
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiosynthesis of a bacterial meroterpenoid reveals a non-canonical class II meroterpenoid cyclase†
Zengyuan
Wang
a,
Tyler A.
Alsup
b,
Xingming
Pan
a,
Lu-Lu
Li
a,
Jupeng
Tian
a,
Ziyi
Yang
a,
Xiaoxu
Lin
a,
Hui-Min
Xu
c,
Jeffrey D.
Rudolf
 b and
Liao-Bin
Dong
*a
b and
Liao-Bin
Dong
*a
aState Key Laboratory of Natural Medicines, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Nanjing, 211198, Jiangsu, China. E-mail: ldong@cpu.edu.cn
bDepartment of Chemistry, University of Florida, Gainesville, Florida 32611-7011, USA
cThe Public Laboratory Platform, China Pharmaceutical University, Nanjing, 211198, China
First published on 15th November 2024
Abstract
Meroterpenoids are hybrid natural products that arise from the integration of terpenoid and non-terpenoid biosynthetic pathways. While the biosynthesis of fungal meroterpenoids typically follows a well-established sequence of prenylation, epoxidation, and cyclization, the pathways for bacterial perhydrophenanthrene meroterpenoids remain poorly understood. In this study, we report the construction of an engineered metabolic pathway in Streptomyces for the production of the bacterial meroterpenoid, atolypene A (1). Our research reveals a novel biosynthetic pathway wherein the structure of 1 is assembled through a distinct sequence of epoxidation, prenylation, and cyclization, divergent from its fungal counterparts. We demonstrate that the noncanonical class II meroterpenoid cyclase (MTC) AtoE initiates cyclization by protonating the epoxide via the E314 residue, which acts as a Brønsted acid within the characteristic xxxE314TAE motif. Additionally, bioinformatic analysis of biosynthetic gene clusters (BGCs) that contain AtoE-like MTCs supports that bacteria have the potential to produce a wide array of meroterpenoids.
Introduction
Meroterpenoids, a well-known class of hybrid natural products, are synthesized by merging terpenoid and non-terpenoid biosynthetic pathways.1–3 These compounds, derived from a broad spectrum of organisms including animals, plants, fungi, and bacteria, are characterized for their structural diversity and extensive biological functions.4–6 Fungi are the most abundant source of meroterpenoids, yielding promising pharmaceutical candidates like pyripyropene A,7 andrastin A,8 and tropolactone B.9 The pioneering elucidation of the biosynthetic pathway for fungal-derived pyripyropene A has unveiled a strategy prevalent in the construction of complex molecular architectures within this class of natural products.1,7 Typically, the biosynthetic pathway of fungal meroterpenoids commences with the formation of a non-terpenoid precursor, followed by prenylation, epoxidation, and a protonation-initiated cyclization cascade, ultimately forming the intricate meroterpenoid cores (Fig. S1†). This model, while instructive for fungal meroterpenoids, may not fully represent the biosynthesis in other organisms, posing an intriguing avenue for future research.While bacterial meroterpenoids are less common than their fungal equivalents, genomic analyses have revealed that bacteria, particularly actinomycetes, are a promising source of meroterpenoids.10,11 Meroterpenoids with perhydrophenanthrene skeletons represent a distinct subset within bacterial meroterpenoids and have attracted increasing attention owing to the discovery of several noteworthy examples (Fig. 1), including diterpenes brasilicardin A,12–15 phenalinolactone A,16–18 tiancilactone A,19 and a unique tetraterpene, longestin (KS-505a).20,21 The intriguing chemical structures and unique biological activities of these compounds indicate their promising potential in pharmaceutical development. However, the chemical synthesis of these bacterial perhydrophenanthrene meroterpenoids poses significant challenges due to the requisite precise control over the ring fusion.15,22,23 Although the biosynthetic gene clusters (BGCs) for these meroterpenoids have been identified and their biosynthetic processes partially studied in vivo (Fig. S2†),12,15–17,19–21 the specifics of their core skeletons formation and the underlying enzymatic mechanisms remains elusive.
 | ||
| Fig. 1 Structures of representative bacterial perhydrophenanthrene meroterpenoids. The perhydrophenanthrene skeleton part is shown in red. | ||
Terpene cyclization, a committed step in the formation of core skeletons in meroterpenoids and typically catalyzed by terpene cyclases (TCs), involves the cyclization of acyclic isoprenoid precursors into (poly)cyclic skeletons.24–26 TCs are traditionally categorized into two canonical classes depending on how the initial carbocation is generated, namely class I and class II TCs.19,25,27 Canonical class I TCs utilize two distinct Asp-rich motifs (DDxxD and NSE/DTE) to bind the trinuclear Mg2+ cluster, facilitating diphosphate abstraction. Canonical class II TCs rely on the central Asp in characteristic DxDD motif as a Brønsted acid to protonate an alkene or oxirane. It is noteworthy that the rapid development of genomics has led to the discovery of an increasing number of noncanonical MTCs, such as Bra4,12 PlaT2,16 TnlT2,19 and Lon15.20 These MTCs, characterized by a consensus atypical motif xxx(E/D)(T/S)xE, which deviates from the canonical DxDDTxx motif, are involved in the biosynthesis of brasilicardin A, phenalinolactone A, tiancilactone A, and longestin (KS-505a), respectively (Fig. S2–S4†).12,16,19,20 Interestingly, the distinct anti/syn/anti and anti/anti/syn core configurations observed in brasilicardin A and phenalinolactone A, respectively, imply that noncanonical MTCs contribute to a diverse range of stereoisomers, highlighting their enzymatic specificity.
Atolypene A (1), isolated by Brady and co-workers, is a rare sesterterpene–amino acid hybrid meroterpenoid, with a perhydrophenanthrene skeleton featuring a unique methyl substitution pattern on the rings and an extended tail (Fig. 1).28 It has also been reported to exhibit moderate cytotoxicity against multiple human cancer cell lines.28 In this work, we successfully reconstituted the biosynthetic pathway of 1 in model Streptomyces strains. By employing a heterologous production system, we systematically established a distinct meroterpenoid biosynthetic pathway in bacteria. Furthermore, through in vitro and targeted mutation experiments with the noncanonical class II MTC AtoE, we uncovered its catalytic function in the biosynthesis of 1. Significantly, our research offers a new insight into the previously enigmatic biosynthetic pathways of bacterial perhydrophenanthrene meroterpenoids.
