DOI:
10.1039/D5RE00080G
(Paper)
React. Chem. Eng., 2025,
10, 1615-1626
New dispersible and low-melting cellulose ester produced with molten adipic acid as a solvent, reagent and catalyst, and its application to improve the mechanical properties of PLA†
Received
17th February 2025
, Accepted 14th April 2025
First published on 16th April 2025
Abstract
The synthesis of a new cellulose ester (CE) from microcrystalline cellulose (MCC) and adipic acid is described. The solvent-free reaction was carried at 155 °C, slightly above the melting point of adipic acid. The molten acid is both the reactant and the Brønsted acid catalyst in this reaction. Cellulose adipate is highly crystalline and the modification of the XRD powder pattern with respect to MCC indicates that the spatial arrangement of the pristine cellulose Iβ and its hydrogen bonding network have drastically changed. According to TGA and XRD, esterified cellulose surrounds a core of intact MCC (25% by weight), however, the new cellulose adipate material has a melting point of 153 °C and is dispersable in chlorinated solvents. The composites with PLA were prepared by the solvent casting method. The best results were obtained using 3 wt% of the cellulose ester, with a maximum elongation increased by 59%, and a reduction of the tensile strength of 25% only. The SEM images of the best PLA composite show the absence of the cracks found in the PLA films. This work demonstrates how value-added materials can be prepared from renewable and readily available resources through green and sustainable technologies.
1. Introduction
The growing awareness of climate change and the push to reduce the emission of pollutants demand new environmentally friendly technologies.1,2 Besides the environmental aspect, our planet's finite resources must be replaced with renewable and sustainable alternatives.3–5 Agro-food waste and forestry biomasses are the best candidates to substitute fossil feedstocks, and several possibilities for innovation in this field, including biofuels, have been actively investigated in the last years.6–9 Furanic compounds such as furfural (FF), hydroxymethyl furfural (HMF) and furandicarboxylic acid (FDCA) are examples of platform molecules derived from lignocellulosic materials for the production of biofuels.8–10 Pharmaceuticals from renewable natural resources often ensure significantly lower production costs, making them affordable for the entire population.11 Bio-plastics can be produced directly or indirectly from natural resources.12,13 Nevertheless, 300 million tons of plastic are produced annually, and the raw materials for polystyrene (PS), polyvinyl chloride (PVC), polyurethanes (PU), polyethylene terephthalate (PET) and so on are still chemicals of fossil origin.14,15 Bio-plastics represent the new era for the environment and the economy and in the last years many materials deriving from renewable sources have appeared on the market,16 even if biodegradation is one of the crucial aspects that is being investigated.17,18 The most common examples of bioplastics are aliphatic esters such as polyhydroxyalkanoate (PHA) and polylactic acid (PLA).19,20 PHAs are produced by bacteria under stress conditions dictated by the absence of important nutrients such as nitrogen, phosphorus and oxygen but with abundant availability of carbon.21 Today's limitation of such materials is the industrial-scale production.22 PLA is produced industrially through the polymerization of lactic acid, a renewable and sustainable resource, or by the ring-opening polymerization of lactide, the cyclic ester of lactic acid.23 Together with its advantages (recyclability, compatibility with the environment, small energetic footprint), PLA has some limitations such as the slow biodegradation rate, its high hydrophobicity, the modest elongation at break (<10%) which translates into poor toughness and high fragility.24,25 PLA composites made with sustainable resources could address these limitations and further increase the potential of this material.26 Song et al. recently used cardanol derivatives for strengthening and toughening PLA–bamboo fiber composites.27 In another work, Peixoto et al. produced PLA composites of graphene through a green methodology with potential future applications in biomedicine.28 Although cellulose and its derivatives have been investigated for decades, they still suffer several limitations, such as: low solubility in water and organic solvents, poor mechanical resistance and high melting point, a network of intramolecular and intermolecular hydrogen bonds which, without appropriate physical or chemical pre-treatment methods, limit the contact with reagents.29–33 Functionalizing cellulose with green and sustainable techniques still represents an open field of study, especially in the direction of creating consumer materials.33–35 Adipic acid is an important precursor in the synthesis of commonly used plastics such as Nylon 66 and polyurethanes.36,37 The industrial production of nearly 3 million tons per year, still heavily relies on oil derivatives as the starting reagents.38 Fortunately, many strategies have been introduced to obtain it directly from biomasses or natural products.39,40 Niu et al. have proved the direct biosynthesis of adipic acid from lignin-derived aromatics.41,42 The esterification reaction of carboxylic acids is generally a low-reactivity system that requires activation through catalysis or the conversion of the –COOH group to acyl chlorides or anhydrides, which are several times more reactive.43,44 Unlike carboxylic acids, which are often harmless molecules of natural origin, anhydrides and acyl chlorides are highly toxic.45 In addition, when the latter are used in chemical synthesis, they require the use of organic solvents and catalysts, resulting in a high environmental impact.46 In a recent work by Ma et al., cellulose was esterified and crosslinked by citric acid and 1,2,3,4-butanetetracarboxylic acid (BTCA), by catalysis with sodium hypophosphite, to prevent the problem of fibers fibrillation.47 In another work, Batta-Mpouma et al. described the covalent crosslinking of colloidal cellulose nanocrystals using glutaraldehyde or epichlorohydrin and hydrochloric acid or sodium hydroxide as the reaction catalyst, to produce nanostructured hydrogels.48 Kar et al. experimented a rapid crosslinking in the reaction between cellulose and adipic anhydride, and attributed this behavior to the presence of amounts of poly(adipic anhydride) in the reagent; the problem of the esterification reaction between cellulose chains was solved through the initial synthesis of benzyl cellulose acetate adipate propionate and the subsequent Pd-catalyzed hydrogenation reaction to deprotect the adipate groups, with no evidence of crosslinking.49 Recently, Li et al. esterified cellulose with oxalic acid in a solvent-free and catalyst-free reaction, obtaining hydrophilic cellulose esters of lower molecular weight, characterized by little or no interchain crosslinking.50 As demonstrated by previous literature references, the use of reagents with highly reactive functional groups or the use of strong catalysts, promotes cross-linking between cellulose chains. On the ground of these experimental results, we decided to investigate a new cellulose functionalization by a mild and green approach, using a dicarboxylic acid and without the use of additional catalysts, in order to verify the possibility to produce a new cellulose derivative with a low degree of crosslinking. In the present work, microcrystalline cellulose (MCC) has been esterified with adipic acid, through a solvent-free and green reaction, being water the only by-product. The obtained cellulose ester, with a melting point of 153 °C, has been characterized by several physico-chemical techniques and, then, tested as an additive in PLA films.
2. Experimental
2.1 Materials
Chloroform (99.5%, Sigma Aldrich, St. Louis, MA, USA), microcrystalline cellulose (MCC, Avicel PH-101, 50 μm particle size, Sigma Aldrich, St. Louis, MA, USA), polylactic acid (PLA, grade PLA2003D, Ingeo Biopolymer, Minneapolis, MN, USA), Adipic acid (AA, 99%, Carl Roth, Karlsruhe, Germany) were used as received, without any further purification.
