
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design of organic diradicals with robust high-spin ground state based on antiaromatic linkers†
Raul
Santiago
 *a,
M. Àngels
Carvajal
a,
Jordi
Poater
bc,
Ibério de P. R.
Moreira
a,
Stefan T.
Bromley
ac,
Mercè
Deumal
a and
Jordi
Ribas-Ariño
*a
*a,
M. Àngels
Carvajal
a,
Jordi
Poater
bc,
Ibério de P. R.
Moreira
a,
Stefan T.
Bromley
ac,
Mercè
Deumal
a and
Jordi
Ribas-Ariño
*a
aDepartament de Ciència de Materials i Química Física & Institut de Química Teòrica i Computacional (IQTC), Universitat de Barcelona, c/ Martí i Franquès 1-11, 08028 Barcelona, Spain. E-mail: raul.sant.1972@gmail.com; j.ribas@ub.edu
bDepartament de Química Inorgànica i Orgànica & Institut de Química Teòrica i Computacional (IQTC), Universitat de Barcelona, c/ Martí i Franquès 1-11, 08028 Barcelona, Spain
cInstitució Catalana de Recerca i Estudis Avançats (ICREA), Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 21st November 2024
Abstract
Fully-organic molecules with high-spin ground states are promising building blocks for new lightweight flexible magnetic materials for emerging technological applications (e.g. spintronics). In this study, we explore the potential of diradicals made of two diphenylmethyl-based open-shell cores covalently linked via different types of pentalene and diazapentalene-based antiaromatic couplers (including dibenzopentalenes and acene-inserted derivatives). Accurate electronic structure calculations have been employed to target non-bonding and non-disjoint frontier molecular orbitals that favor high-spin configurations, leading to the identification of diradicals displaying robust triplet ground states. These candidates exhibit singlet-triplet energy gaps that are up to ten times the thermal energy at room temperature. These substantial gaps emerge from strong interactions between the π-systems of the open-shell centers and the antiaromatic coupler. These interactions not only result in high spin states but are also found to lead to an enhanced stability of the diradicals by drastically dampening their inherent antiaromatic character as compared to the bare couplers, and promoting a high degree of spin density delocalization. These findings highlight the potential of pentalene-based diradicals as building blocks for developing new advanced fully organic magnetic materials.
Introduction
Antiaromatic molecules are attracting increasing interest for applications in chemistry and materials science.1–7 This trend is primarily driven by both advances in synthetic techniques8–11 and innovative designs devoted to enhancing the stability of these molecules. Increased stability of antiaromatic systems can be achieved through the attenuation and dilution of their antiaromatic character (e.g., by dibenzoannulation12–16 or by insertion of nitrogen atoms17–19). Likewise, the inclusion of bulky substituents has also been proposed as an alternative strategy to increase the time and thermal resilience of the unstable precursors.17,20,21 Once stability is not a critical issue, the unique electronic properties of antiaromatic molecules, specifically a low HOMO–LUMO gap, make them promising candidates for organic electronics22–31 and optoelectronic applications.22,32–35 The confluence of strategic designs, accessible synthesis and the inherent electronic attributes of antiaromatic compounds have demonstrated their viability and broadened their practical use. Such advances have led to the successful synthesis of a new class of nanohoops15,19,36–39 and covalent organic frameworks,35 demonstrating their potential for real-world applications.Recent work by Winter and co-workers40 has shown that the strategy of conjugating an antiaromatic moiety to an open-shell unit results in a synergistic stabilization of the whole system. This is because the partial intramolecular charge transfer between the antiaromatic and open-shell moieties dampens the overall antiaromatic character of the combined systems and favors the delocalization of the unpaired electron; a common strategy to stabilize organic radicals. This conceptual approach prompted us to explore whether similar strategies could be exploited to stabilize diradicals and strengthen the magnetic interactions between their unpaired electrons. In particular, we focus on open-shell organic molecules possessing a robust high-spin ground state.
The design and synthesis of organic diradicals displaying a high-spin ground state is a long-standing research goal with recognized potential for spintronics,41,42 memory devices,43 spin filters,44–46 and organic magnets.47,48 The practical application of thermally stable high-spin organic diradicals requires large energy gaps, ΔEST, between the singlet (S) excited state and the triplet (T) ground state.49 Many organic compounds, including recently reported antiaromatic molecules,13,50–56 possess an open-shell singlet ground state with an antiferromagnetic (AFM) interaction between spins. Examples of fully-organic diradicals displaying large ferromagnetic (FM) interactions between spins are still uncommon compared to those exhibiting AFM interactions, with much fewer studies reporting either experimentally characterized compounds57–83 or computational predictions that meet such conditions.84–89
One of the simplest, yet illustrative, examples of the potential of antiaromatic molecules to bolster FM couplings in π-conjugated organic systems is 2,5-dimethylenylpentalene,90My2Pl, which can be considered as a functionalized pentalene coupler with two added terminal methylenyl open-shell cores (R = My in Fig. 1a) that serve as radical centers. My2Pl is a non-alternant and non-Kekulé π-conjugated hydrocarbon that possesses a doubly-degenerate set of non-bonding molecular orbitals (NBMOs), as presented in Fig. 1b. In this case, Pauli repulsion between nearby unpaired electrons promotes their parallel alignment, thus favoring a triplet ground state to a first approximation.91 In addition, the specific carbon scaffold of My2Pl results in a non-disjoint set of Singly-Occupied Molecular Orbitals90 (SOMOs), i.e., the SOMOs cannot be localized on distinct groups of non-overlapping atoms by any linear combination between them.92 Overall, the molecular orbital (MO) topology typified by My2Pl significantly affects the energy stability associated with antiparallel spin alignment (favoring, e.g., bond formation).91 Consequently, the antiparallel spin alignment primarily results in unfavorable electron–electron Coulomb repulsion, favoring a triplet ground state and making My2Pl a notable example of a topological ferromagnetic molecule to a first approximation. As it may be seen in Fig. 1c, the valence-bond structures delocalise each unpaired electron and preserve an open-shell character through two separated conjugation paths that prevent any neutral closed-shell form. Accordingly, a valence-bond approach provides a complementary explanation for the stabilization of the triplet state.
 | ||
| Fig. 1 (a) Schematic overview of the modular diradical design featuring an antiaromatic pentalene coupler (Pl, highlighted in orange) and two open-shell cores R = My, DPM, PDM (highlighted in gray). Some hydrogen atoms in DPM are explicitly shown to highlight the differences with PDM. (b) SOMOs of My2Pl shown at a 0.04 e Å−3 isosurface with blue and red regions indicating positive and negative signs of the orbitals, respectively. (c) Representative Lewis structures of My2Pl. | ||
Herein, we focus on the design of high-spin antiaromatic diradicals featuring non-bonding and non-disjoint SOMOs. Our approach is modular, involving the assembly of diradicals from separate building blocks, i.e., two open-shell cores and an antiaromatic coupler (see Fig. 1a). Concerning the antiaromatic couplers, we will rely on three different strategies proven to enhance the stability of the raw antiaromatic precursors (i.e., before forming the diradical): (i) phenyl ring insertion,93–96 (ii) dibenzannulation16,97 and (iii) substitution of CH by nitrogen atoms.17–19 These strategies give rise to dicyclopenta-[n]acenes (DPA[n]), dibenzopentalenes (DBP), and their respective diaza-dicyclopenta-[n]acenes (DADPA[n]) or diaza-dibenzopentalenes (DADBP). Regarding the open-shell units attached to the antiaromatic coupler, methylenyl (R = My in Fig. 1a) provides the simplest example. However, its anticipated inherent instability (upon favored disproportionation or recombination processes) makes it more suitable as a theoretical proof of concept rather than a realizable choice. Hence, for real-world applications, our selection is inspired by persistent open-shell units that are stable at ambient conditions. In this regard, diphenylmethyl (R = DPM in Fig. 1a) cores offer steric protection to the spin-bearing center.98 We also consider polychloro-diphenylmethylenyl (R = PDM in Fig. 1a) cores, specifically di-(2,6-dichloro)-diphenylmethylenyl, because the chlorine atoms in the ortho positions of PDM further protect the open-shell center.99
Preliminary insights into the molecular orbital topology of these planar π-conjugated hydrocarbon diradicals can be gained from methods like spectral graph theory100 (SGT, further elaborated in ESI Section 1†). Here, we show how these methods can be used in a computationally efficient approach to design new high-spin antiaromatic diradicals with a specific MO topology akin to My2Pl. Extended-Hückel (EH) and simple SGT models lack the capability to fully capture higher-order effects like electron correlation of non-bonding SOMOs with the rest of the π-MOs.101 Therefore, we have employed Density Functional Theory (DFT) and Correlated Wave-Function (CWF) ab initio methods for a rigorous evaluation of ΔEST gaps and magnetic properties.
In our strategic diradical design, we have also considered the chemical stability of the compounds compared to their bare antiaromatic precursors. However, chemical stability is a multifaceted concept that lacks a universally accepted definition or measurable unit. When dealing with fully-conjugated polycyclic hydrocarbons, the (anti)aromatic character can provide a comparative indicator of stability across different chemical systems. Accordingly, we have opted for Bond Length Alternation (BLA), Nuclear Independent Chemical Shifts (NICS),102 Anisotropy of the Induced Current Density (ACID),103,104 magnetically induced current density maps,105–108 and bond current strengths105 as indirect measures of aromaticity. Additionally, we have also employed an analysis based on Radical Stabilization Energy (RSE),109,110 further discussed in the Results and discussion section. These molecular descriptors have provided a robust framework to gauge the stability of the studied pentalene-based diradicals and couplers from their (anti)aromatic character.
Pentalene is a relatively unstable molecule that has been traditionally studied mostly as a formal chemical example of antiaromaticity. The potential for practical applications of pentalene derivatives has been somewhat overlooked until recently.5,6 We underscore that the instability of pentalene, and some of its derivatives, is markedly mitigated when their diradical derivatives are considered. This insight provides the basis for a new class of promising pentalene-based diradicals with robust triplet ground states and large ΔEST gaps, together with a few examples exhibiting singlet-triplet gap inversion. Our results could breathe new life into the practical potential of pentalene-based systems for emerging applications.
