DOI:
10.1039/D5RA06776F
(Paper)
RSC Adv., 2025,
15, 45783-45798
Accelerated discovery of new organic photovoltaic dyes for OPVs using light absorbance as the primary screening criterion via machine learning and DFT
Received
8th September 2025
, Accepted 14th November 2025
First published on 21st November 2025
Abstract
This research provides an analysis of the light absorption properties of organic dyes in different organic solvents. By employing state-of-the-art machine learning (ML) techniques, including multi-output Gaussian process regression and ensemble methods like XGBoost and Random Forest regressors, we successfully predicted solvent-specific absorbance characteristics. XGBoost demonstrated outstanding predictive efficiency, and interpretation via SHapley Additive exPlanations (SHAP) analysis identified the topological polar surface area as the most critical molecular descriptor. For the de novo design of novel dyes, we developed a Transformer-Assisted Orientation (TAO) approach, generating three iterative rounds of new structures. The photovoltaic potential of these newly designed dyes was validated through density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations. Geometry optimizations and electronic property calculations were performed at the ωB97XD/LanL2DZ level, while electronic spectra were simulated using the CAM-B3LYP/6-31G+(d,p) method with a polarizable continuum model (PCM) for acetonitrile. This integrated ML/DFT pipeline yielded dyes with remarkable predicted photovoltaic parameters, including a peak open-circuit voltage (Voc) of 0.96 V, a light harvesting efficiency (LHE) of 95%, a fill factor (FF) of 0.87, and a short-circuit current density (Jsc) of 28.75 mA cm−2. This study establishes a robust, data-driven framework for the rapid discovery and design of high-performance organic photovoltaic materials.
Introduction
Designing and developing novel organic photovoltaic (PV) dyes has become increasingly important in advancing sustainable and highly efficient solar energy technologies.1 With the growing global shift toward renewable energy to address climate change and reduce dependence on fossil fuels, these OPVs present a compelling alternative, thanks to their lightweight design, flexibility, and potential for cost-effective manufacturing.2 The processability of these dyes in solution is crucial for enhancing their potential for large-scale production.3 Employing solution-based techniques, such as spin-coating or inkjet printing, simplifies the fabrication of PV devices, enabling the seamless incorporation of PV materials into diverse substrates, including textiles and construction materials.4 This flexibility expands the range of applications for solar technologies and promotes the adoption of OPVs in various settings, from bustling urban areas to isolated rural locations.5 Additionally, the advancement of new organic dyes with enhanced light absorption and charge transport characteristics can substantially boost the efficiency of OPV cells, increasing their competitiveness with conventional silicon-based solar cells.6 The capacity to precisely adjust the chemical structure of these organic dyes also creates opportunities to optimize their performance across varying lighting conditions and environmental factors.7 As research progresses in this area, the development of new organic dyes that offer high stability, low toxicity, and outstanding solution processability will be vital in advancing the next generation of solar energy technologies, playing a key role in fostering a more sustainable and energy-efficient future.8
The solubility of organic dyes is a critical factor influencing their performance in applications such as OPVs and biological imaging,9 as it is governed by molecular interactions.10 The dye molecules engage in interactions with the solvent molecules, establishing a dynamic equilibrium between the dissolved form and any undissolved particles. When organic dyes are dissolved in a solvent.11 The solubility of a dye is affected by its chemical structure, particularly its functional groups, to interact with the solvent molecules.12 For example, polar dyes typically dissolve efficiently in polar solvents due to favorable interactions, whereas non-polar dyes are more soluble in non-polar solvents.13 In light-harvesting processes within natural systems, the solubility of organic dyes is vital for functions like photosynthesis and cellular respiration. For instance, in mitochondria—the cell energy centers—specific organic dyes can serve as fluorescent probes to investigate mitochondrial function and dynamics.14 When these dyes are taken up into the mitochondrial matrix, they can absorb particular wavelengths (λmax) of light and emit fluorescence, enabling researchers to observe and monitor mitochondrial activity.15 The solubility of these dyes in light-harvesting environments is important for their efficient uptake and proper functionality.16 The dyes such as rhodamine 123, renowned for their ability to accumulate in mitochondria, illustrate how solubility and a specific affinity for cellular compartments can be utilized for imaging and investigating cellular processes.17 Furthermore, the absorption of light by these dyes occurs due to electronic transitions within the dye molecules.18 In the case of OPVs, the solubility of organic dyes directly influences film formation during device fabrication, impacting the morphology and, in turn, the efficiency of light absorption and charge transport.19 Therefore, understanding the solubility of organic dyes is important not only for their use in PVs but also for their function in biological systems, where they can offer valuable insights into cellular mechanisms and energy production.20
Machine learning (ML) significantly advances the field of photovoltaics by accelerating the discovery and optimization of organic dye materials critical for cell efficiency. ML models analyze large datasets to predict key dye properties such as light absorption, electron injection efficiency, and photovoltaic conversion efficiency without extensive lab experiments. This data-driven approach enables rational design of dyes with improved power conversion efficiency (PCE), often validated with complementary computational methods like density functional theory. Furthermore, ML can optimize device parameters and monitor degradation patterns, enhancing the durability and effectiveness of DSSCs. This integration streamlines the development of cost-effective, high-performance solar cells.21–23 Recent advancements in machine learning (ML) have significantly improved dye-sensitized solar cells (DSSCs). Yadav et al.24 review cutting-edge ML methodologies that overcome traditional experimental limitations, enabling rapid material screening and optimization of device architectures. Algorithms like Decision Trees and Convolutional Neural Networks effectively predict photovoltaic characteristics and identify novel materials. In parallel, Yadav et al.25 also investigated vanadium oxide (VO2) nanoparticles as a cost-effective alternative to platinum counter electrodes, demonstrating enhanced stability and comparable photovoltaic performance. Complementing these efforts, Gupta et al.26 conducted a first-principles study of the MnCoSb half-Heusler alloy, revealing its advantageous structural, mechanical, optical, and thermoelectric properties. With a cubic non-centrosymmetric structure and a peak thermoelectric performance (zT = 12.97 at room temperature), MnCoSb shows significant potential for optoelectronic applications.
The integration of machine learning (ML) into the materials discovery pipeline represents a paradigm shift from traditional, often serendipitous, experimentation to a targeted, data-driven approach. ML models excel at decoding complex, non-linear relationships between a material's chemical structure and its properties from large datasets, a task that is intractable for human intuition alone. This capability allows for the rapid virtual screening of vast chemical spaces, orders of magnitude larger than what can be feasibly explored in a laboratory.22 Machine learning (ML) can be highly effective in predicting the solubility of organic dyes by utilizing extensive datasets of chemical properties and their associated solubility metrics, such as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (partition coefficient).27 The logP values, which reflect the hydrophobicity or lipophilicity of a compound, are essential for determining how effectively a dye will dissolve in different solvents.28 By utilizing ML algorithms, researchers can examine the intricate relationships between molecular structure and solubility, uncovering patterns that may not be easily identified using conventional methods.29 For instance, models can be trained on available data that includes molecular descriptors (such as molecular weight, functional groups, and structural features) along with their experimentally obtained logP values.30 The current study aims at predicting the solubility and absorbance properties of approximately 70000 organic dyes, spanning various classes such as indole, benzothiazine, benzophenanthrene, benzothiazole, benzothiazine, benzodithiophene, and carbazole. This research aims to identify optimal solvents for these dyes, improving their performance in photovoltaic applications by linking their structural characteristics to solubility and absorbance traits. By thoroughly examining this large dataset, the study seeks to advance the design of more efficient organic solar cells and contribute to the development of sustainable energy solutions.
