
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of a six-membered heterocyclic 1,3-diphosphaallene†
Mahendra K.
Sharma
 a,
Christoph
Wölper
a,
Hannah
Siera
b,
Gebhard
Haberhauer
b and
Stephan
Schulz
*ac
a,
Christoph
Wölper
a,
Hannah
Siera
b,
Gebhard
Haberhauer
b and
Stephan
Schulz
*ac
aInstitute of Inorganic Chemistry, University of Duisburg-Essen, Universitätsstraße 5-7, D-45141 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Web: https://www.uni-due.de/ak_schulz/index_en.php
bInstitute of Organic Chemistry, University of Duisburg-Essen, Universitätsstraße 5-7, D-45141 Essen, Germany
cCenter for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Carl-Benz-Straße 199, 47057 Duisburg, Germany
First published on 29th November 2024
Abstract
1,3-Diphosphaallenes are a new class of heavier heteroallenes and show a fascinating chemical behavior and reactivity. Herein we report on the room temperature transformation of gallaphosphene LGa(OCP)PGaL 1 (L = HC[C(Me)N(Ar)]2, Ar = 2,6-i-Pr2C6H3) to the six-membered metallaheterocycle LGa(PCP)OGaL 2 featuring a LGa-substituted 1,3-diphosphaallene unit. The possible mechanism of formation of 2 is supported by quantum chemical calculations, which revealed that the formation of 2 is energetically more favorable (ca. 2 kcal mol−1) than the formation of 1 at ambient temperature. Remarkably, 2 reacts with singlet carbenes selectively to new five-membered metallaheterocycles LGa(PC)OGaL(P)NHC (NHC = [CMeN(R)]2C; R = Me 3, iPr 4; C{(NAr)CMe2CH2CMe2 = cAAC (5) featuring a 1,3-diphospha-1,3-butadiene unit. In stark contrast, its reaction with trimethylsilyldiazomethane yields (LGa)2O(P2C2H)SiMe36 featuring a 1,3-diphosphacyclobutene unit. Compounds 2–6 were characterized by heteronuclear NMR (1H, 13C, 31P), UV-vis, and IR spectroscopy. Compounds 2–4 and 6 were also characterized by single crystal X-ray diffraction (sc-XRD) and their bonding nature was investigated by quantum chemical calculations.
Introduction
Allenes with two contiguous C–C double bonds represent a fascinating class of unsaturated hydrocarbons due to the unique orthogonal arrangement of the cumulated bonds, which localizes and directs electron distribution, imparting allenes with distinctive chemical properties and reactivity.1 For instance, they were reported to undergo cycloaddition and nucleophilic addition reactions, oxidation reactions as well as insertion and bond activation reactions.1 Embedding heteroatoms into these allenic systems is particularly attractive in order to modify their electronic structures, reactivity patterns, and photophysical properties, making them valuable reagents in synthetic organic chemistry.2,3 In particular, the incorporation of a phosphorus atom into allenes has received significant interest because of its diagonal relationship with carbon in the periodic table that offers a distinctive influence on the allene framework by varying its electron distribution.4 The calculated weaker P–C (43 kcal mol−1) compared to the C–C double bond energy (65 kcal mol−1) provides notable advantages over all-carbon allenes by altering the energies of their frontier molecular orbitals, expanding their utility to small molecule activation, catalysis, and material science.5In 1984, Yoshifuji,6 Appel,7,8 and Karsch et al.9,10 independently prepared the first stable 1,3-diphosphaallene, ArPCPAr (Ar = 2,4,6-t-BuC6H2). Since then, additional kinetically stabilized 1,3-diphosphaallenes have been reported (Scheme 1).2,3,6–19 However, their chemical reactivity has been relatively less explored, particularly when compared to intensely studied allenes R2CCCR2 as well as lighter heteroallenes.1–3,20 Quantum chemical calculations on the parent 1,3-diphosphaallene revealed that the LUMO of these compounds typically consists of the low-lying π* orbitals of the P–C double bonds, while the HOMO comprises, more or less similar, two sets of quasi-degenerate n and π orbitals of the PCP unit.21 In addition, the calculated charges indicate a negatively charged carbon and positively charged phosphorus atoms.21 As a consequence, 1,3-diphosphaallenes were found to react with Lewis acidic transition metals via η1 or η2-coordination. In addition, they underwent protonation as well as dimerization reactions.2,3 Furthermore, one-electron redox reactions, reduction reactions, and reactions with organo-lithium reagents have also been reported,2,3 whereas reactions of phosphorus-substituted 1,3-diphosphaallenes with Lewis basic N-heterocyclic carbenes (NHCs) remain scarce, in marked contrast to well known reactions of heteroallenes, i.e. CO2, carbodiimides, isocyanates, and isothiocyanates, with NHCs.
 | ||
| Scheme 1 Previously reported synthetic routes to 1,3-diphosphaallenes and synthesis of compound 2 (this work). | ||
We recently prepared L(X)Ga-substituted phosphaketenes, LGa(X)PCO (X = Cl, PCO; L = HC[C(Me)N(Ar)]2; Ar = 2,6-i-Pr2C6H3), which reacted with LGa to gallaphosphenes LGa(X)PGaL (X = Cl, OCP).22,23 These were found to undergo cycloaddition reactions with heteroallenes,24 X–H bond activation reactions,25 and phosphinidene transfer-type reactions with NHCs.26 In addition, the photolysis of LGa(Cl)PCO resulted in the formation of the L(Cl)Ga-substituted diphosphene [L(Cl)GaP]2, which reacted with an NHC followed by one-electron oxidation to the corresponding radical cation.27 Treatment of the bisphosphaketene LGa(PCO)2 with a cyclic alkyl(amino)carbene (cAAC) furthermore yielded the 1,2-diphospha-1,3-butadiene LGa(P2OC)cAAC containing a π-conjugated P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P–CC framework, which by one-electron reduction reaction with potassium graphite (KC8) gave the corresponding radical anion.28 These remarkable findings encouraged us to investigate the reactivity of gallaphosphenes in more detail, and we herein report on the isomerization of LGa(OCP)PGaL 1 to the six-membered metallaheterocycle LGa(PCP)OGaL 2 featuring a 1,3-diphosphaallene unit (Scheme 1). Subsequent reactions with singlet carbenes and trimethylsilyldiazomethane (TMSCHN2) yielded unprecedented five-membered metallaheterocycles featuring 1,3-diphospha-1,3-butadienes LGa(PC)OGaL(P)RNHC (RNHC = [CMeN(R)]2C; R = Me 3, i-Pr 4; C{(NAr)CMe2CH2CMe25) and a fused-ring metallaheterocycle (LGa)2O(P2C2H)SiMe36 featuring a 1,3-diphosphacyclobutene ring.
