh, A. Ramirez, T. Shoinkhorova, E. J. M. Hensen and J. Gascon, A Multi-Parametric Catalyst Screening for CO2 Hydrogenation to Ethanol, ChemCatChem, 2021, 13, 3324–3332 CrossRef.
- K. T. Rommens and M. Saeys, Molecular Views on Fischer-Tropsch Synthesis, Chem. Rev., 2023, 123, 5798–5858 CrossRef.
- P. Bredy, D. Farrusseng, Y. Schuurman and F. C. Meunier, On the link between CO surface coverage and selectivity to CH4 during CO2 hydrogenation over supported cobalt catalysts, J. Catal., 2022, 411, 93–96 CrossRef CAS.
- M. K. Gnanamani, G. Jacobs, H. H. Hamdeh, W. D. Shafer, F. Liu, S. D. Hopps, G. A. Thomas and B. H. Davis, Hydrogenation of Carbon Dioxide over Co–Fe Bimetallic Catalysts, ACS Catal., 2016, 6, 913–927 CrossRef.
- W. Li, X. Nie, H. Yang, X. Wang, F. Polo-Garzon, Z. Wu, J. Zhu, J. Wang, Y. Liu, C. Shi, C. Song and X. Guo, Crystallographic dependence of CO2 hydrogenation pathways over HCP-Co and FCC-Co catalysts, Appl. Catal., B, 2022, 315, 121529 CrossRef.
- P. Gao, L. Zhang, S. Li, Z. Zhou and Y. Sun, Novel Heterogeneous Catalysts for CO2 Hydrogenation to Liquid Fuels, ACS Cent. Sci., 2020, 6, 1657–1670 CrossRef.
- C. Panzone, R. Philippe, A. Chappaz, P. Fongarland and A. Bengaouer, Power-to-liquid catalytic CO2 valorization into fuels and chemicals: focus on the Fischer-Tropsch route, J. CO2 Util., 2020, 38, 314–347 CrossRef.
- X. Nie, H. Wang, M. J. Janik, Y. Chen, X. Guo and C. Song, Mechanistic Insight into C–C Coupling over Fe–Cu Bimetallic Catalysts in CO2 Hydrogenation, J. Phys. Chem. C, 2017, 121, 13164–13174 CrossRef.
- R. P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y. G. Yao, CO2 hydrogenation to high-value products via heterogeneous catalysis, Nat. Commun., 2019, 10, 5698 CrossRef PubMed.
- R. Yao, J. Wei, Q. Ge, J. Xu, Y. Han, Q. Ma, H. Xu and J. Sun, Monometallic iron catalysts with synergistic Na and S for higher alcohols synthesis via CO2 hydrogenation, Appl. Catal., B, 2021, 298, 120556 CrossRef.
- J. Wei, Q. Ge, R. Yao, Z. Wen, C. Fang, L. Guo, H. Xu and J. Sun, Directly converting CO2 into a gasoline fuel, Nat. Commun., 2017, 8, 15174 CrossRef PubMed.
- Y. Xu, P. Zhai, Y. Deng, J. Xie, X. Liu, S. Wang and D. Ma, Highly Selective Olefin Production from CO2 Hydrogenation on Iron Catalysts: A Subtle Synergy between Manganese and Sodium Additives, Angew. Chem., Int. Ed., 2020, 59, 21736–21744 CrossRef PubMed.
- M. K. Khan, P. Butolia, H. Jo, M. Irshad, D. Han, K.-W. Nam and J. Kim, Selective Conversion of Carbon Dioxide into Liquid Hydrocarbons and Long-Chain α-Olefins over Fe-Amorphous AlOx Bifunctional Catalysts, ACS Catal., 2020, 10, 10325–10338 CrossRef.
- Y. H. Choi, Y. J. Jang, H. Park, W. Y. Kim, Y. H. Lee, S. H. Choi and J. S. Lee, Carbon dioxide Fischer-Tropsch synthesis: a new path to carbon-neutral fuels, Appl. Catal., B, 2017, 202, 605–610 CrossRef CAS.
- M. Al-Dossary, A. A. Ismail, J. L. G. Fierro, H. Bouzid and S. A. Al-Sayari, Effect of Mn loading onto MnFeO nanocomposites for the CO2 hydrogenation reaction, Appl. Catal., B, 2015, 165, 651–660 CrossRef CAS.
- M. Albrecht, U. Rodemerck, M. Schneider, M. Bröring, D. Baabe and E. V. Kondratenko, Unexpectedly efficient CO2 hydrogenation to higher hydrocarbons over non-doped Fe2O3, Appl. Catal., B, 2017, 204, 119–126 CrossRef CAS.
- B. Liang, T. Sun, J. Ma, H. Duan, L. Li, X. Yang, Y. Zhang, X. Su, Y. Huang and T. Zhang, Mn decorated Na/Fe catalysts for CO2 hydrogenation to light olefins, Catal. Sci. Technol., 2019, 9, 456–464 RSC.
- P. P. Paalanen and B. M. Weckhuysen, Carbon Pathways, Sodium-Sulphur Promotion and Identification of Iron Carbides in Iron-based Fischer-Tropsch Synthesis, ChemCatChem, 2020, 12, 4202–4223 CrossRef CAS.
- P. P. Paalanen, S. H. van Vreeswijk and B. M. Weckhuysen, Combined In Situ X-ray Powder Diffractometry/Raman Spectroscopy of Iron Carbide and Carbon Species Evolution in Fe(–Na–S)/α-Al2O3 Catalysts during Fischer–Tropsch Synthesis, ACS Catal., 2020, 10, 9837–9855 CrossRef CAS.
- J. Zhang, M. Abbas and J. Chen, The evolution of Fe phases of a fused iron catalyst during reduction and Fischer–Tropsch synthesis, Catal. Sci. Technol., 2017, 7, 3626–3636 RSC.
- J. Wang, S. Huang, S. Howard, B. W. Muir, H. Wang, D. F. Kennedy and X. Ma, Elucidating Surface and Bulk Phase Transformation in Fischer–Tropsch Synthesis Catalysts and Their Influences on Catalytic Performance, ACS Catal., 2019, 9, 7976–7983 CrossRef CAS.