Results and discussion
This section is organized in five parts. In the first part, we focus on the evaluation of ΔEST gaps for diradicals built from pentalene and diaza-pentalene derivatives. We have also assessed the effect on ΔEST of the insertion of poly-acenes within the pentalene core (i.e., dicyclopenta-[n]acenes or DPA[n]) and the replacement of the methylenyl radical units by other protected radical centers. As mentioned in the introduction, we employ My as our theoretical proof of principle, and DPM or PDM as potentially realizable units. In the second part, we gauge the relative stability of DPA[n]-based diradicals in comparison to their bare antiaromatic precursors by means of BLA, aromaticity descriptors (such as NICS, ACID, magnetically induced current density maps, and bond current strengths), and an analysis based on the RSE. In the third part, we explore the impact of inserting standard functional groups to DPA[n]-based diradicals in both the magnetic properties and structural parameters related to the electron correlation. In the fourth part, we carry out similar ΔEST, BLA and NICS analyses for diradicals based on dibenzopentalene couplers (i.e., DBP). To conclude, in the fifth part, we report the ΔEST gaps and discuss the viability of all newly suggested diradicals, leading to the identification of the most promising candidates over the studied set of compounds.ΔEST gap in core-expanded and substituted pentalene diradicals
The set of antiaromatic diradicals explored in this section consists of two open-shell R cores (R = My, DPM, PDM) linked by either DPA[n] or DADPA[n] (X = CH and X = N in Fig. 2a, respectively). Among the six possible constitutional isomers of My2-DPA[n], we have exclusively considered the 2,5-connectivity because the other isomers display non-degenerate SOMOs, thus leading to AFM interactions (see ESI Section S2.1†). The evaluation of ΔEST gap for My2-DPA[n] diradicals at DFT level (PBE0/6-311G*, see Computational details) reveals a triplet ground state with a large gap of ca. 5.6 kcal mol−1 for the first compound of the series (n = 0, My2-DPA[0] colored in blue in Fig. 2b). Now, considering that thermal energy at room temperature (RT) is roughly 0.6 kcal mol−1, the ΔEST gap effectively results in a 100% population of the triplet state at room temperature.111 The nitrogen-doped counterpart, My2-DADPA[0], exhibits an even larger ΔEST gap of ca. 8.5 kcal mol−1, as depicted in Fig. 2c (colored in blue). It should be mentioned that DPA[0] has also been predicted to mediate strong magnetic couplings between metal centers in 2D Cr-based metal organic frameworks.112 Notably, the ΔEST gaps of DPM2-DPA[0] and PDM2-DPA[0] analogues also show comparable values, suggesting that the incorporation of DPM and PDM cores does not significantly alter the magnetic interactions between the unpaired electrons of these diradicals. | ||
| Fig. 2 (a) Chemical structures of diradicals based on dicyclopenta-[n]acenes (R2-DPA[n], X = CH) and diaza-dicyclopenta-[n]acenes (R2-DADPA[n], X = N). Plots (b) and (c) show the singlet-triplet gap (ΔEST in kcal mol−1) as a function of the increasing number of fused 6-membered rings (n from 0 to 3) for R2-DPA[n] and R2-DADPA[n], respectively. Colored data points correspond to different cores: R = My (blue), R = DPM (red), and R = PDM (yellow). Dashed lines are included as guides to the eye. | ||
Interestingly, our DFT results show a reduction of the ΔEST gap upon increasing the number of inserted 6-membered rings (see Fig. 2b and c). Remarkably, a crossover is observed in both series for n > 2, reversing the energetic stability of the singlet and the triplet states. Such a crossover is at odds with the predictions of models based solely on the topology of the frontier orbitals. In all cases the SOMOs of all diradicals of My2-DPA[n] and My2-DADPA[n] series are predicted to be non-disjoint NBMOs, thus, anticipating a triplet ground state for all of them to a first approximation (see ESI Section S2.2† for a visual representation of the SOMOs derived from SGT). Accordingly, our results highlight the significant role of electron correlation in expanded (diaza-)pentalene series and the limitations of rational design based solely on the topology of the SOMOs.
The observed trends can be rationalized in two different ways. First, a close examination of the spin distribution of R2-DPA[0] and R2-DADPA[0], provided by Mulliken atomically-partitioned spin populations, reveals stark contrasts between the singlet and triplet states configurations (see Section S3.1 of the ESI†). In the singlet state, the spin distribution resembles that of the two allylic subunits of the well-known tetramethylenethane diradical.92,113 In contrast, in the triplet state, the spin distribution is delocalized towards the terminal open-shell units, arguably enhancing the triplet stability with respect to the singlet by reducing the electron Coulomb repulsion. This effect is also observed when progressing along the R2-DPA[n] and R2-DADPA[n] series. However, the gain from delocalizing the spin density becomes progressively smaller relative to the total number of atoms. As a result, the impact of spin density delocalization on the ΔEST gap decreases with increasing molecular size. Alternatively, the trends can be understood on the basis of a qualitative analysis of the SOMOs of R2-DPA[0] and R2-DADPA[0]. The shape of the SOMOs (see Fig. 1b) suggests a high degree of overlap density, as the orbitals are confined to the same limited spatial region. This overlap density is generally associated with substantial positive ΔEST gaps and implies significant exchange integrals.114 To further substantiate these qualitative observations, we calculated the exchange integrals across each series (see ESI Section S3.2†). We found that the exchange integral markedly decreases with increasing n in both series, which aligns with our finding that the SOMOs become increasingly delocalized with each increment in the series and, thus, the overlap density drops.
The DFT/PBE0 results in Fig. 2b and c have been validated by other hybrid DFT functionals (reported in ESI Section 4.1†) and also by employing Complete Active Space (CAS) and Difference-Dedicated Configuration Interaction115,116 (DDCI) methods. The good agreement found between DFT/PBE0 and DDCI results (see first two rows of Table 1) demonstrates that PBE0 is a suitable choice of DFT functional to adequately assess ΔEST gaps for this kind of systems. Regarding the CASSCF results, we systematically expanded the active space from the simplest 2-orbitals and 2-electrons, CAS(2,2), up to 10-orbitals and 10-electrons, CAS(10,10), to examine the impact of electron correlation along the series (the CAS(10,10) molecular orbitals are shown in ESI Section S4.2†). Accordingly, we calculated the ΔEST gap in both R2-DPA[n] and R2-DADPA[n] at n = 0 and n = 2, i.e., just after the ΔEST crossover of R2-DPA[n]. We carried out these calculations only for methylenyl (R = My) core as a proof of principle. As observed in Table 1, DDCI and CAS(2,2) results predict a similar ΔEST value for both My2-DPA[0] and My2-DADPA[0]. The good agreement between DDCI and CAS(2,2) demonstrates the small impact of electron correlation at n = 0, as the latter only accounts for the correlation between the SOMOs of the diradical. In contrast, for n = 2, the predicted ordering of singlet and triplet energy states is reversed when going from CAS(2,2) to DDCI: while DDCI predicts an AFM interaction (ΔEST < 0), CAS(2,2) wrongly predicts a FM interaction (ΔEST > 0). Notably, the correct relative energy of the spin states is recovered for CAS(4,4) and CAS(10,10), where a higher degree of electron correlation is allowed by expanding the active space. Overall, the CASSCF and DDCI results further highlight the impact of electron correlation on the ΔEST gap for this type of systems.

|
Stability analysis of expanded pentalene couplers
The relative stability of diradicals belonging to the R2-DPA[n] and R2-DADPA[n] series has been assessed by means of: (i) bond length alternation analysis (BLA), (ii) aromaticity criteria based on the induced effects on the molecule as a response to an external magnetic field, such as NICS, ACID, current density maps and bond current strengths, and (iii) RSE. In the first part of this subsection we focus on the simplest compound of the expanded pentalene series, i.e., My2-DPA[0], to illustrate the overall behavior. Further evidence for the rest of the cases is provided later in the main text and in ESI Section S5.†The bare DPA[0] moiety (i.e., pentalene) can be identified as one of the most unstable couplers against dimerization reactions (among other processes) of the expanded pentalene series.20 In line with previous theoretical work,117 as well as NMR studies on 1,3,5-tri-tert-butylpentalene,8 the isolated DPA[0] adopts a C2h configuration with alternate short and long C–C bonds (see BLA Fig. 3a). This configuration results from a pseudo-Jahn–Teller distortion of the D2h configuration with an even distribution of C–C distances118 and shows a localized π system that helps attenuate the antiaromatic character of the compound.55 The alternating C–C bond pattern of the DPA[0] moiety drastically changes when My units are connected to the DPA[0] coupler and form the My2-DPA[0] diradical. Specifically, My2-DPA[0] features two local π-conjugated fragments (displaying two pairs of adjacent C–C distances of 1.387 Å, colored in green at Fig. 3a) arranged such that the system recovers a D2h symmetry. Another significant structural feature observed for My2-DPA[0] is a short C–C bond length of 1.364 Å between the My and DPA[0] blocks (see the bond lengths colored in red of Fig. 3a).
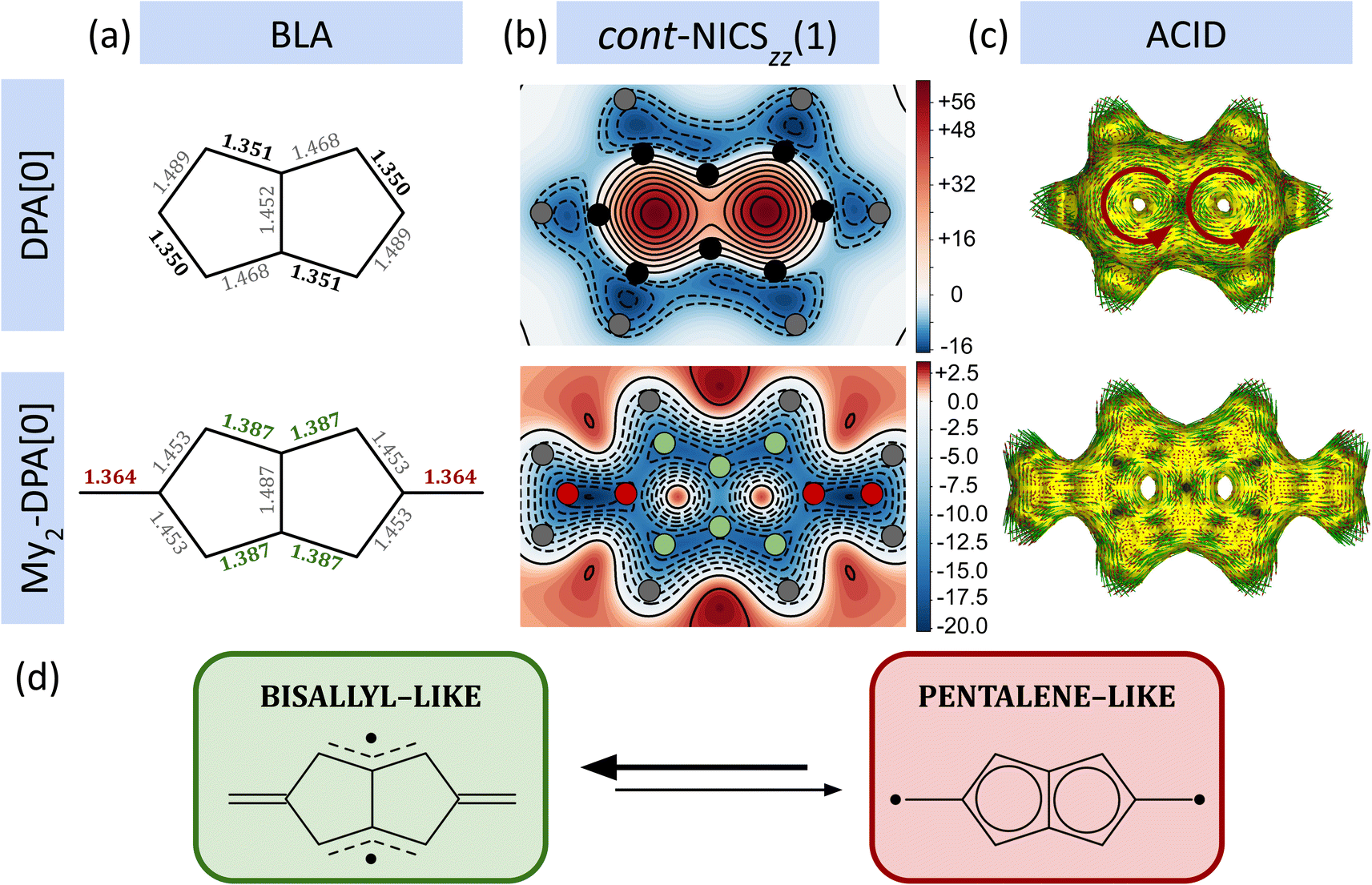 | ||
| Fig. 3 (a) BLA analysis for bare DPA[0] and My2-DPA[0] diradical, with distances (given in Ångstrom) in bold indicating shorter values and distances in gray representing larger values. (b) cont-NICSZZ(1), where the color gradient ranges from red to blue, indicating regions from high deshielding to high shielding, respectively. NICS values are provided in ppm. The position of all carbon atoms is colored in black for the bare DPA[0], while carbon atoms in My2-DPA[0] diradical are colored according to the color scheme used in BLA. The position of hydrogen atoms is colored in gray in both compounds. (c) ACID results for DPA[0] and My2-DPA[0], where red arrows indicate the direction of the current. (d) Lewis resonance structures for My2-DPA[0], with bisallyl-like structures (green) and pentalene-like structures (red), emphasizing the former being more likely. Note that the analyses for DPA[0] and My2-DPA[0] have been carried out for their respective ground electronic states (closed-shell singlet and triplet, respectively). | ||
To assess the (anti)aromatic character of My2-DPA[0], we analyzed the chemical shielding at the center of the five-membered ring of the diradical, using the bare DPA[0] unit as a reference. In an external magnetic field, aromatic systems produce a diatropic electron current, resulting in molecular shielding.119,120 Conversely, antiaromatic systems induce a paratropic current, causing deshielding specially at the center of carbon rings.121–123 We have gauged the tropicity of the current and assessed the (anti)aromatic character of the system by means of the ZZ component of the magnetic shielding tensor (NICSZZ). Here, we focused on NICSZZ, located at 1 Å above the molecular plane, designated as NICSZZ(1), since the major contributions to the chemical shielding arise from the π-system124 and, thus, it is an appropriate indicator of the (anti)aromaticity of planar molecules. In addition, following the methodology of Lampkin et al.,125 we extended the calculation of NICSZZ(1) beyond the ring's centers by evaluating contour levels of NICSZZ(1) across the entire molecular plane. This approach, named herein as cont-NICSZZ(1), not only offers an illustrative NICS map, but can also hint at average resonance structures of planar polycyclic hydrocarbons. Additionally, we have also calculated ACID, current density maps and bond current strengths induced by magnetic field applied perpendicular to the molecular plane, which provides complementary evidence to our analyses.