P–CC framework, which by one-electron reduction reaction with potassium graphite (KC8) gave the corresponding radical anion.28 These remarkable findings encouraged us to investigate the reactivity of gallaphosphenes in more detail, and we herein report on the isomerization of LGa(OCP)PGaL 1 to the six-membered metallaheterocycle LGa(PCP)OGaL 2 featuring a 1,3-diphosphaallene unit (Scheme 1). Subsequent reactions with singlet carbenes and trimethylsilyldiazomethane (TMSCHN2) yielded unprecedented five-membered metallaheterocycles featuring 1,3-diphospha-1,3-butadienes LGa(PC)OGaL(P)RNHC (RNHC = [CMeN(R)]2C; R = Me 3, i-Pr 4; C{(NAr)CMe2CH2CMe25) and a fused-ring metallaheterocycle (LGa)2O(P2C2H)SiMe36 featuring a 1,3-diphosphacyclobutene ring.
Results and discussion
Gallaphosphene LGa(OCP)PGaL 1 undergoes gradual isomerization to the six-membered metallacycle LGa(PCP)OGaL 2 featuring a 1,3-diphosphaallene unit upon storage in solution (toluene, THF) at ambient temperature (Scheme 2). However, heating a toluene solution of 1 to 70 °C was found to significantly accelerate the rate of this isomerization reaction, resulting in quantitative conversion of 1 to 2 within 5 hours. Upon evaporation of the solvent, 2 was isolated as an olive colored solid in 98% yield. Compound 2 is soluble in THF, toluene, fluorobenzene, and n-hexane and stable both in solution and in solid state under inert gas atmosphere at room temperature, whereas it immediately decomposes upon exposure to air and moisture.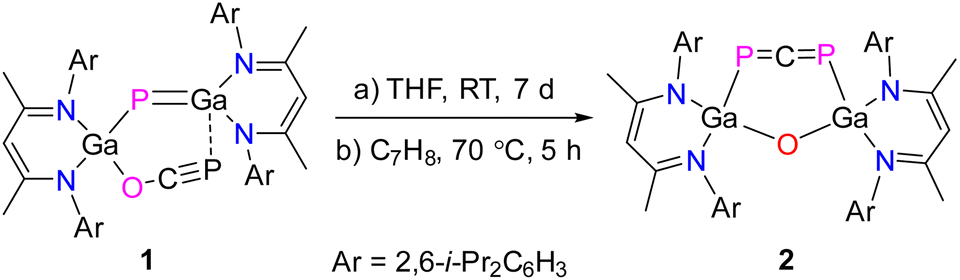 | ||
| Scheme 2 Synthesis of 1,3-diphosphaallene 2. | ||
Its 1H NMR spectrum shows two sets of resonances for the aryl groups of the β-diketiminate ligand, indicating their asymmetric nature in solution (Fig. S1†). This was confirmed by variable-temperature (VT) 1H NMR studies, which showed no dynamic behaviour over the temperature range of −60 °C to +75 °C (Fig. S25 and S26†). The 13C{1H} NMR spectrum of 2 (272.1 ppm, 1JCP = 73.3 Hz) shows the expected triplet for the phosphorus bound carbon atom of the 1,3-diphosphaallene unit, which is in the range of known 1,3-diphosphaallenes (270 ppm to 280 ppm).2,3 However, the 1JPC coupling constant (73.3 Hz) is rather large in comparison to those of other 1,3-diphosphaallenes (1JPC = ∼58 Hz),2,3 which most likely results from the introduction of electropositive LGa-substituents. The 31P{1H} NMR spectrum of 2 (–30.9 ppm) displays a sharp singlet for the phosphorus atoms, which is shifted to higher field compared to known 1,3-diphosphaallenes (140 to 169 ppm), which again reflects the influence of the electropositive LGa-substituents.2,3
2 crystallizes in the monoclinic space group P21/c containing one molecule per asymmetric unit cell or four molecules per unit cell as shown by single-crystal X-ray diffraction (sc-XRD, Fig. 1).29 To the best of our knowlegde, 2 represents only the second structurally characterized 1,3-diphosphaallene with cumulated P–C and C–P double bonds.