P (partition coefficient).27 The logP values, which reflect the hydrophobicity or lipophilicity of a compound, are essential for determining how effectively a dye will dissolve in different solvents.28 By utilizing ML algorithms, researchers can examine the intricate relationships between molecular structure and solubility, uncovering patterns that may not be easily identified using conventional methods.29 For instance, models can be trained on available data that includes molecular descriptors (such as molecular weight, functional groups, and structural features) along with their experimentally obtained logP values.30 The current study aims at predicting the solubility and absorbance properties of approximately 70000 organic dyes, spanning various classes such as indole, benzothiazine, benzophenanthrene, benzothiazole, benzothiazine, benzodithiophene, and carbazole. This research aims to identify optimal solvents for these dyes, improving their performance in photovoltaic applications by linking their structural characteristics to solubility and absorbance traits. By thoroughly examining this large dataset, the study seeks to advance the design of more efficient organic solar cells and contribute to the development of sustainable energy solutions.
Theory and methodology
When light in the form of photons strikes a PV material, absorption occurs when the photon's energy levels are higher than their bandgap energy.31 This excitation leads to the formation of electron–hole pairs. Subsequently, these separated charge carriers are collected at the electrodes to complete a circuit for producing an electric current. The photocurrent density produced by the absorbed light is calculated as by following equation (eqn (1)):| |
 | (1) |
where Jph shows photocurrent density (A m−2), while q is the electronic charge (approximately 1.6 × 10−19 C), Φ is the photonic flux, which is the number of photons per unit area per second. It Eg represents the energy bandgap (Joules) of the material.
The open-circuit voltage (Voc)32 of a solar cell can be estimated by the following equation:
| |
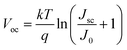 | (2) |
Here, the
k shows Boltzmann's constant (1.38 × 10
−23 J K
−1) and
T is its absolute temperature (K). While
J0 shows their current density (A m
−2). Their power output (
P) for their PV cell (
W) is calculated by using the given equation (
eqn (3)).
where
V shows its voltage across the load (
V) and
J is its current flowing through that load (
A).
The density functional theory (DFT), a quantum mechanical modeling approach, is employed for exploring their electronic structure to determine their bandgap energies. The bandgap energy (Eg) can be derived from the electronic band structure calculated by DFT, particularly by determining their difference between the conduction band minimum (CBM) and the valence band maximum (VBM) energies (eqn (4)).
Bandgap energy was calculated as:
Eg = bandgap energy (eV),
ECBM is the energy of the conduction band minimum (eV), and
EVBM is the energy of the valence band maximum (eV). The Kohn–Sham equation
33 (
eqn (5)), which forms the basis of DFT, can be written as:
| |
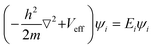 | (5) |
where ℏ is their reduced Planck's constant, while
m is their electronic mass. The
Veff shows its effective potential, which includes the external potential and the electron–electron interaction.
ψi is a Kohn–Sham orbital,
Ei is the energy eigenvalue corresponding to the orbital (
eqn (6)). The exchange-correlation energy functional (
Exc) is a component in DFT to be approximated by various functionals like generalized gradient approximation (GGA) or local density approximations.
| | |
Exc[ρ] = ∫εXC(ρ)ρ(r)dr
| (6) |
Here,
εXC(
ρ) represents the exchange-correlation energy density as a function of their electronic density (
ρ)
. To determine absorbance using ML techniques, especially after calculating the log
P values of organic PV dyes, a systematic approach is employed (
eqn (7)). This process incorporates various theoretical principles and mathematical equations. Absorbance (
A) is defined by the Beer–Lambert Law,
34 which states:
| |
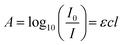 | (7) |
where
A is absorbance and
I0 is their incident light intensity (W m
−2). While
I is their intensity for their transmitted light (W m
−2) and
ε is their molar absorptivity (L (mol
−1 cm
−1)). The concentration of the dye in solution (mol L
−1) is represented by
c, while
l is their path length (cm) of light through their solution.
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P and its relation to absorbance
P and its relation to absorbance
The logP represents the hydrophobicity of a compound, which directly affects its solubility and, in turn, its absorbance characteristics. The connection between logP and absorbance can be explored through empirical methods or ML techniques. To model the relationship between absorbance (A) and logP values, a mathematical framework can be developed to capture how logP impacts the absorbance of organic PV dyes. While the precise relationship depends on the dyes' unique properties and their chemical contexts, linear regression or advanced ML models are commonly used to establish this correlation (eqn (8)). Below are the equations that can be used to express this relationship. A simple linear regression model can be expressed as:| | |
A = β0 + β1·logP +…+ εA
| (8) |
where A is absorbance, β0 is the intercept (constant term), β1 is the coefficient representing the change in absorbance per unit change in logP, and ε is the error term (captures the variability not explained by the model). If additional molecular descriptors are included, the equation can be expanded to (eqn (9)):| | |
A = β0 + β1·logP2 + β2·MW + β3·TPSA +…+ ε
| (9) |
where, MW is the molecular weight, TPSA = topological polar surface area. If the relationship is non-linear, polynomial regression or other non-linear models can be used (10). For example, a quadratic relationship might be expressed as:| | |
A = β0 + β1·logP2 + β2·log·P2 + ε
| (10) |
Data collection and handling
The initial dataset was compiled from publicly available chemical databases, primarily PubChem35 and ChEMBL,36 and supplemented with data from the literature on organic photovoltaic materials.37 The selection criteria for molecules were as follows: the molecule must be an organic compound reported or hypothesized to have light-absorption properties relevant to photovoltaics; it must contain at least one known chromophoric group (e.g., cyanine, porphyrin, carbazole, etc.); experimental or computationally derived absorbance data (λmax and/or molar absorptivity) in at least one common organic solvent (e.g., ethanol, acetonitrile, DMSO) had to be available or calculable; and the SMILES string had to be valid and interpretable by the RDKit cheminformatics library for descriptor calculation.
Input data beyond SMILES
Beyond the SMILES strings, which served as the primary input for generating 2D molecular descriptors, the dataset incorporated several key data points for each molecule–solvent pair. This includes the experimental context with the solvent name for each absorbance measurement as a categorical feature; the target variable, which is the measured or calculated peak absorbance wavelength (λmax) and its corresponding intensity (ε or absorbance value); and pre-calculated properties where available, such as experimentally derived logP values from sources like PubChem for a subset of molecules to supplement the calculated descriptors during the initial analysis phase. Their length of Simplified Molecular-Input Line-Entry System (SMILES) ranged from ∼40–100 (Fig. 1).
 |
| | Fig. 1 SMILES length of the collected dataset. | |
Generalizability
The generalizability of the machine learning models is supported by the strategic design of the dataset. The inclusion of a vast number of dyes (∼70000) spanning multiple, distinct chemical classes prevents the model from overfitting to a narrow chemical space. Furthermore, by incorporating data across six different organic solvents (ethanol, DCM, DMSO, ACN, DMF, and methanol), the model learns solvent-dye interaction patterns, enhancing its predictive capability for new dye–solvent combinations. The use of a 75/25 train-test split, along with validation against external literature data (Table 1), provides a robust assessment of the model's ability to predict properties for entirely new molecules.
Table 1 Solvent-specific logP analysis for top-performing dye candidates
| Solvent |
LogP (solvent) |
LogP (dye) |
ΔLogP (dye – solvent) |
Experimental (logP) |
Reference |
Data points |
| Ethanol |
2.5 |
3.0 |
0.5 |
0.7 |
— |
003714 |
| CCl4 |
1.0 |
3.0 |
2.0 |
— |
— |
000265 |
| DCM |
0.5 |
3.0 |
2.5 |
— |
— |
004212 |
| Water |
9 |
3.0 |
−6.0 |
3.7 |
54 |
008765 |
| DMSO |
0.2 |
2.9 |
2.7 |
3.1 |
55 |
010096 |
| ACN |
1.5 |
3.2 |
1.7 |
— |
— |
018765 |
| DMF |
3 |
3.7 |
0.7 |
— |
— |
015321 |
| Methanol |
3 |
3.7 |
0.7 |
— |
— |
013543 |
Feature extractions
The feature extraction process utilized two complementary approaches to generate molecular descriptors. The primary set of features for machine learning consisted of 2D molecular descriptors calculated directly from the SMILES representations using the RDKit cheminformatics toolkit.38 These descriptors capture topological, constitutional, and group contribution properties. Additionally, to incorporate fundamental electronic structure information into the initial screening, high-throughput quantum chemical calculations were performed on the entire dataset using the PSI4 software.39 From these calculations, key electronic descriptors, most notably the HOMO–LUMO energy gap (Eg), were extracted and included in the initial feature pool for model training.