The cont-NICSZZ(1) map for the bare DPA[0] unit shows a significant positive NICSZZ(1) of ca. 60 ppm at the center of both 5-membered rings (see Fig. 3b), indicating a severe deshielding at each center and, thus, a strong antiaromatic character. Moreover, the shared region between both 5-membered rings features a lower deshielding compared to the ring centers, denoting a certain degree of localized paratropicity where the direction of the currents in both rings counterbalance. ACID plots further support this interpretation, showing pronounced paratropic currents in both DPA[0] rings (see red arrows in Fig. 3c). In line with the BLA results discussed above, cont-NICSZZ(1) also shows alternant shielded regions along the carbon scaffold, revealing localized π–π interactions and further evidence of the characteristic C2h pseudo-Jahn–Teller distortion of the bare DPA[0]. In contrast, cont-NICSZZ(1) displays a residual positive NICSZZ(1) of ca. 2 ppm at the ring centers of My2-DPA[0], which indicates a considerable dampening of the antiaromatic character compared to its precursor. The cont-NICSZZ(1) map depicts two local π-conjugated regions (see Fig. 3b), each consisting of three carbon atoms, which resemble two connected allyl moieties (i.e., ·CH2–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, see atoms colored in green). There is also a significantly shielded region between My and DPA[0] carbon atoms, further proving the strong interaction between the open-shell centers and the coupler. The ACID results for My2-DPA[0] show a weaker and disordered electron current compared to DPA[0] that supports the mitigated antiaromaticity of My2-DPA[0] (refer to Fig. 3c). This current also encompasses the carbon atom of the My core, revealing the strong participation of the My open-shell units to the π-system. Given that BLA, ACID and NICS provide somewhat qualitative insights of electron currents, Fig. 4 shows the results of current density maps and bond current strengths analysis. As it may be seen in the current density maps, DPA[0] features a strong paratropic ring current, irrespective of whether all orbitals or only π-orbitals are considered (see Fig. 4a and c, respectively). Consistent with these results, high values of bond current strength are found (see arrows in Fig. 4e). Conversely, the magnetic field is not able to induce any relevant ring current (either paratropic or diatropic) in My2-DPA[0] and, thus, no significant bond current strengths are found (see Fig. 4b, d and f).
CH2, see atoms colored in green). There is also a significantly shielded region between My and DPA[0] carbon atoms, further proving the strong interaction between the open-shell centers and the coupler. The ACID results for My2-DPA[0] show a weaker and disordered electron current compared to DPA[0] that supports the mitigated antiaromaticity of My2-DPA[0] (refer to Fig. 3c). This current also encompasses the carbon atom of the My core, revealing the strong participation of the My open-shell units to the π-system. Given that BLA, ACID and NICS provide somewhat qualitative insights of electron currents, Fig. 4 shows the results of current density maps and bond current strengths analysis. As it may be seen in the current density maps, DPA[0] features a strong paratropic ring current, irrespective of whether all orbitals or only π-orbitals are considered (see Fig. 4a and c, respectively). Consistent with these results, high values of bond current strength are found (see arrows in Fig. 4e). Conversely, the magnetic field is not able to induce any relevant ring current (either paratropic or diatropic) in My2-DPA[0] and, thus, no significant bond current strengths are found (see Fig. 4b, d and f).
 | ||
| Fig. 4 Current density maps for a perpendicular magnetic field over a plane 1 au above the molecular plane of DPA[0] and My2-DPA[0], at the PBE0/6-311G* level of theory. (a and b) All-electron, and (c and d) π-electron contribution for DPA[0] and My2-DPA[0], respectively. Bond current strengths for a magnetic field perpendicular to the molecular plane for (e) DPA[0] and (f) My2-DPA[0]. Red/blue contours are for ±10−2 au of the current density cross section, delimiting domains for the integration of the bond current strength. Values aside each arrow represent the percentage relationship with respect to a reference current strength of 12 nA T−1. Diatropic/paratropic circulations are clockwise/anticlockwise. | ||
Our findings suggest that the electronic structure of the DPA[0] coupler in My2-DPA[0] diradical resembles that of a non-aromatic, bisallyl-like unit rather than an antiaromatic, pentalene-like (see Fig. 3d, green and red, respectively). Overall, our results unveil a significant dampening of the antiaromatic character of My2-DPA[0] compared to DPA[0] and, consequently, the former can be expected to have an enhanced stability with respect to the precursor based on aromaticity criteria.
We have also performed similar BLA analyses for DPM and PDM cores, as detailed in ESI Section 5.1.† The results for DPM2-DPA[0] and PDM2-DPA[0] align well with our conclusions for My2-DPA[0]. Notably, both sterically protected derivatives display the characteristic bisallyl-like character and display pronounced C–C double-bonds between each core and the DPA[0] coupler. Such a robust core–coupler interaction provides further insights to explain why DPM and PDM units do not alter significantly the ΔEST gap compared to My2-DPA[0] (as observed in Fig. 2b). Namely, promoting a bisallyl-like electronic structure is preferred over diluting the spin density to the terminal phenyl rings of DPM and PDM and, thus, the core choice barely affects the topology of the SOMOs and the ΔEST gap.
In order to gauge the relative stability of the diradicals compared to their bare precursors along the My2-DPA[n] and My2-DADPA[n] series, we conducted a systematic NICSZZ(1) evaluation (see Fig. 5a) at the center of each 5-membered (C5, colored in blue) and both 6-membered ring types (middle and side, i.e., CM6 and CS6, respectively colored in orange and green). For the My2-DPA[n] series, the NICSZZ(1) gap between triplet (T) and open-shell singlet (OSS) states progressively widens (see Fig. 5a) for all C5, CM6 and CS6. This indicates that the OSS electronic structure becomes increasingly more aromatic relative to the T solution across all its rings. In turn, the NICSZZ(1) values of the bare DPA[n] coupler (framed in red at Fig. 5a) decrease notably upon advancing through the series, in line with other studies.126 That is, My2-DPA[0] (2.3 ppm) and My2-DPA[1] (1.2/7.8 ppm for C5/CM6) diradicals exhibit much smaller NICSZZ(1) values compared to their respective bare DPA[0] (61.2 ppm) and DPA[1] (50.9/43.3 ppm for C5/CM6) precursors. It is worth mentioning that the antiaromaticity dampening observed for My2-DPA[1] is in line with the results obtained for diradicals made of two verdazyl radicals linked through indacene,88 although the degree of dampening observed in our case is much larger. Conversely, the NICSZZ(1) for My2-DPA[2] approaches those of DPA[2] and even exceeds them in the case of My2-DPA[3] (see color code for comparison between numerical values in Fig. 5). Essentially, the antiaromatic dampening of diradicals in both T and OSS compared to their bare precursors is clearly mitigated upon progressing through the series. The NICSZZ(1) results are in line with the BLA study carried out for the whole My2-DPA[n] set (see Section S5.1 of the ESI†). We found that the characteristic alternate pseudo-Jahn–Teller distortion from symmetric D2h to C2h point group spotted for the bare DPA[0] disappears when progressing through the series. However, the bisallyl-like character of the corresponding My2-DPA[0] diradicals is preserved as n increases. The trends of NICSZZ(1) for the My2-DADPA[n] series are similar to the trends discussed here (see ESI Section S5.2† for further details).
 | ||
| Fig. 5 (a) NICSZZ(1) values for the constituent rings (C5, CM6, and CS6) of My2-DPA[n] diradicals across the total number of fused [n]acenes (i.e., n in x-axis). NICSZZ(1) values are given for both triplet (T, italic) and open-shell singlet (OSS, bold) states. The gaps between the NICSZZ(1) values of the T and OSS states are given using the following color code: blue (C5), orange (CM6), and green (CS6). The values for bare couplers are framed in red for each ring. Next to each value of n, it is specified whether the triplet state is lower or higher in energy compared to the open-shell singlet. (b) Radical stabilization energy (RSE, in kcal mol−1) for the members of the My2-DPA[n] (blue) and My2-DADPA[n] (red) series. RSE values have been obtained considering the reaction shown on the left. | ||
Although the aromaticity criteria employed so far usually correlate with chemical stability, recent literature40,127 suggests the relationship between (anti)aromaticity and (in)stability for radicals may not be as clear-cut as previously thought. For this reason, a complementary assessment of the relative stability of the compounds of both My2-DPA[n] and My2-DADPA[n] series have been carried out by means of a RSE analysis of the isodesmic109,110 reaction sketched in Fig. 5b.128 The RSE energy is found in all cases to be positive, thus indicating that there is a significant energy gain in the resonance energy of the diradical due to the presence of the extra unpaired electrons in the π-system. The RSE for My2-DPA[0] is ca. 10 kcal mol−1 higher than that of My2-DPA[1] and monotonically decreases as the order of the fused [n]acene increases (see blue curve). Accordingly, My2-DPA[0] is the diradical benefiting the most from the participation of the unpaired electrons in the π-system and its associated dampening of the antiaromatic character of the coupler. Notably, the trends discussed for My2-DPA[n] also hold for My2-DADPA[n], although the RSE values are lower than those of the all-carbon analogs (see red curve) due to the inherently lower antiaromatic character of the diaza-couplers.
In conclusion, all BLA, NICS, ACID, current density maps, bond current strengths, and RSE analyses indicate that the bonding of the radical centers and the antiaromatic coupler result in an enhanced stability of the diradical compounds for all the members of My2-DPA[n] and My2-DADPA[n] series due to a dampening of the antiaromatic character. Notably, the smallest diradicals (i.e., R2-(DA)DPA[0] and R2-(DA)DPA[1]) not only display greater stabilization compared to the bare precursors but also exhibit the most pronounced ferromagnetic interactions.
