
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable site-selective C–C bond cleavage for the divergent synthesis of imidazo[1,5-a]pyridine derivatives
Qiang Wang *,
Xinyu Song,
Shujun Qin,
Wenya Dong,
Pingjing Xue,
Jiayi Wu and
Haibo Wang
*,
Xinyu Song,
Shujun Qin,
Wenya Dong,
Pingjing Xue,
Jiayi Wu and
Haibo Wang
College of Biomedical and Health Sciences, Anhui Science and Technology University, Fengyang 233100, PR China. E-mail: wangqiang@ahstu.edu.cn
First published on 10th December 2025
Abstract
Ethyl 3-oxo-3-phenylpropanoate reacts with phenyl(pyridin-2-yl)methanamine to afford a selective product via the site-selective C–C bond cleavage of ethyl 3-oxo-3-phenylpropanoate by adjusting reaction parameters, and a series of imidazo[1,5-a]pyridines can be effectively prepared using this method. This study enables controllable site-selectivity in both C–C bond cleavage and C–C bond formation, which may offer new insights for chemists in the C–C bond cleavage research field.
Introduction
Organic reactions mediated by C–C bond cleavage have attracted the attention of many researchers in recent years, but compared with conventional C–C bond formation reactions, this area is still in the early stages and remains to be explored.1–4 It is evident from the literature that there are three challenges in organic reactions mediated by C–C bond cleavage.1,3,5–13 Firstly, reactions mediated by C–C bond cleavage are not as extensive as C–C bond formation reactions, and there is no mature theory to guide chemists to apply such reactions. The second problem is the selectivity of C–C bond cleavage, that is, the selectivity between the retained carbon group and the leaving carbon group is difficult to achieve when the C–C bond breaks. To be specific, if the electronic and spatial properties of the bonding carbon group and the leaving carbon group are significantly different, the selectivity is relatively easy to achieve, otherwise, there are greater difficulties. The third problem is the controllability of C–C bond cleavage for directional synthesis. Theoretically, it is possible to realize the switch between the bonding group and the leaving group by adjusting the experimental conditions, while related study is rarely reported.1,3-Dicarbonyl compounds are important synthons widely used in various synthetic reactions, and their selective C–C bond cleavage reactions have been also reported in several papers.8,14–18 These reactions mainly involve transition metal catalyzed C–C bond cleavage mediated bond formation. Besides, only two studies of metal-free selective C–C bond cleavage mediated for heterocycles synthesis have been reported17,19 (Scheme 1), which makes it possible to synthesize carbonyl-derived heterocycles by using 1,3-dicarbonyl compounds as a carbonyl source. Compared with single selectivity of C–C cleavage reactions of 1,3-dicarbonyl compounds above, controllable C–C cleavage reactions at two sites for two different carbonyl derived products have not been reported to the best of our knowledge. 1,3-dicarbonyl compounds are good model substrates, due to the methylene group being connected to two carbon groups, which have similar electronic and spatial properties on both sides, whether the cleavage site could be controlled is worth trying.
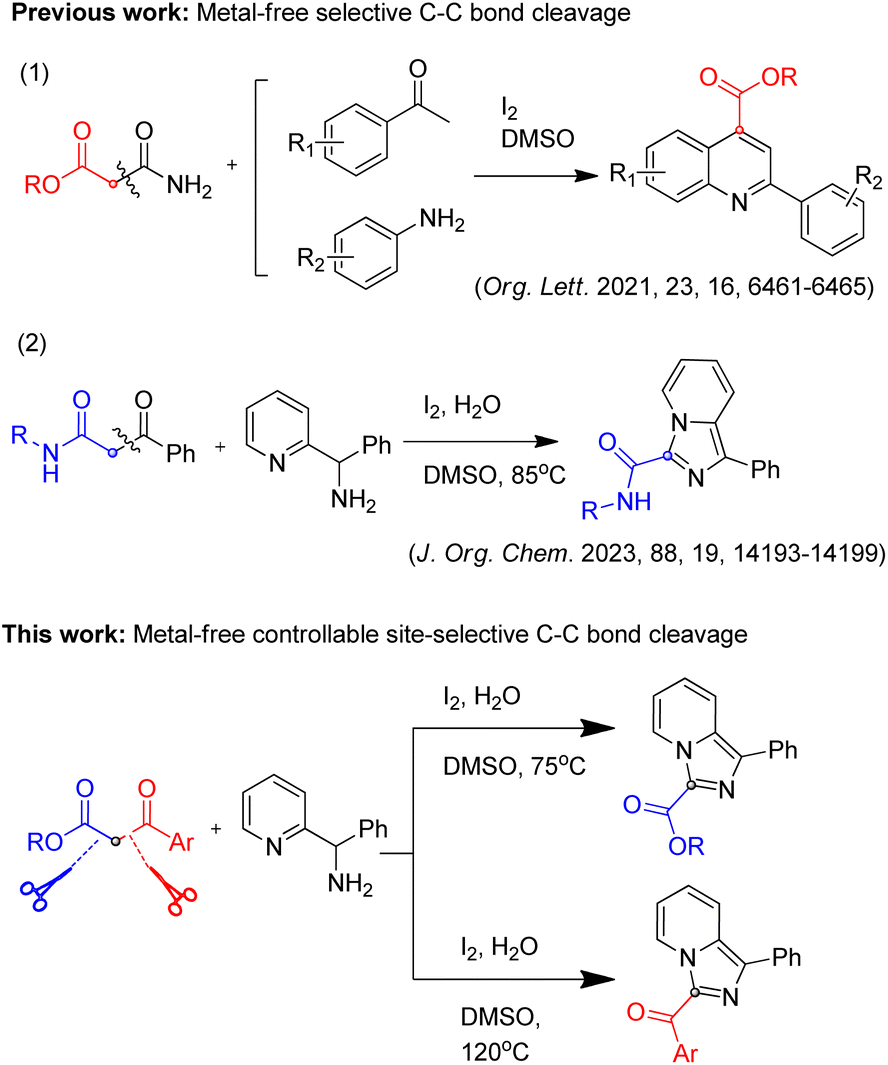 | ||
| Scheme 1 Selective C–C bond cleavage and controllable site-selective C–C bond cleavage mediated reactions. | ||
Based on the reported work and our previous studies,19 we conducted deep exploration and realized controllable two-site C–C cleavage mediated reaction of 1,3-dicarbonyl compounds by adjusting the reaction conditions, and successfully synthesized 3-ester imidazo[1,5-a]pyridines and 3-acyl imidazo[1,5-a]pyridines, respectively (Scheme 1). Such compounds have a wide range of biological activities in the field of medicinal chemistry,20–22 therefore, this method is very useful for medicinal chemists in related areas.