To refine this extensive feature set, a feature selection process was implemented to reduce dimensionality and mitigate multicollinearity. Descriptors with near-zero variance were eliminated, and a Pearson correlation matrix analysis retained only one descriptor from any pair with a correlation coefficient greater than 0.95. This resulted in a final selection of 35 significant and non-redundant descriptors, covering constitutional, topological, electronic, and hydrophobic properties (Table S1). These descriptors provide a comprehensive representation of molecular features relevant to light absorption. The molecular weight (MW) was calculated using the specified equation (eqn (11)).
| |
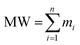 | (11) |
where
mi is their atomic masses for an at
i in the molecule.
The logP measures hydrophobicity by representing a ratio of a substance concentration in octanol to water. It also indicates how well a substance can interact with hydrophobic and hydrophilic environments (eqn (12)).
| |
 | (12) |
The electronegativity (EN) measures an atom (ii) with its tendency to attract electrons in its bonding situation to influence their chemical reactivity/interactions within their molecules (χi) (eqn (13)).
| |
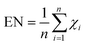 | (13) |
The zero-order molecular valence connectivity index (χvo)40 was calculated by using their hydrogen-suppressed molecular skeletons. It relied on their atomic valence delta (δv)41 to reflect their unique connectivity for relevant non-hydrogen atoms (eqn (14)).
| |
 | (14) |
Similarly, their atomic valence deltas (δv) were calculated for their non-hydrogen atoms by using their atomic number (Z), valence electrons (Zv), and their attached hydrogens (h) to produce their unique δv value of each atom (eqn (15)).
| |
 | (15) |
Python libraries were used for all the ML-related calculations, which included Pandas42 for their data import, NumPy43 and RDKit38 toolkit for their descriptor design, Matplotlib for data visualization, and Scikit-learn44 for their scientific calculations. Their related quantum chemical calculations were performed using PSI4,39 estimating Eb values via density functional theory (DFT) and time-dependent DFT (TD-DFT) for their ground and excited states, respectively.
Correlations and feature scores
The Pearson correlation coefficient (r)45 is used to quantify the linear relationship between two variables XX and YY (eqn (16)).| |
 | (16) |
Calculating the correlation coefficient involves various components like their number of data points (n), the sum of the products of their paired scores (∑XY), and the sums of their individual scores (∑X and ∑Y) with squared scores (∑X2 and ∑Y2). Their feature importance (FIj) for a feature (j) was determined based on their evaluated models (eqn (17)).
| |
 | (17) |
Computational calculations
Initial high-throughput quantum chemical calculations were performed using the PSI4 software39 due to its computational efficiency. The primary objective of this step was to generate fundamental electronic structure descriptors, such as the HOMO–LUMO energy gap (Eg), for the entire dataset of ∼70000 molecules. These computational descriptors supplemented the 2D RDKit descriptors during the initial machine learning phase to provide a more comprehensive representation of molecular properties relevant to light absorption. However, for the detailed electronic structure analysis, geometry optimization, and excited-state calculations of the newly designed dye candidates, we employed Gaussian 09 software46 owing to its extensive validation for organic photovoltaic materials and wider availability of functionals suitable for TD-DFT calculations of chromophores. The ωB97XD functional47 was selected for geometry optimization as it includes empirical dispersion correction and long-range correction, which are crucial for accurately modeling π-conjugated systems and non-covalent interactions prevalent in organic dyes.48 The LanL2DZ basis set49 provides a balanced approach for elements common in organic dyes while being computationally tractable. It is important to note that due to the computational expense, full geometry optimization and subsequent TD-DFT calculations were performed only on the subset of newly designed dyes (approximately 1150 from the three design rounds), not on the entire dataset of ∼70000 molecules.
Frontier molecular orbitals (FMOs)
The HOMO and LUMO energies were obtained directly from the converged SCF calculations at the ωB97XD/LanL2DZ level on the optimized geometries.
Chemical reactivity parameters. Global reactivity descriptors were calculated using the following relationships based on Koopmans' theorem,50 are provided in SI.
Photovoltaic parameters. The light harvesting efficiency (LHE) was calculated as LHE = 1 − 10(−f), where f is the oscillator strength from TD-DFT.51 The open-circuit voltage (Voc) was estimated using eqn (2), while Jsc and FF were computed using established empirical relationships based on the electronic structure parameters.52
TD-DFT calculations. The TD-DFT calculations for electronic spectra were performed using the polarizable continuum model (PCM) to simulate the solvation environment in acetonitrile, which was identified as the optimal solvent from our machine learning analysis. This approach accounts for solvent effects on the excitation energies and provides more realistic absorption maxima compared to vacuum calculations.
Results and discussion
Advancing solar energy technologies relies heavily on the development of efficient PV materials, with organic photovoltaic (OPV) cells showing significant potential. A key aspect of improving their performance and boosting energy conversion efficiency lies in understanding the light absorbance properties of PV dyes across different organic solvents.
Solvent selection
The study centered on assessing solvent selection by analyzing logP values, which play a critical role in determining the solubility and interactions of PV dyes. By calculating the logP values for various solvents in relation to the dyes, the objective was to pinpoint solvent-dye combinations that optimize performance in applications like dye-sensitized solar cells. The results highlighted those solvents such as ethanol and DMSO demonstrated favorable logP differences with the dyes, suggesting their suitability for enhancing solubility and interaction.53–55 The analysis in Table 1 reveals distinct solvation trends by examining the average logP of the top-performing dye candidates in each solvent. A key observation is the consistency of the average dye logP, which clusters around ∼3.0 for most solvents. This indicates that our ML model consistently identifies moderately hydrophobic dyes as high-absorbers across different solvent environments. The critical insight comes from the ΔlogP (dye – solvent) values. Solvents like DCM and DMSO, with large positive ΔlogP values (2.5 and 2.7, respectively), create a strongly solvating environment for these hydrophobic dyes, which is favorable for dissolution. In contrast, water, with a large negative ΔlogP (−6.0), is a poor solvent for this class of dyes. Acetonitrile (ACN) and ethanol present intermediate ΔlogP values (1.7 and 0.5), suggesting a balanced solvation capability. Notably, ACN hosted the largest number of top-performing dye predictions, indicating that its specific dielectric properties and moderate solvation strength provide a uniquely favorable environment for the electronic transitions critical to light harvesting in a wide range of OPV dyes (Fig. 2).
 |
| | Fig. 2 A graphical view of the SlogP distribution in different organic solvents. | |
An ML model, trained on an extensive dataset, was utilized to predict logP values for solvent-dye combinations not yet tested experimentally. These predictions were then validated against available experimental data. This methodology highlights the critical role of solvent selection in optimizing the solubility and performance of PV dyes, paving the way for more efficient solar energy conversion technologies (eqn (18)).
| | |
logPsolvent = logPdye − logPreferencesolvent
| (18) |
After training the model, the predicted logP values for candidate solvents are calculated. The solvent selection can be summarized as (eqn (19)):
| | |
Best solvent = arg·maxsŷs
| (19) |
where
ŷs is the predicted log
P for solvent s.
Light absorbance prediction
The xGBoost56 and Random Forest regression models demonstrated excellent predictive performance for absorbance, with R2 values ranging from 0.87–0.92 and root mean square error (RMSE) values between 0.0021 and 0.026. These results highlight the model with its ability to accurately capture the relationships between input features and absorbance values. An R2 value approaching 1 indicated that a significant portion of the variance in absorbance is explained by the models, demonstrating their robustness. The low RMSE values, reflecting minimal average deviation between predicted and actual absorbance, further confirmed its accuracy and effective tuning (Table 2). It is important to note that while our model identified general solvent trends across the entire dataset, solvent-dye interactions can be family-specific. The high predictive accuracy of our model suggests that the learned relationships, such as the superiority of ACN, are robust across the diverse chemical space explored. Future investigations could focus on deconvoluting these effects for specific dye classes to uncover more nuanced design rules. The xGBoost, with its high-performance, stems from its capacity to model complex interactions and non-linear relationships within the data, while Random Forest leverages its ensemble structure to minimize overfitting and improve generalization. Together, these models provide a robust framework for accurately predicting absorbance, making them invaluable in analyzing PV dyes and their interactions with solvents (Fig. 3). These findings underscore the role of advanced ML methods in enhancing predictive accuracy in chemical and materials science research.
Table 2 Comparative performance of machine learning models for predicting light absorbance
| Model |
R2 (coefficient of determination) |
RMSE (root mean square error) |
| xGBoost |
0.92 |
0.0021 |
| Random Forest |
0.87 |
0.026 |
 |
| | Fig. 3 A scatter plot of calculated and predicted light absorption for Random Forest (left) and xGBoost (right) regression models. | |
The density of the residual scatter plot offers critical insights into the predictive performance of the xGBoost and Random Forest regression models. The xGBoost model appears better suited to handle broader variability across the general dataset, while the Random Forest model demonstrates more consistent but potentially less flexible predictions. For the xGBoost model, residuals ranged from −1 to 0.5, indicating that its predictions closely align with the actual absorbance values, with a slight tendency to underestimate in some cases. This range suggested strong overall performance but also revealed occasional challenges in capturing the full variability of the data. In comparison, the Random Forest model exhibited a narrower residual range of −1.0 to 0.2, suggesting a consistent underestimation of absorbance values. While the tighter range implies greater stability in predictions, it may also indicate that the model is less responsive to certain data variations compared to xGBoost. These observations underline the promising predictive capabilities of both models, with each offering distinct strengths. The xGBoost model appears better suited to handle broader variability, while the Random Forest model demonstrates more consistent but potentially less flexible predictions (Fig. 4). Further analysis of the scatter plots for patterns or systematic errors could provide actionable insights for refining the models or enhancing feature selection, ultimately improving predictive accuracy.
 |
| | Fig. 4 A scatter plot of the density of residuals for predicted light absorption for Random Forest (left) and xGBoost (right) regression models. | |
The SHapley Additive exPlanations (SHAP)57 analysis identified the polar surface area (PSA) as the most influential feature affecting the performance of the models. This highlighted the PSA with its critical role in determining the absorbance of PV dyes, likely due to its significant impact on solubility and molecular interactions. Following PSA, molecular weight (MW) and the logarithm of the partition coefficient (S![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) logP) were recognized as the next most important features. Molecular weight influences the dye behavior in solution, affecting factors such as diffusion and solvent interactions. Similarly, SlogP, which measures hydrophobicity or lipophilicity, is important for understanding the dye solubility in different solvents and its stability in solution. This ranking of features underscored the necessity of incorporating molecular properties into absorbance predictions. By emphasizing these critical features, the models could be refined to improve their predictive accuracy. Furthermore, understanding the influence of these features provided valuable guidance for designing new dyes with optimized properties, advancing their performance in PV applications. The current analysis not only emphasized the most impactful features but could also lay the foundation for future research and development in dye-sensitized solar cells. By pinpointing key factors such as polar surface area, molecular weight, and hydrophobicity, the analysis paves the way for designing more efficient PV dyes (Fig. 5). These insights could also drive further advancements in the performance, stability, and overall efficiency of dye-sensitized solar cells.
logP) were recognized as the next most important features. Molecular weight influences the dye behavior in solution, affecting factors such as diffusion and solvent interactions. Similarly, SlogP, which measures hydrophobicity or lipophilicity, is important for understanding the dye solubility in different solvents and its stability in solution. This ranking of features underscored the necessity of incorporating molecular properties into absorbance predictions. By emphasizing these critical features, the models could be refined to improve their predictive accuracy. Furthermore, understanding the influence of these features provided valuable guidance for designing new dyes with optimized properties, advancing their performance in PV applications. The current analysis not only emphasized the most impactful features but could also lay the foundation for future research and development in dye-sensitized solar cells. By pinpointing key factors such as polar surface area, molecular weight, and hydrophobicity, the analysis paves the way for designing more efficient PV dyes (Fig. 5). These insights could also drive further advancements in the performance, stability, and overall efficiency of dye-sensitized solar cells.
 |
| | Fig. 5 (a) SHAP value beeswarm plot and (b) t-SNE map of the collected dataset. | |
The t-distributed Stochastic Neighbor Embedding (t-SNE)58 maps offer a visual representation of high-dimensional data, with both the x- and y-components ranging from −150 to 150. This range indicated that the data points were well-distributed across the two-dimensional space, allowing for clear clustering and separation of distinct groups within the dataset. The t-SNE map could particularly be effective for visualizing complex data as it preserves the local structure while reducing dimensionality. The broad range of values observed suggested that the model captured substantial variations in the features, which could correspond to different categories of PV dyes. The clustering seen in the t-SNE maps helps reveal patterns and relationships among the dyes, based on key molecular characteristics such as polar surface area, molecular weight, and logP values (Fig. 6). Analyzing these maps provided insights into how these features could influence the dyes with their behavior and interactions with solvents. Additionally, this visualization can help identify outliers or unique compounds that may warrant further exploration.
 |
| | Fig. 6 Pearson correlation heatmap of top descriptors to affect light absorption. | |
New dye designs
The Transformer-Assisted Orientation (TAO)59 method was employed for the de novo design of new dyes. To ensure the validity and practicality of the generated structures, a multi-step validation process was implemented. First, the chemical validity of each generated SMILES string was verified using the RDKit library. Second, the synthetic accessibility (SA)60 of each dye was quantitatively assessed using a score based on fragment contributions and complexity penalties, with scores ranging from 1 (easy to synthesize) to 10 (very difficult to synthesize). The newly designed dyes consistently achieved favorable SA scores between 2.5 and 4.5, indicating high synthetic feasibility. Third, the predictive performance of the generative process was quantitatively validated by comparing the ML-predicted absorbance of the newly generated dyes against the values from subsequent TD-DFT calculations.
This method facilitates the optimization of dye orientations, which play a critical role in determining their electronic properties and absorbance characteristics. In a transformer layer, combining attention and feed-forward transformations involves residual (eqn (20)) connections and layer normalization.61
| |
 | (20) |
Assuming the final output of the transformer model is (H), which encodes the features of the input dye structure (eqn (21)), property prediction can be formulated as:
For its transformer layer, combining their attention and feed-forward transformations involves its residual connections and layer normalization. The concept of “Transformer Assisted Orientation” in molecule design represents an exciting convergence of advanced ML techniques and materials science. By utilizing. By synthesizing dyes based on these model predictions and measuring their absorbance (normalized to a maximum of 1), researchers can enhance the performance of applications such as photovoltaics and sensors. The integration of ML with experimental validation opens avenues for future research into a broader range of dye structures and their associated properties, leading to more efficient materials across various technological fields (Fig. 7). In the first round of development, 50 new dyes were designed, exhibiting an absorbance range from 0.61–0.78. This initial batch provided valuable insights into the relationship between dye structure and optical properties. The second round scaled the process to generate 100 new dyes, with an improved absorbance range of 0.71–0.87, reflecting a positive trend in the design and optimization of these materials. The increase in both the quantity of dyes and their absorbance range suggested that the methodologies employed were successful in enhancing optical performance.
 |
| | Fig. 7 Performance of the Transformer-Assisted Orientation (TAO) method across three design rounds. The structures shown are representative examples of the complete molecules generated de novo, illustrating the progression in predicted light absorption efficiency. These structures utilize known high-performing chemical motifs (e.g., carbazole, triphenylamine) but are novel in their specific architectural combinations and substitutions, as optimized by the TAO model. | |
In the third round, a significant advancement was achieved with the creation of 1000 new dyes, which demonstrated an even wider absorbance range from 0.77–0.91. This substantial increase not only underscores the scalability of the approach but also highlights improvements in the predictive models used to design the dyes. The consistent enhancement of absorbance ranges across the rounds reinforces the potential of iterative design processes in dye development, paving the way for the creation of materials with customized optical properties for diverse applications.
In current study, their activity cliffs were also accessed by focusing on their Structure Activity landscape Index (SALI)62 scores. The highest observed SALI score was 15, indicating significant potential for practical applicability in various domains such as dye production, sensor development, and other areas where dyes are important (Fig. 8). A high SALI score suggested that these dyes could be more sensitive to their small structural changes to make them ideal candidates for further exploration/development.
 |
| | Fig. 8 SALI score of the top 1000 newly predicted dyes. | |
This finding highlighted the importance of structure–activity relationships in selecting dyes for real-world applications, as it directly influences their feasibility for large-scale production. The SALI score of 15 not only reflects the structural simplicity and favorable characteristics of these dyes but also underscores their potential for integration into commercial products. This insight can help guide researchers and industry professionals in prioritizing dyes for further study, ultimately accelerating the development of new materials and technologies. The analysis of the top 30 highest SALI scores among the 973 dyes provided valuable insights into their structure, activity and absorbance properties. The highest SALI score was 14.9, indicating that this dye had a moderate effect of its structural change to activity with an absorbance value of 0.39, highlighting its potential for practical applications in fields such as dye production and sensor development (Table 3). The absorbance values of the top-ranked dyes varied, with the highest being 0.82 for the 8th dye, which had a SALI score of 7.5. This suggested that while many dyes were sensitive to structural changes, only a select few could exhibit both a high SALI score and strong absorbance properties.
Table 3 A comparison of the predicted light absorption with its top 30 dyes with the highest SALI scores
| Dye |
Absorbance |
SALI score |
Dye |
Absorbance |
SALI score |
| 1 |
0.39 |
14.9 |
16 |
0.39 |
7.0 |
| 2 |
0.34 |
8.9 |
17 |
0.33 |
6.9 |
| 3 |
0.77 |
8.8 |
18 |
0.34 |
6.9 |
| 4 |
0.34 |
8.7 |
19 |
0.39 |
6.9 |
| 5 |
0.38 |
8.4 |
20 |
0.32 |
6.5 |
| 6 |
0.41 |
8.0 |
21 |
0.46 |
6.5 |
| 7 |
0.31 |
7.7 |
22 |
0.33 |
6.4 |
| 8 |
0.82 |
7.5 |
23 |
0.31 |
6.4 |
| 9 |
0.40 |
7.4 |
24 |
0.49 |
6.4 |
| 10 |
0.28 |
7.3 |
25 |
0.42 |
6.3 |
| 11 |
0.40 |
7.3 |
26 |
0.46 |
6.2 |
| 12 |
0.30 |
7.3 |
27 |
0.46 |
6.2 |
| 13 |
0.40 |
7.2 |
28 |
0.49 |
6.2 |
| 14 |
0.38 |
7.1 |
29 |
0.31 |
6.2 |
| 15 |
0.38 |
7.0 |
30 |
0.52 |
6.2 |
The data also revealed a noticeable drop in SALI scores after the top few entries, emphasizing the importance of prioritizing these high-scoring dyes for further research and development. The combination of high absorbance and favorable SALI scores in the top dyes highlighted their practical viability for industrial applications. These findings could contribute to advancements in materials science and photonics, potentially leading to the creation of more efficient and accessible dyes for various technologies.
Charge transfer patterns
Charge transfer analysis of dyes is important for understanding their electronic properties and reactivity, particularly through the examination of their Frontier Molecular Orbitals (FMOs),63 which include the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO). The HOMO represented the highest energy orbital occupied by electrons and plays a key role in determining a molecule with its ability to donate electrons. A higher energy HOMO typically indicates a greater propensity for electron donation. To provide a concrete understanding of the structure–property relationships predicted by our ML model, we selected a representative subset of five dyes (dyes 1–5, structures in Table 5) for in-depth charge transfer analysis. These dyes were chosen to represent a range of structural motifs and electronic characteristics found within our newly designed set, thereby offering a broad perspective on the design principles. Their specific structures, provided in Table 5, exhibit diversity in their donor groups and π-conjugated linkers. It is important to note that this selection was made to demonstrate the analytical workflow and diversity of our designed library; they are not necessarily the absolute top performers identified by the ML screening. A full DFT/TD-DFT analysis of all ∼1000 top candidates from Round 3 was computationally prohibitive. The charge transfer analysis of this representative subset is crucial for validating the electronic trends predicted by the ML model. For dyes 1 and 2, the HOMOs are delocalized across the entire moiety, suggesting a broad electron-donating capacity. This delocalized electronic structure enhances their ability to donate electrons effectively. In contrast, the dyes 3, 4, and 5 showed more localized HOMO distributions, concentrated on one side of the moiety (Fig. 9). This suggested a more restricted electron-donating ability, which may influence their charge transfer characteristics when interacting with electron acceptors. The variation in HOMO distribution between these dyes underscores the importance of their electronic structure in determining their charge transfer properties. The dyes with delocalized HOMOs, like 1 and 2, were likely to exhibit more efficient electron donation, which could be important for applications involving charge transfer, such as in PVs and sensors.
 |
| | Fig. 9 A view of the charge transfer patterns, their computed UV-vis analysis and charge density difference cubes. | |
Regarding the LUMO, the distribution is nearly identical across all dyes, suggesting that their electron-accepting abilities are comparable. This indicated that while dyes 1 and 2 might be more effective as electron donors due to their delocalized HOMOs, all dyes had the potential to act as electron acceptors. The ability of these dyes to both donate and accept electrons can open up exciting possibilities for charge transfer complexes, which could exhibit unique photophysical properties. Such properties are particularly relevant in applications like organic solar cells, where efficient charge separation and transport are essential for high-performance devices. The charge transfer analysis, based on the distribution of HOMOs and LUMOs, highlights significant differences in the electron-donating abilities of the dyes, allowing for strategic design of materials with tailored electronic properties. These insights could be used to optimize the performance of materials for a variety of applications in organic electronics and photonics, where efficient charge dynamics are important.
The analysis of the HOMO and LUMO energies provided significant insights into the electronic properties of the dyes. A higher HOMO energy (less negative) indicates a greater propensity for electron donation, as the molecule is more easily oxidized. In this context, dye 3 had the highest HOMO energy at −2.43 eV, indicating a strong electron-donating ability. In contrast, dye 5 had the lowest HOMO energy at −3.67 eV, suggesting a relatively weaker electron-donating capacity. When examining LUMO energies, dye 5 also exhibits the highest LUMO energy at −0.11 eV, making it a strong electron acceptor. The energy gap, calculated as the difference between the HOMO and LUMO energies, offers additional information on the stability and reactivity of the dyes. Dye 5 had the largest energy gap of 3.56 eV, implying a more stable electronic configuration and lower reactivity. In contrast, dyes 1, 2, 3, and 4 showed smaller energy gaps ranging from 1.45 to 2.00 eV, suggesting that they may be more reactive and better suited for charge transfer processes. This energy gap analysis highlights the suitability of these dyes for different applications, particularly in organic electronics. The balance between electron-donating and electron-accepting abilities is important for optimizing the performance of electronic devices. Moreover, the TD-DFT parameters provided valuable insights into the electronic transitions of these dyes, such as excitation energy (E), λmax, oscillator strength (f), and the contributions of specific electronic transitions. These parameters are essential for understanding dye behavior in light-harvesting applications, further informing their potential use in organic electronics and photonics (Table 4).
Table 4 Computed UV-vis parameters of selected newly predicted dyes
| Dye |
E (cm−1) |
λmax (nm) |
f |
Major contribs (%) |
| 1 |
34988 |
285 |
0.0269 |
HOMO → LUMO (28) |
| 2 |
34759 |
287 |
0.0551 |
HOMO → LUMO (94) |
| 3 |
25225 |
396 |
00.002 |
HOMO → LUMO (90) |
| 4 |
30337 |
329 |
0.1079 |
HOMO-1 → LUMO (85) |
| 5 |
26112 |
382 |
0.0001 |
HOMO-4 → LUMO (94) |
The dye 1 exhibited an E of 34988 cm−1 (285 nm) with a relatively low f of 0.0269, suggesting a weak electronic transition primarily from the HOMO to the first LUMO, with this transition contributing 28% of the overall excitation. On the other hand, dye 2 showed a higher f of 0.05 for the HOMO to LUMO transition, with a similar E of 34759 cm−1 (287 nm), indicating a stronger and more favorable transition. The dye 3 had a significantly lower E of 25225 cm−1 (396 nm) and a very low f of 0.002, suggesting a weak transition. Despite this, the HOMO to LUMO transition contributes 90% of the excitation, indicating that it still plays a dominant role in the electronic behavior of the dye. The dye 4, with an E of 30337 cm−1 (329 nm), showed a much higher f of 0.108, with the transition primarily occurring from HOMO-1 to LUMO, contributing 85% of the excitation. This suggested a strong and significant transition. The dye 5 had an E of 26112 cm−1 (382 nm) with an extremely low f of 0.0001, indicating a very weak transition, primarily from HOMO-4 to LUMO, despite the contribution of 94%. This suggested that while the transition was weak, it still significantly influences the dye with its electronic properties. All of their TD-DFT parameters revealed significant variation in the strengths and characteristics of the electronic transitions among the dyes, which are important for their potential applications in photonic and electronic devices. The differences in f values and E could directly affect their light absorption properties and their suitability for use in applications like organic solar cells, photodetectors, and other optoelectronic devices.64
Global chemical reactivity
The global chemical reactivity parameters have provided the necessary focus on the electronic properties and reactivity of the dyes, which include ionization potential (IP), electron affinity (EA), electronegativity (χ), chemical potential (µ), hardness (η), softness (σ), and electrophilicity index (ω). These parameters help predict how the dyes can behave in chemical reactions, particularly in terms of their ability to donate or accept electrons. The dye 1 had an IP of 3.21 eV and an EA of 1.76 eV, resulting in a χ of 2.48 eV and a µ of −2.48 eV. Its η of 0.73 eV indicated moderate stability, while a σ of 0.69 eV suggested that it can easily undergo chemical reactions. The ω of 4.23 eV indicated a relatively high tendency to accept electrons (Table 5). The dye 2, with a slightly higher IP of 3.32 eV and a lower EA of 1.32 eV, had a lower χ of 2.32 eV and a µ of −2.32 eV. Its η of 1.0 eV and σ of 0.50 eV suggested that it was less reactive than dye 1, with an ω of 2.70 eV, indicating a lower tendency to accept electrons.
Table 5 Calculated global chemical reactivity parameters (eV) of selected dyes
| Dye |
IP |
EA |
χ |
µ |
η |
σ |
ω |
| 1 |
3.21 |
1.76 |
2.48 |
−2.48 |
0.73 |
0.69 |
4.23 |
| 2 |
3.32 |
1.32 |
2.32 |
−2.32 |
1.00 |
0.50 |
2.70 |
| 3 |
2.43 |
0.46 |
1.44 |
−1.44 |
0.99 |
0.51 |
1.06 |
| 4 |
2.75 |
1.22 |
1.98 |
−1.98 |
0.77 |
0.65 |
2.56 |
| 5 |
3.67 |
0.11 |
1.89 |
−1.89 |
1.78 |
0.28 |
1.01 |
The dye 3 stood out with a significantly lower IP of 2.43 eV and a very low EA of 0.46 eV, resulting in a low χ of 1.44 eV and a µ of −1.44 eV. Its η of 0.99 eV and σ of 0.51 eV suggested that it was relatively reactive, with an ω of 1.06 eV, indicating a weak tendency to act as an electrophile. The dye 4 had an IP of 2.75 eV and an EA of 1.22 eV, leading to an χ of 1.98 eV and a µ of −1.98 eV. Its η of 0.77 eV and σ of 0.65 eV indicated moderate reactivity, with an ω of 2.56. The dye 5 had the highest IP of 3.67 eV and the lowest EA of 0.11 eV, resulting in an χ of 1.89 and a µ of −1.89 eV. Its η of 1.78 eV indicated high stability, while a σ of 0.28 eV suggested that it could be less reactive, with an ω of 1.01 eV, indicating a low tendency to accept electrons.
Electronic excitation analysis
The transition density matrix (TDM) analysis provides crucial insights into the spatial nature of the electronic excitations in the exemplified dyes.65 The TDM heatmaps depict the electron–hole correlation upon excitation, where the axes represent the atom indices of the molecule. A bright, localized region along the diagonal indicates a local excitation, while significant off-diagonal intensity suggests a charge-transfer character. The strength of an electronic transition, quantified by the oscillator strength, is visually reflected in the Transition Density Matrix (TDM) heatmaps. For dyes 1 and 2, which exhibit high oscillator strengths, the TDM heatmaps (Fig. 10a) display bright, coherent patterns with significant off-diagonal intensity. This pattern indicates that the primary electronic excitation involves an electron moving from one distinct region of the molecule (the donor) to another (the acceptor), confirming a pronounced charge-transfer character. Such a spatial separation of charge in the excited state is highly beneficial for efficient charge separation in photovoltaic devices. In contrast, the TDM for Dye 5 shows a more localized pattern along the diagonal, correlating with its lower oscillator strength and suggesting a more localized excitation with less pronounced charge-transfer character. This analysis validates the structure–property relationships inferred from the UV-vis parameters (Table 4) and frontier molecular orbitals (Fig. 9), confirming that the dyes with more delocalized and coherent TDMs correspond to those with superior light-harvesting potential as quantified by their oscillator strength and LHE values.
 |
| | Fig. 10 (a) Transition Density Matrix (TDM) heatmaps for the exemplified dyes. The bright, coherent patterns indicate the degree of spatial delocalization and charge-transfer character of the primary electronic transition. (b) Molecular electrostatic potential (MEP) surfaces mapped onto the electron density isosurface (red = negative, blue = positive). | |
Photovoltaic parameters
The PV parameters of the dyes offer valuable insights into their potential for use in solar cell applications. The light harvesting efficiency (LHE) indicates the dye's ability to absorb light across different wavelengths. Its higher value suggested that the dye could absorb a greater proportion of incident light, which is important for efficient solar energy conversion (Table 6). The Voc is the maximum voltage a solar cell can produce when no current is flowing. A higher Voc is generally indicative of a dye stronger capacity for charge separation, contributing to better overall device performance. Fill Factor (FF) measures how well a solar cell can convert absorbed light into usable electrical power. A higher FF implied that the dye allowed for efficient charge transport and reduced losses due to recombination or resistance.
Table 6 Calculated photovoltaic parameters of the selected dyes
| Dye |
LHE (%) |
Voc (V) |
FF |
Jsc (mA cm−2) |
| 1 |
95 |
0.96 |
0.34 |
21.80 |
| 2 |
88 |
0.52 |
0.91 |
20.39 |
| 3 |
60 |
0.74 |
0.87 |
19.24 |
| 4 |
78 |
0.42 |
0.39 |
20.70 |
| 5 |
93 |
0.31 |
0.42 |
28.75 |
| 6 |
15 |
0.26 |
0.59 |
04.40 |
The Jsc reflects the amount of current generated when the cell is exposed to light and has its terminals shorted. This parameter is influenced by the dye ability to generate charge carriers and transport them effectively. A higher Jsc indicates better charge generation and collection, contributing to overall higher PV efficiency. Together, these parameters serve as the fundamental metrics for assessing a dye suitability for use in PV devices, guiding the design of more efficient and effective solar energy conversion materials. Dye 1 exhibited the highest LHE at 95%, which indicated its excellent ability to absorb sunlight. Coupled with a high Voc of 0.96 V and a Jsc of 21.80 mA cm−2, dye 1 demonstrated its strong potential for PV applications. However, its fill factor (FF) of 0.34 suggested that there may be some losses in the conversion process, indicating room for improvement in optimizing the device architecture or charge transport properties. The dye 2, while having a slightly lower LHE of 88%, showed a significantly lower Voc of 0.52 V. However, it had a high FF of 0.91, indicating efficient charge collection and minimal losses during the conversion process. The Jsc of 20.39 mA cm−2 was also competitive, suggesting that dye 2 could be effective in applications where high fill factors are critical. The dye 3 had a lower LHE of 60% and a Voc of 0.74 V, which is moderate compared to the others. Its FF of 0.87 indicated good charge collection efficiency, but the Jsc of 19.24 mA cm−2 was lower than that of dyes 1 and 2, suggesting that its overall performance might be limited by its light absorption capabilities. The dye 4 showed an LHE of 78% and a low Voc of 0.42 V, which might limit its overall efficiency. The FF of 0.39 indicated significant losses in the conversion process, while the Jsc of 20.70 mA cm−2 suggested that it could still generate a reasonable current density despite its lower voltage. The dye 5 had a high LHE of 93%, but its Voc of 0.31 V was the lowest among all, indicating limited potential for generating voltage. However, it had a relatively high Jsc of 28.75 mA cm−2, suggesting that it can produce a significant current density, although the low Voc might hinder its overall efficiency. The dye 6 had the lowest LHE at 15%, a Voc of 0.26 V, and a Jsc of only 4.40 mA cm−2, indicating poor performance in PV applications. Its FF of 0.59 suggested some efficiency in charge collection, but it could not be suitable for effective solar energy conversion. The dyes 1 and 2 showed the most promise for PV applications due to their high LHE and competitive Jsc values, while dyes 4 and 5 presented their mixed performance profile. The dye 6, however, demonstrated limited potential for solar energy conversion. Understanding these parameters could be important for optimizing the design and application of these dyes in solar cell technologies. The demonstration that our integrated ML/DFT pipeline can identify dyes with a promising combination of high LHE, VOC, JSC, and FF, as exemplified by the parameters calculated for this representative set, indicates a strong potential for achieving high Power Conversion Efficiency (PCE) in a fully fabricated device. The ML-predicted absorbance and SALI scores for the larger set of ∼1000 candidates provide a clear and computationally efficient roadmap for prioritizing the most promising dyes for subsequent experimental synthesis and device integration.
Conclusion
In conclusion, our comprehensive study on the light absorbance of over 70000 organic photovoltaic (OPV) dyes has demonstrated the powerful application of machine learning (ML) techniques, especially the Gaussian process multi-output models, in predicting solvent absorbance properties. The identification of acetonitrile as a highly promising solvent, based on its association with the highest predicted light absorbance across a diverse set of dye families, underscores the critical role of solvent selection. While the optimal solvent may vary for specific dyes, our large-scale analysis positions ACN as an excellent general-purpose choice for initial experimental validation of novel OPV dyes. While logP provided an initial solubility screen, the absorbance-based criterion offers a more direct link to DSSC functionality. The successful deployment of the xGBoost model, complemented by insights from SHAP value analysis, further emphasizes the critical role of molecular descriptors, particularly polar surface area, in determining dye performance. The introduction of the Transformer-Assisted Orientation (TAO) method enabled the design of new dyes with favorable synthetic accessibility, laying the foundation for practical laboratory synthesis. The integrated ML/DFT pipeline yielded candidate dyes with promising predicted photovoltaic parameters, with the best values in our representative DFT analysis reaching a Voc of 0.96 V, LHE of 95%, FF of 0.87, and Jsc of 28.75 mA cm−2. These results, derived from a subset of structures, indicate the high potential of the overall design strategy. Moving forward, it will be essential to experimentally validate the predicted absorbance and PV properties of these new dyes. Additionally, expanding the dataset to encompass a broader variety of organic solvents and incorporating more intricate molecular descriptors could enhance the predictive capacity of the models. Integrating these dyes into actual PV devices will be vital for assessing their practical performance and scalability. This study establishes a strong foundation for data-driven approaches in designing and optimizing OPV materials, offering the potential for substantial advancements in renewable energy technologies.
Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Data availability
We confirm that the data collected and used in the present research are original and collected by the authors. It can be made public as per the requirement of the journal or may be provided upon a reasonable request to the corresponding authors.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ra06776f.
Acknowledgements
The authors would like to acknowledge Deanship of Graduate Studies and Scientific Research, Taif University for funding this work.
References
- O. A. Al-Shahri, F. B. Ismail, M. A. Hannan, M. S. H. Lipu, A. Q. Al-Shetwi, R. A. Begum, N. F. O. Al-Muhsen and E. Soujeri, J. Clean. Prod., 2021, 284, 125465 CrossRef.
- T. Zeng, L. Meng, L. Cheng, R. Wang, Z. Ran and D. Liu, Adv. Funct. Mater., 2025, 35, 2419278 CrossRef CAS.
- Z. Du, W. Chen, S. Wen, S. Qiao, Q. Liu, D. Ouyang, N. Wang, X. Bao and R. Yang, ChemSusChem, 2014, 7, 3319 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhang, B. Zheng, H. Cui and H. Qi, Renewable Energy, 2025, 242, 122491 CrossRef.
- A. Armin, W. Li, O. J. Sandberg, Z. Xiao, L. Ding, J. Nelson, D. Neher, K. Vandewal, S. Shoaee and T. Wang, et.al., Adv. Energy Mater., 2021, 11, 2003570 CrossRef CAS.
- R. Yu, Q. Lin, S.-F. Leung and Z. Fan, Nano Energy, 2012, 1, 57 CrossRef CAS.
- Z. You, D. Lu, K. K. Kondamareddy, W. Gu, Y. Su and J. Pan, Sep. Purif. Technol., 2025, 361, 131293 CrossRef CAS.
- N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org Chem., 2014, 2014, 7069 CrossRef CAS.
- W.-J. Kwak, S. Chae, R. Feng, P. Gao, J. Read, M. H. Engelhard, L. Zhong, W. Xu and J.-G. Zhang, ACS Energy Lett., 2020, 5(7), 2182–2190 CrossRef CAS.
- R. Zhou, P. Su, J. Liu, L. Jia and X. Lü, Inorg. Chem. Commun., 2017, 85, 56 CrossRef CAS.
- J. Zhang, L. Zhang, X. Wang, Z. Xie, L. Hu, H. Mao, G. Xu, L. Tan and Y. Chen, Adv. Energy Mater., 2022, 12, 2200165 CrossRef CAS.
- A. Tehrani Bagha and K. Holmberg, Materials, 2013, 6, 580 CrossRef CAS PubMed.
- H. O. Tekin, G. ALMisned, S. A. M. Issa, E. S. Kasikci, M. Arooj, A. Ene, M. S. Al-Buriahi, M. Konuk and H. M. H. Zakaly, Front. Phys., 2022, 10, 838725 CrossRef.
- H. Hashimoto, Y. Sugai, C. Uragami, A. T. Gardiner and R. J. Cogdell, J. Photochem. Photobiol., C, 2015, 25, 46 Search PubMed.
- A. Blázquez-Moraleja, I. Sáenz-de-Santa María, M. D. Chiara, D. Álvarez-Fernández, I. García-Moreno, R. Prieto-Montero, V. Martínez-Martínez, I. López Arbeloa and J. L. Chiara, Chem. Sci., 2019, 11, 1052 RSC.
- U. Mahajan, K. Prajapat, M. Dhonde, K. Sahu and P. M. Shirage, Nano-Struct. Nano-Objects, 2024, 37, 101111 CrossRef CAS.
- A. Baracca, G. Sgarbi, G. Solaini and G. Lenaz, Biochim. Biophys. Acta Bioenerg., 2003, 1606, 137 CrossRef CAS PubMed.
- Y. Xia, G. Wang, Y. Lv, C. Shao and Z. Yang, Chem. Phys. Lett., 2024, 836, 141030 CrossRef CAS.
- C. Liu, L. Shao, S. Chen, Z. Hu, H. Cai and F. Huang, Prog. Polym. Sci., 2023, 143, 101711 CrossRef CAS.