- X. Ding, M. Zhu, B. Sun, Z. Yang and Y.-F. Han, An Overview on Dynamic Phase Transformation and Surface Reconstruction of Iron Catalysts for Catalytic Hydrogenation of COx for Hydrocarbons, ACS Catal., 2024, 14, 6137–6168 CrossRef CAS.
- J. Zhu, P. Wang, X. Zhang, G. Zhang, R. Li, W. Li, P. Senftle Thomas, W. Liu, J. Wang, Y. Wang, A. Zhang, Q. Fu, C. Song and X. Guo, Dynamic structural evolution of iron catalysts involving competitive oxidation and carburization during CO2 hydrogenation, Sci. Adv., 2022, 8, eabm3629 CrossRef CAS PubMed.
- S. Chavez, B. Werghi, K. M. Sanroman Gutierrez, R. Chen, S. Lall and M. Cargnello, Studying, Promoting, Exploiting, and Predicting Catalyst Dynamics: The Next Frontier in Heterogeneous Catalysis, J. Phys. Chem. C, 2023, 127, 2127–2146 CrossRef CAS.
- S. A. Chernyak, M. Corda, J. P. Dath, V. V. Ordomsky and A. Y. Khodakov, Light olefin synthesis from a diversity of renewable and fossil feedstocks: state-of the-art and outlook, Chem. Soc. Rev., 2022, 51, 7994–8044 RSC.
- J. Wei, R. Yao, Y. Han, Q. Ge and J. Sun, Towards the development of the emerging process of CO2 heterogenous hydrogenation into high-value unsaturated heavy hydrocarbons, Chem. Soc. Rev., 2021, 50, 10764–10805 RSC.
- F. Zeng, C. Mebrahtu, X. Xi, L. Liao, J. Ren, J. Xie, H. J. Heeres and R. Palkovits, Catalysts design for higher alcohols synthesis by CO2 hydrogenation: trends and future perspectives, Appl. Catal., B, 2021, 291, 120073 CrossRef.
- J. Liu, Y. Song, X. Guo, C. Song and X. Guo, Recent advances in application of iron-based catalysts for COx hydrogenation to value-added hydrocarbons, Chin. J. Catal., 2022, 43, 731–754 CrossRef.
- Y. Nian, X. Huang, M. Liu, J. Zhang and Y. Han, Insight into the Dynamic Evolution of Supported Metal Catalysts by In Situ/Operando Techniques and Theoretical Simulations, ACS Catal., 2023, 13, 11164–11171 CrossRef.
- G. Wan, G. Zhang, J. Z. Chen, M. F. Toney, J. T. Miller and C. J. Tassone, Reaction-Mediated Transformation of Working Catalysts, ACS Catal., 2022, 12, 8007–8018 CrossRef.
- F. Tao and M. Salmeron, In Situ Studies of Chemistry and Structure of Materials in Reactive Environments, Science, 2011, 331, 171–174 CrossRef.
- J. Timoshenko and B. Roldan Cuenya, In Situ/Operando Electrocatalyst Characterization by X-ray Absorption Spectroscopy, Chem. Rev., 2021, 121, 882–961 CrossRef PubMed.
- S. W. Chee, T. Lunkenbein, R. Schlogl and B. Roldan Cuenya, Operando Electron Microscopy of Catalysts: The Missing Cornerstone in Heterogeneous Catalysis Research?, Chem. Rev., 2023, 123, 13374–13418 CrossRef PubMed.
- J. Zhu, G. Zhang, W. Li, X. Zhang, F. Ding, C. Song and X. Guo, Deconvolution of the Particle Size Effect on CO2 Hydrogenation over Iron-Based Catalysts, ACS Catal., 2020, 10, 7424–7433 CrossRef.
- A. S. Skrypnik, Q. Yang, A. A. Matvienko, V. Y. Bychkov, Y. P. Tulenin, H. Lund, S. A. Petrov, R. Kraehnert, A. Arinchtein, J. Weiss, A. Brueckner and E. V. Kondratenko, Understanding reaction-induced restructuring of well-defined FexOyCz compositions and its effect on CO2 hydrogenation, Appl. Catal., B, 2021, 291, 120121 CrossRef.
- H. Chen, Z. Zhao, G. Wang, Z. Zheng, J. Chen, Q. Kuang and Z. Xie, Dynamic Phase Transition of Iron Oxycarbide Facilitated by Pt Nanoparticles for Promoting the Reverse Water Gas Shift Reaction, ACS Catal., 2021, 11, 14586–14595 CrossRef.
- Y. Liu, P. R. Murthy, X. Zhang, H. Wang and C. Shi, Phase transformation of iron oxide to carbide and Fe3C as an active center for the RWGS reaction, New J. Chem., 2021, 45, 22444–22449 RSC.
- J. Zhu, M. Mu, Y. Liu, M. Zhang, G. Zhang, Z. Cheng, B. Hang Yin, A. C. K. Yip, C. Song and X. Guo, Unveiling the promoting effect of potassium on the structural evolution of iron catalysts during CO2 hydrogenation, Chem. Eng. Sci., 2023, 282, 119228 CrossRef CAS.
- Q. Yang, V. A. Kondratenko, A. S. Skrypnik, H. Lund, S. Bartling, J. Weiss, A. Brückner and E. V. Kondratenko, Understanding of the Fate of α-Fe2O3 in CO2 Hydrogenation through Combined Time-Resolved In Situ Characterization and Microkinetic Analysis, ACS Catal., 2023, 13, 9064–9077 CrossRef CAS.
- Y. Zhang, D. Fu, X. Liu, Z. Zhang, C. Zhang, B. Shi, J. Xu and Y.-F. Han, Operando Spectroscopic Study of Dynamic Structure of Iron Oxide Catalysts during CO2 Hydrogenation, ChemCatChem, 2018, 10, 1272–1276 CrossRef CAS.
- A. V. Puga, On the nature of active phases and sites in CO and CO2 hydrogenation catalysts, Catal. Sci. Technol., 2018, 8, 5681–5707 RSC.
- Y. Zhang, C. Cao, C. Zhang, Z. Zhang, X. Liu, Z. Yang, M. Zhu, B. Meng, J. Xu and Y.-F. Han, The study of structure-performance relationship of iron catalyst during a full life cycle for CO2 hydrogenation, J. Catal., 2019, 378, 51–62 CrossRef CAS.
