DOI:
10.1039/D5RA08080K
(Paper)
RSC Adv., 2025,
15, 48969-48977
Enhanced mechanism of a TiO2/g-C3N4 photoanode in photoelectrocatalytic synergistic persulfate activation for antibiotic degradation
Received
21st October 2025
, Accepted 3rd December 2025
First published on 9th December 2025
Abstract
Photocatalysis and persulfate-based advanced oxidation technologies have received considerable attention. In this study, a Z-scheme heterojunction photoanode composed of graphitic carbon nitride and titanium dioxide (TiO2/g-C3N4) was constructed to develop an efficient photoelectrochemical–persulfate (PEC/PMS) activation system for antibiotic degradation. The PEC/PMS system significantly accelerated electron transfer and enhanced the photoresponse current, achieving 96.04% tetracycline removal within 12 min. The optimal operating conditions were a bias voltage of 1.0 V, a PMS concentration of 0.2 mmol L−1, and a neutral to slightly alkaline environment. The system effectively resisted interference from water matrix components and achieved over 88% removal of multiple antibiotics in aquaculture pond water. TiO2/g-C3N4 exhibited a Z-scheme heterostructure, and its photocatalytic activity served as the primary driving force in the synergistic process, in which degradation was dominated by radical pathways (˙OH and ˙SO4−) accompanied by a non-radical 1O2 pathway. The calculated synergy factor further confirmed that the introduction of an external electric field and PMS markedly enhanced electron transfer and produced a pronounced synergistic effect.
1 Introduction
Photocatalytic technology (PC) is a green advanced oxidation method. However, the high recombination rate of photogenerated electron–hole pairs (e−/h+) limits its efficiency. Combining PC with electrocatalysis (EC) to form photoelectrocatalysis (PEC) is an effective strategy to enhance photocatalytic performance. PEC applies an external electric field to promote electron transfer and continuously directs photogenerated electrons to the cathode, thereby suppressing e−/h+ recombination. Electrochemical methods offer strong controllability, high stability, and no secondary pollution, and the conversion of photocatalysts into electrodes eliminates the recovery issues inherent in heterogeneous catalysts. Consequently, PEC has emerged as a promising wastewater treatment technology.1–5
The photoelectrochemical activity of photoanode materials plays a crucial role in the performance of PEC systems. Graphitic carbon nitride (g-C3N4) exhibits visible-light activity, and its band structure is appropriate for redox reactions, rendering it a highly promising semiconductor for PEC applications.6–8 Liang et al. first fabricated a g-C3N4 photoanode for phenol degradation, where the electrocatalysis and photocatalysis exerted a synergistic promotional effect. The applied bias decreased e−/h+ recombination, while photocatalysis mitigated the electrode passivation caused by phenolic polymer films, thereby facilitating electrocatalytic oxidation.9 Numerous studies have focused on enhancing the PEC performance of g-C3N4 using approaches such as nanoporous structure design, heteroatom doping, and heterojunction construction. Among these, coupling g-C3N4 with other semiconductor materials has been proven particularly effective.10–12 TiO2 is widely used because of its low cost, chemical stability, high catalytic activity, and corrosion resistance, and it can be integrated with g-C3N4 to prepare electrodes for photoelectrocatalytic applications. Huang fabricated a core–shell heterostructured photoanode by anchoring g-C3N4 onto TiO2, which exhibited the excellent visible-light responsivity and significantly enhanced photoelectrocatalytic degradation efficiency. This synergistic effect accelerates the separation and migration of photogenerated carriers and the production of reactive species, resulting in a 2.4-fold improvement compared with pristine materials.13,14 Although studies have demonstrated the excellent photocatalytic performance of g-C3N4/TiO2 composites, the underlying mechanism governing the enhanced photoelectrocatalytic behaviour of g-C3N4 coupled with TiO2 requires further investigation.15,16
Under visible-light irradiation, the catalytic performance of PEC systems depends not only on the intrinsic activity of the photoelectrode but also on the electrolyte composition. The presence of strong oxidants, such as persulfate or hydrogen peroxide, enables Fenton-like oxidation, as these species can act as electron scavengers that promote electron–hole separation while being activated to generate reactive radicals.17,18 Currently, studies combining persulfate with PEC systems have primarily focused on metal-based photoanodes, such as Bi- or W-based materials, whereas research employing g-C3N4 as the photoanode remains limited.19
In this study, a composite g-C3N4-TiO2 photoanode with a direct Z-scheme heterojunction structure was fabricated and combined with peroxymonosulfate (PMS) to construct a photoelectrocatalytic–persulfate activation (PEC/PMS) system. The synergistic photoelectrocatalytic activity within the heterostructure was enhanced through improvements in photochemical performance. The effects of bias voltage, PMS concentration, pH, and water matrix components on antibiotic degradation were systematically investigated, and the degradation behaviour of multiple antibiotics in aquaculture pond water was evaluated. The enhancement mechanism of the synergistic system was elucidated through analyses of reactive oxygen species (ROS) generation pathways and reaction kinetics. The results demonstrated that photocatalysis served as the core driving force, while the combined effects of applied bias and PMS promoted electron transfer and ROS generation, enabling efficient photoanode recovery. This study provides a mechanistic basis for understanding photoelectrocatalytic persulfate activation and its potential applications.
2 Experiments and methods
2.1 Preparation of photoanode
Dicyandiamide (2 g) and ammonium oxalate (1 g) were mixed with 80 mL of deionised water, heated to 160 °C, and maintained for 8 h in a 100 mL Teflon-lined autoclave. After naturally cooling to 25 °C, the precipitate was dried at 80 °C for 8 h. The dried material was thoroughly ground and calcined at 550 °C for 4 h in a muffle furnace, followed by natural cooling and further grinding to obtain g-C3N4 powder. The obtained g-C3N4 powder and commercial nano-TiO2 powder were mixed in methanol at a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, ultrasonicated for 30 min in an 80 °C water bath, dried at 100 °C for 12 h, and finally heated at 500 °C for 10 min. After natural cooling, the product was dispersed ultrasonically in anhydrous ethanol and then coated onto ITO conductive glass to form a homogeneous slurry. The coated electrodes were dried at 50 °C, yielding a TiO2/g-C3N4 photoanode for photoelectrocatalytic synergistic catalysis.