Results and discussion
Employing regulatory elements for the reconstruction of ato BGC
The atolypene (ato) BGC from the soil actinomycete Amycolatopsis tolypomycina NRRL B-24205 consists of six genes (atoA–F, Fig. 2A, and Table S1†).28 The putative functions of encoded proteins are generally consistent with the structure of 1 and include a cytochrome P450 enzyme (P450, AtoA), an aminotransferase (AtoB), a geranylfarnesyl diphosphate synthase (GFPPS, AtoC), a UbiA family prenyltransferase (AtoD), a sesterterpene TC (AtoE), and an epoxidase (AtoF). AtoE contains a noncanonical class II MTC motif of xxxE314TAE suggesting that it likely protonates an epoxide;19 similar motifs are also seen in Bra4 and PlaT2 (Fig. S4†). In a previous study, Brady and co-workers achieved the production of 1 by heterologously expressing the ato BGC in S. albus J1074, utilizing a strategy of BGC disassembly and reassembly with inserted synthetic promoters.28 However, the relatively low yield of 1 (approximately 0.2 mg L−1) hindered a comprehensive understanding of its biosynthetic pathway. To effectively enhance the production of 1, we systematically assembled each gene with its designated promoter, RBS, and terminator, resulting in the formation of distinct expression units.29–31 Subsequently, using the atoC gene controlled by the promoter-RBS P3R5 as the foundational element, the other expression units were strategically integrated with it (Fig. S5†). These meticulously-designed combinations were then successfully cloned into the E. coli–Streptomyces integrating vector, pSET152, for heterologous expression (Tables S2–S4†).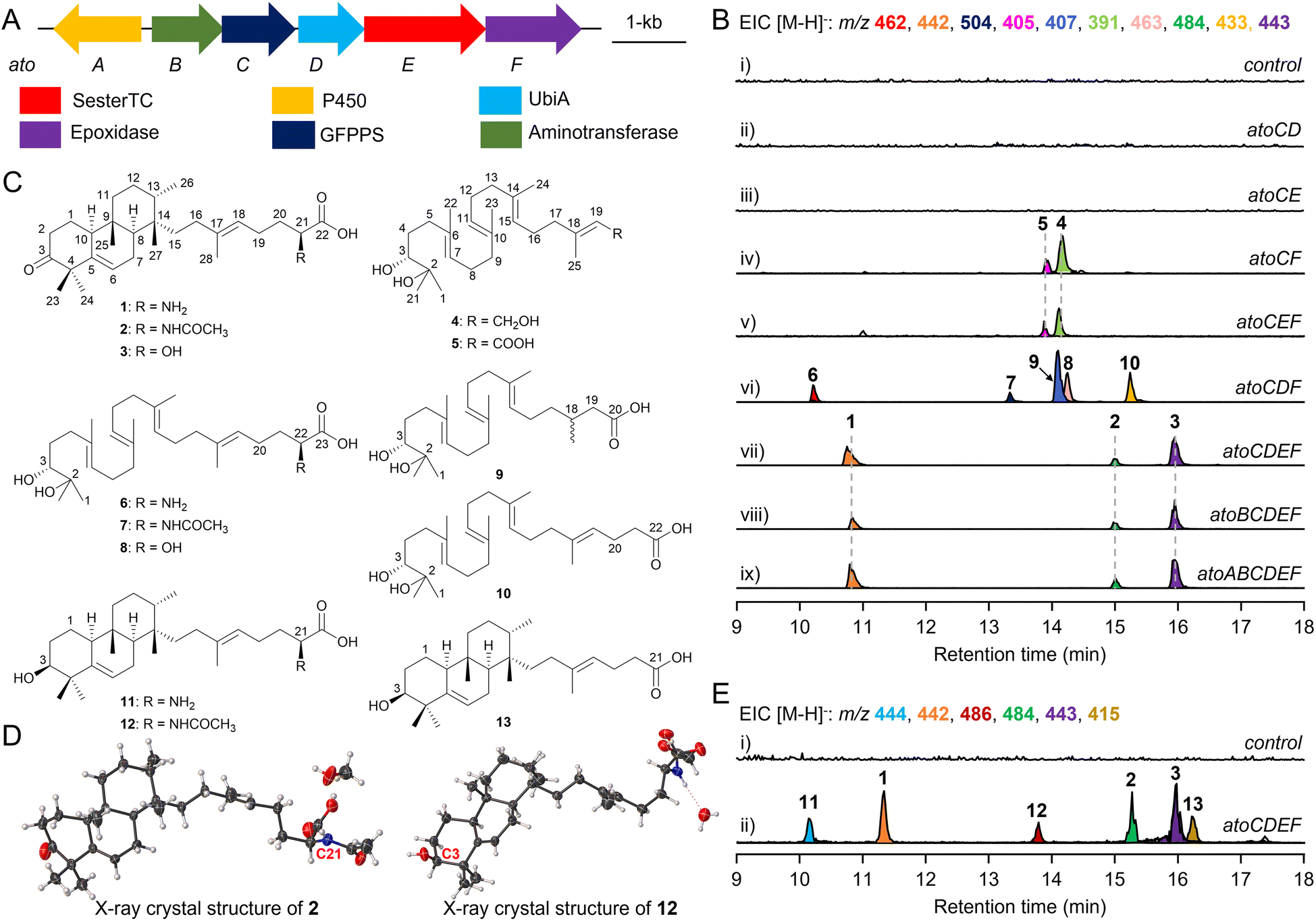 | ||
| Fig. 2 Analysis of atolypene A biosynthesis. (A) The ato BGC from A. tolypomycina NRRL B-24205. (B) EIC (extracted ion chromatogram) analysis of metabolites from engineered Streptomyces strains. S. albus J1074 with empty pSET152 was used as a control. (C) Structures of the metabolites isolated in this study (1–13). (D) X-ray crystal structures of 2 and 12. (E) EIC analysis of metabolites from S. lividans DLW1012 harboring atoCDEF. S. lividans SBT18 with empty pSET152 was used as a control. | ||
To verify the effect of this approach, we cloned and heterologously expressed the ato BGC with promoters, RBS, and terminator in S. albus J1074 to yield S. albus DLW1011, which was cultivated in PTMM, a terpenoid production medium.19,32 Analysis of DLW1011 metabolites using LC-MS identified three primary products, 1–3, with molecular weights (m/z [M − H]− at 442, 484, and 443) corresponding to anticipated cyclized products (Fig. 2B, panel ix, and S6†). Large-scale fermentation of DLW1011 (7.5 L) enabled isolation of these compounds for NMR analysis. The 1H and 13C NMR data for 1 matched previously reported data (Fig. S7 and S8†).28 Notably, optimizing gene expression regulation significantly improved the isolable yield of 1 (∼1.9 mg L−1), and additionally promoted the production of atolypene C (2, ∼1.2 mg L−1) and atolypene D (3, ∼2.1 mg L−1), resulting in a 15-fold increase in the total yield of atolypenes. Atolypene C (2) showed an additional acetyl amino group compared to 1 (Table S5, Fig. 2C and S9–S15†), and atolypene D (3) exhibited hydroxylation at C21 (Table S6, Fig. 2C and S16–S21†). Although the absolute configuration of 1 had been previously confirmed through key NOESY correlations and chemical derivatization methods,28 the crystal structure of 2 (CCDC 2309354† and Fig. 2D) conclusively established C21 as S-configured and supported the configurations of 1 and its analogues.