Results and discussion
As shown in Table 1, phenyl(pyridin-2-yl)methanamine (1a) and ethyl 3-oxo-3-phenylpropanoate (2a) and were used as model substrates to explore the controllability of C–C bond cleavage. The results showed that substrate ratio, the amount of I2, temperature and reaction time all affected the reaction results by selecting C–C cleavage sites. With 1a and 2a in equimolar amounts (0.2 mmol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.2 mmol), 3 equivalents of I2, and the reaction conducted in DMSO at different temperatures, it was found that lower temperature favored the C–C bond cleavage at the benzoyl group side and provided 3-ester imidazo[1,5-a] pyridine (3a′) as single product (entries 1 and 2), with the temperature increased, the C–C bond cleavage site gradually transferred to the ester side and provided 3-benzoyl imidazo[1,5-a] pyridine (3a) and 3-ester imidazo[1,5-a] pyridine (3a′) (entries 1, 2 vs. entries 3–5), and the reaction time was greatly saved to 3 hours (entry 5) and 2 hours (entries 6 and 8–13), also the yield of 3a′ was increased with temperature increasing (entry 5 vs. entries 6 and 8–13). When the temperature was 120 °C, 3-benzoyl imidazo[1,5-a] pyridine (3a) presented as major product (56%) and primary selectivity on another side could be observed (entry 6), these results indicated that temperature was a key factor. With the phenomenon observed, further investigation for controllable C–C bond cleavage at ester side was performed. Firstly, adjusting substrate ratio to 0.25:0.2, and keep the other factors unchanged, satisfactory yield (76%) of 3a′ could be obtained at 75 °C as single product (entry 7). Keeping this substrate ratio and gradually adjusting the amount of I2 provided elevated selectivity for the other product (3a) with modest yields at 120 °C (entries 8–9), while reducing I2 to 0.2 mmol the yield was affected greatly (entry 10). Subsequently, adjusting the substrate ratio to 0.2:0.25 and 0.2:0.3, satisfactory selectivity results were obtained (entries 11–13), 3a (75%) and 3a′ (7%) were obtained in entry 12, more than 10:1 selectivity was obtained, and the reaction was not optimized further.
:0.2 mmol), 3 equivalents of I2, and the reaction conducted in DMSO at different temperatures, it was found that lower temperature favored the C–C bond cleavage at the benzoyl group side and provided 3-ester imidazo[1,5-a] pyridine (3a′) as single product (entries 1 and 2), with the temperature increased, the C–C bond cleavage site gradually transferred to the ester side and provided 3-benzoyl imidazo[1,5-a] pyridine (3a) and 3-ester imidazo[1,5-a] pyridine (3a′) (entries 1, 2 vs. entries 3–5), and the reaction time was greatly saved to 3 hours (entry 5) and 2 hours (entries 6 and 8–13), also the yield of 3a′ was increased with temperature increasing (entry 5 vs. entries 6 and 8–13). When the temperature was 120 °C, 3-benzoyl imidazo[1,5-a] pyridine (3a) presented as major product (56%) and primary selectivity on another side could be observed (entry 6), these results indicated that temperature was a key factor. With the phenomenon observed, further investigation for controllable C–C bond cleavage at ester side was performed. Firstly, adjusting substrate ratio to 0.25:0.2, and keep the other factors unchanged, satisfactory yield (76%) of 3a′ could be obtained at 75 °C as single product (entry 7). Keeping this substrate ratio and gradually adjusting the amount of I2 provided elevated selectivity for the other product (3a) with modest yields at 120 °C (entries 8–9), while reducing I2 to 0.2 mmol the yield was affected greatly (entry 10). Subsequently, adjusting the substrate ratio to 0.2:0.25 and 0.2:0.3, satisfactory selectivity results were obtained (entries 11–13), 3a (75%) and 3a′ (7%) were obtained in entry 12, more than 10:1 selectivity was obtained, and the reaction was not optimized further.