Diradical stability enhancement through coupler functionalization
Having shown that the most promising candidates are R2-(DA)DPA[0] and R2-(DA)DPA[1], we now focus on blocking the potential dimerization reactions commonly occurring in organic open-shell systems. Taking into account that the most vulnerable sites against dimerization processes are those possessing the highest spin density,109 it is clear from the spin density of My2-(DA)DPA[1] that special attention must be paid to Ca, Cb and Cc atoms shown in Fig. 6a. Note that a similar situation arises in Ca and Cb sites of R2-(DA)DPA[0] (see Fig. S4 in ESI Section S3.1†). Since the use of bulky DPM or PDM cores already offers steric protection to Ca and Cb sites, we focus on the functionalization of the remaining vulnerable Cc sites of My2-(DA)DPA[1]. Our selection of functional groups (–R in Fig. 6b) is based on a twofold approach: (i) achieving a higher degree of spin density delocalization through electron-withdrawing (EW) groups such as –NO2 and –CN, or electron-donating (ED) groups like –NH2 and –NMe2; and (ii) providing steric hindrance using bulky groups like mesityl (–Mes) without a particular withdrawing or donating character.15,51 In this section we assess the impact of these functionalizations on our target property, namely the ΔEST gap. | ||
| Fig. 6 (a) Spin density of My2-DPA[1] at a 0.03 e Å−3 isosurface, with purple and orange representing positive and negative density regions, respectively. (b) Scheme reflecting the coupler functionalization strategy applied to DPA[1] and DADPA[1]-based diradicals, where R represents various functional groups (NO2, CN, Mes, NH2, or NMe2). (c) The distribution of ΔEST gaps (y-axis) is shown relative to the functionalization group (R) and the x-axis corresponds to the C–C bond length indicated in (b). Specific ΔEST gaps for functionalized DPM2-DPA[1], PDM2-DPA[1], and their diaza-substituted analogs (DPM2-DADPA[1] and PDM2-DADPA[2]) are highlighted in red, green, orange, and purple circles, respectively, with a colored dashed line serving as a visual guide to the eye. The non-functionalized diradicals are marked with stars, using the same color code. | ||
Fig. 6c depicts the distribution of the ΔEST values with respect to the C–C distance of the core-coupler building blocks (labeled as dCC in Fig. 6b) for all functionalized R2-DPA[1] and R2-DADPA[1] diradicals, only using DPM and PDM cores. Focusing first on the ΔEST values (y-axis), ED substitutions (–NH2 and –NMe2) generally pull the ΔEST gaps in favor of AFM interactions in all cases but for DPM2-DADPA[1], where the ΔEST is still positive but lower in magnitude. This underscores the role of ED groups in attenuating the antiaromaticity of the bare DPA[1] coupler by serving as an alternative electron source to the π-system, thereby reducing the contribution of the core unpaired electrons and, thus, decreasing the ΔEST. On the other hand, EW substitutions (–NO2 and –CN) induce the opposite effect compared to ED functionalization, retaining the FM interactions of the diradical. Interestingly, the initially negative ΔEST gap in the bare PDM-DPA[1] becomes positive upon functionalization with either –NO2 or –CN. This can be attributed to the fact that EW groups pull out the electron density from the coupler, forcing a higher contribution of the open-shell core electrons to the π-system. The different degree of contribution of the open-shell core to the π-system can be evaluated through the C–C bond lengths (see x-axis in Fig. 6c). Indeed, R2-DPA[1] shows a shorter bond length when the diradical is functionalized with EW groups because these force a larger contribution of the core to the π-system. Following an analogous rationale, the C–C bond length of the core-coupler bridge in ED-substituted diradicals displays a larger value. Remarkably, mesityl (–Mes) functionalization results in ΔEST gaps akin to those of bare diradicals, demonstrating that such functionalization has no impact on electronic structure.
ΔEST gap in dibenzoannulated pentalene diradicals
In this part, the focus is shifted to derivatives of dibenzopentalene (see Fig. 7a), which comprise a subset of pentalene-based antiaromatic compounds that have recently gained recognition for their stability and potential applications.15,129 Similarly to the DPA[n] and DADPA[n] series, the stability of dibenzopentalenes arises from the attenuation of the antiaromatic character through the annulation of two benzene rings to the pentalene unit. Notably, dibenzo[a,e]pentalene and dibenzo[a,f]pentalene isomers have been subject of intensive characterization in recent studies,37,129–131 revealing that the latter intrinsically possesses an open-shell character prior to functionalization with open-shell cores.15 The discussion here is limited to dibenzo[a,e]pentalene and its diaza-derivative, 5,10-diaza-dibenzo[a,e]pentalene, referred hereafter as DBP and DADBP, respectively. Following the approach employed in previous parts, our study considers the double functionalization of DBP and DADBP units with My, DPM, and PDM open-shell cores at different positions (collectively referred to as R in Fig. 7a). Note that although 28 different constitutional isomers can be conceived, we have selected four representative cases for an in-depth characterization of their magnetic and electronic properties. | ||
| Fig. 7 Lewis structures and cont-NICSZZ(1) contour plots of selected dibenzo[a,e]pentalene isomers. (a) Chemical structures display bare dibenzo[a,f]pentalene (left) and dibenzo[a,e]pentalene isomers (right, green). Below, some of the possible diradical derivatives of dibenzo[a,e]pentalene (DBP, X = CH) and diaza-dibenzo[a,e]pentalene (DADBP, X = N) are depicted. Cont-NICSZZ(1) contour plots for (b) the bare DBP coupler and (c) 2,7-My2-DBP diradical. The color gradient in (b) and (c) ranges from red to blue, indicating regions from high deshielding to high shielding (in ppm), respectively. | ||
According to both SGT and EH methods, 2,7-R2-DBP and 4,9-R2-DBP diradical isomers are predicted to feature a pair of non-bonding, non-disjoint SOMOs (refer to ESI Section S2.3†). Hence, these isomers are anticipated to be diradicals displaying FM interactions. In contrast, the SOMOs of 1,6-R2-DBP and 3,8-R2-DBP isomers, although still non-disjoint, cannot be classified as NBMOs, which renders them prone to exhibit negative ΔEST values. Their inclusion in this study, however, has served as an evaluation of the consistency of the SGT and EH models compared to the DFT and CWF approaches. The PBE0/6-311G* ΔEST gaps for both My2-DBP and My2-DADBP are summarized in Table 2 and validated with specific CAS and DDCI calculations (see ESI Sections S4.2 and S4.3†). In agreement with their associated molecular orbital topologies, both 2,7-My2-DBP and 4,9-My2-DBP isomers exhibit large ΔEST gaps of ca. 3.5 and 3.0 kcal mol−1, respectively. As in R2-DPA[n], these gaps significantly exceed thermal energy at room temperature (RT ≈ 0.6 kcal mol−1), which place them as robust FM diradicals. Consistent with these findings, their diaza-derivatives, i.e., 2,7-My2-DADBP and 4,9-My2-DADBP, display even larger ΔEST gaps compared to their DBP analogs. In contrast, the 1,6-My2-DBP and 3,8-My2-DBP isomers display considerably smaller ΔEST gaps, with 3,8-My2-DBP undergoing a severe singlet-triplet gap inversion for both DBP and DADBP couplers. This can be understood by the fact that conjugation of radical centers in 3,8 configuration leads to closed-shell resonance forms (see ESI Section S4.4†). Extending upon the previous framework, we have evaluated the ΔEST gap for the isomer having a 2,7-connectivity with DPM and PDM open-shell cores (see last two rows of Table 2). While all these diradicals display positive ΔEST values, our results reveal that PDM derivatives specially retain a robust ΔEST gap, thus positioning 2,7-PDM2-DBP and 2,7-PDM2-DADBP as the most promising and experimentally realizable candidates among this set.
| a Calculations that led to a closed-shell singlet state, otherwise, the open-shell singlet state is reported. |
|---|

|
In line with the methodologies applied to the DPA[n] series in previous parts, cont-NICSZZ(1) and BLA analyses were performed on My2-DBP-based compounds. For the 2,7-My2-DBP isomer, our findings parallel those from the My2-DPA[n] series, i.e. the C–C bond length between the My cores and the DBP coupler is relatively short, indicative of a significant core participation in the π-system (see ESI Section S5.1†). Unlike the My2-DPA[n] series, however, the BLA analysis reveals that the pentalene moiety in the bare DBP undergoes less severe structural deformations upon open-shell functionalization. Furthermore, cont-NICSZZ(1) analysis (see Fig. 7b) indicates that the shielding values in the 5-membered rings of DBP are far less negative compared to isolated DPA[0], confirming a dampened antiaromatic character of the former (further corroborated by cont-NICSZZ(1) results reported in ESI Section S5.3†). Starting from an inherently less antiaromatic base structure such as DBP may reduce the driving force for extensive electronic structure deformations to mitigate antiaromaticity upon diradical formation, such as the emergence of the bisallyl-like average resonance form observed in R2-DPA[n]. Yet, the cont-NICSZZ(1) values at the center of the 5-membered rings in the diradical are substantially lower than in bare DBP (see Fig. 7c).
Promising pentalene-based derivatives as high-spin diradicals
In view of the large number of molecules considered in this study, a classification has been devised based on their potential experimental viability and their magnetic ΔEST gap. The viability criteria was established based on three key factors: (i) whether or not the bare coupler (or a closely related analogue) has been synthesized, (ii) the stability associated with the (anti)aromatic character of the diradical as well as that related to the bare coupler, and (iii) potential steric hindrance concerns between the bare coupler and the open-shell centers when the radical is formed, specifically for R = DPM and PDM. The classification is summarized in Fig. 8, where a radial diagram is employed to categorize the molecules according to their experimental viability and ΔEST. The angular component and color gradient are utilized to represent ΔEST values. The left region, colored in varying shades of blue, includes diradicals possessing AFM interactions. Conversely, the right region, colored in shades of red, encompasses diradicals with strong FM interactions. The radial axis reflects the potential viability of each molecule based on the aforementioned criteria. A discrete color scheme further differentiates specific cases according to their experimental viability, ranging from unlikely (yellow) to possible (green) and feasible (purple). | ||
| Fig. 8 Scheme of the experimental viability and ΔEST gap of key pentalene-based diradicals explored across this work. The scheme aims at classifying the diradicals based on their potential experimental viability (radial axis) and ΔEST gap (angular axis). The background color transitions from blue (left) to red (right), going from the most AFM to the most FM gaps, respectively. The chemical structures are colored according to their expected experimental viability: yellow (unlikely), green (possible), or purple (probable). | ||
For the DPA[n] and DADPA[n] series, our findings reveal that compounds with n ≥ 2 generally display AFM interactions, irrespective of their synthetic feasibility. The DPA[0] and DADPA[0] members of the series exhibit the largest positive ΔEST gaps, but the inherent antiaromatic instability of their precursors limits their viability. Additionally, the high values of spin population on some of the carbon atoms might make them prone to react when the cores are not protected. The most favorable balance between a robust triplet ΔEST gap and experimental viability of the precursors is expected for DPA[1] and DADPA[1] derivatives, especially for the latter. Although these specific precursors have not been experimentally characterized, closely related derivatives have been reported in the literature.10,18,21 Notably, the functionalization of the existing derivatives is mainly based on steric protection, which is unlikely to substantially perturb the topology of the SOMOs, thereby preserving their magnetic ΔEST gap (as we have shown above for mesityl substitutions). For DBP and DADBP derivatives, robust FM interactions are primarily observed in 2,7- and 4,9-substitutions, while 1,6- and 3,8-substitutions have been proven to be less effective. However, 4,9- configuration may face synthetic challenges due to the potential steric hindrance when functionalized by bulky DPM and PDM open-shell cores, thus rendering 2,7-DBP and 2,7-DADBP the most promising candidates devised for this set. As with the DPA[1] and DADPA[1] compounds, protected derivatives of DBP and DADBP have been reported,12,13,131,132 reinforcing the case for their synthetic feasibility. Finally, it is worth noting that our aromaticity analysis based on BLA, NICS, ACID, current density maps, bond current strengths, and RSE firmly reveal that the diradical derivatives show dampened antiaromatic character compared to the bare coupler, which further strengthens the potential viability.