2.2 Characterization
FT-IR spectra through KBr pellets were acquired by a Nicolet Impact 410 FTIR Spectrometer (SpectraLab Scientific Inc., Markham, ON, Canada), in the spectral range 400–4000 cm−1 with a resolution of 1 cm−1, with 48 scans per analysis. The substrate was ground with KBr powder (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 ratio) and reduced to a pellet with a hydraulic press. Solid state NMR spectra were obtained on a Bruker AVANCE 300 wide bore spectrometer operating at 300.130 MHz for 1H. 1H MAS NMR measurements were performed with a 2.5 mm double resonance MAS probe. The spectra were acquired through single pulse excitation with a π/2 pulse of 2.1 μs, spin-rate between 5 and 32 kHz and a delay time of 5 s. 13C CP/MAS NMR was recorded at a resonance frequency of 75.468 MHz with a MAS spinning frequency of 5 kHz. A ramped CP MAS sequence was used with a contact time of 3 ms, and 1H decoupling was performed with a B1 field of 64 kHz. For the acquisition of the 13C CP MAS spectrum 9216 scans were accumulated with a recycle delay of 10 s. Line broadening was 15. Water-soluble by-products (carbohydrates and total organic content) were analyzed by an Evolution 201 UV-vis spectrophotometer (ThermoFisher Scientific, Hillsboro, OR, USA), in a range between 200 and 750 nm. The morphology of MCC, of cellulose adipate and of the composite PLA films was investigated by SEM (LEO 420, Leica Microsystems, Cambridge, UK) with an accelerating voltage of 10 kV, after sputtering with an ultrathin gold layer. The tensile strength and fracture strain of the composite-PLA films were measured on rectangular strips (3 cm long and 5 mm wide) at a strain rate of 0.1 mm min−1 with a Sauter TVO-S tensile tester equipped with a Sauter FH-1 k digital dynamometer and AFH FAST software (Sauter GmbH, Balingen, Germany). X-ray diffraction patterns (XRD) were recorded on a Miniflex 600 diffractometer (Rigaku, Tokyo, Japan), using a Ni filtered Cu Kα wavelength of 1.5418740 Å with a D/teX Ultra2 detector, operating at 40 kV and 15 mA. MCC and CE4, in the form of dry powders, were compacted in the sample holder. PLA (300 mg) and the PLA–3CE composite (300 mg of PLA with 3% weight of cellulose ester CE4, entry 3 in Table 3) dissolved in CHCl3 were poured in the Al holders, the solvent was let to evaporate to dryness, and then the solvent residues were removed in a vacuum oven overnight before the analysis. Differential scanning calorimetry (DSC200 PC, Netzsch, Selb, Germany) was carried out by a standard procedure: about 10.0 mg of each sample were placed in an aluminum pan and closed tightly with a perforated aluminum lid. The thermal analyses were performed under a dry nitrogen atmosphere with a flow rate of 20 mL min−1: i) from 20 to 200 °C (heating rate 10 °C min−1); ii) from 200 °C to room temperature (1 °C min−1); iii) from room temperature to 200 °C (10 °C min−1); iv) from 200 °C to room temperature (1 °C min−1).
:100 ratio) and reduced to a pellet with a hydraulic press. Solid state NMR spectra were obtained on a Bruker AVANCE 300 wide bore spectrometer operating at 300.130 MHz for 1H. 1H MAS NMR measurements were performed with a 2.5 mm double resonance MAS probe. The spectra were acquired through single pulse excitation with a π/2 pulse of 2.1 μs, spin-rate between 5 and 32 kHz and a delay time of 5 s. 13C CP/MAS NMR was recorded at a resonance frequency of 75.468 MHz with a MAS spinning frequency of 5 kHz. A ramped CP MAS sequence was used with a contact time of 3 ms, and 1H decoupling was performed with a B1 field of 64 kHz. For the acquisition of the 13C CP MAS spectrum 9216 scans were accumulated with a recycle delay of 10 s. Line broadening was 15. Water-soluble by-products (carbohydrates and total organic content) were analyzed by an Evolution 201 UV-vis spectrophotometer (ThermoFisher Scientific, Hillsboro, OR, USA), in a range between 200 and 750 nm. The morphology of MCC, of cellulose adipate and of the composite PLA films was investigated by SEM (LEO 420, Leica Microsystems, Cambridge, UK) with an accelerating voltage of 10 kV, after sputtering with an ultrathin gold layer. The tensile strength and fracture strain of the composite-PLA films were measured on rectangular strips (3 cm long and 5 mm wide) at a strain rate of 0.1 mm min−1 with a Sauter TVO-S tensile tester equipped with a Sauter FH-1 k digital dynamometer and AFH FAST software (Sauter GmbH, Balingen, Germany). X-ray diffraction patterns (XRD) were recorded on a Miniflex 600 diffractometer (Rigaku, Tokyo, Japan), using a Ni filtered Cu Kα wavelength of 1.5418740 Å with a D/teX Ultra2 detector, operating at 40 kV and 15 mA. MCC and CE4, in the form of dry powders, were compacted in the sample holder. PLA (300 mg) and the PLA–3CE composite (300 mg of PLA with 3% weight of cellulose ester CE4, entry 3 in Table 3) dissolved in CHCl3 were poured in the Al holders, the solvent was let to evaporate to dryness, and then the solvent residues were removed in a vacuum oven overnight before the analysis. Differential scanning calorimetry (DSC200 PC, Netzsch, Selb, Germany) was carried out by a standard procedure: about 10.0 mg of each sample were placed in an aluminum pan and closed tightly with a perforated aluminum lid. The thermal analyses were performed under a dry nitrogen atmosphere with a flow rate of 20 mL min−1: i) from 20 to 200 °C (heating rate 10 °C min−1); ii) from 200 °C to room temperature (1 °C min−1); iii) from room temperature to 200 °C (10 °C min−1); iv) from 200 °C to room temperature (1 °C min−1).
TGA measurements were performed on a PerkinElmer TGA-6 instrument using ca. 7 mg of powdered samples. The measurements were carried out in the temperature range 30–800 °C, heating rate 10 °C min−1, under nitrogen flow (50 mL min−1).
2.3 Esterification of cellulose with adipic acid
MCC and adipic acid were dried at 80 °C in a vacuum oven overnight. In a typical reaction, 300 mg of MCC were mixed with 1.2 g of adipic acid in a Pyrex vial. The reaction vial was put into an oil bath, under open air conditions and heated to 156 °C for two hours, without stirring. After this time, the obtained solid was crushed and washed with 1 ml of water (see the ESI† for the characterization of water-soluble species), washed with 100 ml of acetone over a sintered glass filter and dried at 80 °C in a vacuum oven overnight. The gravimetric yield of the optimized product (CE4, see Table 1 below) was 105%. The obtained cellulose esters were crushed in a mortar in the form of a fine white powder before use. The degree of substitution of the cellulose esters (DS), the amount of free –COOH groups (FCC) and the total –COOH content (TCC) were determined by conductometric and acid–base titrations,50 as described in detail in the ESI† file.
Table 1 Adipic acid-to-cellulose mass ratio (AA/MCC) and degree of substitution (DS) of cellulose adipate
| Entrya |
Name of sample |
AA/MCCb |
DSc |
|
Reaction conditions: 156 °C, open air, 120 minutes.
Adipic acid-to-cellulose mass ratio.
DS was calculated by titration (see ESI† file).
|
| 1 |
CE0.5 |
0.5 |
0.04 |
| 2 |
CE1 |
1.0 |
0.08 |
| 3 |
CE1.5 |
1.5 |
0.11 |
| 4 |
CE2 |
2.0 |
0.14 |
| 5 |
CE2.5 |
2.5 |
0.17 |
| 6 |
CE3 |
3.0 |
0.19 |
| 7 |
CE3.5 |
3.5 |
0.22 |
| 8 |
CE4 |
4.0 |
0.25 |
| 9 |
CE4.5 |
4.5 |
0.25 |
2.4 Preparation of the composited PLA-esterified cellulose
The films were prepared in Petri dishes (120 mm diameter) through the solvent casting technique. 300 mg of PLA were dissolved in 14 ml of CHCl3 at room temperature, while the cellulose ester, CE (3.0, 6.0, 9.0 and 18.0 mg) was dispersed in 2 mL of CHCl3 at room temperature. Then, the solution and the dispersion were combined and left stirring for 15 minutes before being poured into the Petri dish. The latter was covered with an aluminum foil and the solvent was left to evaporate slowly at 25 °C overnight.