In vivo characterization of the biosynthetic pathway for atolypenes
The function of each gene in the biosynthesis of 1 was investigated through heterologously expressing them in S. albus J1074 using a pre-constructed expression plasmid. Mirroring fungal meroterpenoid biosynthesis, the initial biosynthetic step of 1 might also involve a prenylation that was proposed to be catalyzed by a UbiA-type prenyltransferase, AtoD.28 However, S. albus DLW1003, containing atoC (GFPPS) and atoD, exhibited no detectable product formation (Fig. 2B, panel ii, and S6†). This indicates a potential divergence in the initial biosynthetic steps of 1 from those known in fungal meroterpenoid pathways. Similarly, co-expression of atoC and atoE (MTC) did not result in product formation (Fig. 2B, panel iii). Thus, these findings have directed further research towards AtoF, a putative epoxidase hypothesized to catalyze epoxidation in the biosynthesis of 1, although no homologous enzymes have been reported. When co-expression of atoC and atoF in S. albus DLW1005, two novel hydrophilic compounds, 4 and 5, were produced as shown by LC-MS analysis (Fig. 2B, panel iv). A large-scale fermentation yielded sufficient amounts of 4 and 5 for structural analysis. Structurally, 4 possesses a structure similar to that of geranylfarnesol (GFOH) but with two additional hydroxyl groups (δC 73.8, C2; and 79.1, C3) at the other terminus of the geranylfarnesyl chain (Table S7, Fig. 2C and S22–S27†). It is known that epoxides produced under cellular expression conditions are prone to hydrolysis.7,33 Therefore, the diol moiety is presumed to result from the hydrolysis of an epoxide ring synthesized by AtoF, indicating that the epoxide and diphosphate version of 4 is a nascent intermediate in the biosynthesis of 1. Compound 5 has a carboxyl group at C20, instead of the hydroxyl group in 4 (Table S8, and Fig. 2C and S28–S33†). Additionally, the metabolites of S. albus DLW1007 (harboring genes atoC, atoE, and atoF) are consistent with those of S. albus DL1005, indicating that the cyclization does not occur subsequent to epoxidation (Fig. 2B, panel v, and S6†). Co-expression of genes atoC, atoD, and atoF in S. albus J1074 led to the production of five main metabolites, labeled 6–10 (Fig. 2B, panel vi, 2C, and S6†). Metabolite 6 (Table S9 and Fig. S34–S39†), identified with a molecular formula of C28H49NO4, differed from 5 by an additional 47 mass units. The epoxide form (6′) of 6 is considered a crucial intermediate in the biosynthesis of 1, yet to undergo cyclization (Fig. 3 and S40†). Compounds 7 and 8, in contrast, incorporate acetamide and hydroxyl groups at C22, respectively (Tables S10, S11, and Fig. S41–S52†). The formation of 6–8 is postulated to be driven by AtoD, which is hypothesized to catalyze a prenylation-like addition of three carbon atoms, potentially derived from phosphoenolpyruvate (Fig. 3 and S40†),16 to the C20 carbocation after diphosphate abstraction. Streptomyces endogenous enzymes, including aminotransferase, acetyltransferase, and dehydrogenase, are proposed to modify the proposed ketone at C22 after prenylation. In addition, 10 contains an acetic acid group at C20, whereas 9 shows a reduction of the double bond at C18–C19 in 5 (Tables S12, S13 and Fig. S53–S64†). The above findings suggest that in the biosynthesis of 1, the epoxidation step, catalyzed by AtoF, occurs before prenylation, which is carried out by AtoD. To determine the stereochemistry of the secondary alcohols at C3 in 4–10, the modified Mosher method was applied (Tables S14, S15, and Fig. S65–S79†).34,35 The stereochemistry of C3 was then determined to be R-configured based on the Δδ values (δ(S) − δ(R)) shown in Fig. S80.†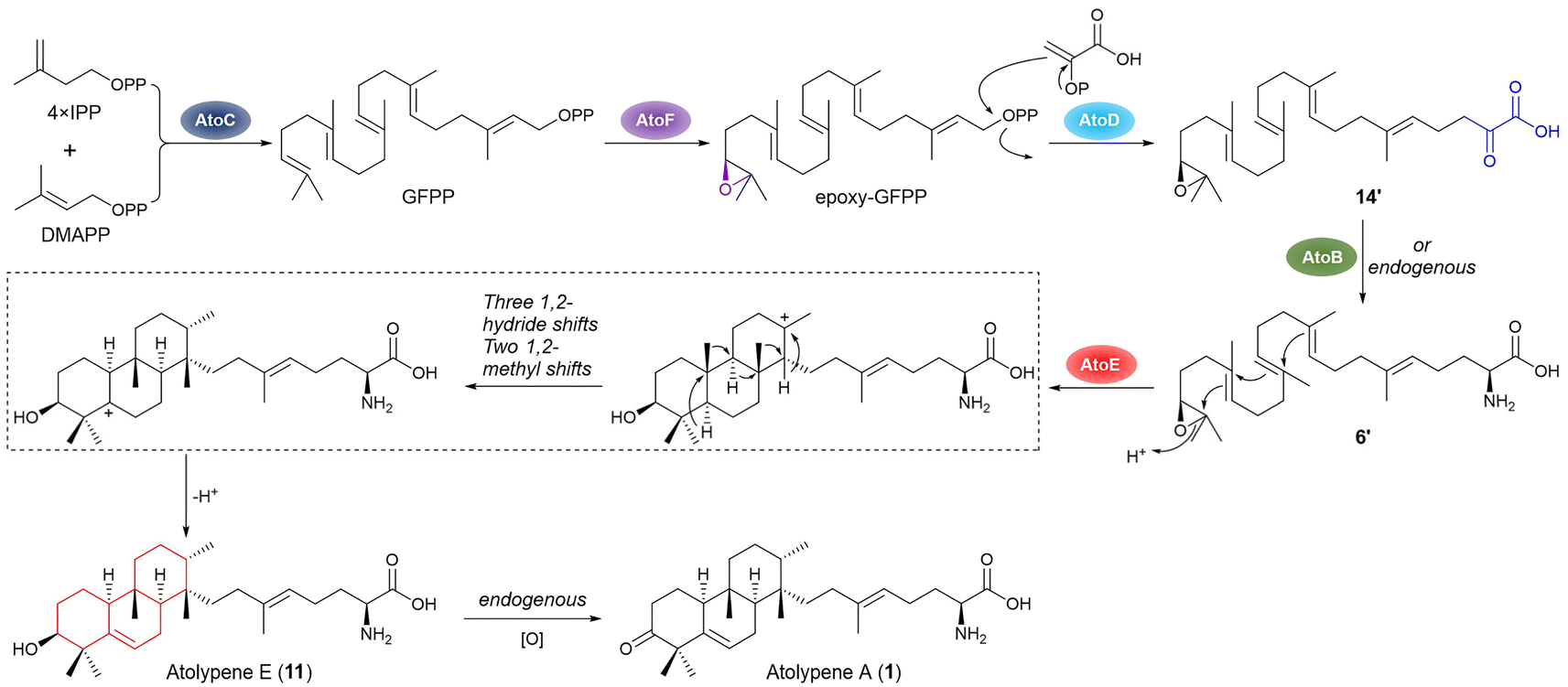 | ||