In summary, we believe that the most promising candidates for achieving room-temperature robust high-spin diradicals across this study are the 2,7-PDM2-DBP and 2,7-PDM2-DADBP derivatives and diaza-substituted PDM2-DPA[1] and PDM2-DADPA[1], due to their significant ΔEST value and expected synthetic viability.
Conclusions
We provide a detailed computational study of various π-conjugated organic diradicals, based on the modular assembly of two open-shell cores covalently linked to an antiaromatic coupler. We primarily focus on pentalene-like couplers displaying an attenuated antiaromatic character, specifically targeting the dicyclopenta-[n]acene series (DPA[n]), dibenzo[a,e]pentalene (DBP), and their diaza-derivatives (DADPA[n] and DADBP, respectively). Our findings reveal a suite of viable compounds with robust singlet-triplet ΔEST gaps, enabling a 100% triplet state population at room temperature.Remarkably, we discovered that chemical or structural variations of the base molecules broadens the attainable spectrum of magnetic interactions. In particular, the introduction of high-order [n]acenes in radicals derived from DPA[n] and DADPA[n] results in a decreasing ΔEST gap, leading to a singlet-triplet energy crossover for compounds with n > 2, which display pronounced AFM interactions. In turn, the magnetic interactions from diradicals derived from DBP and DADBP can be tailored by means of constitutional isomerism.
Our work demonstrates that the strategy of covalently linking two π-radical centers through an antiaromatic moiety results in a strong interaction between their π-systems. This not only leads to strong magnetic interactions, but also to an enhanced stability of the overall diradical, which stems from: (i) a mitigation of the antiaromatic character of the diradicals compared to their bare precursors and, (ii) a large degree of delocalization of the spin density.
Overall, the large ΔEST gaps, together with a dampened antiaromatic character, provide a solid foundation for further exploration into potentially stable organic diradicals. The insights gained from this study suggest that pentalene-based compounds could play a more practical role in emerging technologies (e.g., organic spintronics). Among all the diradicals explored across this work, our results identify 2,7-PDM2-DBP and 2,7-PDM2-DADBP derivatives, as well as diaza-substituted PDM2-DPA[1] and PDM2-DADPA[1] (see diradicals colored in purple in Fig. 8), as the most promising candidates for synthesis and future applications.
Computational details
The molecular orbital structure of all open-shell compounds was pre-screened using SGT methods, providing a computationally-light approximation to evaluate the non-bonding and non-disjoint topology of frontier MOs. Regarding SGT method, eigenvalues and eigenvectors were derived from direct diagonalization of the adjacency matrix (A), represented solely by the carbon scaffold of the diradical. Elements of A, namely Aij, were designated as either 0 (disconnected) or 1 (connected), based on a distance threshold criterion of 2.0 Å. This cutoff served as the appropriate value to render the atomic connectivity matrix according to the usual Lewis representation. Subsequently, the eigenstates of A were visually inspected by projecting the sign of the eigenvectors of interest onto the molecular structure (ESI Section S1†).DFT calculations were performed by means of PBE0,133 B3LYP134–136 and LC-ω-PBE137 exchange-correlation functionals as implemented in the Gaussian09 package,138 using an unrestricted formalism for all the open-shell calculations. A 6-311G* split-valence basis set with integrated polarization functions139,140 has been employed, and MO symmetry not imposed during the calculation (NoSymm keyword). Only PBE0 ΔEST results are reported in the main text as a representative method that compares well with DDCI results and B3LYP and LC-ω-PBE results are reported in ESI Section S4.1.† The ground state structures of diradicals were determined by a five-step protocol: (1) geometry optimization in the triplet state, starting from a distorted diradical structure to avoid highly-symmetric local minima structures. (2) Geometry optimization of the broken-symmetry141 open-shell singlet state, guessing wave-function and atomic positions from the previous triplet (T) state. (3) Evaluation of the ΔadEST = (ES,opt – ET,opt) gap to determine whether the triplet or singlet spin multiplicity state is the ground state (note that the subscript ‘ad’ stands for adiabatic). Single point calculations at the opposite spin alignment were performed by taking the ground state structure and evaluating the vertical singlet-triplet gap, ΔEST. For example, when ΔadEST > 0 the triplet is the ground state and a new ΔEST = EST,opt – ETT,opt would be evaluated. (4) A validation step was performed by repeating the last step with the structure of the excited spin state but with opposite spin alignment. (5) Steps 2–4 were repeated for the closed-shell singlet (CSS) structure. The singlet-triplet gaps were calculated according to ΔEST = 2(EBSS – ET)/(1 + Sab),142 where Sab represents the overlap between SOMOs and EBSS and ET correspond to the energies of the BS singlet and triplet states, respectively, which are calculated using the optimized geometry at the triplet state (referred to as EST,opt and ETT,opt formerly in step 3). Due to significant overlap between SOMOs in all diradicals examined in this work, a value of Sab = 1 has been assumed in all ΔEST calculations, simplifying the former expression to ΔEST = (EBSS – ET) = ΔEBSST. The accuracy of this approximation has been validated by means of DDCI calculations (see below).
The CASSCF143 and DDCI115,116 calculations were performed using the ORCA (version 4.2.1) package,144 a cc-pVTZ basis set145 and the optimized structures at the PBE0 level. All DDCI calculations were carried out using the orbitals resulting from state-average (average between the lowest-energy singlet and lowest-energy triplet) CASSCF(10,10) calculations. The ΔEST gaps at the DDCI level (MR-DDCI3 keyword) reported in the article were obtained using the following values for the TPre and TSel parameters: 1 × 10−4 and 1 × 10−10. As made apparent in ESI Section S4.3,† these values ensure a converged and accurate value for the ΔEST gaps.
NICS calculations for diradicals were performed using Gaussian09 package, utilizing the unrestricted PBE0 formalism with Gauge-Including Atomic Orbitals146 (PBE0-GIAO/6-311G*) and an ultrafine grid for integral evaluations. These calculations were carried out at the ground state spin configuration of each diradical, or at the closed-shell singlet in the bare antiaromatic couplers. NICSZZ(1) values were computed as the ZZ component of the magnetic tensor with a reverse sign at the center of each ring, and positioned 1 Å above the molecular plane by means of inserting a “ghost” atom, X, at the target position. Cont-NICSZZ(1) mapping was performed similarly, employing a multiple evaluation of the magnetic shielding tensor in a 150 × 150 grid of equidistant points in all the cases. Aromaticity has also been evaluated by means of the anisotropy of the induced current density (ACID).103,104 Based on the fact that magnetic properties are suitable to evaluate delocalization and conjugation in aromatic systems, the ACID was proposed in analogy to the anisotropy of the magnetic susceptibility. Similar to the square of the wavefunction which defines the total electron density, the ACID scalar field defines the density of delocalized electrons. ACID plots have been computed for the systems under analysis. Current density maps and bond current strengths have been computed by means of the SYSMOIC package, employing the same level of theory as the rest of the calculations.147–150
Data availability
A data set collection of computational results is available in the ioChem-BD repository151 and can be accessed viahttps://dx.doi.org/10.19061/iochem-bd-6-359. The structure of the data collected is detailed in the Section 6 of the ESI.†Author contributions
R. S. conceived the idea of using pentalene and derivatives of it to promote ferromagnetic interactions between spins. I. P. R. M., S. T. B., M.D. and J. R. A. contributed to the design of the high-spin organic diradicals by proposing stable spin centers that can be linked to pentalene-based linkers. R. S. did the DFT calculations to obtain singlet-triplet gaps, as well as the NICSZZ and RSE calculations to gauge the stability of diradicals. J. R. A. did some DFT calculations to obtain singlet-triplet gaps. M. A. C. carried out the CASSCF and DDCI calculations. J. P. did the calculations to obtain the ACID plots, the current density maps and the bond current strengths. R. S., M. D. and J. R. A. wrote the first draft of the manuscript. All authors contributed to the final version of the manuscript and participated in the discussion of the results.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support from the Spanish Ministerio de Ciencia, Innovación y Universidades (Project Grants: PID2020-117803GB-I00/AEI/10.13039/501100011033, PID2019-109518GB-I00, PID2021-127957NB-I00, PID2022-138861NB-I00, PID2023-149691NB-I00, TED2021-132550B–C21), Spanish Structures of Excellence María de Maeztu program (Grant CEX2021-001202-M), Agència de Gestió d’Ajuts Universitaris i de Recerca of Generalitat de Catalunya (Grants 2021SGR00354 and 2021SGR442). R. S. acknowledges a predoctoral FPI grant under grant agreement PRE2018-084053.References
- M. Rosenberg, C. Dahlstrand, K. Kilså and H. Ottosson, Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations, Chem. Rev., 2014, 114, 5379–5425 CrossRef CAS PubMed.
- Y. Tobe, Non-Alternant Non-Benzenoid Aromatic Compounds: Past, Present, and Future, Chem. Rec., 2014, 15, 86–96 CrossRef PubMed.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Explorations of the Indenofluorenes and Expanded Quinoidal Analogues, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed.
- W. Chen, F. Yu, Q. Xu, G. Zhou and Q. Zhang, Recent Progress in High Linearly Fused Polycyclic Conjugated Hydrocarbons (PCHs, n >6) with Well-Defined Structures, Adv. Sci., 2020, 7, 1903766 CrossRef CAS PubMed.
- A. Konishi and M. Yasuda, Breathing New Life into Nonalternant Hydrocarbon Chemistry: Syntheses and Properties of Polycyclic Hydrocarbons Containing Azulene, Pentalene, and Heptalene Frameworks, Chem. Lett., 2020, 50, 195–212 CrossRef.
- L. J. Karas and J. I.-C. Wu, Antiaromatic compounds: a brief history, applications, and the many ways they escape antiaromaticity. in Aromaticity, ed. I. Fernandez, Elsevier, 2021 Search PubMed.
- C. Hong, J. Baltazar and J. D. Tovar, Manifestations of Antiaromaticity in Organic Materials: Case Studies of Cyclobutadiene, Borole, and Pentalene, Eur. J. Org Chem., 2022, e202101343 CrossRef CAS.
- H. Hopf, Pentalenes—From Highly Reactive Antiaromatics to Substrates for Material Science, Angew. Chem., Int. Ed., 2013, 52, 12224–12226 CrossRef CAS PubMed.
- T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. W. Ng and C. Chi, Antiaromatic Dicyclopenta [b,g]/[a,f]Naphthalene Isomers Showing an Open-Shell Singlet Ground State with Tunable Diradical Character, J. Am. Chem. Soc., 2021, 143, 20562–20568 CrossRef CAS.
- S.-J. Jhang, J. Pandidurai, C.-P. Chu, H. Miyoshi, Y. Takahara, M. Miki, H. Sotome, H. Miyasaka, S. Chatterjee, R. Ozawa, Y. Ie, I. Hisaki, C.-L. Tsai, Y.-J. Cheng and Y. Tobe, S-Indacene Revisited: Modular Synthesis and Modulation of Structures and Molecular Orbitals of Hexaaryl Derivatives, J. Am. Chem. Soc., 2023, 145, 4716–4729 CrossRef CAS.
- E. Pérez-Elvira, A. Barragán and Q. Chen, et al. , Generating Antiaromaticity in Polycyclic Conjugated Hydrocarbons by Thermally Selective Skeletal Rearrangements at Interfaces, Nat. Synth., 2023, 2, 1159–1170 CrossRef.
- Z. U. Levi and T. D. Tilley, Versatile Synthesis of Pentalene Derivatives via the Pd-Catalyzed Homocoupling of Haloenynes, J. Am. Chem. Soc., 2009, 131, 2796–2797 CrossRef CAS PubMed.
- T. Kawase, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata and T. Kubo, An Extremely Simple Dibenzopentalene Synthesis from 2-Bromo-1-ethynylbenzenes Using Nickel(0) Complexes: Construction of Its Derivatives with Various Functionalities, Chem. - Eur. J., 2009, 15, 2653–2661 CrossRef CAS.
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Indeno[1,2-b]Fluorenes: Fully Conjugated Antiaromatic Analogues of Acenes, Angew. Chem., Int. Ed., 2010, 50, 1127–1130 CrossRef.
- A. Konishi, Y. Okada, M. Nakano, K. Sugisaki, K. Sato, T. Takui and M. Yasuda, Synthesis and Characterization of Dibenzo[a,f]Pentalene: Harmonization of the Antiaromatic and Singlet Biradical Character, J. Am. Chem. Soc., 2017, 139, 15284–15287 CrossRef CAS.
- M. Baranac-Stojanović and M. Stojanović, The Effect of Two Types of Dibenzoannulation of Pentalene on Molecular Energies and Magnetically Induced Currents, Phys. Chem. Chem. Phys., 2019, 21, 3250–3263 RSC.
- S. Tanaka, K. Satake, A. Kiyomine, T. Kumagai and T. Mukai, Synthesis and Physical Properties of 3,6-di-Tert-butyl-1,4-diazapentalene, Angew. Chem., Int. Ed., 1988, 27, 1061–1062 CrossRef.
- K. Hanida, J. Kim, N. Fukui, Y. Tsutsui, S. Seki, D. Kim and H. Shinokubo, Antiaromatic 1,5-Diaza-s-indacenes, Angew. Chem., Int. Ed., 2021, 60, 20765–20770 CrossRef CAS.
- J. Usuba and A. Fukazawa, Thiophene-Fused 1,4-Diazapentalene: A Stable C=N-Containing π-Conjugated System with Restored Antiaromaticity, Chem. - Eur. J., 2021, 27, 16127–16134 CrossRef CAS.
- K. Hafner and H. U. Süss, 1,3,5-Tri-Tert-Butylpentalene. A Stabilized Planar 8π-Electron System, Angew. Chem., Int. Ed., 1973, 12, 575–577 CrossRef.
- L. J. Karas, S. Jalife, R. V. Viesser, J. V. Soares, M. M. Haley and J. I. Wu, Tetra-Tert-butyl-S-Indacene Is a Bond-Localized C2h Structure and a Challenge for Computational Chemistry, Angew. Chem., Int. Ed., 2023, 62, e202307379 CrossRef CAS PubMed.
- T. Kawase, T. Fujiwara, C. Kitamura, A. Konishi, Y. Hirao, K. Matsumoto, H. Kurata, T. Kubo, S. Shinamura, H. Mori, E. Miyazaki and K. Takimiya, Dinaphthopentalenes: Pentalene Derivatives for Organic Thin-Film Transistors, Angew. Chem., Int. Ed., 2010, 49, 7728–7732 CrossRef CAS PubMed.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, 6,12-Diarylindeno[1,2-b]Fluorenes: Syntheses, Photophysics, and Ambipolar OFETs, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- J. Nishida, S. Tsukaguchi and Y. Yamashita, Synthesis, Crystal Structures, and Properties of 6,12-Diaryl-Substituted Indeno[1,2-b]Fluorenes, Chem. - Eur. J., 2012, 18, 8964–8970 CrossRef CAS PubMed.
- C. Liu, S. Xu, W. Zhu, X. Zhu, W. Hu, Z. Li and Z. Wang, Diaceno[a,e]Pentalenes: An Excellent Molecular Platform for High-Performance Organic Semiconductors, Chem. - Eur. J., 2015, 21, 17016–17022 CrossRef CAS.
- G. Dai, J. Chang, X. Shi, W. Zhang, B. Zheng, K. Huang and C. Chi, Thienoacene-Fused Pentalenes: Syntheses, Structures, Physical Properties and Applications for Organic Field-Effect Transistors, Chem. - Eur. J., 2014, 21, 2019–2028 CrossRef.
- L. Ren, C. Liu, Z. Wang and X. Zhu, Isomeric Indacenedibenzothiophenes: Synthesis, Photoelectric Properties and Ambipolar Semiconductivity, J. Mater. Chem. C, 2016, 4, 5202–5206 RSC.
- B. Yuan, J. Zhuang, K. M. Kirmess, C. N. Bridgmohan, A. C. Whalley, L. Wang and K. N. Plunkett, Pentaleno[1,2-a:4,5′]Diacenaphthylenes: Uniquely Stabilized Pentalene Derivatives, J. Org. Chem., 2016, 81, 8312–8318 CrossRef CAS PubMed.
- M. Hermann, R. Wu, D. C. Grenz, D. Kratzert, H. Li and B. Esser, Thioether- and Sulfone-Functionalized Dibenzopentalenes as n-Channel Semiconductors for Organic Field-Effect Transistors, J. Mater. Chem. C, 2018, 6, 5420–5426 RSC.
- M. Schmidt, D. Wassy, M. Hermann, M. T. González, N. Agraït, L. A. Zotti, B. Esser and E. Leary, Single-Molecule Conductance of Dibenzopentalenes: Antiaromaticity and Quantum Interference, Chem. Commun., 2021, 57, 745–748 RSC.
- K. Horii, A. Nogata, Y. Mizuno, H. Iwasa, M. Suzuki, K. Nakayama, A. Konishi and M. Yasuda, Synthesis and Characterization of Dinaphtho[2,1-a:2,3-f]Pentalene: A Stable Antiaromatic/Quinoidal Hydrocarbon Showing Appropriate Carrier Mobility in the Amorphous Layer, Chem. Lett., 2022, 51, 325–329 CrossRef CAS.
- Y. Wu, Y. Wang, J. Chen, G. Zhang, J. Yao, D. Zhang and H. Fu, Intramolecular Singlet Fission in an Antiaromatic Polycyclic Hydrocarbon, Angew. Chem., Int. Ed., 2017, 56, 9400–9404 CrossRef CAS PubMed.
- Z. Zhang, H. Fan and X. Zhu, Fast Construction of Dianthraceno[a,e]Pentalenes for OPV Applications, Org. Chem. Front., 2017, 4, 711–716 RSC.
- O. El Bakouri, J. R. Smith and H. Ottosson, Strategies for Design of Potential Singlet Fission Chromophores Utilizing a Combination of Ground-State and Excited-State Aromaticity Rules, J. Am. Chem. Soc., 2020, 142, 5602–5617 CrossRef CAS PubMed.
- J. Sprachmann, T. Wachsmuth, M. Bhosale, D. Burmeister, G. J. Smales, M. Schmidt, Z. Kochovski, N. Grabicki, R. Wessling, E. J. W. List-Kratochvil, B. Esser and O. Dumele, Antiaromatic Covalent Organic Frameworks Based on Dibenzopentalenes, J. Am. Chem. Soc., 2023, 145, 2840–2851 CrossRef CAS.
- B. Esser, J. S. Wössner and M. Hermann, Conjugated Nanohoops with Dibenzo[a,e]Pentalenes as Nonalternant and Antiaromatic π-Systems, Synlett, 2022, 33, 737–753 CrossRef CAS.
- D. Wassy, M. Pfeifer and B. Esser, Synthesis and Properties of Conjugated Nanohoops Incorporating Dibenzo[a,e]Pentalenes: [2]DBP[12]CPPs, J. Org. Chem., 2019, 85, 34–43 CrossRef.
- J. S. Wössner, D. Wassy, A. Weber, M. Bovenkerk, M. Hermann, M. Schmidt and B. Esser, [N]Cyclodibenzopentalenes as Antiaromatic Curved Nanocarbons with High Strain and Strong Fullerene Binding, J. Am. Chem. Soc., 2021, 143, 12244–12252 CrossRef.
- J. S. Wössner, J. Kohn, D. Wassy, M. Hermann, S. Grimme and B. Esser, Increased Antiaromaticity through Pentalene Connection in [n]Cyclo-1,5-Dibenzopentalenes, Org. Lett., 2022, 24, 983–988 CrossRef.
- R. Zhang, A. Ellern and A. H. Winter, Anti-Aromaticity Relief as an Approach to Stabilize Free Radicals, Angew. Chem., Int. Ed., 2021, 60, 25082–25088 CrossRef CAS PubMed.
- S. Sanvito, Molecular Spintronics, Chem. Soc. Rev., 2011, 40, 3336–3355 RSC.
- T. Sugawara, H. Komatsu and K. Suzuki, Interplay between Magnetism and Conductivity Derived from Spin-Polarized Donor Radicals, Chem. Soc. Rev., 2011, 40, 3105–3118 RSC.
- R. Gaudenzi, J. de Bruijckere, D. Reta, I. de P. R. Moreira, C. Rovira, J. Veciana, H. S. J. van der Zant and E. Burzurí, Redox-Induced Gating of the Exchange Interactions in a Single Organic Diradical, ACS Nano, 2017, 11, 5879–5883 CrossRef CAS PubMed.
- C. Herrmann, G. C. Solomon and M. A. Ratner, Organic Radicals As Spin Filters, J. Am. Chem. Soc., 2010, 132, 3682–3684 CrossRef CAS PubMed.
- S. Shil, D. Bhattacharya, A. Misra and D. J. Klein, A High-Spin Organic Diradical as a Spin Filter, Phys. Chem. Chem. Phys., 2015, 17, 23378–23383 RSC.
- D. Cho and J. Y. Lee, Organic Stable Radical Oligomers as Spin Filters, J. Phys. Chem. C, 2023, 127, 8256–8262 CrossRef CAS.
- A. Rajca, From High-Spin Organic Molecules to Organic Polymers with Magnetic Ordering, Chem. - Eur. J., 2002, 8, 4834–4841 CrossRef CAS.
- I. Ratera and J. Veciana, Playing with Organic Radicals as Building Blocks for Functional Molecular Materials, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- C. Shu, Z. Yang and A. Rajca, From Stable Radicals to Thermally Robust High-Spin Diradicals and Triradicals, Chem. Rev., 2023, 123, 11954–12003 CrossRef CAS PubMed.
- Y. Tobe, Quinodimethanes Incorporated in Non-Benzenoid Aromatic or Antiaromatic Frameworks, Top. Curr. Chem., 2018, 376, 12 CrossRef.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Thiophene and Its Sulfur Inhibit Indenoindenodibenzothiophene Diradicals from Low-Energy Lying Thermal Triplets, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS.
- J. E. Barker, J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, Molecule Isomerism Modulates the Diradical Properties of Stable Si†nglet Diradicaloids, J. Am. Chem. Soc., 2019, 142, 1548–1555 CrossRef.
- J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Diindenoanthracene Diradicaloids Enable Rational, Incremental Tuning of Their Singlet-Triplet Energy Gaps, Chem, 2020, 6, 1353–1368 CAS.
- T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. W. Ng and C. Chi, Antiaromatic Dicyclopenta[b,g]/[a,f]Naphthalene Isomers Showing an Open-Shell Singlet Ground State with Tunable Diradical Character, J. Am. Chem. Soc., 2021, 143, 20562–20568 CrossRef CAS PubMed.
- S. Moles Quintero, M. M. Haley, M. Kertesz and J. Casado, Polycyclic Hydrocarbons from [4n]Annulenes: Correlation versus Hybridization Forces in the Formation of Diradicaloids, Angew. Chem., Int. Ed., 2022, 61, e202209138 CrossRef CAS.
- Y. Mizuno, A. Nogata, M. Suzuki, K. Nakayama, I. Hisaki, R. Kishi, A. Konishi and M. Yasuda, Synthesis and Characterization of Dibenzothieno[a,f]Pentalenes Enabling Large Antiaromaticity and Moderate Open-Shell Character through a Small Energy Barrier for Bond-Shift Valence Tautomerization, J. Am. Chem. Soc., 2023, 145, 20595–20609 CrossRef CAS PubMed.