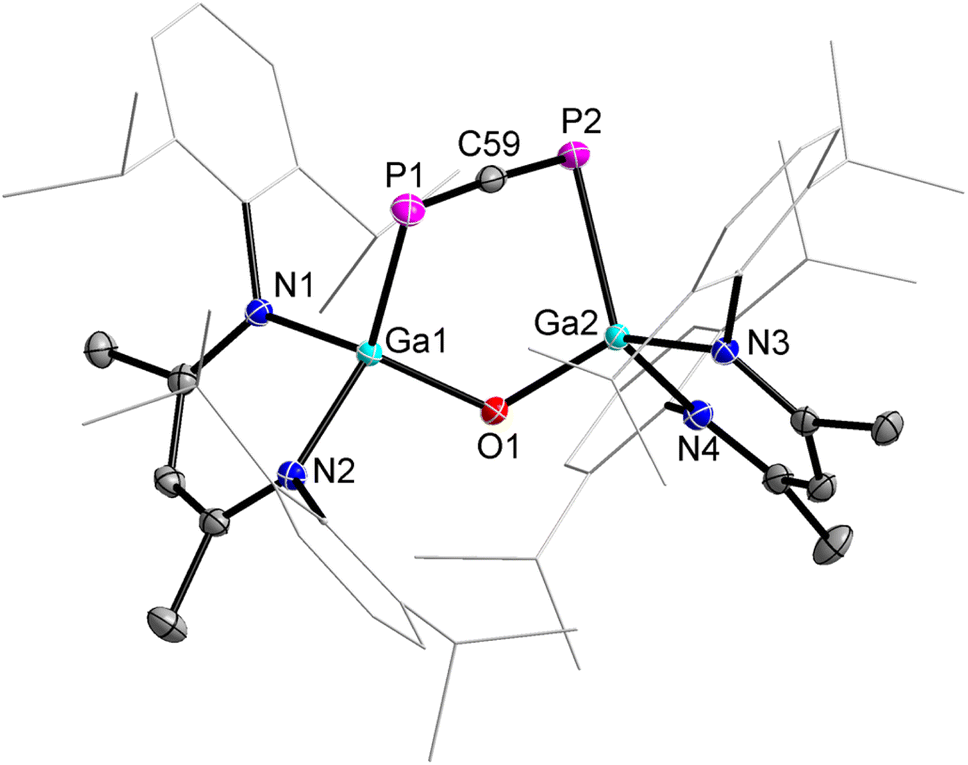 | ||
| Fig. 1 Molecular structure of 1,3-diphosphaallene 2. Ellipsoids set at 50% probability; hydrogen atoms and alternate positions of the disordered parts are omitted for clarity. Selected bond length (Å) and angles (°): P(1)–C(59) 1.6516(13), P(2)–C(59) 1.6506(13), Ga(1)–P(1) 2.3767(3), Ga(2)–P(2) 2.3845(3), Ga(1)–O(1) 1.8082(8), Ga(2)–O(1) 1.8046(8); P(2)–C(59)–P(1) 176.48(8), O(1)–Ga(1)–P(1) 107.78(3), O(1)–Ga(2)–P(2) 107.62(3), Ga(2)–O(1)–Ga(1) 123.63(4). | ||
The solid-state molecular structure of compound 2 revealed a linear P–C–P unit with a bond angle of 176.48(8)°, which is slightly larger than that of the 1,3-diphosphaallene ArPCPAr (Ar = 2,4,6-t-BuC6H2, 172.6(5)°).10 The P atoms are twofold- and the Ga atoms fourfold-coordinated. The P1–C59 (1.6516(13) Å) and P2–C59 (1.6506(13) Å) bond lengths are equidistant and in the typical range of phosphaalkenes (1.65–1.67 Å).30 They agree with the sum of the calculated P–C double bond radii (P 1.02 Å; C 0.67 Å)31 and are comparable to P–C double bond lengths reported for phosphaallenes.2,3 The Ga1–P1 (2.3767(3) Å) and Ga2–P2 (2.3845(3) Å) bond lengths are almost identical but slightly longer than the Ga–P single bonds of gallaphosphene 1 (2.2943(16) Å),22 phosphaalkenes L(Cl)GaP(RNHC) (R = Me, 2.2441(3) Å; R = i-Pr, 2.2538(3) Å),26 L(Cl)GaP(cAAC) (2.289 Å),32 diphosphene [L(Cl)GaP]2 (2.313(3) Å),27 and 1,2-diphospha-1,3-butadiene (2.2844(4) Å),28 respectively, but agree with the sum of the calculated Ga–P single-bond radii (Ga 1.24 Å; P 1.11 Å).33 The IR spectrum of 2 was simulated by DFT calculations, showing strong P–C asymmetric stretching frequencies at 1347 (PBE0) or 1297 (PBE) cm−1 (Fig. S5 and S6†). Unfortunately, C–C and C–H stretching vibrations typically appear in this regime, hence we were unable to assign the P–C asymmetric stretching band in the experimental IR spectrum.
DFT calculations were performed to get a deeper insight into the electronic structure of compound 2 and the reaction mechanism of the transformation of 1 into 2 (for details see ESI†). The DFT-optimized geometry of 2 is in good agreement with the solid-state molecular structure. Mayer and Wiberg bond orders analyses calculated at PBE0-D3BJ level of theory show bond orders for the PCP unit of 1.77 to 1.78, which is in accordance with a phosphaallene (Fig. S41†). The HOMO and LUMO of 2 range over the PCP unit, with their spatial orientation being almost perpendicular to each other (Fig. 2). The HOMO essentially corresponds to the antibonding linear combination of the lone pairs on the phosphorus atoms, while the LUMO is composed of the p orbitals of the three centers of the phosphaallene unit, whereby it exhibits two nodes. The p-orbitals are located almost in one plane, whereas organic allenes typically show p orbitals of the outer centers perpendicular to each other. This is probably due to the fact that the PCP unit is part of a six-membered ring.
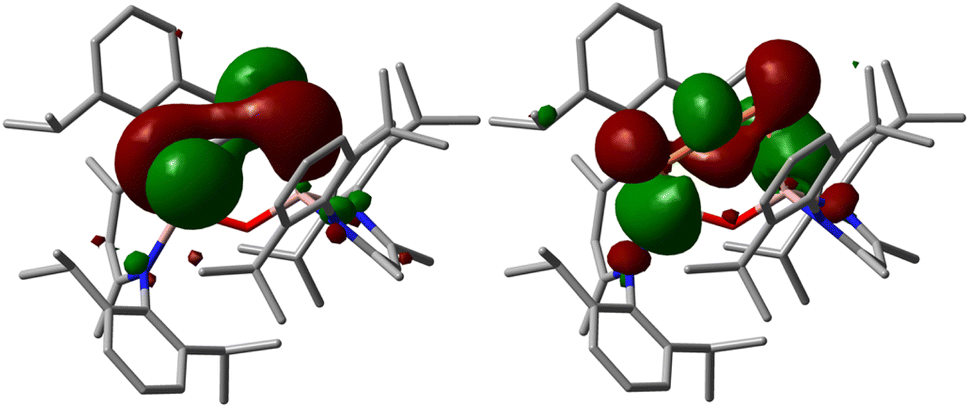 | ||
| Fig. 2 HOMO and LUMO of 1,3-diphosphaallene 2 calculated at PBE0-D3BJ level of theory (isovalue 0.03 a.u.). Hydrogen atoms are omitted for clarity. | ||
The mechanistical calculations reveal that the transformation of 1 into 2 takes place in three steps. In the first one, a P–C bond formation occurs, whereby the bicyclic intermediate Int-1 is formed (Fig. 3). In the next step, the Ga–P bond is broken along with a concerted rotation around the C–P bond and a further Ga–O bond is formed. The final step of the rearrangement represents the opening of the bicycle yielding phosphaallene 2, whereby the C–O bond is broken. The formation of the intermediates Int-1 and Int-2 are endergonic reactions, whereas the generation of product 2 is slightly exergonic. The energies of the transition states of the three steps are in a similar range, with the last step being the rate-determining one. The activation energies calculated using PBE-D3BJ (blue in Fig. 3) and PBE0-D3BJ (red) for the third step amounts to 24.4 and 26.7 kcal mol−1, respectively. From this, half-lives at 70 °C of 0.1 and 2.7 h can be calculated. Within the error limits of DFT calculations, this agrees very well with the experimental observation, which shows a complete conversion of 1 into 2 within 5 h. Calculations for the rearrangement of an analogous system to 1 with sterically less demanding ligands at the gallium atoms reveal that the size of the ligands should not have a significant impact on the reaction rate (Fig. S42†).