3. Results and discussion
3.1 Optimization of the esterification reaction
The target of the present work is the functionalization and the improvement of the chemical–physical properties of cellulose, by a green methodology using adipic acid as a solvent, a reagent and catalyst at the same time. Being the melting point of adipic acid at 152 °C, the reaction temperature was set at 156 °C in order to exploit its double function of solvent and reagent. Initially a 1:4 weight ratio of cellulose-to-acid was used, and the mixture was put in a single-necked round bottom flask connected to a condenser. Despite the visible production of water in the condensation apparatus, the degree of esterification obtained was not satisfactory, since almost all of the used acid was recovered by washing the cellulose with acetone. On the contrary, when an open Pyrex vial system without a condenser was used, after a few minutes water vapors leaving the reaction environment were noticed, just like in a classic Fisher esterification.51 In fact, at 156 °C water evaporates from the reaction environment and, according to the Le Châtelier principle, the equilibrium reaction shifts towards the products, favouring esterification. After 120 min, the obtained product was washed with water, then with acetone to remove any excess of unreacted adipic acid, and dried in an oven at 80 °C overnight. SEM images of MCC and of one cellulose adipate (CE4, see Table 1) at different magnifications are reported in Fig. S13 of the ESI† file. Both samples exhibit particle size in the micron range. The particles of cellulose adipate are 10–50 mm long, on average, and each of them seems to be made of stacked lamellar substructures with no curvatures. MCC instead looks less structured, with smoother and curved surfaces. These observations indicate a more ordered organization of the cellulose adipate chains, in agreement with the much higher crystallinity of the cellulose ester evidenced by X-ray diffraction spectra (see para. 3.4). In Fig. 1 the FT-IR spectra of adipic acid (AA), microcrystalline cellulose (MCC) and produced cellulose ester (CE4, see Table 1) are reported.
 |
| | Fig. 1 Superimposed FT-IR spectra of adipic acid (AA), MCC and cellulose esterified with adipic acid (CE4). | |
In Fig. 1, the O–H stretching at 3351 cm−1 is also present in the esterified cellulose (CE4), demonstrating the survival of non-functionalized primary or secondary OH groups.36 The stretching signal of the ester carbonyl group at 1732 cm−1 in CE4 confirms the successful esterification of MCC, with a slight shift to higher frequencies compared to the stretching vibration of the –COOH group of the acid (AA) at 1698 cm−1. However, a closer look at the FT-IR spectrum shows the overlap of the signals of ester and free –COOH groups. The deconvolution of the IR spectrum between 1600 and 1850 cm−1 (Fig. 2), carried out by fitting the signal with Gaussian functions (Fityk version 0.9), shows the simultaneous presence of the band of the ester groups and –COOH groups, overlapping in a single peak.
 |
| | Fig. 2 Deconvolution of FT-IR spectrum of CE4 between 1600 and 1850 cm−1. | |
Solid state NMR spectroscopy was used to further support this finding. The 1H MAS NMR spectrum of CE4 is dominated by a broad and complex signal centered at 4.1 ppm (Fig. S1†). The network of hydrogen bonds among cellulose chains produces strong homonuclear dipolar interactions, thus leading to broader 1H NMR signals in the solid state. Consequently, the various proton environments in the CE4 sample remain unresolved, even at a MAS spinning frequency of 32 kHz. However, following the typical chemical shifts observed in the liquid state NMR the peak at 5.05 ppm can be reasonably assigned to the protons in the anhydroglucose ring of cellulose (i.e., C–H and –OH)52 whereas the two additional peaks at 1.57 and 2.22 ppm can be ascribed to adipoyl-CH2 protons. Worth to note, the peak at 11.5 ppm provides clear evidence of the presence of protons from carboxylic acid groups upon cellulose esterification. The 13C CP MAS NMR measurements shed light on the chemical structure of the esterification product of cellulose. The spectrum of CE4 in Fig. 3 combines the characteristic peaks of both cellulose (in the range between 60 and 100 ppm) and adipate/adipic acid. In detail, the spectrum exhibits the two adipate methylene peaks at 24.4 and 34.7 ppm, the peak at 64.1 for the C-6 position of the anhydroglucose ring, a poorly resolved resonance at 71.7–73.9 ppm for C2,3,5, the resonance at 86.1 ppm assigned to C-4 and the signal at 104.7 ppm for C-1.53 Notably, the simultaneous presence of both the adipate carbonyl peak (at 172.4 ppm) and the pendant carboxylic acid groups (182.2 ppm) undoubtedly confirms that the synthetic product is cellulose adipate with minor (or no) crosslinking.
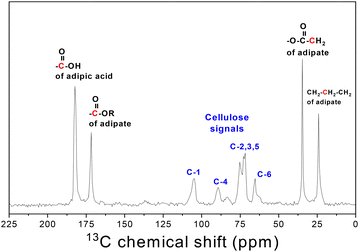 |
| | Fig. 3 The CP/MAS 13C NMR spectrum of CE4, together with the structural assignments, indicated in red (adipate functionality) and blue (cellulose backbone). | |
With the aim of finding the optimal amount of adipic acid, the adipic acid-to-cellulose mass ratio (AA/MCC) in the reaction mixture was varied, maintaining the same reaction time of 120 minutes. The degree of substitution (DS) of the cellulose adipates is reported in Table 1.
The DS increases with the amount of adipic acid used, up to an adipic acid-to-cellulose mass ratio of 4 (entries 1–8 in Table 1). With larger amounts of acid, the DS value does not change, either because there are no more accessible –OH groups available, or because the acid is unable to penetrate deeply into the crystalline domains of the polymer. The DS follows the same trend (Table 2) with respect to time: using a 4:1 adipic acid-to-cellulose mass ratio the highest DS value 0.25 was obtained for a time of 120 min.
Table 2 TCC, FCC, FCC/TCC ratio and DS versus reaction time in the esterification of MCC with adipic acid
| Entrya |
Time (min)b |
TCCc (mmol g−1) |
FCCc (mmol g−1) |
FCC/TCC |
DS |
|
Reaction conditions: 156 °C, open air.
Adipic acid-to-cellulose mass ratio 4:1.
TCC and FCC were calculated by titration (see ESI† file).
|
| 1 |
15 |
0.59 |
0.30 |
0.51 |
0.06 |
| 2 |
30 |
1.08 |
0.55 |
0.51 |
0.12 |
| 3 |
60 |
1.20 |
0.58 |
0.48 |
0.13 |
| 4 |
90 |
2.02 |
0.97 |
0.48 |
0.21 |
| 5 |
120 |
2.29 |
1.06 |
0.46 |
0.25 |
| 6 |
180 |
2.30 |
1.06 |
0.46 |
0.25 |
Table 2 shows the effect of time on the degree of esterification and crosslinking. The decrease of the FCC-to-TCC ratio below 0.5 can be considered as an indicator of the degree of crosslinking.