| Fig. 3 Proposed biosynthetic pathway of atolypene A. 6′ and 14′ are the epoxide forms of 6 and 14, respectively. | ||
Following the regulation and control of promoters and RBS, we cloned and co-expressed atoC–F and atoB–F in S. albus J1074, resulting in S. albus DLW1009 and S. albus DLW1010, respectively. Unexpectedly, these strains produced metabolites 1–3 in a pattern matching that of strains expressing the full atoA–F cluster (Fig. 2B, panels vii–ix). We subsequently cloned and co-expressed atoC–F in a different model strain of S. lividans SBT18 to create DLW1012. LC-MS analysis of the metabolites from DLW1012 showed the production of 1–3 as well as three new compounds 11–13 (atolypenes E–G, Fig. 2B, C, and S81†), each with distinct molecular weights (m/z [M – H]− at 444, 486, and 415, respectively). Atolypene E (11) and atolypene F (12) are the C3 reduced forms of 1 and 2, respectively (Tables S16, S17 and Fig. S82–S93†); atolypene G (13) is the cyclized form of 10 (Table S18 and Fig. S94–S99†). Key ROESY correlations for 11–13 indicate that their C3–OH are all β-oriented (Tables S16–S18†). Crystal structure analysis of 12 (CCDC 2314937† and Fig. 2D) indicated an S-configured stereogenic center at C3. These findings suggest that 11–13 are formed through the cyclization of (S)-epoxide catalyzed by MTC AtoE. The stereochemistry at C3 of 4–10 was R-configured, likely due to an endogenous epoxide hydrolase in Streptomyces,36,37 which hydrolyzes (S)-epoxide via an SN2 mechanism, yielding (R)-diol.
These in vivo results demonstrate that co-expression of four genes, atoC–F, is sufficient for producing 1 in Streptomyces (Fig. 3). Though P450 AtoA and aminotransferase AtoB were previously proposed to oxidize C3 and transfer an amino group at C21 in 1, respectively,28 our results from the atoC–D, atoB–F and atoA–F combinations did not support these roles (Fig. 2B, panels vii–ix). Specifically, feeding 11 and 12 to wild-type S. albus J1074 resulted in the production of 1 and 2, respectively (Fig. S100†). We produced AtoA in E. coli (Fig. S101†) and assayed it with substrates 11 or 12 in the presence of various redox partners, but no expected products were detected (Fig. S102†). Therefore, based on in vivo and in vitro results, we propose that the dehydrogenation of the hydroxyl group at C3 is likely catalyzed by endogenous enzymes in Streptomyces (Fig. 3). Furthermore, we heterologously produced AtoB in E. coli (Fig. S101†), and incubated it with 6, pyridoxal phosphate (PLP), and sodium pyruvate, resulting in the production of 14 (Fig. S103†). A ketone signal (δC 206.0, C22) in the 13C NMR spectrum of 14 supports the role of AtoB as an aminotransferase (Table S19 and Fig. S104–S109†). Additionally, AtoB exhibited reversibility by catalyzing the conversion of 14 into 6 in the presence of L-Ala (Fig. S103†). These findings suggest that, in the biosynthetic pathway of 1, transamination of the amino group at C21 can be catalyzed by AtoB, or by endogenous enzymes in Streptomyces (Fig. 3).