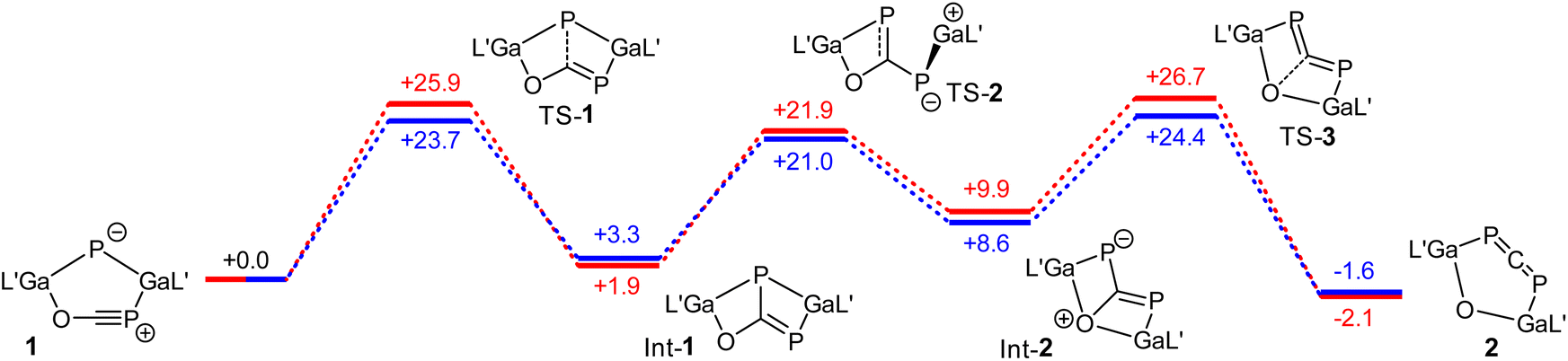 | ||
| Fig. 3 Gibbs energies (G) for the rearrangement from 1 to 2 calculated by means of PBE0-D3BJ (red) and PBE-D3BJ (blue). L′ = HC[C(Me)NDipp]2, Dipp = 2,6-i-Pr2C6H3. The values are given in kcal mol−1. | ||
Encouraged by the results from DFT calculations, we sought to probe the chemical reactivity of the 1,3-diphosphaallene 2 in more detail. Heteroallenes such as CO2, carbodiimides, isocyanates, and isothiocyanates were shown to react with N-heterocyclic carbenes (NHCs) to the corresponding betaine adducts,34 whose electronic properties can be fine-tuned by variation of the carbene. Interestingly, analogous reactions of 1,3-diphosphaallenes with N-heterocyclic carbenes are virtually unknown. To the best of our knowledge, only the 1,3-diphosphaallene ArPCPAr (Ar = 2,4,6-t-BuC6H2)6–10 was reported to react with an in situ generated dichlorocarbene, however this reaction rather yielded a methylenediphosphirane than the corresponding betaine adduct.35 This attracted our interest to explore reactions of 2 with isolable carbenes in more detail. Since numerous singlet carbenes with different stereoelectronic properties are known,36 we initially chose to react compounds 2 with small N-heterocyclic carbenes, RNHC (RNHC = [CMeN(R)]2C; R = Me, i-Pr).
Treatment of 2 with equimolar amounts of RNHC at ambient temperature proceeded with an immediate color change from olive to red and formation of the metallaheterocycles LGa(PC)OGaL(P)RNHC (R = Me 3, i-Pr 4) featuring a 1,3-diphospha-1,3-butadiene unit (Scheme 3). The analogous reaction of 2 with a sterically more demanding cyclic (alkyl)(amino)-carbene (cAAC) to the metallaheterocycle LGa(PC)OGaL(P)cAAC 5 took 6 hours for completion (93%), whereas the sterically even more hindered IPr carbene (IPr = [CHN(Ar)]2C; Ar = 2,6-i-Pr2-C6H3) failed to react, clearly reflecting the effect of steric bulk on the reaction process.