|
||||||
|---|---|---|---|---|---|---|
| Entry | 1a:2a (mmol) |
I2 (mmol) | T (°C) | Reaction time (h) | Yield | |
| 3a | 3a′ | |||||
| a Reaction conditions: specified amount of phenyl(pyridin-2-yl)methanamine (1a), ethyl 3-oxo-3-phenylpropanoate (2a), I2 and H2O (0.6 mmol) were stirred with specified temperature and reaction time in DMSO (2 mL). Isolated yields were shown. n.d. = not detected. | ||||||
| 1 | 0.2:0.2 |
0.6 | 70 | 6 | n.d | 51% |
| 2 | 0.2:0.2 |
0.6 | 75 | 6 | n.d | 56% |
| 3 | 0.2:0.2 |
0.6 | 80 | 6 | 29% | 41% |
| 4 | 0.2:0.2 |
0.6 | 100 | 6 | 43% | 32% |
| 5 | 0.2:0.2 |
0.6 | 110 | 3 | 50% | 23% |
| 6 | 0.2:0.2 |
0.6 | 120 | 2 | 56% | 18% |
| 7 | 0.25:0.2 |
0.6 | 75 | 6 | n.d | 76% |
| 8 | 0.25:0.2 |
0.4 | 120 | 2 | 57% | 13% |
| 9 | 0.25:0.2 |
0.3 | 120 | 2 | 64% | 10% |
| 10 | 0.25:0.2 |
0.2 | 120 | 2 | 43% | 7% |
| 11 | 0.2:0.25 |
0.3 | 120 | 2 | 69% | 10% |
| 12 | 0.2:0.3 |
0.3 | 120 | 2 | 75% | 7% |
| 13 | 0.2:0.4 |
0.3 | 120 | 2 | 61% | 6% |

With the optimized conditions in hand, a series of 1,3-dicarbonyl compounds were employed to investigate the applicability of this controllable C–C bond cleavage mediated reaction. As shown in Table 2, derivatives of ethyl 3-oxo-3-phenylpropanoate were firstly examined. Electron-donating group methyl at ortho-position, meta-position and para-position all provided the target product 3a′ with 73–76% yields under conditions of 75 °C, and exhibited excellent selectivity without detection of 3-acyl imidazo[1,5-a] pyridine. Unsurprisingly, at 120 °C, these three substrates still maintained high selectivity, exhibiting similar selectivity profiles and yields to those of 2a and gave products 3b, 3c and 3d (entries 1–3). Similarly, electron-withdrawing group bromide at ortho-position, meta-position and para-position all provided the target product 3a′ as well with 72–76% yields under conditions of 75 °C, and provided 3e, 3f and 3g (76–79%) under the conditions of 120 °C (entries 4–6). It was found that electron-withdrawing group and electron-donating group both provided similar selectivity profile and yields at 75 °C and 120 °C, indicating electronic effects did not significantly affect the reaction results. Other common substituents were also examined under the optimized conditions of 75 °C and 120 °C respectively, such as halogen group, fluorine, chloride, and iodine substituents all reacted smoothly and provided similar selectivity profile under the optimized condition (entries 7–9). Electron-donating group, methoxy group and other electron-withdrawing group –CF3, –NO2 and –CN all provided similar results (entries 10–13). Besides, multi-substituted ethyl 3-oxo-3-phenylpropanoate derivatives were also investigated and the results were similar as that of 2a (entries 15–17). Moreover, heteroaromatic ring derived 1,3-dicarbonyl compounds, methyl ester and n-butyl ester 1,3-dicarbonyl compounds were still applicable under the optimized condition and exhibited similar selectivity profile (entries 18–20). Results above demonstrated that this controllable C–C bond cleavage reaction was applicable for various analogues of ethyl 3-oxo-3-phenylpropanoate. In addition to the reaction results listed in the Table 2, we also examined 1a analogues 1-(pyridin-2-yl)ethanamine and pyridin-2-ylmethanamine reacting with ethyl 3-oxo-3-phenylpropanoate, but the target products were not detected under 75 °C, and the reaction system were rather chaotic at 120 °C.These results indicated that the benzene ring of phenyl(pyridin-2-yl)methanamine was essential to the reaction.
| a Conditions: reactions were carried out using 1,3-dicarbonyl compounds (0.2 mmol), phenyl(pyridin-2-yl)methanamine (0.25 mmol), I2 (0.6 mmol) and H2O (0.6 mmol) in 2 mL of DMSO and stirred at 75 °C for 6 h.b Reactions were carried out using 1,3-dicarbonyl compounds (0.3 mmol), phenyl(pyridin-2-yl)methanamine (0.2 mmol), I2 (0.3 mmol) and H2O (0.6 mmol) in 2 mL of DMSO and stirred at 120 °C for 2 h. |
|---|
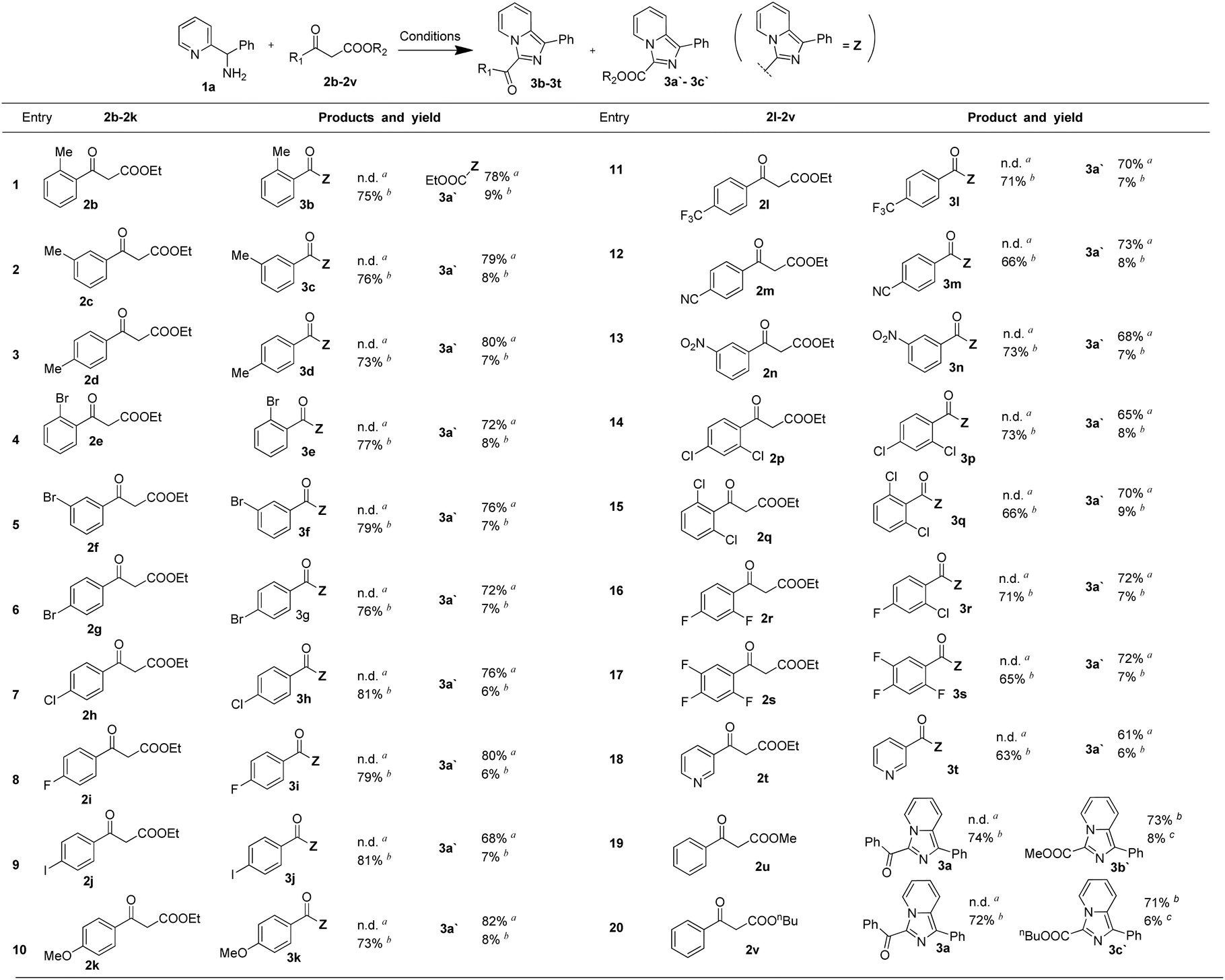 |
To obtain more information about the reaction mechanism, a set of control experiments were performed. As shown in Scheme 2, ethyl 2-iodo-3-oxo-3-phenylpropanoate reacted with phenyl(pyridin-2-yl)methanamine under two standard conditions could provide target product 3a with 77% yield and 3a′ with 73% yield respectively (Scheme 2a). Besides, 2,2,6,6-tetramethylpiperidoxyl (TEMPO) had little effect on the yield under the standard conditions, indicating this reaction did not experience free radical process (Scheme 2b). Moreover, oxygen free experiments demonstrated that oxygen was unnecessary in this transformation (Scheme 2c).
