
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Usage of CoOOH electrodes in a flow channel reactor for the non-alkaline oxidation of 5-(hydroxymethyl)-furfural: an upscaling study†
Marten Niklas
Gey
 ,
Carl
Schneider
and
Uwe
Schröder
*
,
Carl
Schneider
and
Uwe
Schröder
*
Institute of Biochemistry, University of Greifswald, Felix-Hausdorff-Str. 4, 17489 Greifswald, Germany. E-mail: uwe.schroeder@uni-greifswald.de
First published on 22nd November 2024
Abstract
A major barrier for the upscaling of electrosynthetic methods is the transfer of the usually potential-controlled batch experiments to an operation in industry-typical cell designs (i.e. two-electrode flow reactors). To cross this bridge, we here present the implementation of our recently published method for the non-alkaline oxidation of 5-(hydroxymethyl)-furfural (HMF) to 2,5-furandicarboxylic acid (FDCA) in a flow channel reactor, powered by a standard laboratory power supply under cell-voltage/current control. For this purpose, the coating method for the used CoOOH catalyst was adapted to enable an electrochemical deposition in the flow channels devoid of a standard three-electrode setup. HMF oxidations were carried out in an acetate buffer (pH 5) at a current density of 1.0 mA cm−2 and a temperature range between room temperature and 80 °C to provide a direct comparison with the previous batch experiments. The higher electrode surface area of the flow cell thereby allowed a significant reduction of the reaction time while operating under similar (albeit lower) Coulomb efficiencies. Under optimized conditions, the reactor operated at a cell voltage of ca. 2.4 V and yielded 77.1% FDCA at a Coulomb efficiency of 21.0%. Maleic acid was obtained as a side product at a yield of 9.2%.
Introduction
In the past few years, electrosynthesis has been established as a sustainable method for the production of organic compounds.1,2 The use of electricity from renewable sources as a driving force for the conversion of substances is in line with numerous major principles of green chemistry, including energy efficiency, the use of mild reaction conditions and the avoidance of hazardous oxidizing/reducing agents and resulting wastes.3,4 Especially for the production of bulk chemicals from biomass (i.e. platform chemicals), electrochemistry offers an ideal way to perform the necessary de- or refunctionalization steps of the used biogenic molecules – which oftentimes feature a high degree of functionalization.5–8 However, despite the considerable number of electro-organic syntheses published to date, only a few methods have been able to leave the laboratory scale so far.9–11 A general problem here is the transition from the commonly used batch setups (e.g. H-type glass cells) to more yield-efficient reactor designs. Batch reactors allow the precise control of the electrochemical processes via the application of a defined potential (measured and controlled by a reference electrode), which makes them ideal for fundamental research. However, this setup is typically constrained to relatively small electrodes, limiting the possible substrate turnover and thus leading to long reaction times for the production of significant product amounts. In contrast, continuously operated reactors enable larger electrode surface areas (e.g. by using flow channel structures) and consequently significantly increased electrode area-to-reactor volume ratios (Aelectrode/Vreactor). Higher charge transfer rate and space–time yields can thus be achieved.12 Flow channel designs, however, do not allow the implementation of reference electrodes.13 Therefore, a direct transfer of potential-based methods developed in batch cells is not possible. Accordingly, research not only needs to focus on the development of new electrosynthetic methods, but also on their transfer and implementation in industry-typical flow reactor setups, where only the application of either a current or a cell voltage (as opposed to a defined electrode potential) is possible.Recently, we presented a new method for the anodic oxidation of the sugar-based platform chemical 5-(hydroxymethyl)-furfural (HMF) to 2,5-furandicarboxylic acid (FDCA),14 which is a reaction of relevance for the production of biopolymers (e.g. polyethylene furanoate, PEF).15–17 By using CoOOH modified electrodes, we were able to perform this reaction in an acetate buffer (pH 5), which provides several process-related advantages over the conventionally used alkaline systems. For instance, HMF is comparatively stable in this pH-region, whereas in alkaline solutions, it undergoes self-polymerization reactions to humic substances.18,19 Furthermore, alkaline electrolytes need to be neutralized in order to crystallize the poorly water-soluble FDCA,20,21 requiring high amounts of acid and producing equal amounts of salt waste.22 In addition, the acetate electrolyte can also be obtained from biomass, thus strengthening the sustainability aspect of this work. Due to these advantages this system is a suitable benchmark for the use on a larger scale.
In this study, we demonstrate the transfer of our CoOOH-catalyzed HMF oxidation method from the previously used three-electrode batch cell (3E-batch cell) to a two-electrode flow cell (2E-flow cell). As a target reactor, a custom-made flow cell with a spiral-shaped channel design was used. This design exhibited a 100-fold higher Aelectrode/Vreactor ratio than the 3E-batch cell, allowing a substantial decrease of the reaction time (Table 1, columns II. and IV.). Furthermore, a more cost-efficient power supply (ca. 100 €) was used to carry out the electrosyntheses instead of a considerably more expensive potentiostat, whose prices typically range between 5000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 € for single-channel systems. As the HMF oxidation in the batch system was based on the application of a current density,14 the method was directly transferable to the flow system without adaptation. However, for the preparation of the CoOOH modified electrodes, we adapted a procedure from the literature, which relied on the application of potentials.23 This procedure is based on the precipitation of Co(OH)2 from a Co(NO3)2 solution onto a desired electrode support caused by an electrochemically driven alkaline pH shift at the electrode surface (cathodic phase, eqn (1) and (2)), and the subsequent oxidation to CoOOH in a NaOH solution (anodic phase, eqn (3)).