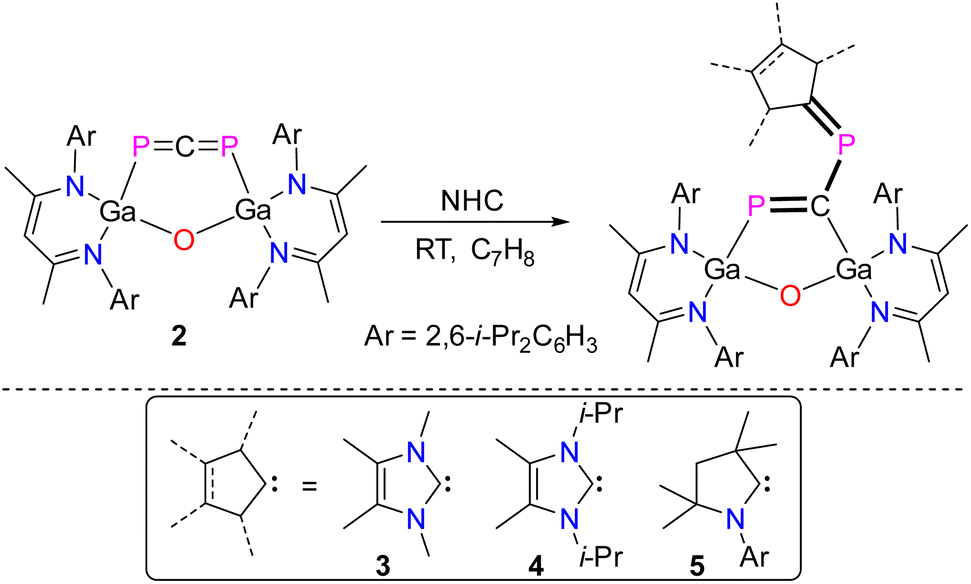 | ||
| Scheme 3 Reactions of 2 with singlet carbenes to 1,3-diphospha-1,3-butadienes 3–5. | ||
Compounds 3–5 are soluble in common organic solvents and stable under argon atmosphere at ambient temperature, but rapidly decompose upon exposure to air. The 1H and 13C{1H} NMR spectra of compounds 3–5 exhibit two distinct sets of resonances for the β-diketiminate ligand as was observed for L(X)Ga-substituted dipnictenes,37 gallapnictenes,22,38 and other complexes.39 The 31P{1H} NMR spectra of 3 (168.59 ppm, 2JPP = 18.0 Hz; 74.73 ppm, 2JPP = 18.0 Hz), 4 (153.34 ppm, 2JPP = 22.1 Hz; 73.94 ppm, 2JPP = 22.1 Hz), and 5 (434.01 ppm, 2JPP = 53.5 Hz; 143.46 ppm, 2JPP = 52.7 Hz) each display two doublets for the electronically nonequivalent phosphorus atoms, which are downfield shifted with respect to starting 1,3-diphosphaallene 2 (–30.9 ppm). This downfield shift in the 31P signals of compound 5 is more pronounced due to the increased π-acidity of the cyclic alkylamino carbene (cAAC) compared to the NHCs in compounds 3 and 4.36 Interestingly, the 2JPP coupling constants in compounds 3–5 (18.0 Hz to 53.4 Hz) are rather small in comparison to the 2JPP coupling constants of the previously reported acyclic 1,3-diphospha-1,3-butadienes, which range from 112 Hz to 122 Hz.40
The molecular structures of compounds 3 and 4 were determined by sc-XRD (Fig. 4). Single crystals were obtained by either storing a saturated n-hexane solution of 3 to −30 °C or by diffusing n-hexane into a saturated benzene solution of 4 at ambient temperature. Compounds 3 and 4 crystallize in the monoclinic space group C2/c.29 The solid-state structures confirmed the formation of the five-membered LGa-containing metallaheterocycles and revealed a planar cis-1,3-diphospha-1,3-butadiene unit with conjugated P–C bonds. The five-membered Ga2OPC metallaheterocycle is twisted, with a dihedral angle (ϕ interplanar angle of the best plane of C60, P2, C59, P1, and Ga2, C59, P1, Ga1, O1) of approximately 17° (3, no reliable value can be given due to the disorder) and 10.46(8)° (4), respectivley, whereas the six-membered C3N2Ga heterocylce is almost perpendicular to this plane.
 | ||
| Fig. 4 Molecular structures of compounds 3 and 4. Ellipsoids set at 50% probability; hydrogen atoms, solvent molecules (n-hexane 3, benzene 4), alternate positions of the disordered parts (3) are omitted for clarity. Selected bond length (Å) and angles (°): 3: Ga(1)–P(1) 2.2926(11), Ga(2)–C(59) 2.092(8), P(1)–C(59) 1.801(6), P(2)–C(59) 1.623(7), P(2)–C(60) 1.847(3); P(2)–C(59)–P(1) 128.9(5), C(59)–P(2)–C(60) 97.5(3); 4: Ga(1)–P(1) 2.3351(4), Ga(2)–C(59) 2.0285(14), P(1)–C(59) 1.7716(15), P(2)–C(59) 1.6990(15), P(2)–C(60) 1.8571(17); P(2)–C(59)–P(1) 129.91(8), C(59)–P(2)–C(60) 105.29(7). | ||
The P1–C59 bond lengths (Table 1) in 3 (1.801(6) Å) and 4 (1.7716(15) Å) are slightly shorter than the P2–C60 bond lengths (1.847(3) Å 3 and 1.8571(17) Å 4) but almost 10 pm elongated compared to P–C double bonds in phosphaalkenes (1.65–1.67 Å)30 as well as the P–C double bonds observed in 2 (1.6516(13) Å, (1.6506(13) Å), respectively. However, they are comparable to the P–C single bonds in LGa-substituted phosphinidenes (1.8099(13) Å)26 and the calculated P–C single-bond radii (P 1.11 Å; C 0.75 Å).33 In marked contrast, the central C59–P2 bonds in 3 (1.623(7) Å) and 4 (1.6990(15) Å) are significantly shorter than the P1–C59 and the P2–C60 bonds but comparable to P–C double bonds in phosphaalkenes (1.65–1.67 Å)30 and the calculated P–C double-bond radii (P 1.02 Å; C 0.67 Å),31 respectively. The structural features point to a highly delocalized P2C2 π-conjugated system.
| P1–C59 | C59–P2 | P2–C60 | Ga1–P1 | Ga2–C59 | |
|---|---|---|---|---|---|
| 2 | 2.038(5) | 2.313(3) | — | 2.313(3) | — |
| 3 | 1.801(6) | 1.623(7) | 1.847(3) | 2.2926(11) | 2.092(8) |
| 4 | 1.7716(15) | 1.6990(15) | 1.8571(17) | 2.3351(4) | 2.0285(14) |
| 6 | 1.827(9) | 1.720(10) | 1.875(6) | 2.503(2) | 1.940(10) |
The Ga1–P1 bond lengths in 3 (2.2926(11) Å) and 4 (2.3351(4) Å) are slightly shorter compared to 2 (2.3767(3)/2.3845(3) Å) but comparable to the sum of the calculated Ga–P single-bond radii (Ga 1.24 Å; P 1.11 Å)33 and Ga–P single bonds in the diphosphene [L(Cl)GaP]2 (2.313(3) Å)27 and the 1,2-diphospha-1,3-butadiene LGa(P2OC)cAAC (2.2844(4) Å), respectively.28 Similarly, the Ga2–C59 bond lengths are comparable to Ga–C single bonds in LGa(C6F5)F (1.991(2) Å),41 LGa(C5H4NO)OH (1.9698(13) Å),42 and the sum of the calculated Ga–C single bond radii (Ga 1.24 Å; C 0.75 Å).33
To check if 1,3-diphosphaallene 2 only reacts as alkylidene carbene transfer reagent as was observed in reactions with NHCs and cAAC or is also capable for cycloaddition reactions at the P–C double bonds, we reacted 2 with an equimolar amount of trimethylsilyldiazomethane (TMSCHN2). This reaction remarkably yielded (LGa)2O(P2C2H)SiMe36 with a 1,3-diphosphacyclobutene unit (Scheme 4).