- A. Szarwaryn, W. Bartkowiak and U. Bazylińska, Colloids Surf., A, 2023, 675, 132083 CrossRef CAS.
- H. A. Maddah, Opt. Mater., 2022, 128, 112343 CrossRef CAS.
- C. Coppola, A. Visibelli, M. L. Parisi, A. Santucci, L. Zani, O. Spiga and A. Sinicropi, npj Comput. Mater., 2025, 11(1), 28 CrossRef CAS.
- H. Michaels, M. Rinderle, R. Freitag, I. Benesperi, T. Edvinsson, R. Socher and M. Freitag, Chem. Sci., 2020, 11(11), 2895–2906 RSC.
- V. Yadav, R. Bhatnagar and U. Kumar, Discov. Electron., 2024, 1(1), 17 CrossRef.
- V. Yadav, R. Bhatnagar and U. Kumar, Adv. Condens. Matter Phys., 2024, 2024(1), 6613380 Search PubMed.
- V. K. Gupta, R. Kumar and U. Kumar, J. Phys. Chem. Solids, 2025, 113071 Search PubMed.
- S. Boobier, D. R. J. Hose, A. J. Blacker and B. N. Nguyen, Nat. Commun., 2020, 11, 5753 CrossRef CAS PubMed.
- A. L. Coutinho, R. Cristofoletti, F. Wu, A. Al Shoyaib, J. Dressman and J. E. Polli, Pharm. Res., 2024, 41, 1121 CrossRef CAS PubMed.
- N. Q. K. Le, T.-X. Tran, P.-A. Nguyen, T.-T. Ho and V.-N. Nguyen, Expert Opin. Drug Metab. Toxicol., 2024, 20, 621 CrossRef CAS PubMed.
- A. Mahmood, A. Irfan and J.-L. Wang, Chin. J. Polym. Sci., 2022, 40, 870 CrossRef CAS.
- B. R. Sutherland, Joule, 2020, 4, 984 CrossRef.
- C. J. Brabec, A. Cravino, D. Meissner, N. S. Sariciftci, T. Fromherz, M. T. Rispens, L. Sanchez and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 374 CrossRef CAS.
- F. Brockherde, L. Vogt, L. Li, M. E. Tuckerman, K. Burke and K. R. Müller, Nat. Commun., 2017, 8(1), 872 CrossRef PubMed.
- S. Pradhan, J. S. Bhattarai, M. Murugavel and O. P. Sharma, ACS Omega, 2025, 10(16), 16597–16601 CrossRef PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He and E. E. Bolton, Nucleic Acids Res., 2023, 51(D1), D1373–D1380 CrossRef PubMed.
- A. Gaulton, A. Hersey, M. Nowotka, A. P. Bento, J. Chambers, D. Mendez and A. R. Leach, Nucleic Acids Res., 2017, 45(D1), D945–D954 CrossRef CAS PubMed.
- G. B. Goh, N. O. Hodas and A. Vishnu, J. Comput. Chem., 2017, 38(16), 1291–1307 CrossRef CAS PubMed.
- V. F. Scalfani, V. D. Patel and A. M. Fernandez, J. Cheminf., 2022, 14, 87 Search PubMed.
- D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus, H. Kruse, R. Di Remigio, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A. Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O'Brien, J. M. Waldrop, A. Kumar, E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, H. F. Schaefer III, A. Yu. Sokolov, K. Patkowski, A. E. DePrince III, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford and C. D. Sherrill, J. Chem. Phys., 2020, 152, 184108 CrossRef CAS PubMed.
- X. H. Li, A. F. Jalbout and M. Solimannejad, J. Mol. Struct., 2003, 663, 81 CrossRef CAS.
- M. Müller, A. Hansen and S. Grimme, J. Chem. Phys., 2023, 159, 164108 CrossRef PubMed.
- W. Mckinney, Python High Performance Science Computer, 2011 Search PubMed.
- C. Schäfer, in Quickstart Python: an Introduction to Programming for STEM Students, ed. C. Schäfer, Springer Fachmedien, Wiesbaden, 2021, pp. 45–53 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, A. Müller, J. Nothman, G. Louppe, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, Journal of machine Learning research, 2011, 12, 2825–2830 Search PubMed.
- K. Okoye and S. Hosseini, in R Programming: Statistical Data Analysis in Research, ed. K. Okoye and S. Hosseini, Springer Nature, Singapore, 2024, pp. 247–277 Search PubMed.
- J. M. Frisch, W. G. Trucks, B. H. Schlegel and et.al., Wallingford CT, 2013.
- H. Farrokhpour and H. Jouypazadeh, Chem. Phys., 2017, 488, 1 CrossRef.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128(8) CrossRef CAS PubMed.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem., 2001, 105, 8111 CrossRef CAS.
- C. Sandoval-Yañez and J. I. Martínez-Araya, Chem. Phys. Lett., 2019, 715, 354–359 CrossRef.
- A. U. Hassan, S. H. Sumrra, M. Zubair, G. Mustafa, S. Noreen and M. Imran, Chem. Pap., 2023, 77(10), 6183–6202 CrossRef CAS.
- H. A. Kyhoiesh, M. A. Awad, K. H. Salem, A. S. Waheeb, A. A. Al-Kubaisi, G. A. Mersal and Z. A. Abass, ChemistrySelect, 2025, 10(37), e03160 CrossRef CAS.
- M. K. Tran, S. Panchal, V. Chauhan, N. Brahmbhatt, A. Mevawalla, R. Fraser and M. Fowler, Int. J. Energy Res., 2022, 46(2), 786–794 CrossRef CAS.
- L. D. Hughes, R. J. Rawle and S. G. Boxer, PLoS One, 2014, 9, e87649 CrossRef PubMed.
- E. Bodio and C. Goze, Dyes Pigm., 2019, 160, 700 CrossRef CAS.
- H. I. Yoon, H. Lee, J. S. Yang, J. H. Choi, D. H. Jung, Y. J. Park and S. H. Park, Agriculture, 2023, 13(8), 1477 CrossRef CAS.
- A. Koushik, M. Manoj and N. Nezamuddin, Transp. in Dev. Econ., 2024, 10, 12 CrossRef.
- C. Güleryüz, S. H. Sumrra, A. U. Hassan, A. Mohyuddin, A. S. Waheeb, M. A. Awad and I. H. El Azab, J. Photochem. Photobiol., A, 2025, 460, 116157 CrossRef.
- A. U. Hassan, C. Güleryüz, I. H. El Azab, A. Y. Elnaggar and M. H. Mahmoud, Mater. Chem. Phys., 2025, 339, 130747 CrossRef CAS.
- H. A. Kyhoiesh, K. H. Salem, A. S. Waheeb, R. A. Hasan, H. R. Salman, A. A. Al-Kubaisi and M. H. Mahmoud, RSC Adv., 2025, 15(28), 22449–22459 RSC.
- H. Wang, S. Ma, L. Dong, S. Huang, D. Zhang and F. Wei, IEEE Trans. Pattern Anal. Mach. Intell., 2024, 46, 6761 Search PubMed.
- R. P. Vivek-Ananth, A. K. Sahoo, S. P. Baskaran, J. Ravichandran and A. Samal, Sci. Total Environ., 2023, 873, 162263 CrossRef CAS PubMed.
- X. Peng, J. Liang, K. Wang, X. Zhao, Z. Peng, Z. Li and D. Huang, npj Comput. Mater., 2024, 10(1), 213 CrossRef CAS.
- K. Liu, Z. Jiang, R. A. Lalancette, X. Tang and F. Jakle, J. Am. Chem. Soc., 2022, 144(41), 18908–18917 CrossRef CAS PubMed.
- A. D. Laurent, C. Adamo and D. Jacquemin, Phys. Chem. Chem. Phys., 2014, 16(28), 14334–14356 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *be,
Hassan E. Abd Elsalamc and
Islam H. El Azabc
*be,
Hassan E. Abd Elsalamc and
Islam H. El Azabc





