In vitro characterization of the AtoE-catalyzed cyclization function
To further confirm the function of AtoE, we attempted to verify its enzymatic activity through in vitro experiments. Since the expression of either wild-type or codon-optimized atoE in E. coli was unsuccessful, we produced AtoE in S. albus J1074 (Fig. S110†).27 As AtoE likely requires an epoxide group to initiate cyclization, and because all of the isolated compounds were diols, we turned to chemical synthesis to obtain a substrate for AtoE. Based on in vivo results, we hypothesized that AtoE may form 13 from the epoxidized analog (10a) of 10 (Fig. 4A). We therefore designed a synthesis of 10a using 10 as the starting material, according to previously reported methodology (Tables S20, S21, and Fig. S111–S118†).38,39 Diol 10 was readily converted to 10a through methylation of the C22 carboxylic acid, mesylation of the secondary alcohol at C3 followed by ring closure, and final deprotection of the C22 ester by hydrolysis with K2CO3 in methanol. Incubating 10a with AtoE in the Tris–HCl buffer (pH 8.0) resulted in its complete conversion to 13 (Fig. 4C and S119†); a boiled control reaction did not yield any new peaks. This indicated that AtoE, a class II-like MTC characterized by its noncanonical sequence motif of xxxE314TAE, cyclizes the merosesterterpenoid via protonation of an epoxide. This result also supports that the other AtoE-like MTCs in the related biosynthetic pathways act similarly.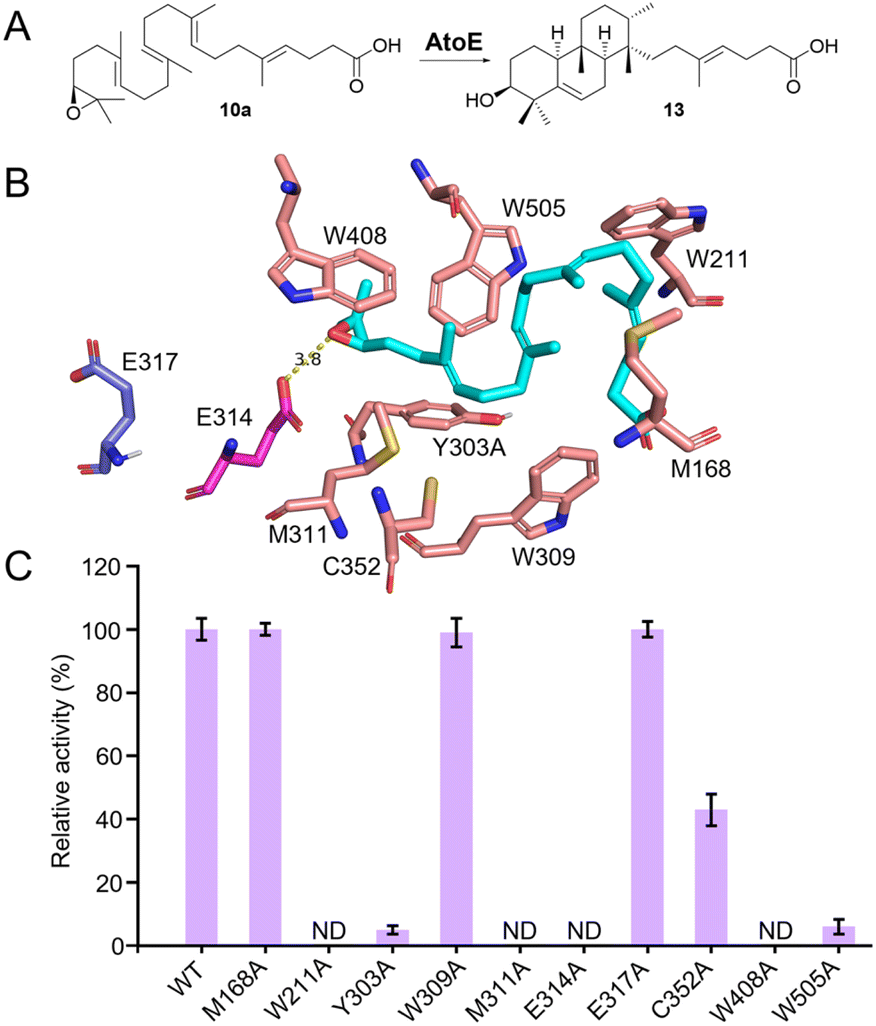 | ||
| Fig. 4 Functional characterization of AtoE. (A) AtoE catalyzes the cyclization of 10a to form 13. (B) Predicted substrate-binding sites in the AlphaFold2-generated model of AtoE. Substrate 10a (shown in cyan) is docked in AtoE. Residues within 4 Å of the substrate are highlighted in salmon; the E314 and E317 residues in the conserved xxxE314TAE317 motif are highlighted in magenta and slate, respectively; dashed lines indicate distances in Å. (C) Relative activities of AtoE variants. | ||
The complex process of terpene cyclization encompasses the dynamic generation and eventual quenching of carbocations.25 As AtoE is only the second MTC to be functionally characterized in vitro,40 we sought to investigate how it controls cyclization. As we were unable to produce crystals suitable for X-ray crystallography analysis, we used an AlphaFold2 model of AtoE to visualize its structure and putative active site (Fig. S120†).41 Although AtoE possesses the noncanonical catalytic motif xxxE314TAE, its model reveals an overall dumbbell-shaped structure, consisting of β and λ domains, resembling those of canonical class II TCs, such as squalene–hopene cyclase (SHC) or oxidosqualene cyclase (OSC).26,42,43 We docked 10a into the AtoE model and recognized that the proposed Brønsted acid E314, from the conserved xxxE314TAE motif, is positioned approximately 3.8 Å away from the epoxide of 10a (Fig. 4B and S120†). Other residues within 4 Å included three aliphatic residues (M168, M311, and C352) and five aromatic residues (W211, Y303, W309, W408, and W505) (Fig. 4B). To investigate the roles of these active site residues in AtoE, we performed site-directed mutagenesis and assessed the impact on the ability of AtoE to cyclize 10a into 13. Similar to the wild-type AtoE, all of these AtoE variants were insoluble when heterologously overproduced in E. coli; therefore S. albus J1074 was used to obtain small amounts of soluble proteins (Fig. S121†).