An FCC/TCC ratio equal to 0.5 after 15 and 30 minutes indicates an equal number of –COOH groups and ester groups on the polymer chains of the esterified cellulose, with the functionalization of only one of the two –COOH groups of adipic acid (entries 1 and 2 in Table 2). After 60, 90 and 180 minutes the FCC/TCC ratio decreases slightly below 0.5, and after 180 minutes the value is 0.46, indicating a modest or negligible crosslinking (entries 3, 4, 5 and 6 in Table 2). The DS increased with time in the first 30 minutes, and reached an upper value of 0.25 after 120 minutes (entries 1–5 in Table 2). Considering that the primary C6 –OH groups are the most reactive, the theoretical substitution degree (DS) for the esterification of pure cellulose fibers, without any other chemical or physical pre-treatment (e.g. swelling), is near 0.05.50 In crystalline cellulose, only half of the primary C6 –OH groups belonging to the cellulose chains on the outer surface of the crystallites are easily accessible, since the other half are hidden within the crystalline particle.33 However, in this study, the obtained maximum degree of substitution of microcrystalline cellulose is 0.25, demonstrating that the molten adipic acid manages to penetrate the innermost parts of the polymer, breaking the intermolecular network of hydrogen bonds and functionalizing a larger fraction of –OH groups. A comparable degree of substitution (up to 0.35) was obtained in the esterification of pure cellulose fibers with oxalic acid, however cellulose fibers contain also disordered amorphous chains, and therefore they are more reactive and prone to hydrolysis than MCC.50
3.2 Solubility tests of the esterified cellulose CE
The solubility in different solvents of the cellulose ester CE4, produced by the optimized reaction (Table 2, entry 5) was investigated at room temperature (Table 3). The ester does not dissolve in any of the solvents tested, and it is not even dispersable in protic and aprotic polar solvents (water, methanol, acetone, THF, ethyl acetate and DMF, entries 1–6). In contrast, the ester (5 mg ml−1) is readily dispersable in chlorinated solvents such as chloroform, dichloromethane and 1,2 dichloroethane (entries 7–9).
Table 3 Dispersability of the AA/MCC 4:1 cellulose ester CE4a in different solvents
| Entrya |
Solvent |
Dispersability |
|
5 mg ml−1, at room temperature.
Dispersion is instantaneous.
|
| 1 |
Water |
Not dispersable |
| 2 |
Methanol |
Not dispersable |
| 3 |
Acetone |
Not dispersable |
| 4 |
THF |
Not dispersable |
| 5 |
Ethyl acetate |
Not dispersable |
| 6 |
DMF |
Not dispersable |
| 7b |
Chloroform |
Dispersable |
| 8b |
Dichloromethane |
Dispersable |
| 9b |
1,2 Dichloroethane |
Dispersable |
3.3 DSC investigation of MCC and esterified cellulose
The analysis of thermal transitions by differential scanning calorimetry (DSC) provides an excellent comparison between the starting microcrystalline cellulose MCC and the functionalized cellulose CE4. From Fig. 4a, the pristine MCC does not show any obvious thermal peak other than a slight loss of water at low temperature, as confirmed by previous studies.54
 |
| | Fig. 4 DSC thermograms of MCC (a) and CE4 (b). First heating run, 10 °C min−1. | |
In contrast, the DSC thermogram of the cellulose adipate CE4 in Fig. 4b presents a clear endothermic peak at 153 °C (150 °C in the second heating run, Fig. S7, ESI†) related to its melting temperature Tm, together with a small endothermic peak at 43.7 °C – a typical glass transition (Tg) of cellulose esters.55 Adipic acid has a melting temperature of 152 °C as evidenced by the endothermic peak in its DSC profile,56 which allows it to work as a solvent of the forming cellulose adipate. In cellulose triacetate (Tm = 203 °C) the short acetyl moieties do not interfere with the packing of the glycosidic main chains, and do not disturb their crystallization, whereas in the analogue cellulose esters tributyrate, triheptanoate and tridecanoate (Tm = 80–83 °C) the longer side chains disturb the crystallization of the cellulose backbone, forming new crystals dominated by the hydrophobic interactions.57 The situation of cellulose adipate looks intermediate between those of cellulose triacetate and of the analogue cellulose esters with longer side chains, notwithstanding that the major part of the –OH groups is still present. It is reasonable to think that the esterification with adipic acid destroys the intermolecular hydrogen bonds network of cellulose, leading to a new packing of the main chains, as suggested by the XRD spectrum (vide infra). Unlike cellulose, cellulose adipate melts, making it an exceptional material for various future industrial applications. More information on the thermal behavior of cellulose adipate is reported in the ESI† file.
3.4 XRD spectra of MCC and esterified cellulose CE
The small width of the cellulose crystallites, together with its variability, make the analysis of the cellulose lattices very complex, due to peak overlapping. In addition, the structure varies according to the different types of cellulose.33,58–61 Avicel PH101 is obtained by HCl hydrolysis of wood pulp, and its cellulose crystallites are reported to be 100–200 nm long and 4–5 nm wide.33,55,57,60,62,63 Its X-ray diffraction pattern (Fig. 5a) nicely reproduces those reported in the literature.56,60,64,65 The five major crystalline peaks of MCC (1−10, 110, 012 – 102, 200, 004) correspond to those of monoclinic cellulose Iβ, and are indexed according to Sugiyama et al.66 The peaks are broad mainly because of the reduced width of the crystalline domains.58–61
 |
| | Fig. 5 XRD spectra of MCC (a) and CE4 (b). | |
In Fig. 5b the XRD spectrum of the synthesized cellulose ester CE4 is characterized by sharp and intense peaks denoting a highly ordered crystalline structure. The presence of two residual broad peaks around 22.3° (the most evident) and about 15.1° (very shallow), corresponding to the main reflections of MCC, suggests that a fraction of crystalline cellulose was not esterified. The main MCC crystalline peaks appear shifted to lower 2θ values in the cellulose adipate, a fact which translates into larger crystallographic spacings for the latter. This finding is reasonable, since some H atoms from the –OH groups on the cellulose rings are substituted by bulky adipoyl units, and the network of intermolecular hydrogen bonds might be weakened.67 The new peaks appear at 2θ values of 13.01, 21.51, 25.32, 25.52, 25.85 and 31.07° (Fig. 4b). The most intense peak at 21.51° corresponds to a spacing of 4.131 Å, and is probably related to the (200) reflection in MCC at 22.16° (4.012 Å). In Fig. 6 the deconvolution of the peaks between the 2θ values of 20° and 24° (Fityk Software vers. 1.3.1) is shown, where the most intense reflections of both microcrystalline cellulose and of the esterified cellulose are evidenced. After integration, the relative areas of the three peaks (the sharper peaks centered around 21.2° and 21.51°, due to cellulose adipate, and the broad one centered around 22.16°, characteristic of the pristine MCC) are in the 16.6:58.8:24.5 ratio, representing the approximate mass ratio of esterified cellulose (75.5%) to non-functionalized innermost MCC (24.5%).
 |
| | Fig. 6 Deconvolution of the XRD spectrum of CE4 in the 2θ range 20–24°. | |
The TGA profiles of MCC and CE4 are displayed in Fig. 7. Microcrystalline cellulose MCC shows a weight loss in the first part of the heating (−5.4% up to 125 °C), very probably due to the loss of moisture, and a large weight loss between 300 and 400 °C (maximum slope around 365 °C, −71.8% between 125 and 391 °C), in agreement with previous data.68 The thermogram of CE instead shows a modest loss of water below 125 °C (−0.72%), probably because the insertion of alkyl chains makes the material hydrophobic, and less prone to absorb humidity. There is a major weight loss (−72.7%) between 125 and 272 °C, probably related to the changes in chemical–physical properties after the esterification, in which the hydrogen bonds network of the polymer is weakened by the elimination of a polar O–H bond and by the insertion of the adipoyl moiety. The minor weight loss of CE between 272 and 800 °C (−24.3%) nicely reproduces the decomposition profile of MCC, therefore this weight loss is attributed to the decomposition of the inaccessible core of cellulose that had not interacted with adipic acid and remained intact. If the major weight loss from 125 to 272 °C is attributed to the decomposition of the fraction of cellulose that underwent esterification by adipic acid, and the minor weight loss from 272 to 800 °C is attributed to the decomposition of the unmodified cellulose, then the mass ratio of the two fractions would be 75:25, in excellent agreement with the 75.5:24.5 ratio obtained from the deconvolution of the XRD spectrum. Both TGA and XRD clearly indicate the presence of intact MCC cores in the CE4 sample, that are not esterified by the treatment with adipic acid. Because of this, it has to be pointed out that the degree of substitution determined experimentally for CE4 (0.25) is an average value.