000 € for single-channel systems. As the HMF oxidation in the batch system was based on the application of a current density,14 the method was directly transferable to the flow system without adaptation. However, for the preparation of the CoOOH modified electrodes, we adapted a procedure from the literature, which relied on the application of potentials.23 This procedure is based on the precipitation of Co(OH)2 from a Co(NO3)2 solution onto a desired electrode support caused by an electrochemically driven alkaline pH shift at the electrode surface (cathodic phase, eqn (1) and (2)), and the subsequent oxidation to CoOOH in a NaOH solution (anodic phase, eqn (3)).
 | (1) |
 | (2) |
 | (3) |
| I. 3E-batch cell (CoOOH coating) | II. 3E-batch cell (HMF oxidation) | III. 3E-flow cell (CoOOH coating) | IV. 2E-flow cell (CoOOH coating & HMF oxidation) | |
|---|---|---|---|---|
| Cell design | Three-electrode, unseparated | Three-electrode, separated | Three-electrode, separated | Two-electrode, separated |
| Electrode design | Planar | Planar | Planar | Flow channels |
| A electrode | 14.0 cm2 | 10.0 cm 2 | 10.0 cm2 | 52.2 cm 2 |
| d WE–CE | 0.9 cm | 7.0 cm | 0.9 cm | 0.4 cm |
| V reactor | 40 mL | 50 mL/half-cell | 4.1 mL/half-cell (50 mL reservoir) | 2.6 mL/half-cell (50 mL reservoir) |
| A electrode/Vreactor | 0.35 cm−1 | 0.20 cm −1 | 2.44 cm−1 | 20.08 cm −1 |
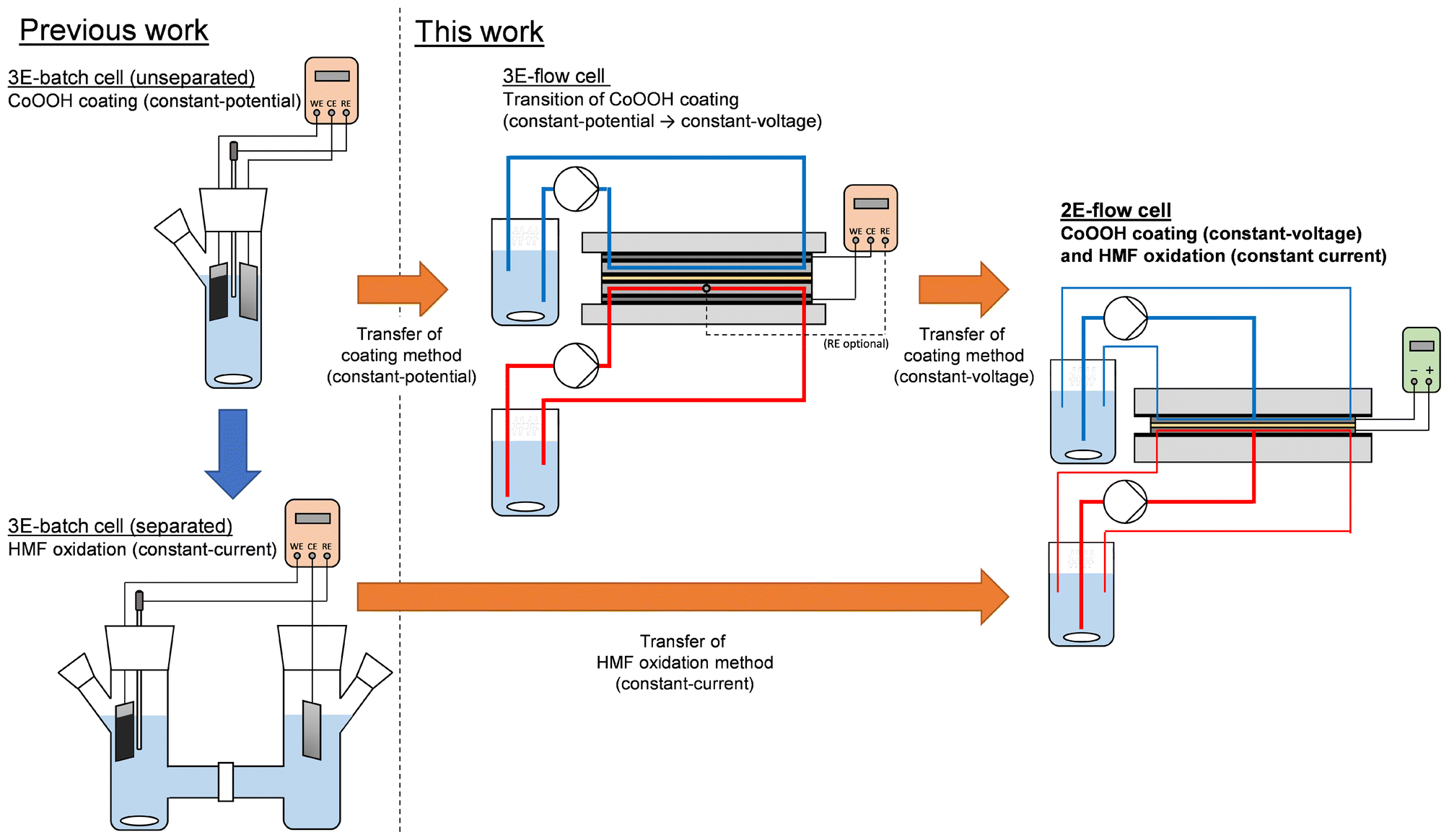 | ||
| Scheme 1 Overview of the transfer methods used for the CoOOH coating and the HMF oxidation. | ||
Results and discussion
Transition of the CoOOH coating method
In order to determine the cell voltages required for the electrode coating in the 2E-flow cell, experiments were carried out in a separated 3E-flow cell equipped with an anion exchange membrane (AEM) to correlate the electrode potential as controlled by the integrated reference electrode and the resulting cell voltage. As an initial experiment, CoOOH electrodes were prepared under application of the same potentials that we used previously in the unseparated 3E-batch cell (Table 1, column I.).14 Thus, for the reductive Co(OH)2 deposition, a potential of −1.00 VAg/AgCl was applied to the titanium electrode in 50 mM Co(NO3)2 solution until a charge density σRed of 0.8 C cm−2 was transferred (σRed = QRed/Aelectrode). Afterwards, the oxidation to CoOOH was performed in 1.0 M NaOH solution at 0.40 VAg/AgCl until the current density dropped below 0.35 mA cm−2. A flow rate of 5 mL min−1 was used. Exemplary current densities and cell voltages of both the cathodic and the anodic phase are shown in Fig. 1 by the black dashed curve (3E-batch cell) and the black solid curve (3E-flow cell). | ||
| Fig. 1 Comparison of the potentiostatic coating experiments in the 3E-batch cell and the 3E-flow cell: a) current density and b) cell voltage curves during reductive Co(OH)2 deposition at different potentials, c) current density, d) cell voltage and e) charge density curves during oxidation to CoOOH at 0.4 VAg/AgCl, f) photographs of the electrodes. | ||
In general, during the cathodic phase slightly lower current density values and at a substantially higher cell voltage was measured in the 3E-flow cell in comparison to the 3E-batch cell (Fig. 1a and b). Since the electrode distance dWE–CE was similar in both cells (Table 1, columns I. and III.), this effect can be attributed to the presence of the AEM in the 3E-flow cell, which increased the cell resistance. Furthermore, an initial phase was observed in both cells, in which the current density and the cell voltage decreased rapidly within the first 2 min of the measurement. A similar behavior was already observed in previous studies, in which the peak was attributed to the formation of titanium hydride species.24 This effect was more pronounced in the 3E-flow cell.