 | ||
| Scheme 2 Control experiments. | ||
Based on our previous reports17,19 and the experimental results, a possible mechanism was depicted in Scheme 3. 2a is iodized on the methylene to form 2a′, 2a′ reacts with 1a via nucleophilic reaction to form intermediate I, which is oxidized by iodine to give intermediate II. At 75 °C, intermediate II could experience an intramolecular nucleophilic attack to give intermediate III, intermediate III experience a iodine and water mediated elimination and dehydrogenation to provide 3a′ and benzoic acid. At 120 °C, Intermediate II mainly experience an intramolecular nucleophilic substitution and provide 3a, carbon dioxide and ethanol directly, and a small part of intermediate II unavoidably experience the reaction process of 75 °C to produce minor product 3a′.
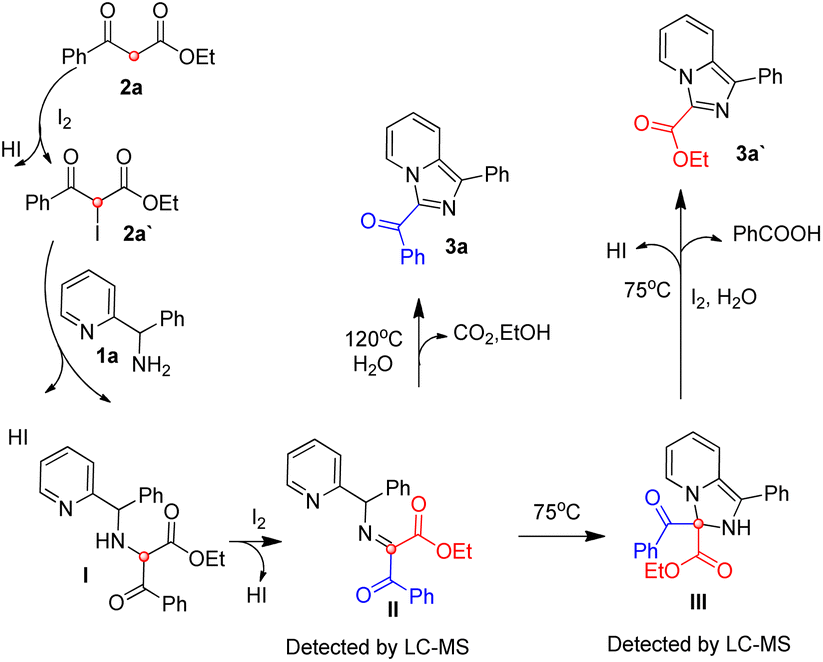 | ||
| Scheme 3 Possible mechanism. | ||
The proposed mechanism was also supported in the further Gaussian calculation modeling, Comparison of the C-ester bond energy and C-benzoyl bond energy in intermediates II and III provides theoretical support for the proposed mechanism. As shown in scheme 4A, the C-ester bond energy (69.68 kcal mol−1) is less than C-benzoyl bond energy (71.01 kcal mol−1), II tends to preferentially remove the ester group under the intramolecular nucleophilic attack at 120 °C. When the reaction temperature is 75 °C, II transformed to III, and the C-ester bond energy and C-benzoyl bond energy in III both decreased significantly (Scheme 4B). Besides, intramolecular hydrogen bond formation made C-ester bond energy (38.95 kcal mol−1) higher than C-benzoyl bond energy (36.59 kcal mol−1). These results provided a relatively rational explanation of experimental phenomenon.