 | ||
| Scheme 4 Reaction of 1,3-diphosphallene 2 with trimethylsilyldiazomethane to 6. | ||
Compound 6 is a yellow crystalline solid, which is soluble in common organic solvents and stable under an inert gas atmosphere at ambient temperature both in solution and in the solid state, but rapidly decomposes when exposed to air. The 1H and 13C{1H} NMR spectra of compound 6 exhibit the expected sets of resonances for the β-diketiminate ligand, and the 31P{1H} NMR spectrum showed two doublets at 386.48 ppm (2JPP = 96.8 Hz) and 89.30 ppm (2JPP = 96.8 Hz) for the magnetically non-equivalent phosphorus atoms. These signals are downfield shifted compared to the signals observed for the 1,3-diphosphaallene 2 and compounds 3–4, respectively, but they are still at higher field compared to compound 5. Interestingly, the 2JPP coupling constants of 6 are considerably larger than those observed for compounds 3–5, but they comparable to those of a 1,3-diphospha-1,3-cyclobutene, (Mes*)2P2H2(Me)t-Bu (2JPP = 92 Hz; Mes* = 2,4,6-t-Bu3C6H2).43
The molecular structure of compound 6 was determined by sc-XRD (Fig. 5). Compound 6 crystallized in the monoclinic space group P21/c.29 The planar four-membered P2C2 ring doesn't adopt a co-planar orientation to the five-membered (LGa)2OPC ring, most likely to reduced steric interactions. The interplanar angle of the best plane of Ga1, P1, C59, Ga2, O1, and P1, C59, P2, C60 is approximately 50° (no reliable value can be given due to the disorder). The refined diffraction data showed a rotational disorder of the four-membered P2CCH(SiMe3) ring (52/48) over two positions, with additional disorder in the solvent molecule (n-hexane). The P1–C59 (1.827(9) Å), P1–C60 (1.888(6) Å), and P2–C60 (1.875(6) Å) single bond lengths in the four-membered P2C2 ring are consistent with previously reported P–C single bond lengths28 and the calculated P–C single-bond radii (P 1.11 Å; C 0.75 Å).33 The P2–C59 (1.720(10) Å) double bond is significantly shorter and comparable to P–C double bonds reported for phosphaalkenes (1.65–1.67 Å)30 and the calculated P–C double-bond radii (P 1.02 Å; C 0.67 Å).31
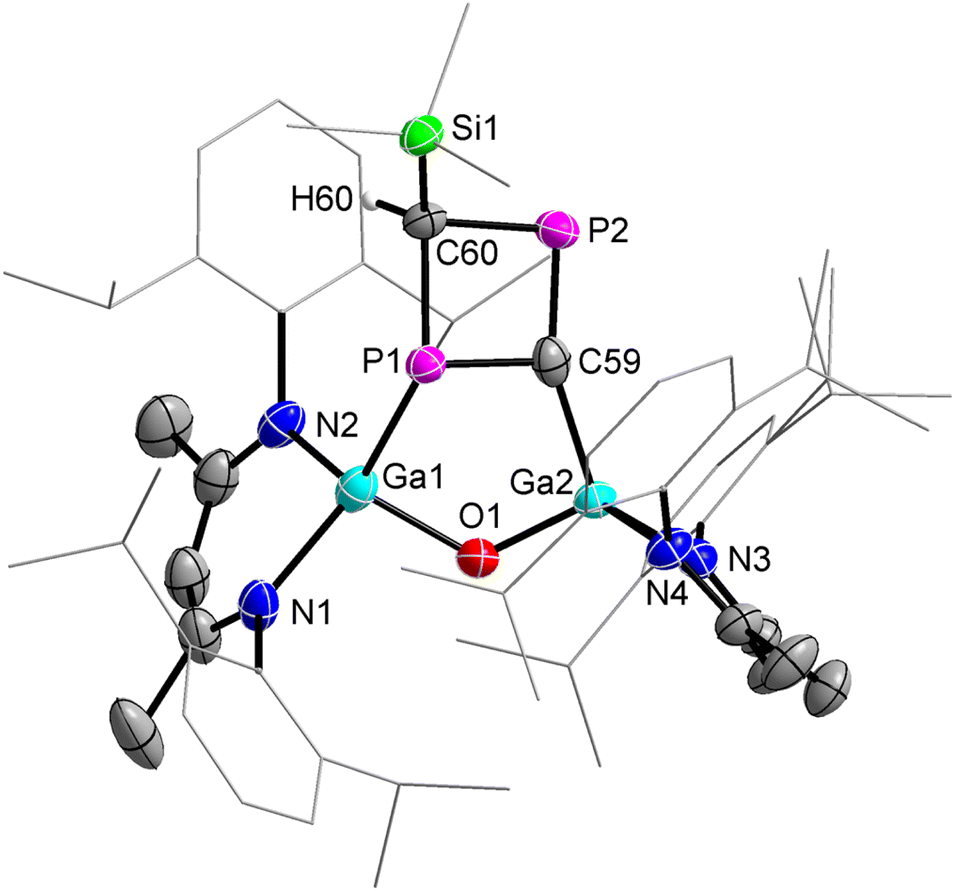 | ||
| Fig. 5 Molecular structure of compound 6. Ellipsoids set at 50% probability; hydrogen atoms, disordered solvent molecules (n-hexane), and alternate positions of the disordered parts are omitted for clarity. Selected bond length (Å) and angels (°): Ga(1)–P(1) 2.503(2), Ga(2)–O(1) 1.815(2), Ga(2)–C(59) 1.940(10), P(1)–C(59) 1.827(9), P(1)–C(60) 1.888(6), P(2)–C(59) 1.720(10), P(2)–C(60) 1.875(6). | ||
The reaction mechanism of the formation of 6 was also investigated by quantum chemical calculations (for details see ESI†). DFT calculations show that the reaction of 2 with trimethylsilyldiazomethanes takes place over several steps (Fig. 6). The initial process (TS-4) is the rate-determining one, and involves the attack of the carbon atom from the carbene on a phosphorus atom of the allene in combination with a concerted rearrangement of the gallium center from the phosphorus to the carbon atom. The stabilizing effect of this attack is the interaction of the free electron pair on the carbene with the LUMO of the phosphaallene. The as-formed intermediate Int-3 can be converted into the diene Int-4via two different routes: The removal of nitrogen can occur in a single step (TS-5), or in two steps via the formation of the bicycle Int-5. The corresponding transition states TS-5 and TS-6 are energetically very similar at 17.3 and 16.6 kcal mol−1, so that neither reaction path can be excluded. The final cyclization of the diene Int-4 then yields bicycle 6.