The E314A variant completely abolished activity (Fig. 4C, S122, and S123†), supporting its critical role as the general acid that initiates cyclization by protonating the epoxide ring of 10a (Fig. 4A). In contrast, the E317A variant maintained wild-type activity, consistent with its side chain being oriented away from the active site (Fig. 4B). Aromatic residues W211, Y303, W408, and W505 are predicted to form crucial interactions with the substrate, such as pi-stacking and hydrophobic contacts. Accordingly, the W211A and W408A variants abolished activity, while the Y303A and W505A variants exhibited only ∼5% and 6% of wild-type activity, respectively (Fig. 4C, S122 and S123†). These results support the essential role of aromatic residues in the AtoE active site for substrate binding and orientation.26 Among aliphatic residues, M311A yielded no cyclized product, whereas C352A retained 43% activity compared to wild-type AtoE (Fig. 4C, S122 and S123†), indicating their importance in catalysis. Notably, the M168A and W309A variants did not substantially impact activity, which could be attributed to the distance and orientation of the side chain of M168 and the aromatic ring of W309 relative to the substrate, suggesting they are less likely to participate in direct interactions (Fig. 4B). Interestingly, the C352A, W309A, M168A, W505, Y303A, and E317A mutants, along with the wild-type, produced new minor peaks (Fig. S123†). Their identical molecular weights (m/z [M − H]− at 415) with 13 suggested that these products may arise from the deprotonation of various carbocation intermediates during cyclization. However, due to the low yield of these peaks, their structures could not be identified. Collectively, these mutagenesis data corroborate our in silico structural model and provide evidence for the specific roles of individual amino acids in the AtoE active site.
BGCs containing AtoE-like MTCs are widespread in bacteria
With the in vivo and in vitro characterization of AtoE confirming that cyclization occurs after both epoxidation and prenylation, we sought to bioinformatically evaluate the entire family of noncanonical class II MTCs from bacteria. Given the structural differences between the atolypenes, brasilicardins, tiancilactones, phenalinolactones, and longestin (i.e., prenyl chain lengths, prenyl acceptors, and stereoconfigurations of the perhydrophenanthrene skeletons; Fig. 1), we suspected that phylogenetic analyses may separate these enzymes into distinct clades. Using AtoE to search for homologous MTCs in other bacterial genomes, we identified 103 non-redundant putative AtoE-like MTCs in the NCBI database with most members originating from Streptomyces, Saccharopolyspora, and Micromonospora (Table S22†). These putative MTCs had an average length of 566.5 amino acids, which is consistent with AtoE and its homologs from characterized BGCs (534–604 amino acids). An unrooted maximum-likelihood phylogenetic tree gave seven main clades, with four clades containing known meroterpenoids with putative MTCs (Fig. 5A). The known PlaT2 and TnlT2 grouped in clade VI, and the Bra4 was in clade IV. Including Bra4, all members of clade IV were from the genus Nocardia and shared between 45.3 and 97.7% identity with Bra4, suggesting a subfamily of MTCs involved in homologous brasilicardin pathways. The AtoE and the Lon15 grouped in clades III and VII, respectively. Clades I, II, and V had no characterized representatives, indicating potentially novel subgroups of MTCs forming chemically distinct skeletons (Fig. 5A). | ||
| Fig. 5 Phylogenetic analysis of AtoE-like MTCs. (A) Maximum-likelihood tree of AtoE homologues in bacteria. MTCs from BGCs with structurally characterized natural products are labeled at the end of their respective branch. (B) Sequence analysis by clade shows variance from the highly conserved DxDDTxx motif present in most canonical class II TCs. Enzymes in clades I–V predominantly contain an xxxExxE motif whereas clades VI and VII contain xxxD/Exxx and xxxD/ExD/ED, respectively. (C) EIC analysis of the in vitro reaction of SerTC with 10a. | ||
To assess the conservation of the acidic motif required for initiating cyclization via epoxide ring opening, we performed a motif analysis for each clade (Fig. 5B). A single conserved acidic residue (E/D) in the first position of the catalytic motif was conserved in every clade. Clades I–V showed nearly absolute conservation of xxxExxE; sequence variance was much greater in clades VI and VII. Clade VI, which contains PlaT2 and TnlT2, had a xxxD/Exxx motif, whereas the Lon15 clade VII had a xxxE/DxD/ED motif. Like the catalytic motif of OSC (VxDC), which protonates an epoxide to spur cyclization,43 only a single conserved acidic residue facilitates cyclization in clades I–VII, supporting that all of these enzymes utilize epoxides for cyclization.
To experimentally validate our bioinformatics analysis, we selected seven bacterial class II MTCs of SerTC, SmaTC, UtaTC, CcrTC, SsyTC, AxyTC, and PshTC (Fig. 5A and Table S22†). Overproduction of these MTCs in E. coli yielded five soluble proteins: SerTC, SmaTC, UtaTC, CcrTC, and SsyTC (Fig. S124†). In vitro reactions with substrate 10a showed that all enzymes formed new products (Fig. S125 and S126†), indicating that these MTCs initiate cyclization through epoxide protonation. In particular, SerTC exhibited the highest activity, fully converting 10a to atolypene H (15) and another unidentified product (Fig. 5C and S125–127†). Structural elucidation revealed 15 differs from 13 in the position of the double bond formed during the final deprotonation step (Table S23 and Fig. S128–133†). These results support the generalization of our bioinformatics analysis and underscore its potential for discovering novel terpene natural products that derived from noncanonical class II MTCs.
To further explore the meroterpenoid biosynthetic diversity of bacteria, we analyzed BGCs containing identified AtoE-like MTCs (Fig. S134†). BGCs harboring putative AtoE homologs showed high conservation of the four core genes required for atolypene biosynthesis: MTC, polyprenyl synthetase, flavin-dependent monooxygenase (i.e., epoxidase), and UbiA prenyltransferase. The conserved terpene core biosynthetic genes appear to be maintained in a single operon in most clusters, with varying organization in some clusters such as in Umezawaea tangerina. Unlike the ato BGC, and more similar to the bra (brasilicardin) and pla (phenalinolactone) BGCs,12,16 other clusters were found to encode additional tailoring enzymes including methyltransferases, sulfotransferases, acyltransferases, and α-ketoglutarate dependent dioxygenases, indicating enormous structural and biochemical diversities in this family of natural products. Interestingly, non-ribosomal peptide synthetases were found in close genomic proximity to the terpene core genes in several clusters, raising the question of whether even more structurally complex meroterpenoid natural products are made in these strains by the joining of non-ribosomal peptide and terpene moieties.