- J. Liu, G. Zhang, X. Jiang, J. Wang, C. Song and X. Guo, Insight into the role of Fe5C2 in CO2 catalytic hydrogenation to hydrocarbons, Catal. Today, 2021, 371, 162–170 CrossRef CAS.
- J. H. Lee, H.-K. Lee, D. H. Chun, H. Choi, G. B. Rhim, M. H. Youn, H. Jeong, S. W. Kang, J.-I. Yang, H. Jung, C. S. Kim and J. C. Park, Phase-controlled synthesis of thermally stable nitrogen-doped carbon supported iron catalysts for highly efficient Fischer-Tropsch synthesis, Nano Res., 2019, 12, 2568–2575 CrossRef.
- Y. Liu, J.-F. Chen, J. Bao and Y. Zhang, Manganese-Modified Fe3O4 Microsphere Catalyst with Effective Active Phase of Forming Light Olefins from Syngas, ACS Catal., 2015, 5, 3905–3909 CrossRef.
- X. Liu, C. Cao, P. Tian, M. Zhu, Y. Zhang, J. Xu, Y. Tian and Y.-F. Han, Resolving CO2 activation and hydrogenation pathways over iron carbides from DFT investigation, J. CO2 Util., 2020, 38, 10–15 CrossRef.
- P. Zhang, F. Han, J. Yan, X. Qiao, M. Zhu, Q. Guan and W. Li, Heteroatom induced synthesis of FeO-Fe3C confined within F-doped graphene shell for efficient CO2 hydrogenation to light olefins, Chem. Eng. J., 2023, 477, 147153 CrossRef.
- B. Zhao, M. Sun, F. Chen, Y. Shi, Y. Yu, X. Li and B. Zhang, Unveiling the Activity Origin of Iron Nitride as Catalytic Material for Efficient Hydrogenation of CO2 to C2+ Hydrocarbons, Angew. Chem., Int. Ed., 2021, 60, 4496–4500 CrossRef PubMed.
- M. Amoyal, R. Vidruk-Nehemya, M. V. Landau and M. Herskowitz, Effect of potassium on the active phases of Fe catalysts for carbon dioxide conversion to liquid fuels through hydrogenation, J. Catal., 2017, 348, 29–39 CrossRef.
- A. Fedorov, H. Lund, V. A. Kondratenko, E. V. Kondratenko and D. Linke, Elucidating reaction pathways occurring in CO2 hydrogenation over Fe-based catalysts, Appl. Catal., B, 2023, 328, 122505 CrossRef.
- X. Nie, L. Meng, H. Wang, Y. Chen, X. Guo and C. Song, DFT insight into the effect of potassium on the adsorption, activation and dissociation of CO2 over Fe-based catalysts, Phys. Chem. Chem. Phys., 2018, 20, 14694–14707 RSC.
- C. Song, X. Liu, M. Xu, D. Masi, Y. Wang, Y. Deng, M. Zhang, X. Qin, K. Feng, J. Yan, J. Leng, Z. Wang, Y. Xu, B. Yan, S. Jin, D. Xu, Z. Yin, D. Xiao and D. Ma, Photothermal Conversion of CO2 with Tunable Selectivity Using Fe-Based Catalysts: From Oxide to Carbide, ACS Catal., 2020, 10, 10364–10374 CrossRef.
- S. Najari, S. Saeidi, A. Sápi, Á. Szamosvölgyi, Á. Papp, A. Efremova, H. Bali and Z. Kónya, Synergistic enhancement of CO2 hydrogenation to C5+ hydrocarbons using mixed Fe5C2 and Na-Fe3O4 catalysts: effects of oxide/carbide ratio, proximity, and reduction, Chem. Eng. J., 2024, 485, 149787 CrossRef.
- Y. Liu, Q. Cheng, S. Xiong, Y. Zhang, L. Tan, S. Song, T. Ding, Y. Tian and X. Li, Enhancing CO2 hydrogenation performance via the synergistic effects of iron carbides and iron oxides, Int. J. Hydrogen Energy, 2024 DOI:10.1016/j.ijhydene.2024.05.272.
- E. de Smit, F. Cinquini, A. M. Beale, O. V. Safonova, W. van Beek, P. Sautet and B. M. Weckhuysen, Stability and Reactivity of ε–χ–θ Iron Carbide Catalyst Phases in Fischer–Tropsch Synthesis: Controlling μC, J. Am. Chem. Soc., 2010, 132, 14928–14941 CrossRef PubMed.
- L. Jiang, K. Li, W. N. Porter, H. Wang, G. Li and J. G. Chen, Role of H2O in Catalytic Conversion of C1 Molecules, J. Am. Chem. Soc., 2024, 146, 2857–2875 CrossRef CAS.
- A. Bordet, J. M. Asensio, K. Soulantica and B. Chaudret, Enhancement of Carbon Oxides Hydrogenation on Iron-Based Nanoparticles by In-Situ Water Removal, ChemCatChem, 2018, 10, 4047–4051 CrossRef CAS.
- S. Najari, G. Gróf and S. Saeidi, Enhancement of hydrogenation of CO2 to hydrocarbons via In-Situ water removal, Int. J. Hydrogen Energy, 2019, 44, 24759–24781 CrossRef CAS.
- Y. Xu, X. Li, J. Gao, J. Wang, G. Ma, X. Wen, Y. Yang, Y. Li and M. Ding, A hydrophobic FeMn@Si catalyst increases olefins from syngas by suppressing C1 by-products, Science, 2021, 371, 610–613 CrossRef CAS PubMed.
- Y. Xu, Z. Zhang, K. Wu, J. Wang, B. Hou, R. Shan, L. Li and M. Ding, Effects of surface hydrophobization on the phase evolution behavior of iron-based catalyst during Fischer-Tropsch synthesis, Nat. Commun., 2024, 15, 7099 CrossRef CAS.
- F. Ding, A. Zhang, M. Liu, X. Guo and C. Song, Effect of SiO2-coating of FeK/Al2O3 catalysts on their activity and selectivity for CO2 hydrogenation to hydrocarbons, RSC Adv., 2014, 4, 8930 RSC.