 |
| | Fig. 7 Thermogravimetric profiles (TGA) of MCC and CE4. | |
3.5 Hypothesis about the reaction mechanism
In the reaction described above, adipic acid acts as a solvent, reagent and acid catalyst, activating the –COOH groups of AA and making them available for the nucleophilic attack by the–OH groups. In addition, thanks to its polarity, AA is able to interpose itself among the glucose chains breaking the intermolecular hydrogen bonds. The esterification reaction with the consequent loss of water is favored by the reaction temperature and the open reaction system. Based on literature studies, and the proven difference in reactivity between primary and secondary –OH groups, the reaction probably occurs easily on the more external primary –OH groups, and then eventually on the inner primary –OH groups.69 The XRD spectrum and the thermogravimetric data indicate an unprecedent internal penetration of the adipic acid, into sites impenetrable until now for a reaction in which there is no pre-treatment of the starting MCC, leaving only the core of the crystallite unchanged. The instantaneous dispersion in chlorinated solvents (Table 3), the presence of both ester and free –COOH signals in the FT-IR spectrum (Fig. 2), as confirmed by the titration, the melting point at 150–153 °C, the 13C and 1H spectra, the TGA and the XRD analyses support the reaction mechanism depicted in Scheme 1.
 |
| | Scheme 1 Hypothesis of reaction mechanism for the formation of cellulose adipate. MCC and adipic acid are mixed together under solvent-free conditions at 156 °C (1). Adipic acid is the reagent and the catalyst by activating the oxygen of the carboxylic groups (2). The reaction temperature and the open reaction apparatus promote water loss and the formation of the cellulose ester (3). | |
It is hypothesized that the small incidence or absence of cross-linking is due to the mild experimental conditions of the synthesis of cellulose adipate presented in this work. The reactivity of carboxylic acids alone in the esterification reactions is low, since no other catalysts are used, except adipic acid itself, which acts as a reagent and weak acid catalyst. The reaction equilibrium is pushed towards the product, thanks to the fact that the reaction occurs in an open container and water, the only by-product of this reaction, once formed, at 165 °C evaporates out of the reaction environment. The core of microcrystalline cellulose fibers is not converted into cellulose adipate, probably because of the well-known resistance of the most ordered domains of cellulose to esterification. Another possible explanation is suggested by the CE4 FCC/TCC value of 0.46 (Table 2), which might indicate a low degree of crosslinking. As discussed in para. 3.4, one effect of esterification is the expansion of the crystalline lattice of MCC. However, only in case this expanded lattice is heavily crosslinked, a stiff shell of cellulose adipate would form, preventing the further esterification and expansion of the MCC core.
3.6 Cellulose adipate as an additive of PLA
The cellulose ester CE4, prepared by the optimized reaction (Table 2, entry 5), readily disperses in chloroform, one of the best solvents of PLA:70 therefore, the CE4 ester was tested as an additive for the preparation of PLA films. The composite films (Fig. S13†) were prepared with different weight percentages of CE4 (see Table 4) as reported in the Experimental section 2.4.
Table 4 PLA composites with the cellulose ester CE4 produced by the optimized method (Table 2, entry 5)
| Entry |
Wt% CE4 |
PLA composites |
| 1 |
1 |
PLA–1CE |
| 2 |
2 |
PLA–2CE |
| 3 |
3 |
PLA–3CE |
| 4 |
6 |
PLA–6CE |
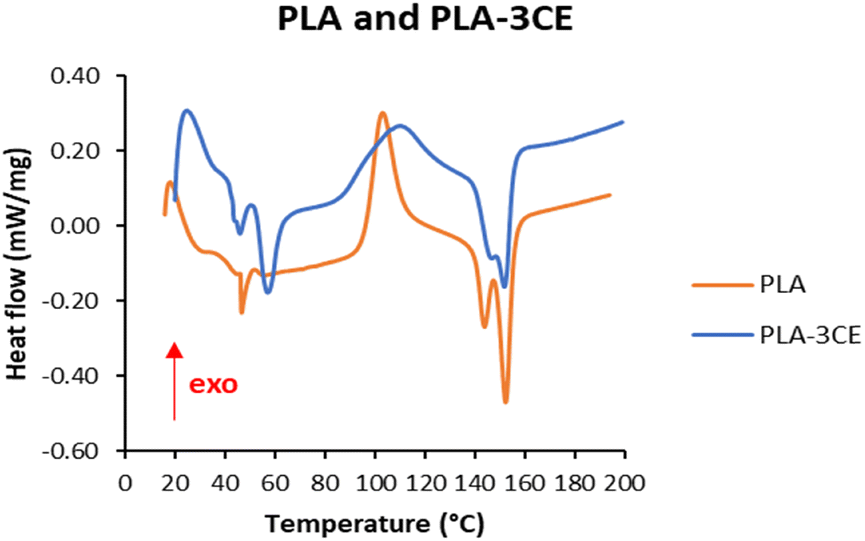
3.7 DSC thermograms of PLA and PLA–3CE composite films
Commercial PLA is a mixture of a higher amount of L-lactide (>95%) and a lower amount of D-lactide (<5%). The DSC thermogram of PLA used in this study (Fig. 8) agrees with the previous literature data.71 The glass transition temperature (Tg) of PLA is clearly visible as an endothermic peak around 46 °C, followed by the exothermic re-crystallization peak between 92 °C and 114 °C. Commercially available PLA structure has a disordered crystalline form α′ and an ordered crystalline form α. The double melting endotherm peak around 144 and 152 °C is probably due to the melting of the α′ phase, while the second peak corresponds to the melting of the α form.67,72
 |
| | Fig. 8 DSC thermograms of PLA and the composite PLA–3CE. | |
The fact that the particles of cellulose adipate CE4 are dispersed in the PLA matrix, but retain their crystalline structure (see infra §3.8), implies that there is an interfacial region in the PLA–3CE composite where PLA chains are in intimate contact with the surface of the particles of CE4. A reasonable hypothesis about the nature of this interface is that hydrogen bonds form between the CE4 surface and the ester groups of PLA, whereas H-bonds are not possible among PLA chains. It has to be considered that the CE4 surface contains both –OH groups along the polymer chain and also the free carboxilic acid groups of the partly esterified adipic acid, and the latter can potentially stick out of the surface and penetrate into the PLA interfacial region. The DSC thermogram of the PLA–3CE composite is similar to the DSC thermogram of pure PLA (Fig. 8), but with some relevant differences, even though the amount of CE in the composite (3 wt%) is small. After the first endothermic peak at around 46 °C, representing the glass transition, a new endothermic peak at 57 °C appears; the first signal might stem from the glass transition of PLA chains far from the interface, whereas the new signal at 57 °C may be the effect of the glass transition of the PLA chains at the interface, more rigid because of the immobilizing effect of the hydrogen bonds with cellulose adipate. The exothermic re-crystallization peak centered around 103.1 °C in PLA becomes a broad peak with a maximum at 110 °C and a shoulder at 93 °C in the composite; the increased width of the peak when cellulose adipate is present is a further hint of the fact that different PLA chains experience different environments. Crystallization requires the ordered re-organization of the amorphous, mobile PLA chains, and the relevant increase of 7 °C of the peak maximum may be a consequence of hydrogen bonds at the interface. The two fusion endotherms at 143.9 and 152.3 °C in PLA get closer in PLA–3CE, with minima at 146.9 and 151.9 °C: the melting of the disordered crystalline form α' is retarded, whereas the ordered crystalline form α melts at a lower temperature in the composite with respect to PLA. Additional information can be inferred from the heats accompanying the transformations in the two materials. The presence of cellulose adipate causes a decrease of both crystallization heats (from 27.6 to 22.4 J g−1) and fusion heats (from −25.6 to −23.0 J g−1) (Fig. S9 and S11†). On the contrary, the heat involved in the glass transition strongly increases (from −2.0 to −(2.85 + 9.30) J g−1) (Fig. S9 and S11†). Both effects indicate a larger amorphous fraction in the PLA–3CE composite material than in the neat PLA, maybe related to the presence of smaller crystalline PLA domains in the composite.