During the anodic phase, a typical thin-film behavior was observed in both cells. Since only the oxidation of Co(OH)2 to CoOOH (eqn (3)) could proceed at the applied potential,14 a complete conversion was observed, causing a drop of the current density to almost zero within seconds (Fig. 1c). Although both cells showed a similar peak current density, the current flow in the 3E-flow cell lasted for a longer period of time so that higher overall charge density values were reached. Thus, a charge density σOx of 0.48 C cm−2 was transferred in the 3E-flow cell as compared to 0.28 C cm−2 in the 3E-batch cell (see black curves in Fig. 1e). According to Faraday's law (eqn (4)), these values correspond to deposited CoOOH mass loadings mCoOOH of 0.46 mg cm−2 (3E-flow cell) and 0.27 mg cm−2 (3E-batch cell).
 | (4) |
σ Ox: charge density transferred during oxidation
M CoOOH: molar mass of CoOOH (91.93 g mol−1)
z: number of electrons transferred per reaction
F: Faraday's constant
Despite the higher mass loading, upon visual inspection of the coated working electrodes, the obtained CoOOH film of the 3E-flow cell showed a higher degree of inhomogeneity as compared to the 3E-batch cell electrode, so that parts were already physically detached after the coating process (Fig. 1f, “−1.00 VAg/AgCl”). We assume that this result was caused by the different cell geometry and convection properties of the 3E-flow cell. Here, the lower current density and the stronger formation of titanium hydride during the cathodic phase indicated that the nitrate reduction (eqn (1)) was suppressed (e.g. by mass transfer limitation), leading to a stronger participation of the hydrogen evolution reaction (HER, eqn (5)).24,25
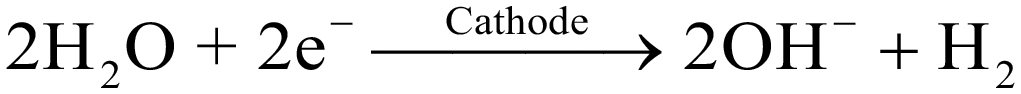 | (5) |
Although the production of hydroxide ions during the HER would also have led to the formation of Co(OH)2 (eqn (2)), the deposition of the film could have been disturbed by the formation of gaseous hydrogen, resulting in a lower degree of attachment to the electrode support. Thus, in the 3E-batch cell, stirring of the Co(NO3)2 solution prevented a mass transfer limitation of the nitrate, leading to a smaller titanium hydride peak and a higher uniformity of the CoOOH film.
In order to decrease the rate of hydrogen formation, coating experiments were conducted at less reductive potentials of −0.95, −0.90 and −0.85 VAg/AgCl in the 3E-flow cell (exemplary curves shown in red, green and blue in Fig. 1). Accordingly, lower absolute current densities and cell voltages were measured during the cathodic phase, meaning that an increasingly longer deposition time was necessary to reach the required charge density σRed of 0.8 C cm−2 (Fig. 1a and b). Additionally, smaller initial current peaks were measured, indicating a decreased formation of the titanium hydride species. During the anodic phase, the films deposited at a less negative reduction potential showed an increased current density, which also resulted in higher cell voltages during the current peak (Fig. 1c and d). Interestingly, the obtained σOx values were similar for the films deposited at −1.00 and −0.95 VAg/AgCl (0.41 and 0.45 C cm−2, respectively) as well as the films deposited at −0.90 and −0.85 VAg/AgCl (both 0.71 C cm−2, Fig. 1e). This indicated that different deposition conditions prevailed in the respective potential areas, with milder reduction potentials leading to a better Co(OH)2 deposition. Accordingly, the film deposited at −0.95 VAg/AgCl also showed some detached fragments, while this was not the case for the films deposited at −0.90 and −0.85 VAg/AgCl (Fig. 1f). Deposition at these potentials resulted in homogenous films, which were also subjectively more resistant to mechanical stress (e.g. scrubbing with a paper towel). We note that replicated experiments resulted in different curve shapes during the anodic phase. It is assumed that this is an effect of different morphologies of the deposited films, caused e.g. by non-identical flow conditions within the cell. However, the coating method proved to be robust to the extent that similar σOx values (and therefore similar CoOOH loadings) were always obtained at the same conditions despite the different shape of the current curves.