- Z. Zhang, B. Chen, L. Jia, W. Liu, X. Gao, J. Gao, B. Meng, Y. Tan, Y. He, W. Tu and Y.-F. Han, Unraveling the role of Fe5C2 in CH4 formation during CO2 hydrogenation over hydrophobic iron catalysts, Appl. Catal., B, 2023, 327, 122449 CrossRef CAS.
- J. Wang, Y. Yang, M. Qing, Y.-p. Bai, H. Wang, C.-x. Hu, H.-w. Xiang and R.-l. Yue, Effect of the promoters on oxidation behavior of Fe-based Fischer-Tropsch catalyst: deciphering the role of H2O, J. Fuel Chem. Technol., 2020, 48, 63–74 CrossRef CAS.
- C. G. Visconti, M. Martinelli, L. Falbo, A. Infantes-Molina, L. Lietti, P. Forzatti, G. Iaquaniello, E. Palo, B. Picutti and F. Brignoli, CO2 hydrogenation to lower olefins on a high surface area K-promoted bulk Fe-catalyst, Appl. Catal., B, 2017, 200, 530–542 CrossRef.
- A. Ramirez, A. Dutta Chowdhury, A. Dokania, P. Cnudde, M. Caglayan, I. Yarulina, E. Abou-Hamad, L. Gevers, S. Ould-Chikh, K. De Wispelaere, V. van Speybroeck and J. Gascon, Effect of Zeolite Topology and Reactor Configuration on the Direct Conversion of CO2 to Light Olefins and Aromatics, ACS Catal., 2019, 9, 6320–6334 CrossRef.
- E. García-Hurtado, A. Rodríguez-Fernández, M. Moliner and C. Martínez, CO2 hydrogenation using bifunctional catalysts based on K-promoted iron oxide and zeolite: influence of the zeolite structure and crystal size, Catal. Sci. Technol., 2020, 10, 5648–5658 RSC.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, M. Chareonpanich, P. Kongkachuichay, N. Yigit, G. Rupprechter, J. Limtrakul and T. Witoon, Tuning Interactions of Surface-adsorbed Species over Fe–Co/K–Al2O3 Catalyst by Different K Contents: Selective CO2 Hydrogenation to Light Olefins, ChemCatChem, 2020, 12, 3306–3320 CrossRef.
- Y. Han, C. Fang, X. Ji, J. Wei, Q. Ge and J. Sun, Interfacing with Carbonaceous Potassium Promoters Boosts Catalytic CO2 Hydrogenation of Iron, ACS Catal., 2020, 10, 12098–12108 CrossRef.
- T. Wu, J. Lin, Y. Cheng, J. Tian, S. Wang, S. Xie, Y. Pei, S. Yan, M. Qiao, H. Xu and B. Zong, Porous Graphene-Confined Fe-K as Highly Efficient Catalyst for CO2 Direct Hydrogenation to Light Olefins, ACS Appl. Mater. Interfaces, 2018, 10, 23439–23443 CrossRef.
- J. Wang, Z. You, Q. Zhang, W. Deng and Y. Wang, Synthesis of lower olefins by hydrogenation of carbon dioxide over supported iron catalysts, Catal. Today, 2013, 215, 186–193 CrossRef.
- J. I. Orege, J. Wei, Y. Han, M. Yang, X. Sun, J. Zhang, C. C. Amoo, Q. Ge and J. Sun, Highly stable Sr and Na co-decorated Fe catalyst for high-valued olefin synthesis from CO2 hydrogenation, Appl. Catal., B, 2022, 316, 121640 CrossRef.
- A. Russkikh, G. Shterk, B. H. Al-Solami, B. A. Fadhel, A. Ramirez and J. Gascon, Turning Waste into Value: Potassium-Promoted Red Mud as an Effective Catalyst for the Hydrogenation of CO2, ChemSusChem, 2020, 13, 2981–2987 CrossRef.
- Z. You, W. Deng, Q. Zhang and Y. Wang, Hydrogenation of carbon dioxide to light olefins over non-supported iron catalyst, Chin. J. Catal., 2013, 34, 956–963 CrossRef.
- B. Liang, H. Duan, T. Sun, J. Ma, X. Liu, J. Xu, X. Su, Y. Huang and T. Zhang, Effect of Na Promoter on Fe-Based Catalyst for CO2 Hydrogenation to Alkenes, ACS Sustainable Chem. Eng., 2019, 7, 925–932 CrossRef.
- Q. Yang, H. Lund, S. Bartling, F. Krumeich, A. S. Skrypnik and E. V. Kondratenko, The role of Na for efficient CO2 hydrogenation to higher hydrocarbons over Fe-based catalysts under externally forced dynamic conditions, J. Catal., 2023, 426, 126–139 CrossRef.
- R. Yao, B. Wu, Y. Yu, N. Liu, Q. Niu, C. Li, J. Wei and Q. Ge, Regulating the electronic property of iron catalysts for higher alcohols synthesis from CO2 hydrogenation, Appl. Catal., B, 2024, 355, 124159 CrossRef.
- A. Ramirez, L. Gevers, A. Bavykina, S. Ould-Chikh and J. Gascon, Metal Organic Framework-Derived Iron Catalysts for the Direct Hydrogenation of CO2 to Short Chain Olefins, ACS Catal., 2018, 8, 9174–9182 CrossRef.
- N. Boreriboon, X. Jiang, C. Song and P. Prasassarakich, Higher Hydrocarbons Synthesis from CO2 Hydrogenation Over K- and La-Promoted Fe–Cu/TiO2 Catalysts, Top. Catal., 2018, 61, 1551–1562 CrossRef.
- Q. Yang, V. A. Kondratenko, S. A. Petrov, D. E. Doronkin, E. Saraci, H. Lund, A. Arinchtein, R. Kraehnert, A. S. Skrypnik, A. A. Matvienko and E. V. Kondratenko, Identifying Performance Descriptors in CO2 Hydrogenation over Iron-Based Catalysts Promoted with Alkali Metals, Angew. Chem., Int. Ed., 2022, 61, e202116517 CrossRef.