 | ||
| Scheme 4 Gaussian calculation of bond energy of intermediate II and III. | ||
Conclusions
In conclusion, an iodine-mediated controllable site-selective C–C bond cleavage of ethyl 3-oxo-3-phenylpropanoate has been achieved for the preparation of 3-carbonyl-substituted imidazo[1,5-a]pyridines. This study provides an alternative method for the efficient synthesis of 3-carbonyl-substituted imidazo[1,5-a]pyridines with satisfactory yields. More importantly, this study has for the first time achieved controllable site-selective C–C bond cleavage of 1,3-dicarbonyl compounds by simply adjusting reaction conditions, and a relatively reasonable reaction mechanism supported by both experimental and theoretical calculation results has been proposed.Experimental section
Reagents and instruments
Unless otherwise noted, all reactions were performed under an air atmosphere. Reagents were purchased from the Tansoole reagent platform and used directly. 1H NMR and 13C NMR spectra were measured at room temperature. High-resolution mass spectra (HRMS) were obtained from Waters UPLC G2-XS QTOF. Flash column chromatography was performed on 200–300 mesh of silica gel.General procedure for the controllable site-selective C–C bond cleavage mediated reactions
Gaussian calculation of bond energy of intermediates II and III
The B3LYP23 density functional method with the D3(BJ)24 dispersion correction was employed in this work to carry out all the computations. The def2 TZVP basis25 set was used for the atoms in geometry optimizations using the PCM model26 with DMSO as the solvent. Vibrational frequency analyses at the same level of theory were performed to characterize stationary points as local minima without any imaginary frequencies. All DFT theoretical calculations have been carried out using the Gaussian program package.NMR spectra data for products
:4) to obtain yellow solid ( 44.7 mg, 75%). 1H NMR (600 MHz, CDCl3) δ 9.93 (d, J = 7.1 Hz), 8.60–8.49 (m), 8.08 (d, J = 9.0 Hz), 8.00–7.94 (m), 7.63–7.57 (m), 7.56–7.48 (m), 7.43–7.35 (m), 7.28 (ddd, J = 8.9, 6.6, 0.9 Hz), 7.11–7.04 (m). 13C (151 MHz, CDCl3) δ 182.2, 138.2, 134.7, 134.0, 133.9, 132.1, 131.2, 130.9, 128.8, 128.0, 127.6, 127.3, 125.1, 118.4, 116.4.:4) to obtain yellow solid (46.8 mg, 75%). 1H NMR (600 MHz, CDCl3) δ 9.92 (d, J = 7.2 Hz), 8.47 (d, J = 8.2 Hz), 8.06 (d, J = 9.0 Hz), 7.98–7.95 (m), 7.51 (t, J = 7.7 Hz), 7.41–7.33 (m), 7.27–7.25 (m), 7.07–7.03 (m), 2.46 (s). 13C NMR (151 MHz, CDCl3) δ 182.0, 153.4, 142.8, 135.5, 134.4, 134.1, 134.0, 131.0, 128.8, 128.7, 127.5, 127.3, 124.9, 118.3, 116.2.:4) to obtain yellow solid (47.4 mg, 76%). 1H NMR (600 MHz, CDCl3) δ 9.92 (d, J = 7.1 Hz), 8.37 (d, J = 7.4 Hz), 8.26 (s), 8.07 (d, J = 9.0 Hz), 7.99–7.93 (m), 7.51 (t, J = 7.7 Hz), 7.45–7.36 (m), 7.28 (ddd, J = 8.9, 6.6, 0.6 Hz), 7.10–7.03 (m), 2.48 (s). 13C NMR (151 MHz, CDCl3) δ 182.5, 138.1, 137.6, 134.5, 134.0, 134.0, 132.9, 131.2, 128.8, 128.4, 127.8, 127.5, 127.3, 125.0, 118.4, 116.4, 21.5.:4) to obtain pale oil (yields shown in Table 1 entries 1–18 for each reaction). 1H NMR (400 MHz, CDCl3) δ 9.41 (d, J = 7.2 Hz, 1H), 8.00–7.86 (m, 3H), 7.47 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.16–7.07 (m, 1H), 6.95 (dd, J = 10.0, 3.7 Hz, 1H), 4.55 (q, J = 7.1 Hz, 2H), 1.49 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 159.8, 134.0, 133.7, 130.7, 128.69, 127.5, 127.4, 126.6, 125.9, 123.1, 118.5, 115.5, 61.3, 14.5.:4) to obtain yellow solid (36.8 mg, 73%). 1H NMR (400 MHz, CDCl3) δ 9.40 (d, J = 7.2 Hz, 1H), 7.98 (d, J = 9.1 Hz, 1H), 7.90 (d, J = 7.6 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.14 (dd, J = 8.9, 6.7 Hz, 1H), 6.97 (t, J = 6.9 Hz, 1H), 4.05 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 160.0, 134.1, 133.5, 130.7, 128.7, 127.5, 127.4, 126.3, 125.8, 123.3, 118.6, 115.7, 52.2.:4) to obtain yellow solid (41.7 mg, 71%). 1H NMR (600 MHz, CDCl3) δ 9.40 (d, J = 7.2 Hz, 1H), 7.96 (d, J = 9.1 Hz, 1H), 7.90 (d, J = 7.3 Hz, 2H), 7.47 (t, J = 7.7 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.17–7.07 (m, 1H), 6.94 (dd, J = 10.0, 3.7 Hz, 1H), 4.48 (t, J = 7.0 Hz, 2H), 1.89–1.83 (m, 2H), 1.53–1.47 (m, 2H), 0.99 (dd, J = 8.6, 6.