- G. Kothe, K. Denkel and W. Sümmermann, Schlenk's Biradical. – A Molecule in the Triplet Ground State, Angew. Chem., Int. Ed., 1970, 9, 906–907 CrossRef CAS.
- J. Veciana, C. Rovira, M. I. Crespo, O. Armet, V. M. Domingo and F. Palacio, Stable Polyradicals with High-Spin Ground States. 1. Synthesis, Separation, and Magnetic Characterization of the Stereoisomers of 2,4,5,6-Tetrachloro-.Alpha.,.Alpha.,.Alpha.’,.Alpha.’-Tetrakis(Pentachlorophenyl)-m-Xylylene Biradical, J. Am. Chem. Soc., 1991, 113, 2552–2561 CrossRef CAS.
- K. Inoue and H. Iwamura, 2-[p(N-tert-butyl-N-oxyamino)Phenyl]-4,4,5,5-tetramethyl-4,5-dihydroimidazol-3-oxide-1-oxyl, a Stable Diradical with a Triplet Ground State, Angew. Chem., Int. Ed., 1995, 34, 927–928 CrossRef CAS.
- D. R. McMasters and J. Wirz, Spectroscopy and Reactivity of Kekulé Hydrocarbons with Very Small Singlet−Triplet Gaps, J. Am. Chem. Soc., 2000, 123, 238–246 CrossRef.
- D. A. Shultz, R. M. Fico, H. Lee, J. W. Kampf, K. Kirschbaum, A. A. Pinkerton and P. D. Boyle, Mechanisms of Exchange Modulation in Trimethylenemethane-Type Biradicals: The Roles of Conformation and Spin Density, J. Am. Chem. Soc., 2003, 125, 15426–15432 CrossRef CAS PubMed.
- A. Rajca, K. Shiraishi, M. Vale, H. Han and S. Rajca, Stable Hydrocarbon Diradical, An Analogue of Trimethylenemethane, J. Am. Chem. Soc., 2005, 127, 9014–9020 CrossRef CAS PubMed.
- H. Quast, W. Nüdling, G. Klemm, A. Kirschfeld, P. Neuhaus, W. Sander, D. A. Hrovat and W. T. Borden, A Perimidine-Derived Non-Kekulé Triplet Diradical, J. Org. Chem., 2008, 73, 4956–4961 CrossRef CAS PubMed.
- A. Rajca, K. Shiraishi and S. Rajca, Stable Diarylnitroxide Diradical with Triplet Ground State, Chem. Commun., 2009, 4372–4374 RSC.
- P. J. Boratyński, M. Pink, S. Rajca and A. Rajca, Isolation of the Triplet Ground State Aminyl Diradical, Angew. Chem., Int. Ed., 2010, 49, 5459–5462 CrossRef PubMed.
- S. Suzuki, T. Furui, M. Kuratsu, M. Kozaki, D. Shiomi, K. Sato, T. Takui and K. Okada, Nitroxide-Substituted Nitronyl Nitroxide and Iminonitroxide, J. Am. Chem. Soc., 2010, 132, 15908–15910 CrossRef CAS PubMed.
- A. Rajca, A. Olankitwanit, Y. Wang, P. J. Boratyński, M. Pink and S. Rajca, High-Spin S = 2 Ground State Aminyl Tetraradicals, J. Am. Chem. Soc., 2013, 135, 18205–18215 CrossRef CAS PubMed.
- Y. Li, K.-W. Huang, Z. Sun, R. D. Webster, Z. Zeng, W. Zeng, C. Chi, K. Furukawa and J. Wu, A Kinetically Blocked 1,14:11,12-Dibenzopentacene: A Persistent Triplet Diradical of a Non-Kekulé Polycyclic Benzenoid Hydrocarbon, Chem. Sci., 2014, 5, 1908–1914 RSC.
- N. M. Gallagher, J. J. Bauer, M. Pink, S. Rajca and A. Rajca, A. High-Spin Organic Diradical with Robust Stability, J. Am. Chem. Soc., 2016, 138, 9377–9380 CrossRef CAS.
- K. Kato, K. Furukawa and A. Osuka, A Stable Trimethylenemethane Triplet Diradical Based on a Trimeric Porphyrin Fused π-System, Angew. Chem., Int. Ed., 2018, 57, 9491–9494 CrossRef CAS.
- W. Wang, C. Chen, C. Shu, S. Rajca, X. Wang and A. Rajca, A S = 1 Tetraazacyclophane Diradical Dication with Robust Stability: A Case of Low-Temperature One-Dimensional Antiferromagnetic Chain, J. Am. Chem. Soc., 2018, 140, 7820–7826 CrossRef CAS.
- S. Tang, L. Zhang, H. Ruan, Y. Zhao and X. A. Wang, Magnetically Robust Triplet Ground State Sulfur-Hydrocarbon Diradical Dication, J. Am. Chem. Soc., 2020, 142, 7340–7344 CrossRef CAS.
- Z. Wang, Y. Dai, L. Ding, B. Dong, S. Jiang, J. Wang and J. Pei, A Stable Triplet-Ground-State Conjugated Diradical Based on a Diindenopyrazine Skeleton, Angew. Chem., Int. Ed., 2021, 60, 4594–4598 CrossRef CAS.
- K. Wang, P. Liu, F. Zhang, L. Xu, M. Zhou, A. Nakai, K. Kato, K. Furukawa, T. Tanaka, A. Osuka and J. Song, A Robust Porphyrin-Stabilized Triplet Carbon Diradical, Angew. Chem., Int. Ed., 2021, 60, 7002–7006 CrossRef CAS PubMed.
- A. Shimizu, T. Morikoshi, K. Sugisaki, D. Shiomi, K. Sato, T. Takui and R. Shintani, Synthesis and Isolation of a Kekulé Hydrocarbon with a Triplet Ground State, Angew. Chem., Int. Ed., 2022, 61, e202205729 CrossRef CAS PubMed.
- A. Rajca, A. Olankitwanit and S. Rajca, Triplet Ground State Derivative of Aza-m-Xylylene Diradical with Large Singlet−Triplet Energy Gap, J. Am. Chem. Soc., 2011, 133, 4750–4753 CrossRef CAS.
- N. Gallagher, H. Zhang, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. Pink, S. Rajca, M. B. Casu and A. Rajca, Thermally and Magnetically Robust Triplet Ground State Diradical, J. Am. Chem. Soc., 2019, 141, 4764–4774 CrossRef CAS.
- C. Shu, H. Zhang, A. Olankitwanit, S. Rajca and A. Rajca, High-Spin Diradical Dication of Chiral π-Conjugated Double Helical Molecule, J. Am. Chem. Soc., 2019, 141, 17287–17294 CrossRef CAS.
- S. Arikawa, A. Shimizu, D. Shiomi, K. Sato and R. Shintani, Synthesis and Isolation of a Kinetically Stabilized Crystalline Triangulene, J. Am. Chem. Soc., 2021, 143, 19599–19605 CrossRef CAS.
- C. Shu, M. Pink, T. Junghoefer, E. Nadler, S. Rajca, M. B. Casu and A. Rajca, Synthesis and Thin Films of Thermally Robust Quartet (S = 3/2) Ground State Triradical, J. Am. Chem. Soc., 2021, 143, 5508–5518 CrossRef CAS.
- P. Saha, N. Chrysochos, B. J. Elvers, S. Pätsch, S. I. Uddin, I. Krummenacher, M. Nandeshwar, A. Mishra, K. V. Raman, G. Rajaraman, G. Prabusankar, H. Braunschweig, P. Ravat, C. Schulzke and A. Jana, Bis-Olefin Based Crystalline Schlenk Hydrocarbon Diradicals with a Triplet Ground State, Angew. Chem., Int. Ed., 2023, 62, e202311868 CrossRef CAS PubMed.
- Z. Zhu, D. Zhang, T. Xiao, Y. Fang, X. Xiao, X. Wang, S. Jiang and D. Zhao, Rational Design of an Air-Stable, High-Spin Diradical with Diazapyrene, Angew. Chem., Int. Ed., 2023, 62, e202314900 CrossRef CAS PubMed.
- S. Kapuściński, J. Szczytko, D. Pociecha and P. Kaszyński, Mesogenic Behavior of a 6-Oxoverdazyl Diradical: Towards Organic High-Spin Liquid Crystals, Mater. Chem. Front., 2024, 8, 1112–1119 RSC.
- Md. E. Ali and S. N. Datta, Broken-Symmetry Density Functional Theory Investigation on Bis-Nitronyl Nitroxide Diradicals: Influence of Length and Aromaticity of Couplers, J. Phys. Chem. A, 2006, 110, 2776–2784 CrossRef CAS PubMed.
- D. Cho, K. C. Ko and J. Y. Lee, Organic Magnetic Diradicals (Radical–Coupler–Radical): Standardization of Couplers for Strong Ferromagnetism, J. Phys. Chem. A, 2014, 118, 5112–5121 CrossRef CAS PubMed.
- R. Khurana, A. Bajaj and Md. E. Ali, Tuning the Magnetic Properties of a Diamagnetic Di-Blatter’s Zwitterion to Antiferro- and Ferromagnetically Coupled Diradicals, Phys. Chem. Chem. Phys., 2022, 24, 2543–2553 RSC.
- H. Yu and T. Heine, Magnetic Coupling Control in Triangulene Dimers, J. Am. Chem. Soc., 2023, 145, 19303–19311 CrossRef CAS PubMed.
- S. Shil, D. Bhattacharya, A. Misra and L. Bytautas, Antiaromatic Molecules as Magnetic Couplers: A Computational Quest, J. Phys. Chem. A, 2024, 128, 815–828 CrossRef CAS PubMed.
- G. Gibu, A. J. Stasyuk and M. Solà, Prediction of the ground state for indenofluorene-type systems with Clar’s π-sextet model, Chem. Sci., 2024, 15, 13676–13687 RSC.
- J. R. Dias, Disjoint Molecular Orbitals in Nonalternant Conjugated Diradical Hydrocarbons, J. Chem. Inf. Comput. Sci., 2003, 43, 1494–1501 CrossRef CAS PubMed.
- H. Iwamura, High-Spin Organic Molecules and Spin Alignment in Organic Molecular Assemblies, Adv. Phys. Org. Chem., 1990, 26, 179–253 CrossRef CAS.
- W. T. Borden and E. R. Davidson, Effects of electron repulsion in conjugated hydrocarbon diradicals, J. Am. Chem. Soc., 1977, 99, 4587–4594 CrossRef CAS.
- R. W. A. Havenith, J. J. Engelberts, P. W. Fowler, E. Steiner, J. H. van Lenthe and P. Lazzeretti, Localisation and Reversal of Paratropic Ring Currents in Molecules with Formal Anti-Aromatic Electron Counts, Phys. Chem. Chem. Phys., 2004, 6, 289–294 RSC.
- M. Makino and J. Aihara, Aromaticity and Magnetotropicity of Dicyclopenta-Fused Polyacenes, Phys. Chem. Chem. Phys., 2008, 10, 591–599 RSC.
- S. Motomura, M. Nakano, H. Fukui, K. Yoneda, T. Kubo, R. Carion and B. Champagne, Size Dependences of the Diradical Character and the Second Hyperpolarizabilities in Dicyclopenta-Fused Acenes: Relationships with Their Aromaticity/Antiaromaticity, Phys. Chem. Chem. Phys., 2011, 13, 20575–20583 RSC.