- P. Zhai, C. Xu, R. Gao, X. Liu, M. Li, W. Li, X. Fu, C. Jia, J. Xie, M. Zhao, X. Wang, Y. W. Li, Q. Zhang, X. D. Wen and D. Ma, Highly Tunable Selectivity for Syngas-Derived Alkenes over Zinc and Sodium-Modulated Fe5C2 Catalyst, Angew. Chem., Int. Ed., 2016, 55, 9902–9907 CrossRef PubMed.
- N. Fischer, R. Henkel, B. Hettel, M. Iglesias, G. Schaub and M. Claeys, Hydrocarbons via CO2 Hydrogenation over Iron Catalysts: The Effect of Potassium on Structure and Performance, Catal. Lett., 2015, 146, 509–517 CrossRef.
- A. Ramirez, S. Ould-Chikh, L. Gevers, A. D. Chowdhury, E. Abou-Hamad, A. Aguilar-Tapia, J. L. Hazemann, N. Wehbe, A. J. Al Abdulghani, S. M. Kozlov, L. Cavallo and J. Gascon, Tandem Conversion of CO2 to Valuable Hydrocarbons in Highly Concentrated Potassium Iron Catalysts, ChemCatChem, 2019, 11, 2879–2886 CrossRef.
- S.-M. Hwang, C. Zhang, S. J. Han, H.-G. Park, Y. T. Kim, S. Yang, K.-W. Jun and S. K. Kim, Mesoporous carbon as an effective support for Fe catalyst for CO2 hydrogenation to liquid hydrocarbons, J. CO2 Util., 2020, 37, 65–73 CrossRef.
- J. Huang, S. Jiang, M. Wang, X. Wang, J. Gao and C. Song, Dynamic Evolution of Fe and Carbon Species over Different ZrO2 Supports during CO Prereduction and Their Effects on CO2 Hydrogenation to Light Olefins, ACS Sustainable Chem. Eng., 2021, 9, 7891–7903 CrossRef.
- J. Liu, A. Zhang, X. Jiang, M. Liu, J. Zhu, C. Song and X. Guo, Direct Transformation of Carbon Dioxide to Value-Added Hydrocarbons by Physical Mixtures of Fe5C2 and K-Modified Al2O3, Ind. Eng. Chem. Res., 2018, 57, 9120–9126 CrossRef.
- L. Torrente-Murciano, R. S. Chapman, A. Narvaez-Dinamarca, D. Mattia and M. D. Jones, Effect of nanostructured ceria as support for the iron catalysed hydrogenation of CO2 into hydrocarbons, Phys. Chem. Chem. Phys., 2016, 18, 15496–15500 RSC.
- M. Xu, X. Liu, G. Song, Y. Cai, B. Shi, Y. Liu, X. Ding, Z. Yang, P. Tian, C. Cao and J. Xu, Regulating iron species compositions by Fe-Al interaction in CO2 hydrogenation, J. Catal., 2022, 413, 331–341 CrossRef.
- M. Lopez Luna, J. Timoshenko, D. Kordus, C. Rettenmaier, S. W. Chee, A. S. Hoffman, S. R. Bare, S. Shaikhutdinov and B. Roldan Cuenya, Role of the Oxide Support on the Structural and Chemical Evolution of Fe Catalysts during the Hydrogenation of CO2, ACS Catal., 2021, 11, 6175–6185 CrossRef.
- L. M. Chew, P. Kangvansura, H. Ruland, H. J. Schulte, C. Somsen, W. Xia, G. Eggeler, A. Worayingyong and M. Muhler, Effect of nitrogen doping on the reducibility, activity and selectivity of carbon nanotube-supported iron catalysts applied in CO2 hydrogenation, Appl. Catal., A, 2014, 482, 163–170 CrossRef.
- S. Wang, T. Wu, J. Lin, Y. Ji, S. Yan, Y. Pei, S. Xie, B. Zong and M. Qiao, Iron–Potassium on Single-Walled Carbon Nanotubes as Efficient Catalyst for CO2 Hydrogenation to Heavy Olefins, ACS Catal., 2020, 10, 6389–6401 CrossRef.
- R. Zhao, X. Meng, Q. Yin, W. Gao, W. Dai, D. Jin, B. Xu and Z. Xin, Effect of Precursors of Fe-Based Fischer–Tropsch Catalysts Supported on Expanded Graphite for CO2 Hydrogenation, ACS Sustainable Chem. Eng., 2021, 9, 15545–15556 CrossRef.
- J. H. Kwak, L. Kovarik and J. Szanyi, CO2 Reduction on Supported Ru/Al2O3 Catalysts: Cluster Size Dependence of Product Selectivity, ACS Catal., 2013, 3, 2449–2455 CrossRef.
- J. C. Matsubu, V. N. Yang and P. Christopher, Isolated metal active site concentration and stability control catalytic CO2 reduction selectivity, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef PubMed.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Tuning the Selectivity of Catalytic Carbon Dioxide Hydrogenation over Iridium/Cerium Oxide Catalysts with a Strong Metal-Support Interaction, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef PubMed.
- H. C. Wu, Y. C. Chang, J. H. Wu, J. H. Lin, I. K. Lin and C. S. Chen, Methanation of CO2 and reverse water gas shift reactions on Ni/SiO2 catalysts: the influence of particle size on selectivity and reaction pathway, Catal. Sci. Technol., 2015, 5, 4154–4163 RSC.
- J. Zheng, K. Lebedev, S. Wu, C. Huang, T. Ayvali, T. S. Wu, Y. Li, P. L. Ho, Y. L. Soo, A. Kirkland and S. C. E. Tsang, High Loading of Transition Metal Single Atoms on Chalcogenide Catalysts, J. Am. Chem. Soc., 2021, 143, 7979–7990 CrossRef CAS PubMed.
- W. Meng, B. C. A. d. Jong, H. v. d. Bovenkamp, G.-J. Boer, G. Leendert Bezemer, A. Iulian Dugulan and J. Xie, Selectivity control between reverse water-gas shift and Fischer-Tropsch synthesis in carbon-supported iron-based catalysts for CO2 hydrogenation, Chem. Eng. J., 2024, 489, 151166 CrossRef CAS.
- D. V. Leybo, A. A. Ryzhova, A. T. Matveev, K. L. Firestein, P. A. Tarakanov, A. S. Konopatsky, A. L. Trigub, E. V. Sukhanova, Z. I. Popov, D. V. Golberg and D. V. Shtansky, Iron phthalocyanine derived Fe1/h-BN single atom catalysts for CO2 hydrogenation, J. Mater. Chem. A, 2023, 11, 11874–11888 RSC.
- T. Xie, J. Wang, F. Ding, A. Zhang, W. Li, X. Guo and C. Song, CO2 hydrogenation to hydrocarbons over alumina-supported iron catalyst: effect of support pore size, J. CO2 Util., 2017, 19, 202–208 CrossRef CAS.
- Q. Yang, E. A. Fedorova, S. A. Petrov, J. Weiss, H. Lund, A. S. Skrypnik, C. R. Kreyenschulte, V. Y. Bychkov, A. A. Matvienko, A. Brueckner and E. V. Kondratenko, Activity and selectivity descriptors for iron carbides in CO2 hydrogenation, Appl. Catal., B, 2023, 327, 122450 CrossRef CAS.
- M. Al-Dossary and J. L. G. Fierro, Effect of high-temperature pre-reduction in Fischer–Tropsch synthesis on Fe/ZrO2 catalysts, Appl. Catal., A, 2015, 499, 109–117 CrossRef CAS.
- W. K. Jozwiak, E. Kaczmarek, T. P. Maniecki, W. Ignaczak and W. Maniukiewicz, Reduction behavior of iron oxides in hydrogen and carbon monoxide atmospheres, Appl. Catal., A, 2007, 326, 17–27 CrossRef CAS.
- A. S. Skrypnik, S. A. Petrov, V. A. Kondratenko, Q. Yang, H. Lund, A. A. Matvienko and E. V. Kondratenko, Descriptors Affecting Methane Selectivity in CO2 Hydrogenation over Unpromoted Bulk Iron(III)-Based Catalysts, ACS Catal., 2022, 12, 11355–11368 CrossRef CAS.
- W. Wang, X. Jiang, X. Wang and C. Song, Fe–Cu Bimetallic Catalysts for Selective CO2 Hydrogenation to Olefin-Rich C2+ Hydrocarbons, Ind. Eng. Chem. Res., 2018, 57, 4535–4542 CrossRef CAS.
- N. Boreriboon, X. Jiang, C. Song and P. Prasassarakich, Fe-based bimetallic catalysts supported on TiO2 for selective CO2 hydrogenation to hydrocarbons, J. CO2 Util., 2018, 25, 330–337 CrossRef CAS.
- P. Du, R. Qi, Y. Zhang, Q. Gu, X. Xu, Y. Tan, X. Liu, A. Wang, B. Zhu, B. Yang and T. Zhang, Single-atom-driven dynamic carburization over Pd1–FeOx catalyst boosting CO2 conversion, Chem, 2022, 8, 3252–3262 Search PubMed.
- A. Halder, M. Kilianová, B. Yang, E. C. Tyo, S. Seifert, R. Prucek, A. Panáček, P. Suchomel, O. Tomanec, D. J. Gosztola, D. Milde, H.-H. Wang, L. Kvítek, R. Zbořil and S. Vajda, Highly efficient Cu-decorated iron oxide nanocatalyst for low pressure CO2 conversion, Appl. Catal., B, 2018, 225, 128–138 CrossRef.
- A. Aitbekova, E. D. Goodman, L. Wu, A. Boubnov, A. S. Hoffman, A. Genc, H. Cheng, L. Casalena, S. R. Bare and M. Cargnello, Engineering of Ruthenium-Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons, Angew. Chem., Int. Ed., 2019, 58, 17451–17457 CrossRef.
- Y. Wang, Y. Zhou, X. Zhang, M. Wang, T. Liu, J. Wei, G. Zhang, X. Hong and G. Liu, PdFe alloy-Fe5C2 interfaces for efficient CO2 hydrogenation to higher alcohols, Appl. Catal., B, 2024, 345, 123691 CrossRef.
- W. Wang, X. Wang, G. Zhang, K. Wang, F. Zhang, T. Yan, J. T. Miller, X. Guo and C. Song, CO2 Hydrogenation to Olefin-Rich Hydrocarbons over Fe-Cu Bimetallic Catalysts: An Investigation of Fe-Cu Interaction and Surface Species, Front. Chem. Eng., 2021, 3, 708014 CrossRef.
- J. Liu, A. Zhang, X. Jiang, M. Liu, Y. Sun, C. Song and X. Guo, Selective CO2 Hydrogenation to Hydrocarbons on Cu-Promoted Fe-Based Catalysts: Dependence on Cu–Fe Interaction, ACS Sustainable Chem. Eng., 2018, 6, 10182–10190 CrossRef.
- S.-M. Hwang, S. J. Han, J. E. Min, H.-G. Park, K.-W. Jun and S. K. Kim, Mechanistic insights into Cu and K promoted Fe-catalyzed production of liquid hydrocarbons via CO2 hydrogenation, J. CO2 Util., 2019, 34, 522–532 CrossRef.
- Z. Li, W. Wu, M. Wang, Y. Wang, X. Ma, L. Luo, Y. Chen, K. Fan, Y. Pan, H. Li and J. Zeng, Ambient-pressure hydrogenation of CO2 into long-chain olefins, Nat. Commun., 2022, 13, 2396 CrossRef.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Bimetallic Fe–Co catalysts for CO2 hydrogenation to higher hydrocarbons, J. CO2 Util., 2013, 3–4, 102–106 CrossRef.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Light olefin synthesis from CO2 hydrogenation over K-promoted Fe–Co bimetallic catalysts, Catal. Today, 2015, 251, 34–40 CrossRef.
- L. Guo, X. Gao, W. Gao, H. Wu, X. Wang, S. Sun, Y. Wei, Y. Kugue, X. Guo, J. Sun and N. Tsubaki, High-yield production of liquid fuels in CO2 hydrogenation on a zeolite-free Fe-based catalyst, Chem. Sci., 2022, 14, 171–178 RSC.
- W. Wang, E. Toshcheva, A. Ramirez, G. Shterk, R. Ahmad, M. Caglayan, J. L. Cerrillo, A. Dokania, G. Clancy, T. B. Shoinkhorova, N. Hijazi, L. Cavallo and J. Gascon, Bimetallic Fe–Co catalysts for the one step selective hydrogenation of CO2 to liquid hydrocarbons, Catal. Sci. Technol., 2023, 13, 1527–1540 RSC.
- F. Jiang, B. Liu, S. Geng, Y. Xu and X. Liu, Hydrogenation of CO2 into hydrocarbons: enhanced catalytic activity over Fe-based Fischer–Tropsch catalysts, Catal. Sci. Technol., 2018, 8, 4097–4107 RSC.
- J. Liang, J. Liu, L. Guo, W. Wang, C. Wang, W. Gao, X. Guo, Y. He, G. Yang, S. Yasuda, B. Liang and N. Tsubaki, CO2 hydrogenation over Fe-Co bimetallic catalysts with tunable selectivity through a graphene fencing approach, Nat. Commun., 2024, 15, 512 CrossRef.
- F. Yuan, G. Zhang, J. Zhu, F. Ding, A. Zhang, C. Song and X. Guo, Boosting light olefin selectivity in CO2 hydrogenation by adding Co to Fe catalysts within close proximity, Catal. Today, 2021, 371, 142–149 CrossRef.
- N. Liu, J. Wei, J. Xu, Y. Yu, J. Yu, Y. Han, K. Wang, J. I. Orege, Q. Ge and J. Sun, Elucidating the structural evolution of highly efficient Co–Fe bimetallic catalysts for the hydrogenation of CO2 into olefins, Appl. Catal., B, 2023, 328, 122476 CrossRef.
- F. Yuan, G. Zhang, M. Wang, J. Zhu, M. Zhang, F. Ding, Z. Cheng, C. Song and X. Guo, Boosting the Production of Light Olefins from CO2 Hydrogenation over Fe–Co Bimetallic Catalysts Derived from Layered Double Hydroxide, Ind. Eng. Chem. Res., 2023, 62, 8210–8221 CrossRef.
- K. Y. Kim, H. Lee, W. Y. Noh, J. Shin, S. J. Han, S. K. Kim, K. An and J. S. Lee, Cobalt Ferrite Nanoparticles to Form a Catalytic Co–Fe Alloy Carbide Phase for Selective CO2 Hydrogenation to Light Olefins, ACS Catal., 2020, 10, 8660–8671 CrossRef.
- S.-M. Hwang, S. J. Han, H.-G. Park, H. Lee, K. An, K.-W. Jun and S. K. Kim, Atomically Alloyed Fe–Co Catalyst Derived from a N-Coordinated Co Single-Atom Structure for CO2 Hydrogenation, ACS Catal., 2021, 11, 2267–2278 CrossRef.
- L. Zhang, Y. Dang, X. Zhou, P. Gao, A. Petrus van Bavel, H. Wang, S. Li, L. Shi, Y. Yang, E. I. Vovk, Y. Gao and Y. Sun, Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts, Innovation, 2021, 2, 100170 Search PubMed.
- H. Chen, C. Wang, M. Zheng, C. Liu, W. Li, Q. Yang, S. Zhou and X. Feng, Reactive ball-milling synthesis of Co-Fe bimetallic catalyst for efficient hydrogenation of carbon dioxide to value-added hydrocarbons, J. Energy Chem., 2023, 84, 210–218 CrossRef.
- B. Yao, T. Xiao, O. A. Makgae, X. Jie, S. Gonzalez-Cortes, S. Guan, A. I. Kirkland, J. R. Dilworth, H. A. Al-Megren, S. M. Alshihri, P. J. Dobson, G. P. Owen, J. M. Thomas and P. P. Edwards, Transforming carbon dioxide into jet fuel using an organic combustion-synthesized Fe-Mn-K catalyst, Nat. Commun., 2020, 11, 6395 CrossRef PubMed.
- Y. H. Choi, E. C. Ra, E. H. Kim, K. Y. Kim, Y. J. Jang, K. N. Kang, S. H. Choi, J. H. Jang and J. S. Lee, Sodium-Containing Spinel Zinc Ferrite as a Catalyst Precursor for the Selective Synthesis of Liquid Hydrocarbon Fuels, ChemSusChem, 2017, 10, 4764–4770 CrossRef PubMed.
- S. Yang, H.-J. Chun, S. Lee, S. J. Han, K.-Y. Lee and Y. T. Kim, Comparative Study of Olefin Production from CO and CO2 Using Na- and K-Promoted Zinc Ferrite, ACS Catal., 2020, 10, 10742–10759 CrossRef.
- X. Cui, P. Gao, S. Li, C. Yang, Z. Liu, H. Wang, L. Zhong and Y. Sun, Selective Production of Aromatics Directly from Carbon Dioxide Hydrogenation, ACS Catal., 2019, 9, 3866–3876 CrossRef.
- X. Gong, Y. Liu, R. He, X. Xu, Z. Han, J. Chen, B. Feng, Z. j. Wang and A. Xing, Insights into the Structural Evolution Process of Na/ZnFe2O4 Spinel Catalyst in CO2 Hydrogenation, ChemCatChem, 2024, 16, e202301341 CrossRef CAS.
- W. Tu, C. Sun, Z. Zhang, W. Liu, H. S. Malhi, W. Ma, M. Zhu and Y.-F. Han, Chemical and structural properties of Na decorated Fe5C2-ZnO catalysts during hydrogenation of CO2 to linear α-olefins, Appl. Catal., B, 2021, 298, 120567 CrossRef CAS.
- C. Zhang, C. Cao, Y. Zhang, X. Liu, J. Xu, M. Zhu, W. Tu and Y.-F. Han, Unraveling the Role of Zinc on Bimetallic Fe5C2–ZnO Catalysts for Highly Selective Carbon Dioxide Hydrogenation to High Carbon α-Olefins, ACS Catal., 2021, 11, 2121–2133 CrossRef CAS.
- T. Liu, D. Xu, M. Song, X. Hong and G. Liu, K–ZrO2 Interfaces Boost CO2 Hydrogenation to Higher Alcohols, ACS Catal., 2023, 13, 4667–4674 CrossRef CAS.