 | ||
| Fig. 6 Gibbs energies (G) for the reaction of 2 with Me3SiCHN2 to 6 calculated by means of PBE0-D3BJ. L′ = HC[C(Me)NDipp]2, Dipp = 2,6-i-Pr2C6H3. The values are given in kcal mol−1. | ||
The UV-vis spectra of the five-membered metallaheterocycles 3 (345, 496 nm), 4 (345, 489 nm), and 5 (345, 498 nm) featuring a 1,3-diphospha-1,3-butadiene unit exhibit two main absorptions (Fig. 7) which based on the TD-DFT calculations at TD-PBE0-D3BJ(SMD,THF)/def2-SVP level of theory, comprise dominant contributions of the π → π* transitions of the β-diketiminate ligand backbone and the 1,3-diphospha-1,3-butadiene π-systems, respectively (Fig. S35–S39 and S46–S51†). In contrast, the UV-vis spectra of compounds 2 and 6 showed only one strong absorption band at 345 nm (Fig. 7) that corresponds to the π → π* transitions of the β-diketiminate ligand backbone π-system (Fig. S33, S34, S40 and S43–S45†).
 | ||
| Fig. 7 UV-vis spectra of compounds 2–6 in THF. | ||
Conclusions
We report on a unique and atom economic method for the synthesis of the LGa-substituted 1,3-diphosphaallene 2, which to the best of our knowledge represents only the second structurally characterized 1,3-diphosphaallene. Quantum chemical calculations provided profounds insights into its electronic structure and the energetics of the thermal rearrangement of gallaphosphene 1 to 1,3-diphosphaallene 2. 1,3-Diphosphaallene 2 was found to react as alkylidene carbene transfer reagent in reactions with NHCs and cAAC, yielding the corresponding five-membered metallaheterocycles 3–5 featuring a unique 1,3-diphospha-1,3-butadiene unit, whereas a [3 + 2] cycloaddition reaction to the metallaheterocycle 6 featuring a diphosphacyclobutene unit occurred with trimethylsilyldiazomethane. These interesting results clearly demonstrate the promising potential of gallaphosphene 1 for the synthesis of P-containing 1,3-diphosha-1,3-butadienes with distinct electronic structures, since a variety of singlet carbenes with different stereoelectronic properties are available. Furthermore, the formation of compounds 6 confirms the promising potential of compound 2 in cycloaddition reactions, which may allow for the synthesis of novel P-containing heterocycles.Data availability
The data supporting this article have been included as part of the Supplementary Information. Crystallographic data has been deposited at the CCDC under CCDC 2362185 (2), 2362186 (3), 2362187 (4), and 2362188 (6) and can be obtained from https://doi.org/10.1039/d4sc06371fAuthor contributions
M. K. S., conceptualization, experimentation, characterization, writing – original draft; C. W., sc-XRD data acquisition and processing; H. S., quantum chemical calculations; G. H. quantum chemical calculations, supervision; S. S., supervision, final writing, funding acquisition, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the DFG (SCHU 1069/27-1) and the University of Duisburg-Essen (S. S.) is gratefully acknowledged.References
- (a) D. R. Taylor, Chem. Rev., 1967, 67, 317–359 CrossRef CAS; (b) Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, Germany, 2004 Search PubMed; (c) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783–816 RSC; (d) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS PubMed; (e) B. Alcaide and P. Almendros, Chem. Soc. Rev., 2014, 43, 2886–2887 RSC.
- J. Escudié, H. Ranaivonjatovo and L. Rigon, Chem. Rev., 2000, 100, 3639–3696 CrossRef and references therein..
- J. Escudié and H. Ranaivonjatovo, Organometallics, 2007, 26, 1542–1559 CrossRef and references therein..
- (a) M. C. Simpson and J. D. Protasiewicz, Pure Appl. Chem., 2013, 85, 801–815 CrossRef CAS; (b) D. A. Pantazis, J. E. McGrady, J. M. Lynam, C. A. Russell and M. Green, Dalton Trans., 2004, 2080–2086 RSC; (c) Phosphorus: The Carbon Copy, ed. K. B. Dillon, F. Mathey and J. F. Nixon, John Wiley & Sons, New York, 1998 Search PubMed.
- M. W. Schmidt, P. N. Truong and M. S. Gordon, J. Am. Chem. Soc., 1987, 109, 5217–5227 CrossRef CAS.
- M. Yoshifuji, K. Toyota and N. Inamoto, J. Chem. Soc., Chem. Commun., 1984, 689–690 RSC.
- R. Appel, P. Fölling, B. Josten, M. Siray, V. Winkhaus and F. Knoch, Angew. Chem., Int. Ed. Engl., 1984, 23, 619–620 ( Angew. Chem. , 1984 , 96 , 620–621 ) CrossRef.
- R. Appel, P. Fölling, L. Krieger, M. Siray and F. Knoch, Angew. Chem., Int. Ed. Engl., 1984, 23, 970–971 ( Angew. Chem. , 1984 , 96 , 620–621 ) CrossRef.
- H. H. Karsch, F. H. Köhler and H.-U. Reisacher, Tetrahedron Lett., 1984, 25, 3687–3690 CrossRef CAS.
- H. H. Karsch, F. H. Köhler and H.-U. Reisacher, Angew. Chem., Int. Ed. Engl., 1984, 23, 618–619 ( Angew. Chem. , 1984 , 96 , 619–620 ) CrossRef.
- M. Yoshifuji, S. Sasaki and N. Inamoto, Tetrahedron Lett., 1989, 30, 839–842 CAS.
- M. Gouygou, C. Tachon, R. El Ouatib, O. Ramarijaona, G. Etemad-Moghadam and M. Koenig, Tetrahedron Lett., 1989, 30, 177–178 CrossRef CAS.
- M. Yoshifuji, S. Sasaki, T. Niitsu and N. Inamoto, Tetrahedron Lett., 1989, 30, 187–188 CrossRef CAS.
- M. Yoshifuji, S. Sasaki and N. Inamoto, J. Chem. Soc., Chem. Commun., 1989, 1732–1733 RSC.