:1, ultrasonicated for 30 min in an 80 °C water bath, dried at 100 °C for 12 h, and finally heated at 500 °C for 10 min. After natural cooling, the product was dispersed ultrasonically in anhydrous ethanol and then coated onto ITO conductive glass to form a homogeneous slurry. The coated electrodes were dried at 50 °C, yielding a TiO2/g-C3N4 photoanode for photoelectrocatalytic synergistic catalysis.
2.2 Material characterization
Scanning electron microscopy (SEM) was used to observe the micromorphology. X-ray diffraction (XRD) was employed to analyse the crystal structure. Fourier-transform infrared spectroscopy (FT-IR) was adopted to examine the surface functional groups. X-ray photoelectron spectroscopy (XPS) was used to determine the surface elemental composition and valence states as well as to measure the valence band spectra. A nitrogen adsorption analyser (BET) was adopted to determine the specific surface area, pore volume, pore size, and adsorption–desorption isotherms. UV-visible diffuse reflectance spectroscopy (UV-vis DRS) equipped with an integrating sphere was used to analyse the light absorption properties of the catalyst. Photoluminescence (PL) spectra were recorded using a fluorescence spectrophotometer. Electrochemical impedance spectroscopy (EIS), Mott–Schottky plots, transient photocurrent density measurements, linear sweep voltammetry (LSV), and light on/off photocurrent–voltage tests, as well as open-circuit potential measurements under light on/off conditions, were performed on an electrochemical workstation. The TiO2/g-C3N4 electrode served as the working electrode, Ag/AgCl was the reference electrode, and a platinum plate was the counter electrode. The electrolyte was 0.5 mol per L Na2SO4, and the light source had a wavelength of 375 nm.
2.3 Catalytic activity test
The PEC/PMS experiments were conducted in a standard three-electrode system under the same basic conditions used for photoelectrochemical measurements. A 300 W xenon lamp equipped with a 420 nm cutoff filter served as the light source. The antibiotic concentration was 1 mg L−1, and the PMS concentration was 0.2 mmol L−1. Both the light source and bias potential (1.0 V) were applied simultaneously. The applied bias voltages were 0.5, 1.0, 1.5, and 2.0 V. The PMS concentrations of 0.1, 0.2, and 0.3 mmol L−1 and pH values of 3, 5, 7, 9, and 11 were investigated. To examine the effect of background water matrices, 10 mmol L−1 of humic acid (HA), Cl−, SO42−, CO32−, and NO3− were added separately. The detailed degradation procedures are described in our previous work.20
2.4 Quenching experiments and capture of reactive species
Tert-butanol (TBA), methanol (MeOH), p-benzoquinone (BQ), β-carotene, disodium ethylenediaminetetraacetate (EDTA-2Na), and K2Cr2O7 were employed as the quenching agents for ˙OH, ˙OH and ˙SO4−, ˙O2−, 1O2, h+, and e−, respectively, to identify the types of ROS involved. Electron spin resonance (ESR) spectroscopy was used for further ROS analysis. 5,5-Dimethyl-1-pyrroline-N-oxide (DMPO) served as a trapping agent for ˙OH and ˙O2− radicals. Notably, ˙OH was trapped in the H2O–DMPO mixture, while ˙O2− was trapped in the MeOH–DMPO mixture. 2,2,6,6-Tetramethylpiperidine (TEMP) was adopted to trap singlet oxygen (1O2). MeOH acted as the electron scavenger, and silver nitrate (AgNO3) was used as the hole (h+) scavenger.
3 Results and discussion
3.1 Material characteristics
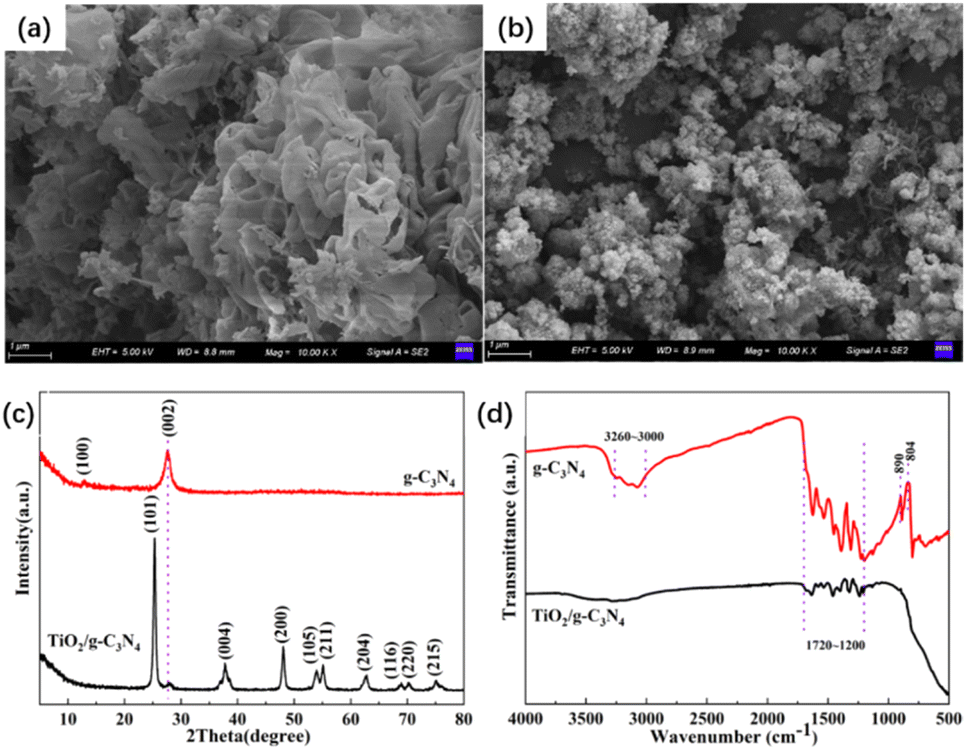
From the SEM images of g-C3N4 and TiO2/g-C3N4 shown in Fig. 1(a) and (b), g-C3N4 was observed to consist of irregularly shaped, wrinkled, and aggregated nanosheets with a smooth two-dimensional layered structure, confirming the formation of a graphitic phase. On the surface of TiO2/g-C3N4, TiO2 nanoparticles were immobilised and aggregated on the g-C3N4 nanosheets, which increased the specific surface area of the composite material.21 The XRD pattern in Fig. 1(c) demonstrates that the characteristic diffraction peak of g-C3N4 at 27.8° corresponded to the (002) plane associated with the stacking of graphitic layers. In contrast, the peak at 12.9° corresponded to the (100) plane, representing the in-plane structural packing of aromatic segments and confirming the successful synthesis of g-C3N4. In TiO2/g-C3N4, aside from the 27.8° peak of g-C3N4, all remaining diffraction peaks matched those of anatase TiO2, demonstrating the coexistence of both precursors within the composite. Because TiO2 possesses high crystallinity, the diffraction intensity of g-C3N4 is suppressed.22 In the FT-IR spectra shown in Fig. 1(d), the absorption peak of g-C3N4 at 804 cm−1 was assigned to the bending vibration of tri-s-triazine units, whereas the peak at 890 cm−1 was attributed to the C–O stretching vibration induced by oxygen doping. The broad absorption band between 1720 and 1200 cm−1 corresponded to C–N heterocyclic stretching, and the broad band from 3260 to 3000 cm−1 was attributed to the N–H stretching vibrations of bridging or terminal amino groups. After coupling with TiO2, interfacial bonding resulted in masking of several intrinsic absorption peaks of g-C3N4. However, the absorption features between 1720 and 1200 cm−1 indicated that the C–N heterocyclic framework of g-C3N4 remained intact.23