The obtained results demonstrate that a higher CoOOH loading and film stability can be achieved, if the Co(OH)2 deposition is performed at a lower cell voltage. To counter-check this insight, coating experiments were conducted in the 3E-flow cell under the application of constant cell voltages. For the cathodic phase, cell voltages of −2.75 V and −3.50 V (with respect to the working electrode) were applied, as these were the approximative cell voltages that resulted from the potentiostatic depositions at −1.00 to −0.95 VAg/AgCl and −0.90 to −0.85 VAg/AgCl, respectively (Fig. 1b). Accordingly, similar current densities to the respective potential-based experiments were measured for both voltages (see dashed curves in Fig. 2a). For the oxidation of CoOOH, a cell voltage of 1.60 V was applied, which corresponds to the value obtained for all deposited films after the decrease of the current peak (Fig. 1d). This ensured that no overoxidation (e.g. to CoO2) could occur. Accordingly, the resulting current densities were considerably lower and the oxidation required a longer duration (Fig. 2b). However, comparable charge densities σOx of 0.50 C cm−2 at −3.30 V and 0.75 C cm−2 at −2.75 V were reached, with the film deposited at −3.30 V showing a higher inhomogeneity (Fig. 2c and d).
 | ||
| Fig. 2 Comparison of the constant-voltage coating experiments in the 3E-flow cell and the 2E-flow cell: a) current density curves during reductive Co(OH)2 deposition and b) oxidation to CoOOH, c) resulting charge densities during the oxidation, d) photographs of the electrodes. | ||
As a result of these experiments, the CoOOH coating of the channel-type 2E-flow cell was performed at cell voltages of −2.75 V (cathodic phase) and 1.6 V (anodic phase). The resulting current densities during both phases are depicted as red solid curves in Fig. 2a and b.
While the reductive deposition proceeded at a similar current density compared to the 3E-flow cell, the oxidation showed a significantly lower current density of <0.4 mA cm−2. As the previously used limit value of 0.35 mA cm−2 was considered too high, the oxidation was stopped only at current densities of <0.1 mA cm−2. Despite this different behavior, a charge density σOx of 0.49 C cm−2 was transferred, showing that the CoOOH loading was in a comparable order of magnitude to the films of the 3E-flow cell (Fig. 2c). As expected from the previous results, the CoOOH film showed no uncoated areas and was distributed homogeneously over the channel structure (Fig. 2d). However, the effect on the current density was very interesting to observe, since the change of the electrode shape alone – and consequently the flow conditions – led to this considerably slower oxidation of the Co(OH)2 film. In addition, the electrode distance dWE–CE of the 2E-flow cell was smaller than in the 3E-flow cell (Table 1, columns III. and IV.), so that the opposite effect would have been expected due to the decrease of electrolyte resistance. For further investigation, we measured voltage-based cyclic voltammograms of deposited Ti/Co(OH)2 electrodes in the 3E-flow cell and the 2E-flow cell to compare the current response of the oxidation to CoOOH (Fig. S4 in the ESI†). These measurements showed, that in the 2E-flow cell the Co(OH)2 oxidation started at higher cell voltages and with a smaller current slope than in the 3E-flow cell, indicating an inhibition of the Co(OH)2 oxidation. Furthermore, the respective current peak did not show a clear separation from the subsequent current increase of the OER in the 2E-flow cell, while this was the case in the 3E-flow cell. Therefore, it was not possible to accelerate the oxidation by increasing the cell voltage, since the competing OER would have affected the calculation of the CoOOH loading mCoOOH by the charge density σOx.
With the adapted coating method, a CoOOH loading of 0.44 ± 0.04 mg cm−2 was yielded in the 2E-flow cell at a deposition charge density σRed of 0.8 C cm−2. In order to obtain additional data on the change of the CoOOH loading mCoOOH, further coating experiments were carried out by varying the deposition charge density σRed (Table 2). A linear dependence between the two values was observed in the investigated range, which was in agreement with the results from the 3E-batch cell.