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 160.0, 134.1, 133.9, 130.8, 128.8, 127.6, 127.5, 126.8, 126.0, 123.2, 118.7, 115.6, 65.2, 30.9, 19.2, 13.9.:4) to obtain yellow solid (45.6 mg, 73%). 1H NMR (600 MHz, CDCl3) δ 9.92 (d, J = 7.2 Hz), 8.47 (d, J = 8.2 Hz), 8.06 (d, J = 9.0 Hz), 7.98–7.95 (m), 7.51 (t, J = 7.7 Hz), 7.41–7.33 (m), 7.27–7.25 (m), 7.07–7.03 (m), 2.46 (s). 13C NMR (151 MHz, CDCl3) δ 182.0, 153.4, 142.8, 135.5, 134.4, 134.1, 134.0, 131.0, 128.8, 128.7, 127.5, 127.3, 124.9, 118.3, 116.2.:4) to obtain light yellow solid (58.1 mg, 77%). 1H NMR (600 MHz, CDCl3) δ 9.92 (dt, J = 7.1, 1.1 Hz), 8.08–8.06 (m), 7.87–7.80 (m), 7.69 (td, J = 7.9, 1.3 Hz), 7.47–7.41 (m), 7.37–7.32 (m), 7.14 (td, J = 6.9, 1.2 Hz). 13C NMR (151 MHz, CDCl3) δ 183.6, 140.3, 135.9, 133.6, 133.5, 133.2, 132.0, 131.0, 130.8, 128.7, 127.7, 127.4, 127.2, 126.6, 125.8, 120.6, 118.5, 116.9. HRMS (ESI) m/z [M + H+] calcd for C20H13BrN2O, 376.0211, found. 376.0218.:4) to obtain light yellow solid (59.6 mg, 79%). 1H NMR (600 MHz, CDCl3) δ 9.92 (dt, J = 7.2, 1.0 Hz), 8.67 (t, J = 1.8 Hz), 8.55–8.49 (m), 8.10 (dt, J = 9.0, 1.1 Hz), 7.99–7.93 (m), 7.70 (ddd, J = 7.9, 2.0, 1.0 Hz), 7.56–7.49 (m), 7.43–7.38 (m), 7.32 (ddd, J = 9.0, 6.6, 1.0 Hz), 7.11 (td, J = 7.0, 1.2 Hz). 13C NMR (151 MHz, CDCl3) δ 180.1, 139.9, 135.1, 134.8, 133.7, 133.7, 131.5, 129.6, 129.5, 128.9, 127.8, 127.4, 127.3, 125.5, 122.1, 118.5, 116.8. HRMS (ESI) m/z [M + H+] calcd for C20H13BrN2O, 376.0211, found. 376.0217.:4) to obtain light yellow solid (57.3 mg, 76%). 1H NMR (600 MHz, CDCl3) δ 9.91 (dt, J = 7.1, 1.1 Hz), 8.47–8.43 (m), 8.08 (dt, J = 9.0, 1.2 Hz), 7.97–7.92 (m), 7.69–7.64 (m), 7.54–7.49 (m), 7.40 (ddd, J = 7.4, 4.1, 1.2 Hz), 7.30 (ddd, J = 9.0, 6.6, 1.0 Hz), 7.08 (td, J = 6.9, 1.2 Hz). 13C NMR (151 MHz, CDCl3) δ 180.6, 136.9, 134.9, 133.7, 132.5, 131.4, 131.2, 128.8, 127.7, 127.3, 127.2, 127.2, 125.4, 118.4, 116.6.:4) to obtain light yellow solid (53.8 mg, 81%). 1H NMR (600 MHz, CDCl3) δ 9.92 (d, J = 7.2 Hz), 8.56–8.51 (m), 8.09 (d, J = 9.0 Hz), 7.98–7.93 (m), 7.55–7.49 (m), 7.40 (d, J = 7.4 Hz), 7.31 (ddd, J = 8.9, 6.6, 0.8 Hz), 7.10 (dd, J = 6.8, 0.8 Hz). 13C NMR (151 MHz, CDCl3) δ 180.5, 138.4, 136.4, 134.9, 133.8, 133.8, 132.4, 131.4, 128.9, 128.3, 127.7, 127.3, 127.3, 125.4, 118.4, 116.6.:4) to obtain light yellow solid (49.9 mg, 79%). 1H NMR (600 MHz, CDCl3) δ 9.91 (d, J = 7.1 Hz), 8.65–8.59 (m), 8.08 (d, J = 9.0 Hz), 7.95 (d, J = 7.2 Hz), 7.52 (t, J = 7.7 Hz), 7.40 (d, J = 7.4 Hz), 7.29 (ddd, J = 9.0, 6.6, 0.6 Hz), 7.21 (t, J = 8.7 Hz), 7.10–7.04 (m). 13C NMR (151 MHz, CDCl3) δ 180.4, 166.1, 164.4, 134.7, 134.3, 134.3, 133.8, 133.5, 133.5, 131.2, 128.8, 127.7, 127.3, 127.3, 125.2, 118.4, 116.5, 115.1, 115.0.:4) to obtain light yellow solid (68.7 mg, 81%).1H NMR (600 MHz, CDCl3) δ 9.91 (d, J = 7.1 Hz), 8.28 (d, J = 8.4 Hz), 8.08 (d, J = 9.0 Hz), 7.94 (d, J = 7.3 Hz), 7.88 (d, J = 8.4 Hz), 7.51 (t, J = 7.7 Hz), 7.39 (t, J = 7.4 Hz), 7.30 (dd, J = 8.4, 7.1 Hz), 7.08 (t, J = 6.8 Hz). 13C NMR (151 MHz, CDCl3) δ 180.9, 137.4, 137.2, 134.9, 133.7, 132.4, 131.4, 128.8, 127.7, 127.3, 127.2, 125.4, 118.4, 116.6, 100.0. HRMS (ESI) m/z [M + H+] calcd for C20H13IN2O, 424.0073, found 424.0076.:4) to obtain light yellow solid (47.9 mg, 73%). 1H NMR (600 MHz, CDCl3) δ 9.90 (d, J = 7.1 Hz), 8.67–8.61 (m), 8.05 (d, J = 9.0 Hz), 7.97 (dd, J = 8.1, 0.9 Hz), 7.51 (t, J = 7.7 Hz), 7.38 (t, J = 7.4 Hz), 7.26–7.22 (m), 7.05–7.00 (m), 3.91 (s). 13C NMR (151 MHz, CDCl3) δ 180.8, 162.9, 134.1, 134.1, 134.0, 133.2, 130.8, 130.8, 128.8, 127.4, 127.2, 124.7, 118.3, 116.1, 113.3, 55.4.:4) to obtain yellow solid (52.0 mg, 71%). 1H NMR (600 MHz, CDCl3) δ 9.95 (dt, J = 7.2, 1.2 Hz), 8.66–8.60 (m), 8.11 (dt, J = 9.1, 1.3 Hz), 7.99–7.92 (m), 7.85–7.75 (m), 7.56–7.50 (m), 7.49–7.36 (m), 7.35 (ddd, J = 9.1, 6.6, 1.2 Hz), 7.16–7.10 (m). 13C (151 MHz, CDCl3) δ 180.6, 141.2, 135.4, 133.7, 133.6, 133.2, 133.0, 131.7, 131.1, 128.9, 127.9, 127.4, 127.3, 125.7, 124.9, 124.9, 124.9, 124.9, 118.5, 116.9.:4) to obtain yellow solid (42.6 mg, 66%). 1H NMR (600 MHz, CDCl3) δ 9.93 (dd, J = 7.1, 0.8 Hz), 8.67–8.53 (m), 8.11 (d, J = 9.0 Hz), 7.93 (dd, J = 8.1, 0.9 Hz), 7.83–7.77 (m), 7.53 (dd, J = 10.7, 4.8 Hz), 7.45–7.38 (m), 7.37 (ddd, J = 8.8, 6.6, 0.7 Hz), 7.19–7.10 (m). 