 |
| | Fig. 1 SEM images of g-C3N4(a) and TiO2/g-C3N4 (b), along with XRD patterns (c) and FT-IR spectra (d) of g-C3N4 and TiO2/g-C3N4. | |
Fig. 2(a) illustrates the XPS full survey spectrum of g-C3N4, with photoelectron peaks at 285, 398, and 532 eV, corresponding to C 1s, N 1s, and O 1s, respectively. For TiO2/g-C3N4, characteristic peaks appeared at 459 and 566 eV, corresponding to Ti 2p and Ti 2s. A significant decrease in the N content of the composite indicated the presence of nitrogen vacancies within the C–N framework. Meanwhile, the electron spin resonance signal in Fig. 2(f) indicated unpaired electrons in the conjugated aromatic structure, with a g value of 2.003, typical of N-vacancy sites.24 In the C 1s spectrum of TiO2/g-C3N4, a sub-peak assigned to C–Ti bonding appeared, demonstrating the electronic interaction between g-C3N4 and TiO2. No Ti–N peak was detected in the N 1s spectrum, likely due to the low nitrogen content. In the Ti 2p spectrum shown in Fig. 2(g), six characteristic peaks were present, among which the peaks at 457.7, 457.3 eV and 463.6, 463.3 eV correspond to Ti 2p3/2 and Ti 2p1/2, respectively. The energy difference of 5.6 eV indicates Ti4+ as the dominant oxidation state.25
 |
| | Fig. 2 XPS survey spectrum (a); high-resolution spectra of (b) C 1s and N 1s (c) for g-C3N4; and high-resolution spectra of C 1s (d), N 1s (e), EPR spectra (f), and Ti 2p (g) for TiO2/g-C3N4. | |
Fig. 3(a) presents that both materials exhibited an H3-type hysteresis loop in their nitrogen adsorption–desorption isotherms, corresponding to a type-IV isotherm characteristic of lamellar cracks and micropores. The specific surface area of the composite was much larger than that of pure g-C3N4. As shown in Fig. 3(b), the average pore diameter of the composite was smaller than that of g-C3N4, consistent with the increase in the specific surface area.26
 |
| | Fig. 3 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of g-C3N4 and TiO2/g-C3N4. | |
Fig. 4(a) shows that the UV-vis DRS spectrum of the composite material exhibited a significantly stronger absorption intensity and a considerably broader absorption range than those of the single-component materials, which contributed to the enhanced photocatalytic performance. Using the Kubelka–Munk function (Fig. 4(b)), the band gaps of g-C3N4, TiO2, and TiO2/g-C3N4 were calculated as 2.47, 3.05, and 2.93 eV, respectively. The electronic coupling between TiO2 and g-C3N4 enhanced the absorption in both the UV and visible regions. The valence-band edges determined by VB–XPS (Fig. 4(c)) were located at 2.30 eV for g-C3N4, 3.01 eV for TiO2, and 2.86 eV for TiO2/g-C3N4. The band structure alignment after conversion to the NHE scale is illustrated in Fig. 4(d).27 The Mott–Schottky plots in Fig. 4(e) present that all materials exhibited the typical n-type semiconductor behaviour, as evidenced by their positive slopes. The intercepts corresponded to flat-band potentials, further confirming the accuracy of the derived band structure. The flat-band potentials of g-C3N4 and TiO2/g-C3N4 (vs. Ag/AgCl) were −0.10 and −0.03 eV, respectively, which converted to −0.32 and −0.22 eV vs. NHE, slightly lower than the conduction-band potentials obtained earlier.28
 |
| | Fig. 4 UV-vis DRS spectra (a), estimated band gap (b), VB–XPS spectra (c), band gap structure alignments (d), and Mott–Schottky curves (e) of g-C3N4 and TiO2/g-C3N4. | |
To further verify that the binary composite structure could enhance the photoelectrocatalytic performance, a series of photoelectrochemical measurements were conducted. Fig. 5(a) shows that the PL intensity of TiO2/g-C3N4 was significantly lower than that of g-C3N4, indicating more efficient separation of photogenerated electron–hole pairs. The absence of the 460 nm emission peak in TiO2/g-C3N4 may be due to the potential difference between TiO2 and g-C3N4, which prevents electron transfer between the conduction bands of the coupled semiconductors and indirectly supports the presence of a Z-scheme heterojunction with superior catalytic activity.29 Fig. 5(b) shows that the impedance arc radius of TiO2/g-C3N4 was much smaller than that of g-C3N4, confirming the reduced interfacial electron resistance and enhanced charge transfer.30 The LSV curves under continuous and chopped light illumination, shown in Fig. 5(c) and (d), indicate significantly improved reaction kinetics for TiO2/g-C3N4, with faster electron transfer, stronger photoresponse capability, and higher concentrations of photogenerated charge carriers. Fig. 5(e) shows that TiO2/g-C3N4 exhibited a higher photocurrent under visible light irradiation, which was attributed to efficient charge separation and transport. The open-circuit potential curves in Fig. 5(f) further demonstrate that the TiO2/g-C3N4 heterostructure possessed markedly enhanced photocurrent responsiveness and accelerated charge transfer at the interface.31 Together, these characterisations confirm that TiO2/g-C3N4 enables faster migration and separation of photogenerated carriers and exhibits a stronger photoresponse behaviour, providing a robust foundation for its photoelectrocatalytic performance.
 |
| | Fig. 5 PL spectra (a), EIS Nyquist plots (b), photocurrent densities vs. bias potential (c), current–potential curves under chopped light (d), photocurrent density plots (e), and open circuit potential under chopped light (f) for g-C3N4 and TiO2/g-C3N4. | |
3.2 Analysis of photoelectrocatalytic–persulfate activation performance
To investigate the degradation performance of photoelectrocatalytic persulfate activation on tetracycline (TC) in water and to elucidate the enhancement effect of PMS on the photoelectrocatalytic system, as well as the influence of the material's photocatalytic and electrocatalytic properties on the synergistic process, a series of experiments were conducted, including PEC/PMS, electrocatalysis under visible light (PEC), photocatalysis coupled with PMS (PC/PMS), electrocatalysis coupled with PMS under dark conditions (EC/PMS), photocatalysis (PC), and electrocatalysis under dark conditions (EC). As shown in Fig. 6(a), the PEC/PMS system achieved the highest removal efficiency and the fastest degradation rate, removing 96.04% of TC within 12 min. In contrast, EC exhibited the weakest degradation performance with only 50.72% removal after 30 min. The oxidative degradation efficiencies for TC among the other systems followed the trend: PC/PMS ≈ PEC > PC > EC/PMS. Notably, the degradation efficiency under illuminated conditions was significantly higher than that in the absence of light, with removal rates exceeding 90% when irradiation was applied. These results indicate that the photocatalytic activity of the material plays a decisive role in accelerating photoelectrocatalytic reactions within the synergistic system.
 |
| | Fig. 6 TC removal efficiency using TiO2/g-C3N4 under various operating conditions (a) and corresponding first-order kinetic fitting results (b). (E = 1.0 V, c0 = 1 mg L−1, cPMS = 0.2 mM, pH = 7). | |
The promoting effect of PMS was also evident. From an electrocatalytic perspective, the introduction of PMS increases the ionic strength of the solution and enhances mass-transfer efficiency. During the photoelectrocatalytic process, PMS undergoes electron transfer with photoexcited electrons on the surface of the photoanode, generating sulfate radicals (˙SO4−). This increased both the diversity and concentration of ROS compared with the PEC process alone, thereby greatly enhancing the degradation capacity and overall reaction efficiency towards TC.
Kinetic analysis further clarified the differences between reaction processes. The fitting results demonstrated that the degradation reactions followed a pseudo-first-order kinetic model and that both light irradiation and PMS addition significantly increased the apparent rate constant. These findings indicate that the excellent photoelectrochemical properties of the material can provide essential conditions for the operation of the synergistic activation system and that the introduction of PMS can exert a remarkable enhancement effect. However, the PEC/PMS process is inherently complex, and its synergistic mechanism, as well as the types and generation pathways of ROS involved, will be further elucidated in the following sections.