| σ Red | m CoOOH (3E-batch cell)14 | m CoOOH (2E-flow cell) |
|---|---|---|
| 0.4 C cm−2 | 0.17 ± 0.02 mg cm−2 | 0.27 ± 0.02 mg cm−2 |
| 0.8 C cm−2 | 0.25 ± 0.02 mg cm−2 | 0.44 ± 0.04 mg cm−2 |
| 1.6 C cm−2 | 0.48 ± 0.02 mg cm−2 | 0.84 ± 0.07 mg cm−2 |
| 2.4 C cm−2 | 0.76 ± 0.05 mg cm−2 | 1.19 ± 0.01 mg cm−2 |
HMF oxidation
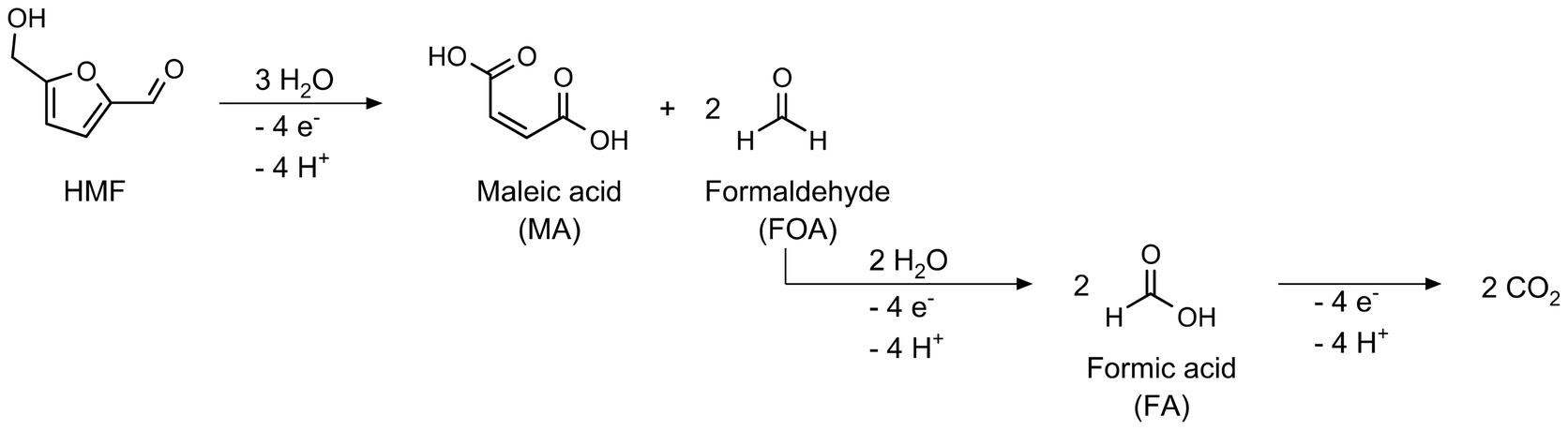
The oxidation of HMF to FDCA proceeds via three intermediate products. Depending on which reaction path is favored, the hydroxy group or the aldehyde group of the HMF molecule becomes oxidized first, leading to 2,5-diformylfuran (DFF) or 5-(hydroxymethyl)-2-furancarboxylic acid (HMFCA). Both products are then further oxidized to 5-formyl-2-furancarboxylic acid (FFCA), which is afterwards oxidized to FDCA (Scheme 2). | ||
| Scheme 2 Reaction pathways and intermediates of the HMF oxidation to FDCA.14 | ||
In our previous study, we demonstrated the use of CoOOH electrodes for the galvanostatic oxidation of HMF to FDCA in acetate-buffered solutions (pH 5).14 For this system, we found that the HMF oxidation proceeds mainly over the DFF pathway, whereby the consecutive oxidation steps became increasingly slower. In optimization reactions, the highest Coulomb efficiencies CE were reached at high CoOOH loadings (0.76 ± 0.05 mg cm−1, Table 2), low current densities (1.0 mA cm−1) and high temperatures (80 °C). Since the latter parameter had the greatest influence on the CE, we used it to directly compare the HMF oxidation in the 3E-batch cell and the 2E-flow cell. Thus, oxidation experiments of 20 mM HMF solutions were carried out in the 2E-flow cell at room temperature (RT), 40, 60 and 80 °C, with an analogous CoOOH loading (0.27 ± 0.02 mg cm−2, Table 2) and current density (1.0 mA cm−1) as used in the batch experiments. As this current density was not sufficient to provide a complete turnover in a single pass of the reactor, the reaction solutions were pumped in cycles with a flow rate of 5 mL min−1. The resulting cell voltages of the reactions ranged between 3.3 V at RT and 2.1 V at 80 °C (Fig. S6a in the ESI†). The HMF solution was analyzed by HPLC before and after the reaction to determine the respective performance parameters (HMF conversion X, product yields Y, Coulomb efficiencies CE and mole balance MB) according to eqn (S1)–(S5) in the ESI.† The resulting performance parameters obtained after a transferred charge of 6 Faraday equivalents (F) – the theoretical charge necessary to fully oxidize HMF to FDCA – are shown in Fig. 3. To consider the entire HMF-to-FDCA process for the efficiency analysis of the system, the single Coulomb efficiencies of DFF, HMFCA, FFCA and FDCA were summed up to a total Coulomb efficiency (CEtot, eqn (S4) in the ESI†). The graphical depiction of the single Coulomb efficiencies can be found in Fig. S7 in the ESI.† Numerical values are stated in Table S1 in the ESI.†
 | ||
| Fig. 3 Performance parameters of the HMF oxidation in the 3E-batch cell and the 2E-flow cell at different temperatures: a) HMF conversion, total Coulomb efficiency, mole balance and b) product yields. | ||
While it was possible to decrease the reaction time from ca. 16 h to ca. 3 h due to the larger electrode surface of the flow cell, a similar CEtot value of 48.7% to the batch cell experiment (51.1%) was obtained at 40 °C. However, the Coulomb efficiencies at the other temperatures were considerably lower. This was particularly evident in the 80 °C experiment, in which the CEtot decreased by 26.9% compared to the batch cell. Thus, the highest CEtot in the flow cell was obtained at 60 °C (being 49.7%). This resulted in lower FDCA yields and higher yields of the less oxidized products (e.g. DFF, HMFCA). Interestingly, the measurement at 40 °C was also an exception here, as the FDCA yield was 4.8% higher than in the batch cell. All flow cell reactions also showed higher yields of maleic acid (MA), which is a ring-opening product of HMF (Scheme 3).
 | ||
| Scheme 3 Oxidation of HMF to maleic acid (MA).14 | ||
These results showed a generally stronger preference for the competing side reactions in the flow cell. Since similar mole balances of 88.1–91.2% were obtained over the entire temperature range in this cell, it can be assumed that these side reactions did not involve HMF (except for the formation of MA, which was taken into account for the mole balance). Therefore, the observed decrease in Coulomb efficiency was more likely caused by a higher rate of the OER or the oxidation of acetate (e.g. by Kolbe electrolysis to ethane).26 A reason for this could be the different convection behavior of both cells. For the given flow rate of 5 mL min−1 (2.5 mL min−1 per channel) very low Reynolds numbers were calculated for the flow cell (Table 3), which correspond to a laminar flow. Accordingly, the HMF oxidation could have been limited by the mass transport to the electrode.27
| Flow rate | RT | 60 °C |
|---|---|---|
| 5 mL min−1 | 27.7 | 58.2 |
| 10 mL min−1 | 55.3 | 116.5 |
Unfortunately, this limitation was a problem that could not be solved with the given setup, as significantly higher flow rates would have been required to achieve a turbulent flow. This would possibly also have led to negative side effects, such as the erosion of the CoOOH coating. Nevertheless, we examined the influence of a higher Reynolds number by performing a HMF oxidation experiment at 60 °C at a flow rate of 10 mL min−1 (Table 3). However, we obtained similar performance parameters as in the 5 mL min−1 experiment (data not shown), which indicated that the influence of flow rate in this range is minimal. We also considered the influence of an excessive HMF turnover during a single pass of the cell. As we observed in the batch experiments that the CEtot decreases during the reaction process,14 it was possible that a too high turnover during a single pass was leading to a local decrease of the Coulomb efficiency at the end of the reactor and therefore to the decrease of CEtot. However, since only a charge of ca. 0.33 F was transferred during a single pass of the reactor, this option can be excluded. Furthermore, the flow cell was heated via pipes in the end plates, which provided a more direct heat transfer than in the batch cell. This could have led to the promotion of side reactions which was especially observable by the strong decrease of CEtot at 80 °C. Another explanation for the decreased efficiency lies in a corrosion effect of the stainless steel-based end and contacting plates of the flow cell that we discovered during the experiments. We recognized a small degree of corrosion in the regions that were in contact with the electrolyte, which caused the release of iron ions into the solutions. It is therefore conceivable that iron was also incorporated in the catalytic coating. Since it is known that iron doping of cobalt oxide electrocatalysts strongly enhance their OER activity,28 this effect could explain the increased proportion of side reactions. A comparison of pure and iron-doped cobalt oxide electrodes (Fig. S5 in the ESI†) supports this assumption, as it shows a shift of the redox potential of the iron doped cobalt oxide redox system towards the OER.
To further investigate the complete conversion of HMF to FDCA, we performed a long-term reaction in the 2E-flow cell, in which a charge of 24 F (four times the theoretically required charge) was transferred. HPLC samples were taken during the reaction in order to study the progression of the consecutive oxidation mechanism. An analogous reaction was carried out during our batch experiments under optimized parameters (mCoOOH = 0.81 ± 0.01 mg cm−1, j = 1.0 mA cm−1, 80 °C).14 For comparability, we chose the same parameters for the 2E-flow cell experiment, however, we changed the temperature to 60 °C due to the different CEtot optimum. This resulted in a cell voltage of ca. 2.4 V (Fig. S6b in the ESI†). The performance parameters of both reactor systems are depicted in Fig. 4, numerical values can be found in Tables S2 and S3 in the ESI.†
 | ||
| Fig. 4 Performance parameters of the long-term experiments under optimized conditions in a) and b) the 3E-batch cell (80 °C, data redrawn from ref. 14) and c) and d) the 2E-flow cell (60 °C). | ||
In the 2E-flow cell, an initial CEtot value of 61.3% was determined after 1 F, which decreased to 52.0% during the transfer of 6 F. Despite these considerably lower Coulomb efficiencies in comparison to the batch cell experiment, the yields of the early intermediate products DFF and HMFCA showed a similar trend. The highest yields were only slightly higher with 16.5% DFF and 3.7% HMFCA obtained after 3 F, whereas the highest yields of 13.0% DFF and 3.5% HMFCA in the batch cell were obtained after 2 F. In contrast, the slower oxidation steps to FFCA and to FDCA were affected to a greater extent. The highest FFCA yield of 44.2%, obtained after 6 F, was significantly lower than in the batch cell (52.2% after 5 F). The last oxidation step also showed a deviation from the behavior observed in the batch cell. The highest FDCA yield of 77.1% was obtained after 18 F (CEFDCA = 21.0%), in which the solution still contained a low amount of FFCA. A continuation of the oxidation led to a decrease of the FDCA yield (71.5% after 24 F). This did not correspond to the expectation, as in the batch experiment an increase of the FDCA yield was observed at the end of the reaction, even in the absence of FFCA (Fig. 4b). Due to the decolorization of the yellow solution, we concluded that humic substances formed during the initial phase of the reaction were oxidized to FDCA. This effect could not be observed in the flow cell, in which an orange-brown solution was obtained after 24 F. The increased involvement of the side reactions was also observed in the higher production rate of MA, which yielded 9.2% after 18 F. As in the batch cell, the MA yield increased linearly over the entire course of the reaction, showing that not only HMF but also its oxidation products can undergo ring-opening reactions. Thus, the decomposition of FDCA in this late oxidation phase could have occurred either electrochemically by C–C-bond cleavage reactions, e.g. via the MA pathway or non-electrochemically by the formation of humic substances.
Conclusion
In this study, we transferred our previously reported non-alkaline HMF oxidation method from a classical three-electrode batch setup to a two-electrode flow setup. This allowed us to power the reaction with a more cost-efficient electrical setup (i.e. a power supply instead of a potentiostat) and carry out the reaction in a fraction of the time needed in the batch cell. We therefore adapted the potential-based CoOOH coating method to make it feasible for the application of voltages in the flow setup. Here it was shown, that higher CoOOH loadings and a more stable film can be obtained by performing the reductive Co(OH)2 deposition at a lower potential or voltage value. Interestingly, in the flow cells, a different behavior of the Co(OH)2 oxidation to CoOOH was observed, whereby in the channels of the 2E-flow cell the reaction proceeded slower than on the planar electrode of the 3E-flow cell.A comparison of the HMF oxidation in both reactor setups showed lower Coulomb efficiencies in the 2E-flow cell over the investigated temperature range of RT – 80 °C. This showed a promotion of side reactions, which however did not include HMF (except for the formation of MA). A mass transfer limitation was attributed as a reason for this, as a strictly laminar flow prevails in the flow channels. However, this problem could not be solved due to the channel dimensioning. Corrosion of the stainless steel-based components of the flow cell was identified as a further problem, as the release of iron ions into the solution (and consequently incorporation into the cobalt catalyst) could have caused a substantial loss of Coulomb efficiency. This showed a technical challenge, which only became apparent through the scale-up from the laboratory-type glass reactor.
A long-term experiment showed that the decreased Coulomb efficiency is mainly due to a slower second and third oxidation step to FFCA and FDCA, which showed significantly lower yields compared to the batch reaction. An increased formation of the side product maleic acid was also observed. Nevertheless, an FDCA yield of 77.1% could be obtained after the transfer of 18 F.
These results show the need for further optimization of the reaction in order to increase the selectivity of the HMF oxidation with respect to the side reactions. Furthermore, due to the necessity of a low current density (and thus a low reaction rate), a complete turnover of HMF within a single reactor pass was not possible, so that the full potential of a flow cell application could not be exploited, yet. An optimal system should enable both the continuous production of FDCA and downstream separation of the product from the solution, as was already shown for an alkaline system by Latsuzbaia et al.29 At the present time, this is not possible with this non-alkaline system. Nonetheless, this study represented an important step, as it showed the transferability of this catalytic system into the flow cell and also the potential for larger scale applications. Thus, future modifications of the cobalt oxide catalyst can be tested in batch systems and then transferred to the flow cell by means of the method established here.
Experimental section
Chemicals
The following chemicals were purchased commercially and used without further purification: Co(NO3)2·6 H2O (≥99%, Carl Roth), NaOH (≥99%, Carl Roth), HMF (98%, abcr), DFF (98%, TCI), HMFCA (98%, TCI), FFCA (98%, TCI), FDCA (98%, Alfa Aesar), MA (>99.0%, Fisher Scientific), CH3COOH (≥99%, Carl Roth), CH3COONa (≥99%, Carl Roth), H2SO4 (≥95%, Fisher Scientific), HNO3 (65%, Fisher Scientific) and HCl (≥37%, Honeywell). Deionized water was used as solvent in all experiments. Acetate buffer solution (CH3COOH/CH3COO−, pH 5) with a total anion concentration of 1.0 mol L−1 was prepared from the respective acid and sodium salt.Batch cell experiments
All batch cell experiments were conducted using an Autolab PGSTAT20 or PGSTAT302 potentiostat/galvanostat (METROHM GmbH, Germany) with a three-electrode arrangement (3E-batch). Titanium and platinum sheets (2 × 5 cm, chemPUR, Germany) were used as working and counter electrodes, respectively. Potentials were measured against Ag/AgCl sat. KCl glass electrodes (Sensortechnik Meinsberg, Germany).The CoOOH coating was performed in an unseparated glass cell with a filling volume of 40 mL. The electrode was coated using a reductive deposition method of Co(OH)2 from an aqueous Co(NO3)2 solution (50 mM) and a subsequent oxidation in an aqueous NaOH solution (1.0 M). HMF oxidation was performed in a H-type batch cell, which was separated with a fumasep FKE-50 cation exchange membrane (FumaTech, Germany). Electrode surface areas and filling volumes of the cell compartments are stated in Table 1. Oxidations of 20 mM HMF solutions were carried out by applying a constant current density of 1.0 mA cm−2 at different temperatures (RT, 40 °C, 60 °C and 80 °C). Details of both methods have been described in our previous work.14
Flow cell experiments
For the transition of the coating method, a MicroFlowCell (ELECTROCELL, Denmark) with titanium working and counter electrodes and an Ag/AgCl 3.4 M KCl capillary reference electrode was used (3E-flow cell). A detailed description of the cell design is given in the ESI.† A fumasep FTAM-E anion exchange membrane (FumaTech, Germany) was used to separate the WE and CE reaction compartment, as the use of a cation exchange membrane would have led to the migration of Co2+ cations during the reductive Co(OH)2 deposition. Electrode surface areas and filling volumes of the cell compartments are stated in Table 1. The experiments were carried out using a VIONIC potentiostat/galvanostat (METROHM GmbH, Germany).For the CoOOH coating, the titanium working and counter electrode were stored in 20% HCl solution for 30 min to remove the passivating TiO2 layer. Furthermore, the planar working electrode was polished with fine-grained sandpaper (grad 400, 800 and 1200) and afterwards roughened with coarse-grained sandpaper (grad 180) to enable better adhesion of the CoOOH coating. For Co(OH)2 deposition, a 50 mM Co(NO3)2 solution (WE compartment) and a 100 mM NaNO3 solution (CE compartment) were cycled through the cell with a flow rate of 5 mL min−1. Reductive potentials (−1.00, −0.95, −0.90 or −0.85 VAg/AgCl) were applied until a deposition charge density σRed of 0.8 C cm−2 had been transferred (cathodic phase). For the oxidation to CoOOH, 1.0 M NaOH solutions were cycled through both electrode compartments with a flow rate of 5 mL min−1. An oxidative potential of 0.40 VAg/AgCl was applied to the working electrode until a current density of <0.35 mA cm−2 was measured (anodic phase). Between the coating steps and at the end of the method, each of the two electrode compartments were rinsed with 80 mL of DI water at a flow rate of 5 mL min−1. During both phases, the resulting counter electrode potential ECE was measured by the potentiostat over the CE-sense cable. This allowed the calculation of the cell voltage ECell according to eqn (6).
 | (6) |
Before the first experiment, the working electrode was roughened through acidic etching using an established method.30 The electrode was stored into a 48% H2SO4 solution at 60 °C for 1 h. Before each experiment, the working and counter electrode was stored in 20% HCl solution for 30 min to remove the passivating TiO2 layer. For the CoOOH coating, the same procedure as for the 3E-flow cell was used with two exceptions: I) constant voltages (resulting from the transition experiments) of −2.75 V or −3.50 V (cathodic phase) and 1.60 V (anodic phase) were applied instead of constant potentials. II) The deposition charge density σRed was varied to obtain the CoOOH loadings stated in Table 2. The electrodes were freshly coated before each experiment.
For the HMF oxidations, a 20 mM HMF solution in acetate buffer (pH 5) and a pure acetate buffer solution (of a total anion concentration of 1.0 mol L−1) were cycled through the WE and CE compartment with a flow rate of 5 or 10 mL min−1, respectively. A constant current density of 1.0 mA cm−2 was applied until the respective charge (6 F or 24 F) had been transferred. Exemplary cell voltage curves of all experiments are depicted in Fig. S6 in the ESI.† Samples of 1.0 mL were taken from the HMF solution before, (during) and after the experiment, filtered and used for HPLC analysis. All reactions were conducted in duplicates, of which the mean values are shown in Fig. 3 and 4. Error bars were not depicted to maintain clarity. Numeric mean values and standard deviations of all reactions are stated in Tables S1–S3 in the ESI.†
HPLC analysis
The concentrations of HMF, DFF, HMFCA, FFCA, FDCA and MA were analyzed with a 1260 Infinity II HPLC system (Agilent, USA) in a REZEX ROA Organic Acid column (300 × 7.8 mm, Phenomenex, USA). An aqueous H2SO4 solution (2.5 mM) was used as eluent with a flow rate of 0.6 mL min−1 at 70 °C. The substances were detected with a refractive index detector and a diode array detector. For quantitative analysis, calibration curves were recorded with standard solutions made from the pure substances specified above. The presence of formaldehyde and formic acid was checked qualitatively.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was funded by the German Federal Ministry for Economic Affairs and Climate Change under the ElektroSyn project (Project ID 03EN2071D). In particular, the authors acknowledge the work of their project partners from Whitecell Eisenhuth GmbH & Co. KG for the development and fabrication of the used 2E-flow cell.References
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- S. Cembellín and B. Batanero, Chem. Rec., 2021, 21, 2453–2471 CrossRef PubMed.
- F. Harnisch and U. Schröder, ChemElectroChem, 2019, 6, 4126–4133 CrossRef CAS.
- M. Guschakowski and U. Schröder, ChemSusChem, 2021, 14, 5216–5225 CrossRef CAS PubMed.
- T. Lenk, V. Rueß, J. Gresch and U. Schröder, Green Chem., 2023, 25, 3077–3085 RSC.
- P. Drögemüller, T. Stobbe and U. Schröder, ChemSusChem, 2024, 17, e2023009 CrossRef PubMed.
- C. A. C. Sequeira and D. M. F. Santos, J. Braz. Chem. Soc., 2009, 20, 387–406 CrossRef CAS.
- D. S. P. Cardoso, B. Šljukić, D. M. F. Santos and C. A. C. Sequeira, Org. Process Res. Dev., 2017, 21, 1213–1226 CrossRef CAS.
- M. C. Leech, A. D. Garcia, A. Petti, A. P. Dobbs and K. Lam, React. Chem. Eng., 2020, 5, 977–990 RSC.
- D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- M. N. Gey and U. Schröder, RSC Sustainability, 2024, 2, 2256–2266 RSC.
- R. J. Van Putten, J. C. Van Der Waal, E. De Jong, C. B. Rasrendra, H. J. Heeres and J. G. De Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- O. Simoska, Z. Rhodes, S. Weliwatte, J. R. Cabrera-Pardo, E. M. Gaffney, K. Lim and S. D. Minteer, ChemSusChem, 2021, 14, 1674–1686 CrossRef CAS PubMed.
- L. Guo, X. Zhang, L. Gan, L. Pan, C. Shi, Z. Huang, X. Zhang and J. Zou, Adv. Sci., 2023, 10, 2205540 CrossRef CAS PubMed.
- K. R. Vuyyuru and P. Strasser, Catal. Today, 2012, 195, 144–154 CrossRef CAS.
- H. A. Rass, N. Essayem and M. Besson, ChemSusChem, 2015, 8, 1206–1217 CrossRef PubMed.
- Y. Zhang, X. Guo, P. Tang and J. Xu, J. Chem. Eng. Data, 2018, 63, 1316–1324 CrossRef CAS.
- H. B. Rose, T. Greinert, C. Held, G. Sadowski and A. S. Bommarius, J. Chem. Eng. Data, 2018, 63, 1460–1470 CrossRef CAS.
- S. R. Kubota and K.-S. Choi, ChemSusChem, 2018, 11, 2138–2145 CrossRef CAS PubMed.
- B. J. Taitt, D. H. Nam and K.-S. Choi, ACS Catal., 2019, 9, 660–670 CrossRef CAS.
- J. M. McEnaney, S. J. Blair, A. C. Nielander, J. A. Schwalbe, D. M. Koshy, M. Cargnello and T. F. Jaramillo, ACS Sustainable Chem. Eng., 2020, 8, 2672–2681 CrossRef CAS.
- M. Li, C. Feng, Z. Zhang and N. Sugiura, Electrochim. Acta, 2009, 54, 4600–4606 CrossRef CAS.
- S. Glasstone and A. Hickling, J. Chem. Soc., 1934, 1878–1888 RSC.
- T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858–2869 CrossRef PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- R. Latsuzbaia, R. J. M. Bisselink, A. Anastasopol, H. van der Meer, R. van Heck, M. S. Yagüe, M. Zijlstra, M. Roelands, M. Crockatt, E. Goetheer and E. Giling, J. Appl. Electrochem., 2018, 48, 611–626 CrossRef CAS.
- S. Ban, Y. Iwaya, H. Kono and H. Sato, Dent. Mater., 2006, 22, 1115–1120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00511b |
| This journal is © The Royal Society of Chemistry 2025 |