- T. Nagami, J. Fujiyoshi, T. Tonami, K. Watanabe, M. Yamane, K. Okada, R. Kishi, M. Nakano, B. Champagne and V. Liégeois, Evaluation of Aromaticity for Open-Shell Singlet Dicyclopenta-Fused Acenes and Polyacenes Based on a Magnetically Induced Current, Chem. - Eur. J., 2018, 24, 13457–13466 CrossRef CAS PubMed.
- R. Ayub, O. E. Bakouri, K. Jorner, M. Solà and H. Ottosson, Can Baird's and Clar's Rules Combined Explain Triplet State Energies of Polycyclic Conjugated Hydrocarbons with Fused 4nπ- and (4n + 2)π-Rings?, J. Org. Chem., 2017, 82, 6327–6340 CrossRef CAS PubMed.
- T. T. Tidwell, Triarylmethyl and Related Radicals, in Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, John Wiley & Sons, Ltd, Chichester, UK., 2010, pp 1–31 Search PubMed.
- I. Ratera, J. Vidal-Gancedo, D. Maspoch, S. T. Bromley, N. Crivillers and M. Mas-Torrent, Perspectives for Polychlorinated Trityl Radicals, J. Mater. Chem. C, 2021, 9, 10610–10623 RSC.
- R. B. Mallion and D. H. Rouvray, Molecular Topology and the Aufbau Principle, Mol. Phys., 1978, 36, 125–128 CrossRef CAS.
- W. T. Borden, H. Iwamura and J. A. Berson, Violations of Hund's Rule in Non-Kekule Hydrocarbons: Theoretical Prediction and Experimental Verification, Acc. Chem. Res., 1994, 27, 109–116 CrossRef CAS.
- P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization, Chem. Rev., 2005, 105, 3758–3772 CrossRef CAS PubMed.
- R. Herges and D. Geuenich, Delocalization of Electrons in Molecules, J. Phys. Chem. A, 2001, 105, 3214–3220 CrossRef CAS.
- J. Jusélius, D. Sundholm and J. Gauss, Calculation of current densities using gauge-including atomic orbitals, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Calculations of magnetically induced current densities: theory and applications, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- R. J. F. Berger, M. Dimitrova, R. T. Nasibullin, R. R. Valiev and D. Sundholm, Integration of global ring currents using the Ampère–Maxwell law, Phys. Chem. Chem. Phys., 2022, 24, 624–628 RSC.
- A. Rabe, Q. Wang and D. Sundholm, Unraveling the enigma of Craig-type Möbius-aromatic osmium compounds, Dalton Trans., 2024, 53, 10938–10946 RSC.
- D. Griller and K. U. Ingold, Persistent Carbon-Centered Radicals, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS.
- M. L. Coote and A. B. Dickerson, The Measurement and Meaning of Intrinsic Radical Stability: Are Chemical Questions Just Problems in Applied Mathematics?, Aust. J. Chem., 2008, 61, 163–167 CrossRef CAS.
- In a two-state system (singlet and triplet), the temperature-dependent Boltzmann population analysis of the triplet state, pTrip(T) in% , is defined as pTrip(T) = 100(3/Z), where 3 stands for the spin multiplicity and Z is the system's partition function, calculated as Z(T) = 3 + exp(−ΔEST/RT), where the energy origin is set to the triplet state energy. With a ΔEST value of 5.7 kcal mol−1, Z(T) approaches 3 + 6.5 × 10−5 at room temperature, leading to pTrip (T=298.15 K) ≈ 100%.
- X. Li and J. Yang, Realizing Two-Dimensional Magnetic Semiconductors with Enhanced Curie Temperature by Antiaromatic Ring Based Organometallic Frameworks, J. Am. Chem. Soc., 2018, 141, 109–112 CrossRef.
- Z. D. Pozun, X. Su and K. D. Jordan, Establishing the Ground State of the Disjoint Diradical Tetramethyleneethane with Quantum Monte Carlo, J. Am. Chem. Soc., 2013, 135, 13862–13869 CrossRef CAS.
- O. Kahn, Molecular Magnetism. John Wiley & Sons. 1993 Search PubMed.
- P. De Loth, P. Cassoux, J. P. Daudey and J. P. Malrieu, Ab Initio Direct Calculation of the Singlet-Triplet Separation in Cupric Acetate Hydrate Dimer, J. Am. Chem. Soc., 1981, 103, 4007–4016 CrossRef CAS.
- J. P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihéry, Magnetic Interactions in Molecules and Highly Correlated Materials: Physical Content, Analytical Derivation, and Rigorous Extraction of Magnetic Hamiltonians, Chem. Rev., 2013, 114, 429–492 CrossRef PubMed.
- I. García Cuesta, S. Coriani, P. Lazzeretti and A. M. J. Sánchez de Merás, From Pentalene to Dicyclopenta[b,g]Naphthalene, or the Change towards Delocalized Structures, ChemPhysChem, 2006, 7, 240–244 CrossRef PubMed.
- A. Toyota and S. Koseki, Energy Component Analysis of the Pseudo-Jahn−Teller Effect in the Bicyclic Nonalternant Hydrocarbons: The Pentalenoid and Heptalenoid Systems, J. Phys. Chem., 1996, 100, 2100–2106 CrossRef CAS.
- V. I. Minkin, M. N. Glukhovtsev and B. Y. Simkin, Aromaticity and Antiaromaticity. Electronic and Structural Aspects, Wiley-Interscience, 1994 Search PubMed.
- G. Merino, T. Heine and G. Seifert, The Induced Magnetic Field in Cyclic Molecules, Chem. - Eur. J., 2004, 10, 4367–4371 CrossRef CAS.
- R. Islas, T. Heine and G. Merino, The Induced Magnetic Field, Acc. Chem. Res., 2012, 45, 215–228 CrossRef CAS.
- M. Orozco-Ic, M. Dimitrova, J. Barroso, D. Sundholm and G. Merino, Magnetically Induced Ring-Current Strengths of Planar and Nonplanar Molecules: New Insights from the Pseudo-π Model, J. Phys. Chem. A, 2021, 125, 5753–5764 CrossRef CAS.
- R. Kumar Jinger, H. Fliegl, R. Bast, M. Dimitrova, S. Lehtola and D. Sundholm, Spatial Contributions to Nuclear Magnetic Shieldings, J. Phys. Chem. A, 2021, 125, 1778–1786 CrossRef PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von R. Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS.
- B. J. Lampkin, P. B. Karadakov and B. VanVeller, Detailed Visualization of Aromaticity Using Isotropic Magnetic Shielding, Angew. Chem., Int. Ed., 2020, 59, 19275–19281 CrossRef CAS.
- A. Stanger, G. Monaco and R. Zanasi, NICS-XY-Scan Predictions of Local, Semi-Global, and Global Ring Currents in Annulated Pentalene and s-Indacene Cores Compared to First-Principles Current Density Maps, ChemPhysChem, 2019, 21, 65–82 CrossRef PubMed.
- L. Lin and J. Zhu, Antiaromaticity-Promoted Radical Stability in α-Methyl Heterocyclics, J. Org. Chem., 2021, 86, 15558–15567 CrossRef CAS PubMed.
- Note that RSE assessments are contingent upon a balanced chemical equation, which inherently reflects the stability of the reference species used upon the same process experimented by the radical. Consequently, the potential RSE measurements are as varied as the reference species available. Although it is unlikely that a genuine RSE measure exists, the specific isodesmic reaction selected here is recognized for minimizing the influence of the reference species, thereby serving as a valuable practical approximation of intrinsic radical stability.
- M. Hermann, D. Wassy, J. Kohn, P. Seitz, M. U. Betschart, S. Grimme and B. Esser, Chiral Dibenzopentalene-Based Conjugated Nanohoops through Stereoselective Synthesis, Angew. Chem., Int. Ed., 2021, 60, 10680–10689 CrossRef CAS PubMed.
- F. Xu, L. Peng, A. Orita and J. Otera, Dihalo-Substituted Dibenzopentalenes: Their Practical Synthesis and Transformation to Dibenzopentalene Derivatives, Org. Lett., 2012, 14, 3970–3973 CrossRef CAS.
- L. Qiu, X. Zhuang, N. Zhao, X. Wang, Z. An, Z. Lan and X. Wan, Benzo[f]Benzo[5,6]Indolo[3,2-b]Indole: A Stable Unsubstituted 4nπ-Electron Acene with an Antiaromatic 1,4-Diazapentalene Core, Chem. Commun., 2014, 50, 3324–3327 RSC.
- M. Nakano, I. Osaka, K. Takimiya and T. Koganezawa, Novel Dibenzo[a,e]Pentalene-Based Conjugated Polymers, J. Mater. Chem. C, 2014, 2, 64–70 RSC.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- O. A. Vydrov and G. E. Scuseria, Assessment of a Long-Range Corrected Hybrid Functional, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. C. Hariharan and J. A. Pople, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, Self-Consistent Molecular Orbital Methods. XXIII. A Polarization-Type Basis Set for Second-Row Elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- L. Noodleman, Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers, J. Chem. Phys., 1981, 74, 5737–5743 CrossRef CAS.
- R. Caballol, O. Castell, F. Illas, I. de P. R. Moreira and J. P. Malrieu, Remarks on the Proper Use of the Broken Symmetry Approach to Magnetic Coupling, J. Phys. Chem. A, 1997, 101, 7860–7866 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, A Complete Active Space SCF Method (CASSCF) Using a Density Matrix Formulated Super-CI Approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- F. Neese, Software update: the ORCA program system, version 4.0, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- T. H. Dunning Jr, Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors, J. Chem. Phys., 1996, 104, 5497–5509 CrossRef CAS.
- G. Monaco, SYSMOIC Version 1.0, 2020, on–line manual http://SYSMOIC.chem.unisa.it/MANUAL/ Search PubMed.
- G. Monaco, F. F. Summa and R. Zanasi, Program Package for the Calculation of Origin-Independent Electron Current Density and Derived Magnetic Properties in Molecular Systems, J. Chem. Inf. Model., 2021, 61, 270–283 CrossRef CAS.
- G. Monaco, F. F. Summa and R. Zanasi, Atomic size adjusted calculation of the magnetically induced current density, Chem. Phys. Lett., 2020, 745, 137281 CrossRef CAS.
- R. J. F. Berger, G. Monaco and R. Zanasi, On the topology of total and diamagnetic induced electronic currents in molecules, J. Chem. Phys., 2020, 152, 194101 CrossRef CAS PubMed.
- M. Álvarez-Moreno, C. de Graaf, N. López, F. Maseras, J. M. Poblet and C. Bo, Managing the Computational Chemistry Big Data Problem: The ioChem-BD Platform, J. Chem. Inf. Model., 2014, 55, 95–103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral graph theory applied to predict trends in ΔEST: general description of the theory and its application to molecular magnetism. Spectral graph theory results: SGT results obtained for R2-DPA[n] and R2-DBP diradicals. Mulliken spin population and Electron Repulsion Integrals (ERIs): Population analysis results obtained for R2-DPA[n] and R2-DBP diradicals. The calculated ERIs are provided for the My2-DPA[n] and My2-DADPA[n] series. Further results related to ΔEST gaps: singlet-triplet energy gaps obtained for My2-DPA[n] and My2-DADPA[n] diradicals at the B3LYP and LC-ω-PBE level of theory. CASSCF results for a CAS(10,10) active space, molecular orbitals, weights and occupations for My2-DPA[0], My2-DPA[2], My2-DADPA[0], My2-DADPA[2], 2,7-My2-DBP and 2,7-My2-DADBP. Convergence of the DDCI parameters for My2-DPA[0]. Further stability results: BLA and NICS results for R2-DPA[n], R2-DADPA[n], R2-DBP and R2-DADBP diradicals. Structure of the data collected in the repository. See DOI: https://doi.org/10.1039/d4sc05225k |
| This journal is © The Royal Society of Chemistry 2025 |