3.3 Analysis of influencing factors
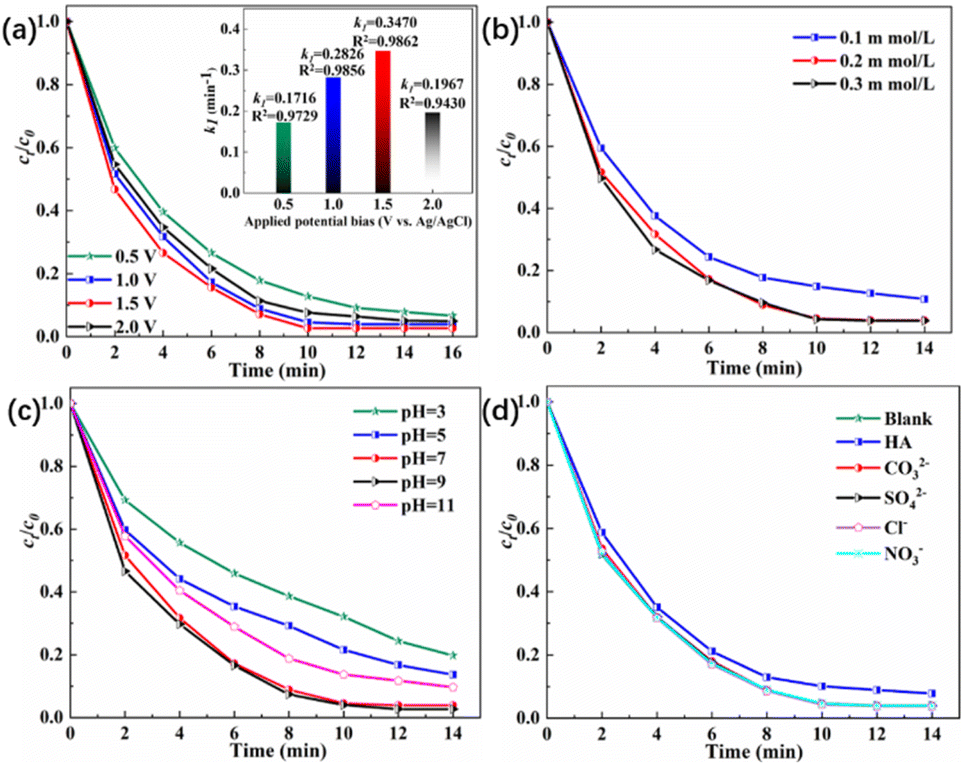
Fig. 7(a) demonstrates that as the applied voltage increased, the current increased accordingly, requiring greater electron transfer to maintain the charge balance. This enhanced the separation efficiency of the e−/h+ pairs during the photocatalytic process and improved the overall degradation performance. When the voltage reached 1.5 V (vs. Ag/AgCl), the TC removal efficiency reached 97.33% within 10 min. The inset in Fig. 7(a) shows that k1 increased more than twofold as the voltage increased from 0.5 to 1.5 V. At 2.0 V, the removal efficiency was lower than that at 1.0 V because excessive voltage induced side reactions on the electrode surface and hindered visible-light absorption and ROS generation. Energy consumption is a vital factor in electrochemical processes, and the specific energy consumption (EC) was calculated using eqn (1).32| |
 | (1) |
where EC represents the unit energy consumption (kWh m3), U is the voltage (V), I is the average current (A), t is the reaction equilibrium time (h), V is the volume of the solution (m3), and c0 and ct are the pollutant concentrations at the beginning and at equilibrium of the reaction (mg L−1), respectively. The calculated EC at 1.0 V was 0.35 kWh m3, whereas at 1.5 V it increased to 0.58 kWh m3. With comparable removal efficiency, the higher voltage consumed 65.71% more energy. Therefore, 1.0 V was identified as the optimal operating condition for balancing degradation efficiency and energy consumption.
 |
| | Fig. 7 (a) TC removal efficiency under various parameters. (a) Applied potential bias (c0 = 1 mg L−1, cPMS = 0.2 mM, pH = 7); (b) PMS concentration (E = 1.0 V, c0 = 1 mg L−1, pH = 7); (c) pH (E = 1.0 V, c0 = 1 mg L−1, cPMS = 0.2 mM); (d) background matrix in water (E = 1.0 V, c0 = 1 mg L−1, cPMS = 0.2 mM, pH = 7). | |
Fig. 7(b) shows that increasing PMS concentration gradually enhanced the TC degradation. The promoting effect of PMS was discussed earlier. When the PMS concentration increased to 0.3 mmol L−1, the degradation efficiency did not exhibit significant improvement. This was because excessive PMS addition could hinder timely electron transfer, restrict the generation of diverse ROS, and cause self-quenching among accumulated radicals within the system.
Fig. 7(c) shows that pH exerted a significant influence on the PEC/PMS degradation of TC, which was directly related to the ionic form of TC under different pH conditions. When the pH was below 5, the degradation efficiency was markedly inhibited because the positively charged TC tended to migrate towards the cathodic region under the electric field, preventing photogenerated holes from directly participating in oxidation. As the pH increased, TC transformed into a zwitterionic form, which was rapidly degraded by various ROS near the photoanode. However, at pH 11, the degradation efficiency decreased again. Although alkaline conditions can be favourable for PMS activation, excessive accumulation of OH− on the photoanode surface leads to competing side reactions, such as O2 evolution or the consumption of ˙OH to form less oxidative 1O2, thereby weakening the overall degradation efficiency.33
Fig. 7(d) shows that HA slightly inhibited the TC removal. In contrast, Cl−, SO42−, CO32−, and NO3− had almost no effect on degradation performance. As an electron-rich organic substance, HA can compete for active sites on the photoanode, consequently suppressing the generation of ˙SO4− and ˙OH radicals and increasing the total amount of organic pollutants in the system, thus reducing the TC degradation rate.34 Some studies have reported that CO32− can generate weakly oxidative ˙HCO3− radicals, whereas Cl− can generate highly oxidative species such as HClO and ˙Cl. However, in this study, the anions did not significantly affect TC degradation. This was because both the weakly oxidative carbonate radicals and strongly oxidative chlorine species contributed to the propagation reactions that generated more ROS, and the opposing effects, radical quenching versus ROS generation, offset each other, resulting in a negligible overall influence on the degradation performance.35
3.4 Application in aquaculture pond water
To further evaluate the practical applicability of the PEC/PMS system, pond aquaculture water was used as the testing medium, to which TC, sulfamethoxazole (SMZ), levofloxacin (LVX), penicillin (PEN), chloramphenicol (CHL), and clindamycin (CLI) were added to assess their degradation under real aquatic conditions. The results suggest that the degradation efficiencies for all antibiotics exceeded 88%, with the degradation efficiency order of CLI > CHL > PEN > TC > LVX > SMZ. The similar removal efficiencies and corresponding k1 values indicated that the PEC/PMS system possessed strong degradation capability towards antibiotics with different molecular structures (Fig. 8). Notably, Na2SO4 was not added as an electrolyte during the simulated real-wastewater experiment, rendering the treatment conditions more representative of actual environmental systems. As shown in Table 2, organic substances similar to HA present in pond water acted as quenching agents. Despite this, the PEC/PMS system still achieved favourable degradation performance, demonstrating its strong practical potential for antibiotic removal in real aquatic environments.