The second heating profiles of PLA and PLA–3CE (Fig. S10 and S12†) were recorded after the slow cooling (1 °C min−1) from 200 °C to room temperature. They are very similar, and this indicates that the heating and cooling cycle reduced the interactions between cellulose adipate and PLA, increasing the segregation of the two phases in the annealed composite.
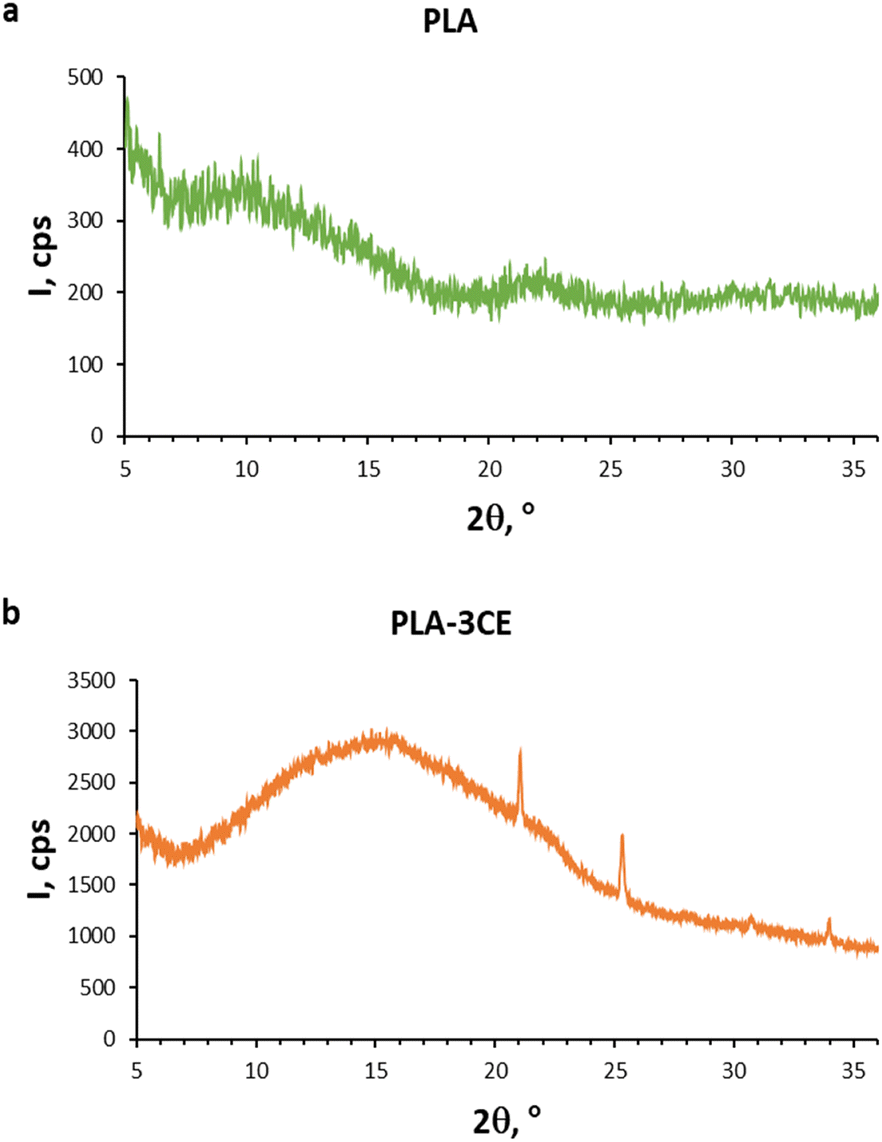
3.8 XRD spectra of PLA and PLA–3CE
The XRD spectrum of PLA in Fig. 9a represents the classic spectrum of an amorphous material.68 In the XRD spectrum of the PLA–3CE composite in Fig. 9b, however, in addition to a broad band between 5 and 25°, due to the amorphous portion, the crystalline peaks of the esterified cellulose CE4 appear. This fact indicates that no homogeneous blend is formed, since cellulose adipate particles retain their structural features in the composite.
 |
| | Fig. 9 XRD spectra of PLA (a) and the composite PLA–3CE (b). | |
3.9 Mechanical properties of PLA–CE composites
The mechanical properties of PLA–CE composites were evaluated and compared with neat PLA films in order to investigate the effect of cellulose ester on PLA tensile strength and elongation at break (Table 5). The tensile strength and Young modulus of the pure PLA films are 28 ± 2 MPa and 1.9 ± 0.1 GPa, respectively, in agreement with literature data.73,74 The introduction of more than 1% cellulose adipate in PLA results in a reduction of tensile strength of PLA–CE composites, which is roughly proportional to the weight percentages of CE4 in the films, reaching the minimum value (9 ± 3 MPa) for PLA–6CE. In turn, a rise in the percentage of elongation at breaking point, and thus in elasticity of PLA–CE composites, is observed. In this study, the combination of PLA and 3 wt% CE achieves the highest value of elasticity, exceeding of approximately 59% that of neat PLA films (3.5% vs. 2.2%, respectively). Therefore, with a tensile strength of only 25% below that of neat PLA films, the PLA–3CE composite represents the best compromise between a lower tensile strength and a higher elongation at break, reducing the brittle behavior of PLA. Results obtained in this work clearly confirm previous literature findings, exhibiting tensile strength values very close to those found in other studies with similar elastic properties.72,73,75 Little differences can be ascribed to the different chemical nature of dopant used, which clearly defines the best compromise in terms of doping amounts and mechanical values obtained.
Table 5 Mechanical properties of PLA–CE composites
| Entry |
Tensile strength (MPa) |
Elongation at break (%) |
| PLA |
28 ± 2 |
2.2 ± 0.4 |
| PLA–1CE |
30 ± 3 |
2.6 ± 0.5 |
| PLA–2CE |
19 ± 2 |
2.9 ± 0.5 |
| PLA–3CE |
21 ± 3 |
3.5 ± 0.3 |
| PLA–6CE |
9 ± 3 |
3.2 ± 0.3 |
3.10 Morphology analysis of pure PLA and PLA–3CE films by SEM
The SEM observation of the pristine PLA polymer (Fig. 10a) evidences the presence of several fractures (some millimeters long) in the polymer surface. Instead, such fractures are absent in the PLA–3CE composite (Fig. 10c). This indicates a reduction of the typical brittleness of PLA, compatible with the formation of strong interactions, such as hydrogen bonds, between PLA and cellulose adipate, in which the latter acts as a binder of PLA chains. As evidenced by the SEM micrographs, PLA and PLA–3CE films exhibit a smoother surface morphology on the side in contact with the Petri dish (Fig. 10b and d) compared to the side exposed to air (Fig. 10a and c). A lesser homogeneity of the PLA–3CE surfaces is in agreement with the findings of thermal analysis, stating that PLA experiences different environments in the composite.
 |
| | Fig. 10 SEM images of PLA and PLA–3CE film surfaces, exposed to air (a and c) and in contact with the Petri dish (b and d). | |
Conclusions
A green and sustainable procedure for the synthesis of a new dispersable and low melting cellulosic material via esterification with adipic acid has been devised, implemented and optimized. The solvent-free reaction is fast, has a low environmental impact and takes place at atmospheric pressure. The simplicity of the proposed process could allow an easy scale-up from the laboratory to the industrial level. Adipic acid plays the role of both reagent and catalyst in this reaction, which allows to avoid the use of either strong catalysts or more reactive reagents. Titrations, IR and solid-state NMR experiments show that the new cellulosic material is characterized by little or no crosslinking. According to XRD and TGA, the new cellulosic material is made of highly crystalline cellulose adipate, surrounding an intact cellulose core, in a 3:1 mass ratio. It is dispersable in common organic chlorinated solvents at room temperature and was used as an additive in the preparation of PLA films. The new cellulosic material is compatible with PLA and mitigates its brittleness, since even a small amount of this additive results in a notable enhancement in the elastic properties of the composite. The maximum elongation of all the composite films improved with respect to pure PLA, with an increase of 59% when 3 wt% of additive was added. The same composite still maintained a good tensile strength of 21 ± 3 MPa, only 25% less than that of pure PLA. The new green and sustainable synthetic procedure disclosed by this work allows to produce a low melting and dispersible cellulose ester, paving the way for more uses of cellulose adipate in different applications, as such or as a platform for further modifications. Once again new materials with improved performance can be produced from renewable resources and readily available reagents by simple and green procedures.
Data availability
The data underlying this study are available in the published article and its online ESI.†
Author contributions
MariaFrancesca Baratta: software, validation, writing – reviewing and editing; Fabrizio Olivito: methodology, conceptualization, supervision; writing – original draft preparation, writing – reviewing and editing; Cataldo Simari: software; validation; visualization; Wan Abd Al Qadr Imad Wan-Mohtar: software; validation; visualization; Isabella Nicotera: software; validation; visualization; Fiore Pasquale Nicoletta: software; validation; visualization; Giovanni De Filpo: software; validation; visualization; Giovanni Golemme: supervision, conceptualization, writing – original draft preparation, writing – reviewing and editing.
Conflicts of interest
There are no conflicts to declare.
References
- S. Adhikari, V. Kelkar, R. Kumar and R. U. Halden, Polym. Int., 2022, 71, 543–551 CrossRef CAS.
- C. Elgin, G. Özgür and K. Cantekin, Sustainable Development, 2023, 31(1), 1–11 CrossRef.
- V. G. Zuin, I. Eilks, M. Elschami and K. Kümmerer, Green Chem., 2021, 23, 1594–1608 RSC.
- F. Olivito, V. Algieri, A. Jiritano, M. A. Tallarida, P. Costanzo, L. Maiuolo and A. De Nino, Polymer, 2023, 15, 1785 CAS.
- C. Especel, G. Lafaye and F. Epron, ChemCatChem, 2023, 15, e202201478 CrossRef CAS.
- J. Winfield, L. D. Chambers, J. Rossiter, A. Stinchcombe, X. A. Walter, J. Greenman and I. Ieropoulos, ChemSusChem, 2015, 8, 2705–2712 CrossRef CAS PubMed.
-
A. Tursi and F. Olivito, 1 - Biomass conversion: general information, chemistry, and processes, in Advances in Bioenergy and Microfluidic Applications, ed. M. R. Rahimpour, R. Kamali, M. A. Makarem and M. K. Dehghan Manshadi, Elsevier, The Netherlands, 2021, pp. 3–39 Search PubMed.
- F. Olivito, P. Jagdale and G. Oza, ACS Sustainable Chem. Eng., 2023, 11(50), 17595–17599 CrossRef CAS.
- S. Fulignati, C. Antonetti, T. Tabanelli, F. Cavani and A. M. Raspolli Galletti, ChemSusChem, 2022, 15, 1–14 CrossRef PubMed.
- F. Olivito, V. Algieri, M. A. Tallarida, A. Jiritano, P. Costanzo, L. Maiuolo and A. De Nino, Green Chem., 2023, 25, 1679–1689 RSC.
- C. Espro, E. Paone, F. Mauriello, R. Gotti, E. Uliassi, M. L. Bolognesi, D. Rodríguez-Padrón and R. Luque, Chem. Soc. Rev., 2021, 50, 11191–11207 RSC.
- C. G. Otoni, H. M. C. Azeredo, B. D. Mattos, M. Beaumont, D. S. Correa and O. J. Rojas, Adv. Mater., 2021, 33, 2102520 CrossRef CAS PubMed.
- B. Wang, S. Ma, Q. Li, H. Zhang, J. Liu, R. Wang, Z. Chen, X. Xu, S. Wang, N. Lu, Y. Liu, S. Yan and J. Zhu, Green Chem., 2020, 22, 1275–1290 RSC.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- S. N. Idris, T. S. M. Amelia, K. Bhubalan, A. M. M. Lazim, N. A. M. A. Zakwan, M. I. Jamaluddin, R. Santhanam, A. A. Amirul, S. Vigneswari and S. Ramakrishna, Environ. Res., 2023, 231(Pt 1), 115988 CrossRef CAS PubMed.
- D. Al-Khairy, W. Fu, A. S. Alzahmi, J. C. Twizere, S. A. Amin, K. Salehi-Ashtiani and A. Mystikou, Microorganisms, 2022, 23, 2320 CrossRef PubMed.
- M. S. Kim, H. Chang, L. Zheng, Q. Yan, B. F. Pfleger, J. Klier, K. Nelson, E. L.-W. Majumder and G. W. Huber, Chem. Rev., 2023, 123(16), 9915–9939 CrossRef CAS PubMed.
- F. Olivito, P. Jagdale and G. Oza, Toxics, 2023, 11, 698 CrossRef CAS PubMed.
- D. Tan, Y. Wang, Y. Tong and G. Q. Chen, Trends Biotechnol., 2021, 39, 953–963 CrossRef CAS PubMed.
- Y. Boonluksiri, B. Prapagdee and N. Sombatsompop, Polym. Degrad. Stab., 2021, 188, 109562 CrossRef CAS.
- P. B. Patil, D. Sarkar, K. Poddar and A. Sarkar, J. Chem. Technol. Biotechnol., 2023, 98, 615–624 CrossRef CAS.
- V. V. Andhalkar, S. Y. Foong, S. H. Kee, S. S. Lam, Y. H. Chan, R. Djellabi, K. Bhubalan, F. Medina and M. Constantí, Macromol. Mater. Eng., 2023, 308, 2300100 CrossRef CAS.
- H. R. Dana and F. Ebrahimi, Polym. Eng. Sci., 2023, 63(1), 22 CrossRef.
- T. A. Swetha, A. Bora, K. Mohanrasu, P. Balaji, R. Raja, K. Ponnuchamy, G. Muthusamy and A. Arun, Int. J. Biol. Macromol., 2023, 234, 123715 CrossRef CAS PubMed.
- X. Li, Y. Lin, M. Liu, L. Meng and C. Li, J. Appl. Polym. Sci., 2023, 140(7), e53477 CrossRef CAS.
- B. Wang, Z. Qi, X. Chen, C. Sun, W. Yao, H. Zheng, M. Liu, W. Li, A. Qin, H. Tan and Y. Zhang, Int. J. Biol. Macromol., 2022, 217, 792–802 CrossRef CAS PubMed.
- X. Song, C. Zhang, Y. Yang, F. Yang and Y. Weng, Polym. Compos., 2023, 44(9), 5675–5688 CrossRef CAS.
- T. Peixoto, J. Nunes, M. A. Lopes, E. Marinho, M. F. Proença, P. E. Lopes and M. C. Paiva, Polym. Compos., 2022, 43(11), 2518 Search PubMed.
- B. Y. Gul, E. Pekgenc, V. Vatanpour and I. Koyuncu, Carbohydr. Polym., 2023, 321, 121296 CrossRef PubMed.
- J. Antti Sirviö, M. Visanko and H. Liimatainen, Green Chem., 2015, 17, 3401–3406 RSC.
- R. Khan, R. Jolly, T. Fatima and M. Shakir, Polym. Adv. Technol., 2022, 33(7), 2069–2090 CrossRef CAS.
- A. A. Abe, C. Oliviero Rossi and P. Caputo, Molecules, 2022, 12, 8826 CrossRef PubMed.
- Y. Habibi, L. A. Lucia and O. J. Rojas, Chem. Rev., 2010, 110(6), 3479–3500 CrossRef CAS PubMed.
- I. Romeo, F. Olivito, A. Tursi, V. Algieri, A. Beneduci, G. Chidichimo, L. Maiuolo, E. Sicilia and A. De Nino, RSC Adv., 2020, 10, 34738–34751 RSC.
- Z.-M. Xu, J.-Y. Luo and Y.-B. Huang, Green Chem., 2022, 24, 3895–3921 RSC.
- M. Wu, J. Di, L. Gong, Y.-C. He, C. Ma and Y. Deng, J. Chem. Eng., 2023, 452(2), 139320 CrossRef CAS.
- N. M. Noor, A. Sendijarevic, V. Sendijarevic, I. Sendijarevic, T. N. M. Tuan Ismail, M. A. Mohd Noor, Y. Shoot Kian and H. Abu Hassan, J. Am. Oil Chem. Soc., 2016, 93, 1529–1540 CrossRef.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- M. Lang and H. Li, ChemSusChem, 2022, 15, e202101531 CrossRef CAS PubMed.
- B. Priya, A. Kumar, P. Garg, U. Deshpande and S. K. Singh, ChemCatChem, 2023, 15, e202300863 CrossRef CAS.
- W. Niu, H. Willett, J. Mueller, X. He, L. Kramer, B. Ma and J. Guo, Metab. Eng., 2020, 59, 51–161 CrossRef PubMed.
- E. Skoog, J. H. Shin, V. Saez-Jimenez, V. Mapelli and L. Olsson, Biotechnol. Adv., 2018, 36(8), 2248–2263 CrossRef CAS PubMed.
- L. Mestrom, J. G. R. Claessen and U. Hanefeld, ChemCatChem, 2019, 11, 2004 CrossRef CAS.
- S. Blohm and T. Heinze, Macromol. Chem. Phys., 2020, 221, 2000264 CrossRef CAS.
- K. M. Venables, Br. J. Ind. Med., 1989, 46(4), 222–232 CAS.
- Z. Khan, F. Javed, Z. Shamair, A. Hafeez, T. Fazal, A. Aslam, W. B. Zimmerman and F. Rehman, J. Ind. Eng. Chem., 2021, 103, 80–101 CrossRef CAS.
- Y. Ma, X. You, M. Rissanen, I. Schlapp-Hackl and H. Sixta, ACS Sustainable Chem. Eng., 2021, 9(49), 16749–16756 CrossRef CAS.
- J. Batta-Mpouma, G. Kandhola, J. Sakon and J.-W. Kim, Biomacromolecules, 2022, 23(10), 4085–4096 CrossRef CAS PubMed.
- N. Kar, H. Liu and K. J. Edgar, Biomacromolecules, 2011, 12(4), 1106–1115 CrossRef CAS PubMed.
- D. Li, J. Henschen and M. Ek, Green Chem., 2017, 19, 5564–5567 RSC.
- Z. Khan, F. Javed, Z. Shamair, A. Hafeez, T. Fazal, A. Aslam, W. B. Zimmerman and F. Rehman, J. Ind. Eng. Chem., 2021, 103, 80–101 CrossRef CAS.
- P. B. Groszewicz, P. Mendes, B. Kumari, J. Lins, M. Biesalski, T. Gutmann and G. Buntkowsky, Cellulose, 2020, 27, 1239–1254 CrossRef CAS.
- P. T. Larsson, E. L. Hult, K. Wickholm, E. Pettersson and T. Iversen, Solid State Nucl. Magn. Reson., 1999, 15, 31–40 CrossRef CAS PubMed.
- F. Barboza, D. D. Vecchia and M. P. Tagliari,
et al.
, Pharm. Chem. J., 2009, 43, 363–368 CrossRef CAS.
- L. Crépy, V. Miri, N. Joly, P. Martin and J.-M. Lefebvre, Carbohydr. Polym., 2011, 83(4), 1812–1820 CrossRef.
- W. K. Ng, J. W. Kwek, A. Yuen, C. L. Tan and R. Tan, AAPS PharmSciTech, 2010, 11(1), 159–167 CrossRef CAS PubMed.
- X. Zhao, I. Anwar, X. Zhang, A. Pellicciotti, S. Storts, D. A. Nagib and Y. Vodovotz, ACS Omega, 2021, 6(38), 24700–24708 CrossRef CAS PubMed.
- A. Thygesen, J. Oddershede, H. Lilholt, A. B. Thomsen and K. Stahl, Cellulose, 2005, 12, 563–576 CrossRef CAS.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels Bioprod., 2010, 3, 10 CrossRef PubMed.
- J. He, S. Cui and S.-Y. Wang, J. Appl. Polym. Sci., 2008, 107, 1029–1038 CrossRef CAS.
- M. L. Pitcher, R. Koshani and A. Sheikhi, J. Polym. Sci., 2024, 62(1), 9 CrossRef CAS.
- C. J. Garvey, I. H. Parker and G. P. Simon, Macromol. Chem. Phys., 2005, 206, 1568–1575 CrossRef CAS.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J. L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9(1), 57–65 CrossRef CAS PubMed.
- Ju. Xiaohui, M. Bowden, E. E. Brown and X. Zhang, Carbohydr. Polym., 2015, 123, 476–481 CrossRef PubMed.
- B. Lindner, L. Petridis, P. Langan and J. C. Smith, Biopolymers, 2015, 103, 67–73 CrossRef CAS PubMed.
- J. Sugiyama, R. Vuong and H. Chanzy, Macromolecules, 1991, 24, 4168–4175 CrossRef CAS.
- Q. Chen,
et al.
, BioResources, 2016, 11(1), 159–173 CrossRef CAS.
- Z. Wang, J. Yu, L. Zhang, Y. Zhou, Y. Yang and Y. Jin, Cellulose, 2017, 24, 5069–5078 CrossRef CAS.
-
J. M. Sugihara, Relative Reactivities of Hydroxyl Groups of Carbohydrates, in Advances in Carbohydrate Chemistry, ed. C. S. Hudson and M. L. Wolfrom, Academic Press, 1953, vol. 8 Search PubMed.
- A. Hadjizadeh, H. Savoji and A. Ajji, BioMed Res. Int., 2016, 8921316 Search PubMed.
- T. Tábi, I. E. Sajó, F. Szabó, A. S. Luyt and J. G. Kovács, eXPRESS Polym. Lett., 2010, 4(10), 659–668 CrossRef.
- P. Pan, B. Zhu, W. Kai, T. Dong and Y. Inoue, Macromolecules, 2008, 41, 4296–4304 CrossRef CAS.
- N. Jamaluddin, T. Kanno, T.-A. Asoh and H. Uyama, Mater. Today Commun., 2019, 21, 10058 Search PubMed.
- A. Boarino, A. Schreier, Y. Leterrier and H.-A. Klok, ACS Appl. Polym. Mater., 2022, 4(7), 4808–4817 CrossRef CAS PubMed.
- W. Ren, X. Pan, G. Wang, W. Cheng and Y. Liu, Green Chem., 2016, 18, 5008–5014 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Fabrizio
Olivito
a,
Fabrizio
Olivito