- M. Gouygou, M. Koenig, J. Escudié and C. Couret, Heteroat. Chem., 1991, 2, 221–227 CrossRef CAS.
- M. Yoshifuji, T. Niitsu, D. Shiomi and N. Inamoto, Tetrahedron Lett., 1989, 30, 5433–5436 CrossRef CAS.
- R. El Ouatib, D. Ballivet-Tkatchenko, G. Etemad-Moghadam and M. Koenig, J. Organomet. Chem., 1993, 453, 77–84 CrossRef.
- A. Alberti, M. Benaglia, M. A. Della Bona, M. Guerra, A. Hudson and D. Macciantelli, Res. Chem. Intermed., 1996, 22, 381–392 CrossRef CAS.
- K. Toyota, A. Nakamura and M. Yoshifuji, Chem. Commun., 2002, 3012–3013 RSC.
- (a) H. Ulrich, Cumulenes In Click Reactions, Wiley, New York, 1st edn, 2010 Search PubMed; (b) S. Braverman, M. Cherkinsky and M. L. Birsa, in Carbon dioxide, carbonyl sulfide, carbon disulfide, isocyanates, isothiocyanates, carbodiimides, and their selenium, tellurium, and phosphorus analogues. Science of Synthesis, ed. J. G. Knight, Thieme, Stuttgart, Germany, 2005, vol. 18, pp. 65–320 Search PubMed.
- M. T. Nguyen and A. F. Hegarty, J. Chem. Soc., Perkin Trans., 1985, 2, 2005–2012 RSC.
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed. Engl., 2021, 60, 6784–6790 ( Angew. Chem. , 2021 , 133 , 6859–6865 ) CrossRef CAS PubMed.
- M. K. Sharma, P. Dhawan, C. Helling, C. Wölper and S. Schulz, Chem.–Eur. J., 2022, 28, e202200444 CrossRef CAS PubMed.
- M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 21784–21788 ( Angew. Chem. , 2021 , 40 , 21953–21957 ) CrossRef CAS PubMed.
- M. K. Sharma, C. Wölper and S. Schulz, Dalton Trans., 2022, 51, 1612–1616 RSC.
- M. K. Sharma, H. M. Weinert, C. Wölper and S. Schulz, Chem.–Eur. J., 2024, 30, e202400110 CrossRef CAS.
- M. K. Sharma, S. Chabbra, C. Wölper, H. M. Weinert, E. J. Reijerse, A. Schnegg and S. Schulz, Chem. Sci., 2022, 13, 12643–12650 RSC.
- M. K. Sharma, H. M. Weinert, B. Li, C. Wölper, J. T. Henthorn, G. E. Cutsail III, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2023, 62, e202309466 ( Angew. Chem. , 2023 , 135 , e202309466 ) CrossRef CAS PubMed.
- Full crystallographic data of all structurally characterized compounds described herein as well as central bond lengths and angles (Tables S1–S2 and Fig. S29–S34) are given in the ESI.†.
- (a) L. Weber, Eur. J. Inorg. Chem., 2000, 2425–2441 CrossRef CAS; (b) L. Weber, Eur. J. Inorg. Chem., 2007, 4095–4117 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. Eur. J., 2009, 15, 12770–12779 CrossRef.
- B. Li, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 1986–1991 ( Angew. Chem. , 2021 , 133 , 2014–2019 ) CrossRef CAS.
- P. Pyykkö and M. Atsumi, Chem. Eur. J., 2009, 15, 186–197 CrossRef.
- (a) J. R. Lamb, C. M. Brown and J. A. Johnson, Chem. Sci., 2021, 12, 2699–2715 RSC; (b) L. Delaude, Eur. J. Inorg. Chem., 2009, 1681–1699 CrossRef CAS.
- M. Yoshifuji, K. Toyota, H. Yoshimura, K. Hirotsu and A. Okamoto, J. Chem. Soc., Chem. Commun., 1991, 124–125 RSC.
- (a) O. Back, M. Henry-Ellinger, C. D. Martin, D. Martin and G. Bertrand, Angew. Chem. Int. Ed., 2013, 52, 2939–2943 ( Angew. Chem. , 2013 , 125 , 3011–3015 ) CrossRef CAS; (b) T. Krachko and J. C. Slootweg, Eur. J. Inorg. Chem., 2018, 2734–2754 CrossRef CAS.
- (a) L. Song, J. Schoening, C. Wölper, S. Schulz and P. R. Schreiner, Organometallics, 2019, 38, 1640–1647 CrossRef CAS; (b) J. Krüger, J. Schoening, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Z. Anorg. Allg. Chem., 2018, 644, 1028–1033 CrossRef; (c) L. Tuscher, C. Helling, C. Wölper, W. Frank, A. S. Nizovtsev and S. Schulz, Chem. Eur. J., 2018, 24, 3241–3250 CrossRef CAS.
- (a) C. Helling, C. Wölper and S. Schulz, J. Am. Chem. Soc., 2018, 140, 5053–5056 CrossRef CAS; (b) J. Krüger, C. Ganesamoorthy, L. John, C. Wölper and S. Schulz, Chem. Eur. J., 2018, 24, 9157–9164 CrossRef PubMed; (c) J. Schoening, L. John, C. Wölper and S. Schulz, Dalton Trans., 2019, 48, 17729–17734 RSC.
- C. Ganesamoorthy, J. Schoening, C. Wölper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020, 12, 608–614 CrossRef CAS PubMed.
- R. Appel, P. Fölling, W. Schuhn and F. Knoch, Tetrahedron Lett., 1986, 27, 1661–1664 CrossRef CAS.
- O. Kysliak, H. Görls and R. Kretschmer, J. Am. Chem. Soc., 2021, 143, 142–148 CrossRef CAS.
- A. Kassymbek, S. F. Vyboishchikov, B. M. Gabidullin, D. Spasyuk, M. Pilkington and G. I. Nikonov, Angew. Chem., Int. Ed., 2019, 58, 18102–18107 ( Angew. Chem. , 2019 , 131 , 18270–18275 ) CrossRef CAS.
- S. Ito, J. Miura, N. Morita, M. Yoshifuji and A. J. Arduengo, Inorg. Chem., 2009, 48, 8063–8065 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2362185 (2)–2362188 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06371f |
| This journal is © The Royal Society of Chemistry 2025 |