 |
| | Fig. 8 Degradation performance of different antibiotics in pond water and their corresponding first-order kinetic constants. (E = 1.0 V, c0 = 1 mg L−1, cPMS = 0.2 mM, pH = 7). | |
3.5 Analysis of PEC/PMS activation pathways and reaction enhancement mechanism
Multiple reaction modes coexisted in the PEC/PMS system. From Fig. 9(a), the quenching experiment results showed that the oxidation contribution followed the order: ˙OH and ˙SO4− > ˙OH > h+ > e− > 1O2 >˙O2−. The contribution rates of different reactive oxygen species were further clarified using kinetic parameters.where R (%) represents the contribution rate of free radicals in the system. kapp (min−1) indicates the reaction rate constant without a quencher. kr (min−1) represents the reaction rate constant after adding a quencher. The contribution rates of ˙OH and ˙SO4−, ˙OH, h+, e−, 1O2 and ˙O2− were 91.26%, 88.46%, 85.88%, 74.84%, 53.40%, and 38.71%, respectively.
 |
| | Fig. 9 Quenching experiments (a), first-order kinetic fitting results (b), and ESR spectra of ˙OH and ˙SO4− (c), ˙O2− (d), and 1O2 (e), and cyclic experiment (f). (E = 1.0 V, c0 = 1 mg L−1, cPMS = 0.2 mM, pH = 7). | |
This indicated that the system was a composite oxidation process involving both radical and non-radical pathways, with ˙OH and ˙SO4− serving as the primary reactive species. The main ROS reaction pathways involved in this system include photocatalytic, electrochemical, and PMS-driven reactions.36,37
| | |
TiO2/CN + hv → e− + h+
| (3) |
| | |
2H2O → O2 + 4H+ + 4e−
| (9) |
| | |
2H2O + 2e− → H2 + 2OH−
| (10) |
| | |
h+ + HSO5− → ˙SO5− + H+
| (11) |
| | |
HSO5− + e− → ˙SO4− + OH−
| (12) |
| | |
S2O82− + e− → ˙SO4− + SO42−
| (13) |
| | |
˙SO4− + H2O → ˙OH + SO42− + H+
| (14) |
| | |
˙SO5− + ˙SO5− → O12 + 2SO4−
| (15) |
Overall, ROS generation pathways can be classified into three categories. (1) Under light irradiation, the catalyst undergoes photocatalytic reactions (eqn (3)–(8)) to generate ˙O2−, ˙OH, and 1O2. (2) Under simultaneous light irradiation and applied bias, electrochemical reactions occur (eqn (9) and (10)) and generate O2 and H2. (3) After PMS addition, reactions associated with PMS generate SO4−, ˙OH, and 1O2 (eqn (11)–(15)). Meanwhile, cross-linked chain reactions also occur among these ROS pathways. For example, ˙OH can originate from both photocatalytic processes and persulfate activation, and both routes can further contribute to the formation of 1O2, resulting in more diversified ROS species and a stronger overall oxidation capability.
EPR trapping experiments also confirmed the presence of these ROS species. In addition to the characteristic ˙OH signals, a comparison with the literature revealed additional ˙SO4− peaks38 (Fig. 9(b)). Therefore, the addition of PMS introduced ˙SO4− radicals into the system. The presence of ˙O2− peaks in Fig. 9(c) further verified the formation of a Z-scheme heterojunction structure. From the band structure shown in Fig. 4(d), if a type-II heterojunction was present, electrons transferred from the conduction band of g-C3N4 to that of TiO2 would not possess sufficient potential to reduce O2 to ˙O2−. Only in the Z-scheme configuration could electrons migrate from the conduction band of TiO2 to the valence band of g-C3N4 and subsequently to its conduction band, where the reduction of O2 to ˙O2− occurred. The trapping of 1O2, shown in Fig. 9(d), further confirmed the coexistence of a non-radical oxidation pathway.
Based on the kinetic data in Table 1, the synergistic activation and enhancement mechanism were analysed, and the synergistic effect of the PEC/PMS system was quantitatively evaluated using the synergy index.39
| |
 | (16) |
where
S represents the synergy index,
kcombined represents the reaction rate constant of the synergistic system, and
ki is the reaction rate constant of the individual system included in the synergistic system.
S > 0 denotes a synergistic effect,
S < 0 denotes a competitive effect, and
S = 0 denotes an cumulative effect. First, the synergy index for PEC was calculated. Compared with PC and EC, the
S value was 0.14, demonstrating that the applied bias voltage promoted photocatalysis. The synergy indices of the PEC/PMS system were evaluated from two perspectives. (1) Considering PEC/PMS as the synergy of PC/PMS and EC/PMS subsystems, the
S value was 0.42. (2) Considering it as a combination of PEC and PMS, the
S value was 0.54. These variations clearly indicate that the synergistic effect in the PEC/PMS system increases significantly with enhanced coupling. To further evaluate system stability, five degradation cycles were conducted. As shown in
Fig. 9(f), after five cycles, the system maintained high tetracycline removal efficiency, demonstrating that the photoanode exhibited strong stability.
Table 1 First-order kinetic model parameters for different degradation modes
| Degradation method |
k1 (min−1) |
t1/2 |
R2 |
| PEC/PMS |
0.2826 |
2.43 |
0.9856 |
| PEC |
0.1314 |
5.27 |
0.9504 |
| PC/PMS |
0.1203 |
5.76 |
0.9537 |
| EC/PMS |
0.0433 |
16.00 |
0.9571 |
| PC |
0.0883 |
7.85 |
0.9082 |
| EC |
0.0247 |
28.06 |
0.9827 |
Table 2 Water quality parameters of pond water
| DO (mg L−1) |
8.45 |
pH |
7.93 |
| COD (mg L−1) |
12.58 |
DOC (mg L−1) |
6.88 |
| SO42− (mM) |
0.34 |
Cl− (mM) |
1.41 |
| Conductivity (µS cm−1) |
218.79 |
Turbidity |
0.87 |
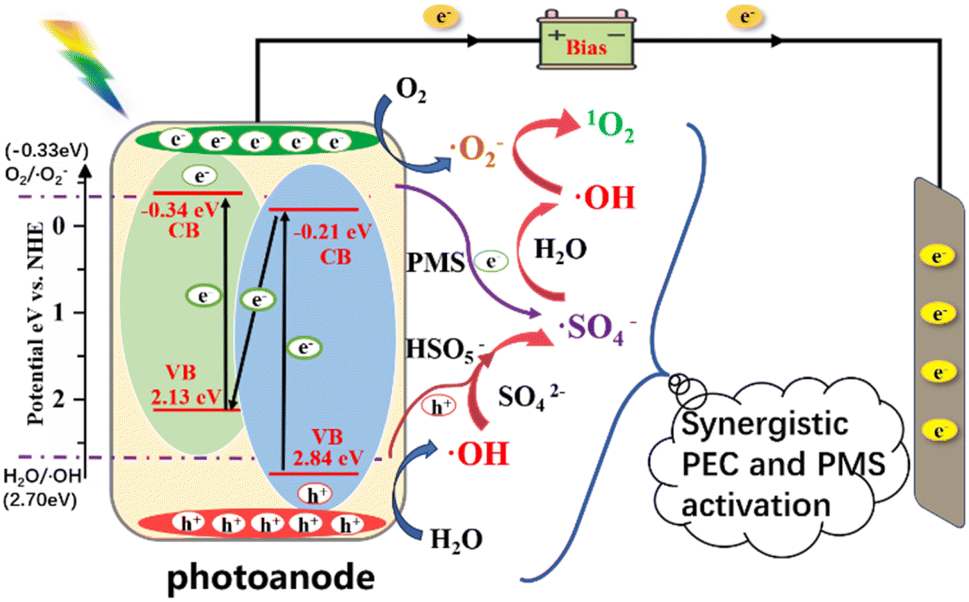
Based on the above analysis, a reaction enhancement mechanism for the PEC/PMS system was proposed (Fig. 10). Under visible-light irradiation, photogenerated holes (h+) and electrons (e−) are produced within the photoanode. According to the direct Z-scheme heterojunction structure, electrons are transferred from the valence band to the conduction band of TiO2, to the valence band of g-C3N4, and finally to its conduction band. This leaves strongly oxidative holes in the valence band of TiO2, whereas electrons accumulate in the conduction band of g-C3N4. The photocatalytic reaction initiated under illumination serves as the primary driving force for the entire synergistic process. Under an applied bias voltage, the electron–hole pairs are rapidly separated, accelerating electron migration. The photogenerated holes directly oxidise H2O to ˙OH and activate HSO5− to generate ˙SO4− radicals. Meanwhile, electrons reduce O2 to ˙O2−. PMS, acting as an electron acceptor, also generates ˙SO4− radicals, demonstrating that PMS addition enhances both the diversity and oxidative strength of ROS in the system. Cross-chain reactions also occur among different radicals. For example, ˙OH can convert ˙O2− into 1O2, ˙SO4− can convert H2O into ˙OH, and ˙OH can further activate PMS by converting SO42− into ˙SO4−, thereby enhancing the oxidation capability of the PEC/PMS system.
 |
| | Fig. 10 Possible band structure of TiO2/g-C3N4 photoanode and schematic diagram of its synergistic PEC activation mechanism for sulfate. | |
4 Conclusion
In this study, g-C3N4 was combined with TiO2 to enhance the synergistic effect between the heterogeneous semiconductors, thereby improving their oxidation and reduction capabilities. The photocatalytic performance was further strengthened under applied external bias voltage. Moreover, efficient and rapid degradation of antibiotics was achieved through photoelectrocatalytic synergistic activation of PMS. The TiO2/g-C3N4 composite exhibited a Z-scheme heterojunction structure, which provided an efficient pathway for photogenerated charge transfer and enhanced the electron migration efficiency. The PEC/PMS system using TiO2/g-C3N4 as the photoanode accelerated electron migration and increased the photoresponse current, achieving 96.04% removal of TC within 12 min. The optimal operating conditions were identified as a bias voltage of 1.0 V, a PMS concentration of 0.2 mmol L−1, and a slightly alkaline environment, which effectively resisted interference from background water matrices. In aquaculture pond water, more than 88% of various antibiotics were successfully removed, demonstrating excellent practical applicability. The oxidation process was dominated by radical species (such as ˙OH and ˙SO4−), with the coexistence of a non-radical 1O2 pathway. The calculated synergy indices further indicated that the introduction of an electric field and PMS significantly enhanced electron transfer, exhibiting a remarkable synergistic enhancement effect.
Author contributions
Xue Zhang: conceptualization, data curation, formal analysis, writing – original draft. Xiangpeng Gao: data curation. Baowei Zhao: writing – review & editing. Nan Wu: formal analysis, resources, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
All relevant data are within the paper.
Acknowledgements
This study was supported by Young Scholars Science Foundation of Lanzhou Jiaotong University (2022045) and the Key Research and Development Program of Gansu Province – Social Development Project under Grant No. 25YFFA014.
Notes and references
- W. Li, G. Lv, M. Liu, F. Zhao, P. Shuai, Y. Feng, D. Chen and L. Liao, J. Energy Chem., 2025, 108, 332–360 CrossRef CAS.
- J. Sun, C. Fu, Z. Hu, I. Sirés and M. Zhou, Crit. Rev. Environ. Sci. Technol., 2025, 55(11), 783–804 CrossRef CAS.
- Y. Liu, X. Xu, X. Sun, W. Li and J. Duan, J. Water Process Eng., 2025, 75, 107911 CrossRef.
- L. Ju, P. Wu, Y. Ju, M. Chen, S. Yang and H. Zhu, Surf. Interfaces, 2021, 23, 100967 CrossRef CAS.
- T. Yusuf, B. Orimolade, D. Masekela, B. Mamba and N. Mabuba, RSC Adv., 2022, 12, 26176–26191 RSC.
- A. Navidpour, D. Hao, X. Li, D. Li, Z. Huang and J. Zhou, Catal. Rev.:Sci. Eng., 2024, 66(5), 1665–1736 CrossRef CAS.
- E. Zandy, A. Bakhtiari and H. Hosseini, Langmuir, 2025, 41(21), 13013–13027 CrossRef CAS PubMed.
- H. Zhuang, l. Lin, M. Xu, W. Xu and X. Li, J. Alloys Compd., 2023, 969, 172302 CrossRef CAS.
- F. Liang and Y. Zhu, Appl. Catal., B, 2016, 180, 324–329 CrossRef CAS.
- J. Dong, Z. Gong, Y. Chen, G. Hao, W. Zhou, J. Li, M. Yang, R. Deng and L. Wang, Sci. China Mater., 2023, 66, 3176–3188 CrossRef CAS.
- M. Kumar, P. Haripriya and D. Kumar, Chem. Pap., 2024, 78, 8465–8472 CrossRef CAS.
- M. Saeed, A. Waseem and A. Intisar, Catal. Surv. Asia, 2025, 29, 11–25 CrossRef CAS.
- M. Mansha, T. Ahmad, N. Ullah, S. A. Khan, M. Ashraf, S. Ali, B. Tan and I. Khan, Chem. Rec., 2022, 22(7), e202100336 CrossRef CAS PubMed.
- K. Huang, C. Li, X. Zhang, X. Meng, L. Wang, W. Wang and Z. Li, Appl. Surf. Sci., 2020, 518, 146219 CrossRef CAS.
- T. Bui, H. Le, H. Nguyen and V. Pham, Mol. Catal., 2025, 577, 114960 CAS.
- Y. Luo, W. Yang, J. Liu, W. Cai, Z. Liao, X. Xiang, B. Li and Y. Fang, Int. J. Hydrogen Energy, 2025, 168, 151065 CrossRef CAS.
- X. Du, X. Bai, L. Xu, L. Yang and P. Jin, Chem. Eng. J., 2020, 384, 123245 CrossRef CAS.
- J. Sun, Y. Guo, Y. Wang, D. Cao, S. Tian, K. Xiao, R. Mao and X. Zhao, Chem. Eng. J., 2018, 332, 312–320 CrossRef CAS.
- M. Petruleviciene, I. Savickaja, J. Jarosevic, J. Juodkazyte, A. Padarauskas, A. Griguceviciene and A. Ramanavicius, ACS Omega, 2025, 10(8), 8538–8550 CrossRef CAS PubMed.
- N. Wu, X. Zhang, X. Zhang, K. Yang and Y. Li, Mater. Res. Express, 2022, 9, 065601 CrossRef CAS.
- Q. Li, T. Zhang, D. Cuia and F. Li, Dalton Trans., 2024, 53, 7742–7750 RSC.
- Y. Qi, H. Zhou and G. Han, RSC Adv., 2025, 15, 21300–21310 RSC.
- A. Beizavi and M. Boroujerdni, Ceram. Interfaces, 2024, 50(23), 50317–50326 CrossRef CAS.
- J. Xu and S. Ma, J. Cryst. Growth, 2023, 671, 127290 CrossRef.
- R. Paul, A. Das, B. Gururajan and S. Parthiban, Diamond Relat. Mater., 2025, 159, 112766 CrossRef CAS.
- L. Yang, Y. Liu, P. Tan, Y. Lu, Q. Ding and J. Pan, Diamond Relat. Mater., 2023, 133, 109727 CrossRef CAS.
- X. Chen, J. Wu, X. Wang, R. Jia, L. Li, Y. Wang, Y. Cai, Z. Chen, C. Jin, X. Wang, P. Qi, R. Wang and N. Zhang, J. Colloid Interface Sci., 2025, 684, 1049–1056 CrossRef PubMed.
- W. Shi, W. Sun, Y. Liu, K. Zhang, H. Sun, X. Lin, Y. Hong and F. Guo, J. Hazard. Mater., 2022, 436, 129141 CrossRef CAS PubMed.
- F. Zhang, J. Liu, H. Yue, G. Cheng and X. Xue, J. Mater. Sci., 2023, 58, 2676–2688 CrossRef CAS.
- F. Liu, M. Fan, X. Liu and J. Chen, Nanomaterials, 2024, 14(13), 1141 CrossRef CAS PubMed.
- C. Yang, X. Zhang, Y. Zhou and S. Hao, Appl. Surf. Sci., 2023, 616, 156471 CrossRef CAS.
- Z. Liu, H. Ding, C. Zhao, T. Wang, P. Wang and D. Dionysiou, Water Res., 2019, 159, 111–121 CrossRef CAS PubMed.
- Q. Yang, Y. Ma, F. Chen, F. Yao, J. Sun, S. Wang, K. Yi, L. Hou, X. Li and D. Wang, Chem. Eng. J., 2019, 378, 122149 CrossRef CAS.
- A. Son, J. Lee, C. Lee, K. Cho, J. Lee and S. Hong, Water Res., 2021, 191, 116803 CrossRef CAS PubMed.
- G. Zhang, X. Zhu, M. Yu and F. Yang, Sep. Purif. Technol., 2023, 318, 124002 CrossRef CAS.
- A. Son, J. Lee, M. Seid, E. Rahman, J. Choe, K. Cho, J. Lee and S. Hong, Appl. Catal., B, 2022, 315, 121543 CrossRef CAS.
- H. Hu, H. Lin, X. Chen, Y. Pan, X. Li, Z. Zhuang, H. Chen, X. Wang, M. Luo, K. Zheng, L. Zhang and F. Chen, Chem. Eng. J., 2023, 476, 146682 CrossRef CAS.
- M. Zhang, Y. Gong, N. Ma and X. Zhao, Chem. Eng. J., 2020, 399, 125088 CrossRef CAS.
- R. Dhawle, S. Giannakopoulos, Z. Frontistis and D. Mantzavinos, Catal. Today, 2023, 413–415, 114026 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Nan Wu
*a and
Nan Wu