- Y. Wang, W. Wang, R. He, M. Li, J. Zhang, F. Cao, J. Liu, S. Lin, X. Gao, G. Yang, M. Wang, T. Xing, T. Liu, Q. Liu, H. Hu, N. Tsubaki and M. Wu, Carbon-Based Electron Buffer Layer on ZnOx-Fe5C2-Fe3O4 Boosts Ethanol Synthesis from CO2 Hydrogenation, Angew. Chem., Int. Ed., 2023, 62, e202311786 CrossRef CAS PubMed.
- H. Yang, Z. Wei, J. Zhang, Y. Dang, S. Li, X. Bu, Z. Zhou, C. Gong, H. Wang, J. Li, Y. Liu, Y. Yang, T. Xiao, C. Liu, Y. Sun and P. Gao, Tuning the selectivity of CO2 hydrogenation to alcohols by crystal structure engineering, Chem, 2024, 10, 2245–2265 Search PubMed.
- P. Wang, W. Chen, F.-K. Chiang, A. I. Dugulan, Y. Song, R. Pestman, K. Zhang, J. Yao, B. Feng, P. Miao, W. Xu and E. J. M. Hensen, Synthesis of stable and low-CO2 selective ε-iron carbide Fischer-Tropsch catalysts, Sci. Adv., 2018, 4, eaau2947 CrossRef CAS PubMed.
- S. Lyu, L. Wang, Z. Li, S. Yin, J. Chen, Y. Zhang, J. Li and Y. Wang, Stabilization of ε-iron carbide as high-temperature catalyst under realistic Fischer–Tropsch synthesis conditions, Nat. Commun., 2020, 11, 6219 CrossRef PubMed.
- S. Li, J. Yang, C. Song, Q. Zhu, D. Xiao and D. Ma, Iron Carbides: Control Synthesis and Catalytic Applications in COx Hydrogenation and Electrochemical HER, Adv. Mater., 2019, 31, e1901796 CrossRef.
- G. J. A. Mannie, L. Lammich, Y.-W. Li, J. W. Niemantsverdriet and J. V. Lauritsen, Monolayer Iron Carbide Films on Au(111) as a Fischer–Tropsch Model Catalyst, ACS Catal., 2014, 4, 3255–3260 CrossRef CAS.
- Y. Li, Z. Li, A. Ahsen, L. Lammich, G. J. A. Mannie, J. W. H. Niemantsverdriet and J. V. Lauritsen, Atomically Defined Iron Carbide Surface for Fischer–Tropsch Synthesis Catalysis, ACS Catal., 2018, 9, 1264–1273 CrossRef.
- D. G. Rodríguez, M. A. Gleeson, J. V. Lauritsen, Z. Li, X. Yu, J. W. Hans Niemantsverdriet and C. J. Kees-Jan Weststrate, Iron carbide formation on thin iron films grown on Cu(100): FCC iron stabilized by a stable surface carbide, Appl. Surf. Sci., 2022, 585, 152684 CrossRef.
- D. Kordus, J. Jelic, M. Lopez Luna, N. J. Divins, J. Timoshenko, S. W. Chee, C. Rettenmaier, J. Krohnert, S. Kuhl, A. Trunschke, R. Schlogl, F. Studt and B. Roldan Cuenya, Shape-Dependent CO2 Hydrogenation to Methanol over Cu2O Nanocubes Supported on ZnO, J. Am. Chem. Soc., 2023, 145, 3016–3030 CrossRef CAS PubMed.
- X. Nie, G. Han, C. Song and X. Guo, Computational identification of facet-dependent CO2 initial activation and hydrogenation over iron carbide catalyst, J. CO2 Util., 2022, 59, 101967 CrossRef CAS.
- T. Chen, W. Jiang, X. Sun, W. Ning, Y. Liu, G. Xu and G. Han, Size-controlled Synthesis of Hematite α-Fe2O3 Nanodisks Closed with (0001) Basal Facets and {11-20} Side Facets and Their Catalytic Performance for CO2 Hydrogenation, ChemistrySelect, 2020, 5, 430–437 CrossRef CAS.
- W. Wu, J. Luo, J. Zhao, M. Wang, L. Luo, S. Hu, B. He, C. Ma, H. Li and J. Zeng, Facet sensitivity of iron carbides in Fischer-Tropsch synthesis, Nat. Commun., 2024, 15, 6108 CrossRef CAS.
- L. de Souza Caldas, M. J. Prieto, L. C. Tanase, A. Tiwari, T. Schmidt and B. Roldan Cuenya, Correlative In Situ Spectro-Microscopy of Supported Single CuO Nanoparticles: Unveiling the Relationships between Morphology and Chemical State during Thermal Reduction, ACS Nano, 2024, 18, 13714–13725 CrossRef CAS PubMed.
- W. Guo, J. Yin, Z. Xu, W. Li, Z. Peng, C. J. Weststrate, X. Yu, Y. He, Z. Cao, X. Wen, Y. Yang, K. Wu, Y. Li, J. W. Niemantsverdriet and X. Zhou, Visualization of on-surface ethylene polymerization through ethylene insertion, Science, 2022, 375, 1188–1191 CrossRef CAS.
- D. Degerman, M. Shipilin, P. Lömker, M. Soldemo, C. M. Goodwin, M. Wagstaffe, M. Börner, C. Schlueter, P. Amann and A. Nilsson, Effect of CO2-Rich Syngas on the Chemical State of Fe(110) during Fischer–Tropsch Synthesis, J. Phys. Chem. C, 2024, 128, 5542–5552 CrossRef CAS.
- Q. Y. Liu, C. Shang and Z. P. Liu, In Situ Active Site for CO Activation in Fe-Catalyzed Fischer-Tropsch Synthesis from Machine Learning, J. Am. Chem. Soc., 2021, 143, 11109–11120 CrossRef CAS PubMed.
- Q. Y. Liu, D. Chen, C. Shang and Z. P. Liu, An optimal Fe-C coordination ensemble for hydrocarbon chain growth: a full Fischer-Tropsch synthesis mechanism from machine learning, Chem. Sci., 2023, 14, 9461–9475 RSC.
| This journal is © The Royal Society of Chemistry 2025 |