13C NMR (151 MHz, CDCl3) δ 179.6, 141.7, 135.6, 133.6, 133.4, 131.8, 131.7, 131.2, 128.9, 128.0, 127.4, 127.3, 126.0, 118.6, 118.4, 117.1, 114.9.:4) to obtain yellow solid (50.1 mg, 73%). 1H NMR (600 MHz, CDCl3) δ 9.94 (dt, J = 7.0, 0.9 Hz), 9.55–9.50 (m), 8.89–8.84 (m), 8.42 (ddd, J = 8.2, 2.3, 1.0 Hz), 8.13 (dt, J = 9.0, 1.1 Hz), 7.96 (dd, J = 8.3, 1.2 Hz), 7.71 (t, J = 7.9 Hz), 7.53 (t, J = 7.8 Hz), 7.44–7.35 (m), 7.16 (td, J = 6.9, 1.2 Hz). 13C NMR (151 MHz, CDCl3) δ 178.6, 147.9, 139.4, 136.4, 135.6, 133.4, 133.4, 131.8, 129.0, 129.0, 128.0, 127.4, 127.2, 126.2, 126.2, 126.0, 118.6, 117.2. HRMS (ESI) m/z [M + H+] calcd for C20H13N3O3 343.0957, found 343.0952.:4) to obtain yellow solid (53.6 mg, 73%). 1H NMR (600 MHz, CDCl3) δ 9.90 (dt, J = 7.1, 1.1 Hz), 8.08 (dt, J = 9.0, 1.2 Hz), 7.87–7.83 (m), 7.69 (d, J = 8.2 Hz), 7.52 (d, J = 1.9 Hz), 7.49–7.43 (m), 7.40–7.34 (m), 7.16 (td, J = 6.9, 1.2 Hz). 13C NMR (151 MHz, CDCl3) δ 181.5, 136.8, 136.3, 136.0, 133.6, 133.4, 133.2, 132.1, 131.8, 130.0, 128.8, 127.9, 127.4, 127.2, 126.5, 126.0, 118.5, 117.1.:4) to obtain yellow solid (48.4 mg, 66%).1H NMR (600 MHz, CDCl3) δ 9.89 (d, J = 7.0 Hz), 8.07 (d, J = 9.0 Hz), 7.82 (d, J = 8.1 Hz), 7.44 (t, J = 7.6 Hz), 7.41–7.31 (m), 7.17 (t, J = 6.9 Hz). 13C NMR (151 MHz, CDCl3) δ 180.7, 138.2, 136.5, 133.5, 132.5, 132.4, 131.1, 130.3, 128.7, 127.9, 127.8, 127.5, 127.1, 126.1, 118.6, 117.2. HRMS (ESI) m/z [M + H+] calcd for C20H12Cl2N2O 366.0327, found 366.0330.:4) to obtain light yellow solid (47.4 mg, 71%). 1H NMR (600 MHz, CDCl3) δ 9.87 (d, J = 7.1 Hz), 8.07 (d, J = 9.0 Hz), 7.98 (td, J = 8.2, 6.7 Hz), 7.91–7.85 (m), 7.48 (t, J = 7.7 Hz), 7.40–7.30 (m), 7.13 (td, J = 7.0, 1.0 Hz), 7.03–6.98 (m), 6.98–6.91 (m). 13C NMR (151 MHz, CDCl3) δ 179.5, 165.3, 163.6, 162.2, 160.5, 135.6, 133.8, 133.6, 133.3, 133.3, 131.9, 128.8, 127.8, 127.3, 127.1, 125.7, 124.0, 118.5, 116.9, 111.1, 110.9, 104.8, 104.6, 104.4. HRMS (ESI) m/z [M + H+] calcd for C20H12F2N2O 334.0918, found 334.0915.:4) to obtain light yellow solid (45.8 mg, 65%). 1H NMR (600 MHz, CDCl3) δ 9.85 (d, J = 7.1 Hz), 8.09 (d, J = 9.0 Hz), 7.91–7.80 (m), 7.50 (t, J = 7.7 Hz), 7.42–7.34 (m), 7.16 (td, J = 7.0, 0.8 Hz), 7.07 (td, J = 9.7, 6.3 Hz). 13C NMR (151 MHz, CDCl3) δ 177.8, 157.21, 157.1, 155.5, 155.4, 152.7, 152.6, 150.9, 150.9, 147.1, 147.0, 145.4, 145.3, 136.0, 133.5, 133.4, 132.2, 128.9, 128.0, 127.3, 127.1, 126.1, 123.7, 123.6, 119.9, 119.8, 118.6, 117.2, 106.5, 106.3, 106.3, 106.2. HRMS (ESI) m/z [M + H+] calcd for C20H11F3N2O, 352.0823, found 352.0827.:2) to obtain light yellow solid (37.6 mg, 63%). 1H NMR (600 MHz, CDCl3) δ 10.05–9.94 (m), 9.93 (dd, J = 7.2, 0.9 Hz), 9.78–9.69 (m), 8.86–8.68 (m), 8.12–8.01 (m), 7.99–7.77 (m), 7.49 (ddd, J = 10.6, 7.9, 6.3 Hz), 7.40 (d, J = 7.4 Hz), 7.36–7.30 (m), 7.13 (d, J = 6.9 Hz). 13C NMR (151 MHz, CDCl3) δ 179.8, 152.0, 151.9, 138.2, 135.4, 133.8, 133.6, 133.5, 131.7, 128.9, 127.8, 127.3, 127.2, 125.8, 123.0, 118.5, 117.0. HRMS (ESI) m/z [M + H+] calcd for C19H13N3O, 299.1059, found 299.1064.Author contributions
Xinyu Song, Shujun Qin, Wenya Dong, Pingjing Xue and Jiayi Wu contributed equally to the experimental work, Haibo Wang contributed to the modification of the manuscript. All authors reviewed and approved the submitted manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support this study are available in the supplementary information (SI). Supplementary information: other experimental procedures, characterization data, and copies of NMR spectra for the compounds. See DOI: https://doi.org/10.1039/d5ra07797d.Acknowledgements
We are grateful for the financial support from the Supporting Program for Outstanding Young Talents in universities (gxyqZD2022068), Innovation and Entrepreneurship Project for College Students (S202310879210), Matching funding of Key Research and Development Plan of Anhui Province (811114) and Enterprise cooperation project (Synthesis and preparation process development of osimertinib-881792).Notes and references
- P. Peng, X. X. Yan, K. Zhang, Z. Liu, L. Zeng, Y. X. Chen, H. Zhang and A. W. Lei, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- Y. Cohen, A. Cohen and I. Marek, Chem. Rev., 2021, 121, 140–161 CrossRef CAS PubMed.