Conclusions
Bacterial meroterpenoids, constituting a small proportion of natural products, have garnered extensive research interest owing to their intricate structures and diverse bioactivities. Particularly, meroterpenoids featuring a perhydrophenanthrene skeleton, exemplified by compounds such as brasilicardin A, phenalinolactone A, tiancilactone A, and longestin (KS-505a), represent a unique subclass in bacteria meroterpenoids. However, the biosynthetic pathways of these bacterial meroterpenoids have been enigmatic. In this study, we systematically reconstructed the biosynthetic pathway of the bacterial meroterpenoid atolypene A (1) in heterologous hosts through gene regulation, heterologous expression, and in vitro experiments. Meanwhile, we have demonstrated that the perhydrophenanthrene core skeleton of 1 is accomplished via sequential epoxidation, prenylation, and cyclization. While the biosynthetic pathway of the fungal meroterpenoid pyripyropene A provides a paradigm for meroterpenoid biosynthesis,7 our research presents a distinct biosynthetic pathway of meroterpenoids in bacteria. In parallel with this study, similar investigations into the early-stage biosynthesis of phenalinolactone diterpenoids were also conducted in vivo, suggesting a hypothesis consistent with the biosynthetic pathway of fungal meroterpenoids, wherein prenylation occurs prior to epoxidation and cyclization.44 Additionally, our site-directed mutagenesis experiments revealed that the conserved residue E314 is a crucial acid required for driving the cyclization reaction of the noncanonical class II MTC AtoE. Being the first enzyme of its kind in bacterial perhydrophenanthrene meroterpenoid biosynthesis to undergo in vitro studies, it opens the door for detailed investigations into its catalytic mechanism. Phylogenetic analysis of AtoE-like MTCs and subsequent BGC analysis supports that bacteria possess the biosynthetic potential for an even broader scope of meroterpenoid natural products than is currently known. Overall, these insights not only augment our understanding of bacterial meroterpenoid biosynthesis but also illuminate the catalytic functions of noncanonical class II MTCs, thereby laying a foundational groundwork for the exploration and potential pharmaceutical application of these bioactive molecules.Data availability
Crystallographic data for compound 2 and 12 has been deposited at the CCDC under 2309354 and 2314937, respectively. The other datasets supporting this article have been uploaded as part of the ESI.†Author contributions
L.-B. D. conceived the project; Z. W. and L.-B. D. designed the experiments; Z. W., T. A. A., X. P., L.-L. L., J. T., Z. Y., X. L. and H.-M. X. performed the experiments; Z. W., J. D. R. and L.-B. D. analyzed the results; Z. W., J. D. R. and L.-B. D. wrote the paper with inputs from all co-authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We wish to thank Prof. Hui Ming Ge from Nanjing University for generous access to the HRESIMS equipment. This work is supported in part by the National Natural Science Foundation of China Grant 82073746 and 82473823 (L.-B. D.), the National Institutes of Health Grants R00 GM124461 (J. D. R.) and R35 GM142574 (J. D. R.), the Thousand Youth Talents Program of China (L.-B. D.), the Jiangsu Specially Appointed Professor Program (L.-B. D.), the Natural Science Foundation of Jiangsu Province for Distinguished Young Scholars (BK20240097, L.-B. D.), the Project Program of State Key Laboratory of Natural Medicines at China Pharmaceutical University (SKLNMZZ2024JS44, L.-B. D.), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (Z. W.). T. A. A. was supported in part by an NIH Chemistry–Biology Interface Research Training Program Grant T32 GM136583.Notes and references
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- M. Nazir, M. Saleem, M. I. Tousif, M. A. Anwar, F. Surup, I. Ali, D. Wang, N. Z. Mamadalieva, E. Alshammari, M. L. Ashour, A. M. Ashour, I. Ahmed, Elizbit, I. R. Green and H. Hussain, Biomolecules, 2021, 11, 957 CrossRef CAS.
- L. A. M. Murray, S. M. K. McKinnie, B. S. Moore and J. H. George, Nat. Prod. Rep., 2020, 37, 1334–1366 RSC.
- N. K. Fuloria, R. K. Raheja, K. H. Shah, M. J. Oza, Y. A. Kulkarni, V. Subramaniyan, M. Sekar and S. Fuloria, Front. Pharmacol., 2022, 13, 830103 CrossRef CAS PubMed.
- Y.-M. Yan, H.-X. Zhang, H. Liu, Y. Wang, J.-B. Wu, Y.-P. Li and Y.-X. Cheng, Org. Let., 2019, 21, 8523–8527 CrossRef CAS.
- Z. Luo, F. Yin, X. Wang and L. Kong, Chin. J. Nat. Med., 2024, 22, 195–211 CAS.
- T. Itoh, K. Tokunaga, Y. Matsuda, I. Fujii, I. Abe, Y. Ebizuka and T. Kushiro, Nat. Chem., 2010, 2, 858–864 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa and I. Abe, Tetrahedron, 2013, 69, 8199–8204 CrossRef CAS.
- M. Cueto, J. B. MacMillan, P. R. Jensen and W. Fenical, Phytochemistry, 2006, 67, 1826–1831 CrossRef CAS PubMed.
- M. Gozari, M. Alborz, H. R. El-Seedi and A. R. Jassbi, Eur. J. Med. Chem., 2021, 210, 112957 CrossRef CAS PubMed.
- E. V. Tarasova, N. A. Luchnikova, V. V. Grishko and I. B. Ivshina, Pharmaceuticals, 2023, 16, 872 CrossRef CAS PubMed.
- Y. Hayashi, N. Matsuura, H. Toshima, N. Itoh, J. Ishikawa, Y. Mikami and T. Dairi, J. Antibiot., 2008, 61, 164–174 CrossRef CAS PubMed.
- H. Komaki, A. Nemoto, Y. Tanaka, H. Takagi, K. Yazawa, Y. Mikami, H. Shigemori, J. Kobayashi, A. Ando and Y. Nagata, J. Antibiot., 1999, 52, 13–19 CrossRef CAS PubMed.
- H. Shigemori, H. Komaki, K. Yazawa, Y. Mikami, A. Nemoto, Y. Tanaka, T. Sasaki, Y. In, T. Ishida and J. i. Kobayashi, J. Org. Chem., 1998, 63, 6900–6904 CrossRef CAS.
- A. Botas, M. Eitel, P. N. Schwarz, A. Buchmann, P. Costales, L. E. Núñez, J. Cortés, F. Morís, M. Krawiec, M. Wolański, B. Gust, M. Rodriguez, W.-N. Fischer, B. Jandeleit, J. Zakrzewska-Czerwińska, W. Wohlleben, E. Stegmann, P. Koch, C. Méndez and H. Gross, Angew. Chem., Int. Ed., 2021, 60, 13536–13541 CrossRef CAS PubMed.
- C. Dürr, H. J. Schnell, A. Luzhetskyy, R. Murillo, M. Weber, K. Welzel, A. Vente and A. Bechthold, Chem. Biol., 2006, 13, 365–377 CrossRef.
- T. M. Binz, S. C. Wenzel, H.-J. Schnell, A. Bechthold and R. Müller, Chembiochem, 2008, 9, 447–454 CrossRef CAS.
- K. Gebhardt, S. W. Meyer, J. Schinko, G. Bringmann, A. Zeeck and H.-P. Fiedler, J. Antibiot., 2011, 64, 229–232 CrossRef CAS.
- L.-B. Dong, J. D. Rudolf, M.-R. Deng, X. Yan and B. Shen, ChemBioChem, 2018, 19, 1727–1733 CrossRef CAS.
- Y. Hayashi, H. Onaka, N. Itoh, H. Seto and T. Dairi, Biosci. Biotech. Bioch., 2007, 71, 3072–3081 CrossRef CAS.
- T. Ozaki, S. S. Shinde, L. Gao, R. Okuizumi, C. Liu, Y. Ogasawara, X. Lei, T. Dairi, A. Minami and H. Oikawa, Angew. Chem., Int. Ed., 2018, 57, 6629–6632 CrossRef CAS PubMed.
- F. Yoshimura, R. Itoh, M. Torizuka, G. Mori and K. Tanino, Angew. Chem., Int. Ed., 2018, 57, 17161–17167 CrossRef CAS PubMed.
- M. Anada, T. Hanari, K. Kakita, Y. Kurosaki, K. Katsuse, Y. Sunadoi, Y. Jinushi, K. Takeda, S. Matsunaga and S. Hashimoto, Org. Lett., 2017, 19, 5581–5584 CrossRef CAS PubMed.
- Y. Matsuda, T. Awakawa, T. Mori and I. Abe, Curr. Opin. Chem. Biol., 2016, 31, 1–7 CrossRef CAS.
- J. D. Rudolf and C. Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- X. Pan, J. D. Rudolf and L.-B. Dong, Nat. Prod. Rep., 2024, 41, 402–433 RSC.
- Z. Wang, Q. Yang, J. He, H. Li, X. Pan, Z. Li, H.-M. Xu, J. D. Rudolf, D. J. Tantillo and L.-B. Dong, Angew. Chem., Int. Ed., 2023, 62, e202312490 CrossRef CAS PubMed.
- S.-H. Kim, W. Lu, M. K. Ahmadi, D. Montiel, M. A. Ternei and S. F. Brady, ACS Synth. Biol., 2019, 8, 109–118 CrossRef CAS.
- S.-Y. Hsu, J. Lee, A. Sychla and M. J. Smanski, Metab. Eng., 2023, 77, 1–11 CrossRef CAS PubMed.
- S.-Y. Hsu, D. Perusse, T. Hougard and M. J. Smanski, ACS Synth. Biol., 2019, 8, 2397–2403 CrossRef CAS.
- Q. Xu, H. Zou, C. Pan, H. Wang, Y. Shen and Y. Li, Chin. J. Nat. Med., 2023, 21, 454–458 CAS.
- L.-B. Dong, X. Zhang, J. D. Rudolf, M.-R. Deng, E. Kalkreuter, A. J. Cepeda, H. Renata and B. Shen, J. Am. Chem. Soc., 2019, 141, 4043–4050 CrossRef CAS PubMed.
- W.-G. Wang, L.-Q. Du, S.-L. Sheng, A. Li, Y.-P. Li, G.-G. Cheng, G.-P. Li, G. Sun, Q.-F. Hu and Y. Matsuda, Org. Chem. Front., 2019, 6, 571–578 RSC.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092–4096 CrossRef CAS.
- L.-B. Dong, J. He, Y.-Y. Wang, X.-D. Wu, X. Deng, Z.-H. Pan, G. Xu, L.-Y. Peng, Y. Zhao, Y. Li, X. Gong and Q.-S. Zhao, J. Nat. Prod., 2011, 74, 234–239 CrossRef CAS.
- G. P. Horsman, A. Lechner, Y. Ohnishi, B. S. Moore and B. Shen, Biochemistry, 2013, 52, 5217–5224 CrossRef CAS.
- B. Wang, F. Guo, J. Ren, G. Ai, B. Aigle, K. Fan and K. Yang, Nat. Commun., 2015, 6, 7674 CrossRef PubMed.
- X.-T. Liang, J.-H. Chen and Z. Yang, J. Am. Chem. Soc., 2020, 142, 8116–8121 CrossRef CAS.
- E. J. Corey, M. C. Noe and W.-C. Shieh, Tetrahedron Lett., 1993, 34, 5995–5998 CrossRef CAS.
- P. Moosmann, R. Ueoka, L. Grauso, A. Mangoni, B. I. Morinaka, M. Gugger and J. Piel, Angew. Chem., Int. Ed., 2017, 56, 4987–4990 CrossRef CAS.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- K. U. Wendt, A. Lenhart and G. E. Schulz, J. Mol. Biol., 1999, 286, 175–187 CrossRef CAS PubMed.
- R. Thoma, T. Schulz-Gasch, B. D'Arcy, J. Benz, J. Aebi, H. Dehmlow, M. Hennig, M. Stihle and A. Ruf, Nature, 2004, 432, 118–122 CrossRef CAS.
- T. A. Alsup, Z. Li, C. A. McCadden, A. Jagels, D. Łomowska-Keehner, E. Marshall, L.-B. Dong, S. Loesgen and J. D. Rudolf, RSC Chem. Biol., 2024, 5, 1010–1016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2309354 and 2314937. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc06010e |
| This journal is © The Royal Society of Chemistry 2025 |