- S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed.
- B. Wang, M. A. Perea and R. Sarpong, Angew. Chem., Int. Ed., 2020, 59, 18898–18919 CrossRef CAS PubMed.
- Y. Ano, D. Takahashi, Y. Yamada and N. Chatani, ACS Catal., 2023, 2234–2239 CrossRef CAS.
- G. M. Dal Forno, E. Latocheski, A. B. Machado, J. Becher, L. Dunsmore, A. L. St John, B. L. Oliveira, C. D. Navo, G. Jiménez-Osés, R. Fior, J. B. Domingos and G. J. L. Bernardes, J. Am. Chem. Soc., 2023, 145, 10790–10799 CrossRef CAS PubMed.
- L. Ge, C. Zhang, C. K. Pan, D. X. Wang, D. Y. Liu, Z. Q. Li, P. K. Shen, L. F. Tian and C. Feng, Nat. Commun., 2022, 13, 5938 CrossRef CAS PubMed.
- C. He, S. Guo, L. Huang and A. Lei, J. Am. Chem. Soc., 2010, 132, 8273–8275 CrossRef CAS PubMed.
- L. Huang, T. F. Ji and M. Rueping, J. Am. Chem. Soc., 2020, 142, 3532–3539 CrossRef CAS PubMed.
- S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS PubMed.
- C. Ning, Z. Q. Yu, M. Shi and Y. Wei, Chem. Sci., 2024, 15, 9192–9200 RSC.
- P. Rahman, N. Chakraborty, B. K. Patel and K. K. Rajbongshi, J. Org. Chem., 2024, 89, 10472–10484 CrossRef CAS PubMed.
- X. Y. Yu, J. R. Chen and W. J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed.
- Q. Gao, Y. Zhu, M. Lian, M. Liu, J. Yuan, G. Yin and A. Wu, J. Org. Chem., 2012, 77, 9865–9870 CrossRef CAS PubMed.
- L. Li, W. Huang, L. Chen, J. Dong, X. Ma and Y. Peng, Angew. Chem., Int. Ed., 2017, 56, 10539–10544 CrossRef CAS PubMed.
- C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257–15262 CrossRef CAS PubMed.
- Y. Zhou, P. Zhao, L.-S. Wang, X.-X. Yu, C. Huang, Y.-D. Wu and A.-X. Wu, Org. Lett., 2021, 23, 6461–6465 CrossRef CAS PubMed.
- L.-H. Zou, D. L. Priebbenow, L. Wang, J. Mottweiler and C. Bolm, Adv. Synth. Catal., 2013, 355, 2558–2563 CrossRef CAS.
- Q. Wang, X. Yao, P. L. Zhao, W. X. Liu, W. Y. Zhao, X. Fang, Y. Zhu and G. Q. Dai, J. Org. Chem., 2023, 88, 14193–14199 CrossRef CAS PubMed.
- O. Delgado, F. Delgado, J. A. Vega and A. A. Trabanco, Eur. J. Med. Chem., 2015, 97, 719–731 CrossRef CAS PubMed.
- R. Nirogi, A. R. Mohammed, A. K. Shinde, S. R. Gagginapally, D. M. Kancharla, V. R. Middekadi, N. Bogaraju, S. R. Ravella, P. Singh, S. R. Birangal, R. Subramanian, R. C. Palacharla, V. Benade, N. Muddana and P. Jayarajan, J. Med. Chem., 2018, 61, 4993–5008 CrossRef CAS PubMed.
- G. Popova, M. J. G. W. Ladds, L. Johansson, A. Saleh, J. Larsson, L. Sandberg, S. H. Sahlberg, W. Qian, H. Gullberg, N. Garg, A.-L. Gustavsson, M. Haraldsson, D. Lane, U. Yngve and S. Lain, J. Med. Chem., 2020, 63, 3915–3934 CrossRef CAS PubMed.
- Z. S. Safi and N. Wazzan, J. Comput. Chem., 2021, 42, 1106–1117 CrossRef CAS PubMed.
- C. Ramos, J. Muehlbrad and B. G. Janesko, J. Comput. Chem., 2021, 42, 1974–1981 CrossRef CAS PubMed.
- A. A. Khairbek, M. I. Al-Zaben, R. Puchta, M. A. Badawi and R. Thomas, Mol. Catal., 2024, 566, 114411 Search PubMed.
- J. Heo, Comput. Theor. Chem., 2024, 1238, 114288 Search PubMed.
- Q. Wang, X. Yao, X.-J. Xu, S. Zhang and L. Ren, ACS Omega, 2022, 7, 4305–4310 CrossRef CAS PubMed.
- J. Zhang, M. Y. She, L. Liu, M. D. Liu, Z. H. Wang, H. Liu, W. Sun, X. G. Liu, P. Liu, S. Y. Zhang and J. L. Li, Chin. Chem. Lett., 2021, 32, 3083–3086 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |