
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid reductive catalytic fractionation for holistic valorization of lignocellulose through precise acid tuning and temporal window identification
Peidong Liab,
Wangyue Xieab,
Muhammad Fayyaz Faridab,
Tianyu Ren b,
Cheng Tung Chongc,
Zhuo Heab,
Yuhe Liaoab,
Yanbin Cui*ab and
Chenguang Wang*ab
b,
Cheng Tung Chongc,
Zhuo Heab,
Yuhe Liaoab,
Yanbin Cui*ab and
Chenguang Wang*ab
aSchool of Energy Science and Engineering, University of Science and Technology of China, Hefei 230026, China. E-mail: cuiyb@ms.giec.ac.cn
bGuangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou 510640, China. E-mail: wangcg@ms.giec.ac.cn
cChina-UK Low Carbon College, Shanghai Jiao Tong University, Shanghai 201306, China
First published on 11th November 2025
Abstract
Reductive catalytic fractionation (RCF) is an effective technique that enables lignin depolymerization into aromatic monomers while preserving cellulose for downstream valorization. However, the typically prolonged reaction durations (>3 h) required for conventional RCF, stemming from slow kinetics of lignin extraction and depolymerization, impeding its industrial scalability. This study systematically investigated solvent composition, acid concentration, and reaction parameters to accelerate RCF kinetics and modulate monomer distribution. Through rapid benchmark reactions measured immediately at target temperature (0 h) using Ni/C, we demonstrated that a phosphoric acid-assisted RCF process in ethanol–water solvent achieves efficient lignin depolymerization. Remarkably, by finely tuning the acid concentration in the reaction system, this approach achieved a high monomer yield of 31% at 0 h, with a remarkably high production efficiency of 3.44 mg monomers per g lignin per min, exceeding most previously reported rates. Time course analysis further established that precise acid tuning and identifying the optimal temporal window are critical for maximizing monomer yield while preserving carbohydrates. Under optimized conditions employing 0.3 wt.‰ H3PO4, an aromatic monomer yield of 43% was attained within only 30 minutes. The resulting cellulose-rich solid residue, containing the RCF catalyst, was directly converted to ethylene glycol (35% yield) via an integrated one-pot aqueous-phase hydrolysis-hydrogenolysis process. This simultaneous valorization strategy also facilitated effective catalyst recovery and reuse. Our work establishes an efficient and potentially scalable rapid RCF paradigm, offering a promising route for the holistic utilization of lignocellulosic biomass.
1 Introduction
The sustainable valorization of lignocellulosic biomass relies on fractionation strategies that maximize the utility of all major components. While cellulose and hemicellulose conversion routes are well-established, lignin remains underexploited due to its recalcitrant and heterogeneous nature.1–4 Conventional biorefining often degrades lignin into intractable wastes through irreversible condensation reactions during prolonged and severe pretreatment processing.5–7 Reductive catalytic fractionation (RCF) has emerged as a transformative lignin-first paradigm, prioritizing the extraction and catalytic depolymerization of lignin under reductive conditions while generating a carbohydrate-rich pulp.8–11 This approach preserves lignin's aromatic nature for value-added chemical production and enables parallel valorization streams. However, achieving sufficient lignin removal and high monomer yields typically necessitates extended reaction times (often > 3 hours) at elevated temperatures, significantly impeding its production efficiency toward industrial scale deployment.8,9,12,13Dilute acid pretreatments (e.g., H2SO4, H3PO4) represent a well-established strategy for lignocellulose fractionation, primarily effective in hydrolyzing hemicellulose and enhancing cellulose accessibility for enzymatic saccharification.14,15 Regarding lignin conversion, acid additives promote the polarization and cleavage of β-O-4 ether linkages while accelerating the disruption of lignin-carbohydrate complexes (LCCs).16 This effect is evident in organosolv fractionation utilizing mildly acidic aqueous-organic solvent mixtures.17 In recent decades, innovative approaches employing acid catalysis with capturing agents (e.g., aldehydes by Luterbacher,18 diols by Barta19,20) demonstrate the potential to efficiently recover well-preserved lignin within shortened processing times. Such lignin provides a high-quality feedstock for depolymerization, enabling yields approaching theoretical maxima. Collectively, these observations underscore acidic catalysis as a key lever to accelerate critical solvolysis and depolymerization kinetics in lignin conversion.
Building on the lignin-first context, the product distribution of lignin monomers is intimately associated with reaction kinetics governed by the solvent system and catalytic environment.21–23 Debecker et al. showed that the employment of H3PO4 under lower RCF reaction temperature could improve the lignin-derived monomer yield and recovery of cellulose in the pulp.24 Sels and coworkers found that the addition of small quantities of acid promoted both delignification and alcoholysis of hemicellulose, leaving behind a cellulose-rich pulp, and such simultaneous acid-catalyzed fractionation of the carbohydrates into separate cellulose and hemicellulose streams provides opportunities for more efficient downstream conversion.25 However, boosting RCF rates involves inherent trade-offs, with increased reaction severity often compromises carbohydrate integrity, crucial for downstream cellulose-centralized valorization. Besides, prolonged acid treatment often leads to significant lignin repolymerization and low monomer production efficiency, undermining the economic feasibility of such production mode. Therefore, the reaction condition and operation window to achieve the optimal balance for both carbohydrate and lignin components need delicate control. We hypothesized that deliberately introducing a targeted acidic catalyst under mild, time-constrained conditions could overcome the kinetic limitations of conventional RCF while preserving the optimum balance between lignin depolymerization and carbohydrate integrity.
In this work, we introduce a rapid phosphoric acid-assisted RCF process that accelerates lignin solvolysis and depolymerization kinetics in water alcohol co-solvents. The monomer yield and distribution throughout the time resolved RCF study was comprehensively studied. By finely tuned reaction condition and temporal window, efficient lignin solubilization and depolymerization was achieved within short reaction times (0–30 min), achieving high recovery and high purity carbohydrate component. The solid carbohydrate residue, mixed with reusable RCF catalyst, is directly valorized through aqueous-phase catalytic hydrogenolysis to platform chemicals such as ethylene glycol. This integrated, acid-assisted RCF process establishes a rapid and effective route for the holistic valorization of lignocellulosic biomass, overcoming key kinetic and selectivity barriers that have hindered conventional approaches.
2 Experimental section
2.1 Materials
Poplar wood chips was dried, pulverized and grounded, and sieved through 50–80 meshes. The woodmeal sample was stored in a desiccator until use. Nickel nitrate (II) hexahydrate, activated carbon, phosphoric acid (85 wt%), sulfuric acid (95 wt%), ethyl acetate (99.5 wt%), tetrahydrofuran (HPLC purity), alcohols (methanol, ethanol, isopropanol, ethylene glycol, isobutanol) and DMSO-d6 were procured from Aladdin and used without further purification unless specified otherwise.2.2 Catalyst preparation
Ni/C catalyst was prepared by a simple incipient-wetness impregnation method. Typically, 16.56 g Ni(NO3)2·6H2O was added into 300 mL pure water in a beaker, and the solution was added to 30 g grounded activated carbon (100 mesh). The suspension covered by tinfoil with holes was stirred for 24 hours at the room temperature. Then the sample was dried into the oven at 100 °C to remove water. The dried sample was ground and sieved with 100 mesh again, and then reduced in a horizontal furnace in a 50 mL min−1 10% H2/Ar flow for 3 h at 450 °C with 5 °C min−1 heating rate.Ni content was quantified by ICP-OES following microwave-assisted aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1, v/v) digestion. ICP-OES measurements were performed on an OPTIMA 8000 DV (PerkinElmer, USA) with dual-view and automatic background correction. Morphology was examined by cold-field-emission cryogenic SEM (HITACHI, Japan) under high vacuum at low beam current and 2–5 kV accelerating voltage (working distance 8–10 mm) with SE.
:HNO3 = 3:1, v/v) digestion. ICP-OES measurements were performed on an OPTIMA 8000 DV (PerkinElmer, USA) with dual-view and automatic background correction. Morphology was examined by cold-field-emission cryogenic SEM (HITACHI, Japan) under high vacuum at low beam current and 2–5 kV accelerating voltage (working distance 8–10 mm) with SE.
2.3 Development of acid assisted RCF process in alcohol–water solvent
In a batch RCF reaction, 0.8 g of 100 mesh poplar, 0.16 g 10 wt% Ni/C, and varied amount of H3PO4 were added into 20 mL of alcohol/H2O solvent for process optimization. The reactor was sealed and purged with H2 three times and then pressurized to designated pressures. The reactor was heated to the target temperature with 3.5 °C min−1 heating rate and maintained for designated reaction times at a stirring speed of 700 rpm. For the time course study, 6 g biomass woodmeal, 1.2 g Ni/C, 75 mL alcohol solvent, 75 mL H2O, varied amount of H3PO4 were added into a 300 mL of stainless steel pressure reactor. The reactor was sealed, then purged with low pressure H2 three times and pressurized to 2 MPa H2, heated to pre-set reaction temperatures with 5 °C min−1 heating rate and maintained for designated reaction time at a stirring speed of 700 rpm. When the reaction end, the reactors were cooled down to room temperature.The liquid products were analyzed by GC-MS (Thermo Trace 1300 ISQ QD) as well as GC-FID (Shimadzu 2014C, Agilent HP-5 column) and then quantified through effective carbon number method with o-isopropylphenol as the internal standard. The flow rate of carrier gas and hydrogen gas was 30 mL min−1, the flow rate of the air was 400 mL min−1. The heating process was set as below: inlet temperature was 300 °C and oven temperature was held at 60 °C for 2 min and then increased to 110 °C at 5 °C min−1, at last increased to 300 °C at 10 °C min−1 and held for 3 min. The molecular weight distribution of lignin in the organic phase were measured by gel permeation chromatography (GPC, 1260 LC Agilent, USA). Before GPC analysis, the liquid sample was extracted with ethyl acetate for 4 times, and organic phase was vacuum-distilled at 45 °C. The concentrated lignin oil was redissolved in tetrahydrofuran and filtrated with a 0.22 μm nylon filtrate pad. The GPC was calibrated by the polystyrene standard. 2D 1H–13C heteronuclear single-quantum coherence (HSQC) NMR spectra were measured on AVANCE III 400 MHz spectrometer (BRUKER, Germany). The HSQC NMR sample was prepared by vacuum distillation of the organic phase at 35 °C to remove the solvent followed by dissolving the dried solid with DMSO-d6. The number of collected complex points was 1024 for the 1H dimension with a delay time of 1.5 s. The number of transients was 64-, and 256-time increments were recorded in the 13C dimension. The coupling constant J1 of C–H used was 147 Hz.
2.4 Compositional analysis and hydrogenolysis of solid residue
The composition of the biomass residue was measured according to the NREL standard hydrolysis procedure.26 Specifically, 2 mL 72% H2SO4 was added to 0.1 g residue and mixed thoroughly in a glass vial at room temperature for 2 h, with the suspension were regularly stirred every 20 min. Then 56 mL of deionized H2O was added, and the samples was transferred to a pressurized bottle, heated in an autoclave at 121 °C for 120 min. The final aqueous mixture containing solubilized products was filtered through a syringe filter with 0.2 μm PTFE and subjected to Waters HPLC analysis to quantify soluble sugars, and the insoluble fraction was filtered from the hydrolysis liquid then dried for 24 h at 60 °C. Liquid product analysis using a Waters e2695 UPLC system equipped with a Shodex SUGAR SH1011 column (300 mm × 8 mm). The mobile phase consisted of 5 mmol L−1 H2SO4 at a flow rate of 0.5 mL min−1, with the column temperature maintained at 50 °C and an injection volume of 20 μL. The quantification of products was performed using external calibration curves, which were prepared from corresponding standard compounds under identical chromatographic conditions. Crystallinity of cellulose was examined with a Rigaku SmartLab multifunction X-ray diffractometer (Cu Kα, 40 kV, 40 mA, 2θ 5–50°, 0.02° step, 2° min−1). The crystallinity index CrI (%) was calculated from the Segal equation: CrI = [(I200 − Iam)/I200] × 100, where I200 (≈22.5°) and Iam (≈18.0°) are the intensities of the (002) reflection and the amorphous valley, respectively.Hydrogenolysis of solid residue was conducted in 100 mL of batch reactor, with 0.2 g solid residue (with catalyst), 20 mL H2O and varied content of H2SO4 were added into a reactor. The reactor was sealed, then purged with H2 three times and pressurized to designated pressure, heating to target temperatures and maintain for 5 h at a stirring speed of 700 rpm. The weight of the catalyst recovered in the residue was measured through a control experiment using merely catalyst in the identical reaction condition.
3 Results and discussion
3.1 Acceleration of RCF process in various alcohol-water solvent systems
EtOH/water mixtures at (a) 3 h and (b) 0 h. Reaction conditions: 210 °C, 20 mL of solvent mixture, 0.8 g of poplar woodmeal, 0.16 g of Ni/C, and 2 MPa H2Solvent composition exerted profound effects on lignin extraction efficiency and monomer production patterns during RCF processing, exhibiting a critical interplay between lignin solvation and catalytic depolymerization.27 Preliminary screening of alcohol–water systems under standard conditions (210 °C, 2 MPa H2, 3 h) confirmed ethanol–water's superiority for monomer production (Fig. S1). Subsequent investigation across differing ethanol:water (EtOH:H2O) ratios uncovered a distinct volcano-shaped dependency for both lignin oil and monomer yields. Pure water afforded limited delignification (26.4 wt% lignin oil) with low monomer yield (8.6 wt%), predominantly generating propyl-substituted species (Fig. 1a). Introduction of ethanol markedly enhanced lignin extraction and monomer formation, culminating in peak lignin oil yield (58.0%) and monomer yield (38.4%) at EtOH:H2O = 50:50. Beyond this optimum, further ethanol enrichment progressively diminished both extraction efficiency and monomer yield, falling to 37.2% lignin oil and 25.3% monomer yield in neat EtOH, aligning with literature observations on reduced depolymerization effectiveness in pure organic solvents.28
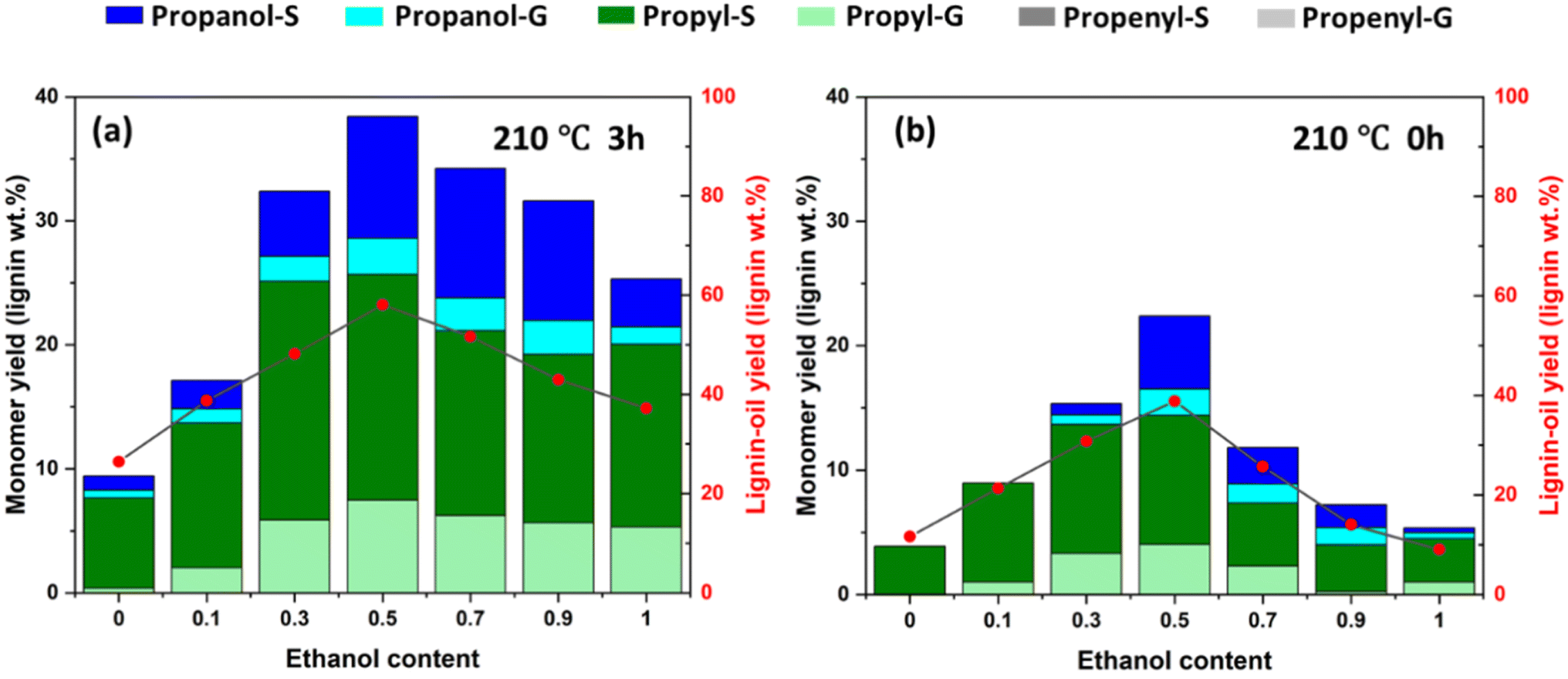 | ||
| Fig. 1 Monomer yield and lignin oil yield for the RCF using varied. | ||
To elucidate the influence of solvent composition on reaction kinetics, complementary short-time experiments (0 h reaction time after 0.5 h heating) further contrasted ethanol ratios. Both lignin oil accumulation and monomer generation at this initial stage (Fig. 1b) exhibited similar volcanic profiles across solvent compositions to those observed at 3 h reaction time (Fig. 1a). This trend highlights that the EtOH:H2O = 50:50 binary system enabled rapid monomer production (22.38 wt% yield at 0 h), demonstrating a pronounced acceleration effect in binary ethanol–water RCF reaction systems. Interestingly, elevated ethanol fractions fundamentally shifted monomer speciation from propyl-substituted to 4-n-propanol derivatives, and then drop to propyl-dominant in pure ethanol (Table S1). This is due to the enhanced stabilization of intermediate γ-hydroxyl groups through ethanol co-solvation effects,29 while the pure alcohol solvent afford extra hydrogen supply capacity to generate saturated monomer product.30
3.2 Monomeric product distribution in acid-assisted rapid RCF process
Building upon solvent optimization benchmarks established at 0 h reaction time, we systematically examined the coupled effects of phosphoric acid concentration and reaction temperature on lignin-derived monomer profiles (Fig. 2). For all tested temperatures, lignin oil yields increased monotonically with rising acid concentration, showing an elevated acid-assisted lignin extraction capacity.25 Conversely, monomer production universally exhibited volcano-shaped relationships versus acid concentration. While higher acid levels continuously promoted lignin fragmentation and solubilization (increasing lignin oil), excessive acid simultaneously diverted reactive intermediates toward acid-catalyzed condensation.31 | ||
| Fig. 2 The dependence of lignin oil and monomer yield on acid concentration at (a) 190 °C, (b) 210 °C and (c) 230 °C. Reaction conditions: 20 mL ethanol:H2O = 1:1, 0.8 g of poplar wood 0.16 g of Ni/C, and 2 MPa H2, 0 h. | ||
Temperature fundamentally modulated the acid concentration generating the highest monomer yield, as well as the magnitude of these volcanic maxima. At 190 °C, peak monomer yield (21.1%) required 1.0‰ H3PO4 addition, and propenyl monomers only present in the product till the concentration reaches 10‰. Elevating the temperature to 210 °C shifts the maximum monomer yield (30.9 wt%) to a lower acid concentration (0.3‰ H3PO4), with propenyl monomers show up at acid concentration of 1.0‰. Further temperature increases to 230 °C shifted maximal monomer yield (26.1%) to 0.1‰, stimulating both propenyl monomers and phenol generation via dehydration and dealkylation pathways,32,33 even at lower acid concentrations (Table S2). Gel permeation chromatography (GPC) analysis of selected samples at varied acid ratios showed a consistent molecular weight distribution pattern. Compared with acid-free condition, 0.3‰ acid addition generates product profiles with decreased molecular weight distribution, while higher acid (10‰) causes considerable amount of oligomer formation (Fig. S2–S4). The dependence of monomer yield and distribution on acid/temperature reflecting a kinetic balance where optimal acid enhanced catalytic cleavage while excess acid drove condensation.5,34 On the other hand, the impact of acid concentration on crystallinity of cellulose component in solid pulp was compared (Fig. S5). The CI value for all samples maintained at a high level (>88%), and the crystallinity level decreases slightly as the acid concentration increases. These results demonstrate an effective approach to maintain well-preserved cellulose while achieving high monomer yield at short retention time.
Quantitative analysis of monomer production rates (yield per unit time, Table 1) revealed production efficiency under varied conditions, alongside the rates achieved in other studies. Crucially, the optimal 0.3‰ H3PO4 assisted RCF system achieved the highest production rate (3.44 mg monomers per g lignin per min) when considering heating and cooling times (90 min). This rate surpassed those obtained under both acid-free conditions (2.33 mg g−1 min−1) and high-acid conditions (2.89 mg g−1 min−1). In comparison, the majority of previously reported RCF processes generating high monomer yields from various feedstocks employ substantially longer reaction durations (2–6 h), resulting in lower production rates (typically 0.38–2.33 mg per g lignin per min). Beckham et al. emphasized that minimizing the process turnaround time is essential for scalable RCF production, proposing a shift from batch to continuous operation as an effective strategy.35 The present study highlights an alternative approach for enhancing scalability, the synergy achieved by precisely modulated acid catalysis combined with optimized solvent composition and reaction temperature significantly accelerated monomer synthesis kinetics, and its applicability was also demonstrated on different types of lignocellulosic feedstocks (Fig. S6).
| Biomass | Reaction condition | Catalyst | Monomer yield (lignin wt%) | Production rate mg g−1 lignin/min | Ref. |
|---|---|---|---|---|---|
| a Note: production rate was calculated based on full reaction time, which includes heating, reaction and cooling durations, and the heating and cooling durations of all cases were unanimously assumed as 90 min according to the case in our study. | |||||
| Eucalyptus | 260 °C, 3 MPa H2, methanol, 4 h | MoOx/SBA-15 | 43.4 | 0.76 | 11 |
| Sawdust | |||||
| Poplar | 230 °C, 3 MPa H2, methanol: H2O = 1:1, 4 h |
Ru/C | 32.6 | 0.99 | 12 |
| Switchgrass | 31.6 | 0.96 | |||
| Corn stover | 32.1 | 0.97 | |||
| Pine | 12.7 | 0.38 | |||
| Eucalyptus sawdust | 200 °C, 3 MPa H2, n-butanol: H2O = 1:1, 2 h |
Ru/C | 49 | 2.33 | 23 |
| Wheat straw | 200 °C, 3 MPa N2, H2O, 0.1 M H3PO4, 6 h | Ru/C | 22 | 0.49 | 24 |
| Poplar | 200 °C, 2 MPa H2 methanol, 3 h | Pd/C | 41 | 1.52 | 25 |
| Poplar | 200 °C, 2 MPa H2, ethanol: H2O = 7:3, 3 h |
Pd/C | 42 | 1.56 | 29 |
| Corn stover | 200 °C, methanol, 6 h | Ni/C | 38 | 0.84 | 36 |
| Birch | 200 °C, 0.2 MPa N2, methanol, 6 h | Ni/C | 32 | 0.71 | 37 |
| Poplar | 225 °C, 3.5 MPa H2, methanol, 12 h | Ni/C | 31 | 0.38 | 38 |
| Poplar | 210 °C, 2 MPa H2, ethanol: H2O = 1:1, 0.3‰ H3PO4, 0 h |
Ni/C | 31 | 3.44 | This work |
3.3 Time course study of acid-assisted RCF process
Extending the investigation of phosphoric acid catalysis under the temperature-optimized condition (210 °C, 2 MPa H2, ethanol:H2O = 1:1), detailed time-course studies were performed to elucidate transient reaction kinetics. Using a mechanically stirred reactor equipped with in situ sampling, the impact of acid concentration on lignin depolymerization pathways was temporally resolved. Critically, without acid addition (Fig. 3a), monomer generation displayed sluggish kinetics. The monomers formation was initiated only above 200 °C, with the monomer yield benchmarked at 0 h only reaches 10%. The monomer formation gradually proceeds throughout the 4-hour isothermal phase, reaching maximal yields of 48% of lignin weight. Monomer profiles were dominated equally by propanol and propyl guaiacol/syringol derivatives (Table S3), similar to the product profile in binary alcohol-water among other works.12
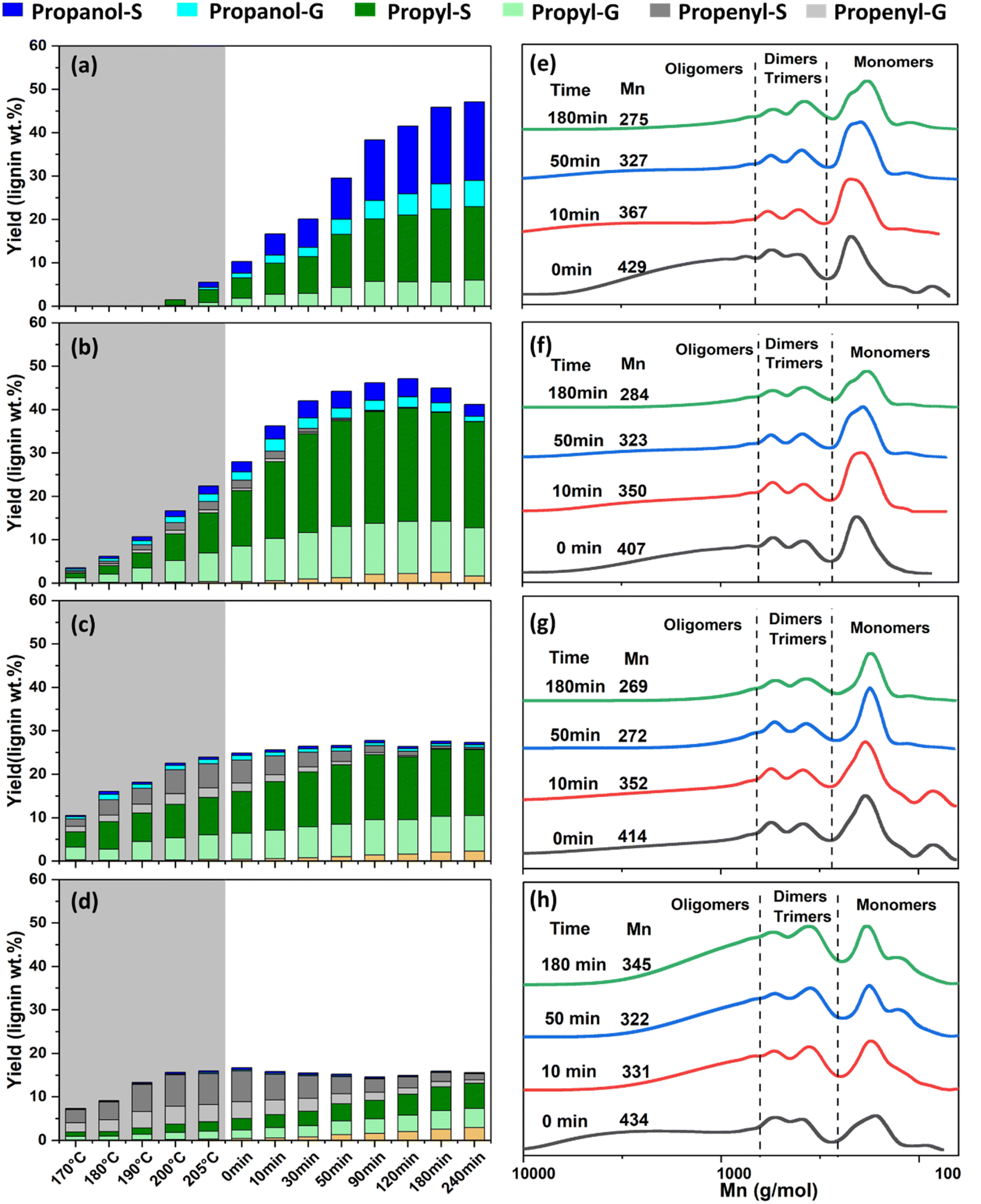 | ||
| Fig. 3 Time course study and GPC analysis of RCF process with varied H3PO4 concentration (a and e) none, (b and f) 0.3‰, (c and g) 3‰ and (d and h) 10‰, reaction condition: 6 g poplar biomass, 1.2 g 10% Ni/C, 150 mL ethanol:H2O = 1:1, 2 MPa H2, 210 °C. | ||
In stark contrast, introduction of the optimal 0.3‰ H3PO4 concentration dramatically accelerated depolymerization while substantially directing selectivity (Fig. 3b). Monomer yields outpaced the acid-free system during the initial heating ramp, with monomer yield approached around 30% at the reaction time of 0 h. Depolymerization rapidly achieved 43% yield after 30 min reaction, and plateaued at a maximum yield of 47% within just 120 minutes of isothermal reaction, demonstrating a considerably accelerated monomer generation kinetics. Notably, the selectivity of propenyl and propanol monomer drops progressively as the reaction proceed, with the selectivity of propyl derivatives dominated in the late phase (Table S4). This might be associated with acid-facilitated dehydration and hydrogenation saturation thereafter.36 Employing gradiently higher acid concentrations (3‰ and 10‰ H3PO4) considerably accelerated monomer generation during heating ramp (Fig. 3c and d), with the monomer yield plateaued at 26.4% and 16.7%, respectively. Noticeably, such condition induced a distinct monomer distribution toward propenyl derivatives in early stage (Table S5), indicating acid-catalyzed dehydration pathways was kinetically favored over metal-catalyzed hydrogenation.23 The dropping monomer yield with increasing acid concentration demonstrated that Ni/C was inevitably deactivated through leaching, destruction, or poisoning, etc.33 Luo et al. reported a similar trend with decreasing yield and increasing propenyl ratio as the catalyst was deactivated.39 The double-sided effect highlights the importance of tuning of acid concentration in RCF process, to the extent to accelerate the reaction rate for efficient lignin extraction and monomer generation, while avoid the undesirable catalyst deactivation.
Time-resolved GPC analysis of liquid products elucidated the molecular weight distribution of acid-driven lignin transformation (Fig. 3d–f). For all acid concentrations, oligomers with large Mw dominated in the early stage, confirming the less fragmented state of lignin upon initial extraction from biomass.40 For non-acidic and low acidic (0.3‰) conditions, the Mw weight consistently decreases as reaction time increases due to ether bond cleavage, matching well with progressive monomer generation. In contrast, increasing acid to 10‰ cause a rapid and sharp Mw declination in the early stage, consistent with the monomer yield trend in Fig. 3c. However, as the reaction proceeded to late stage, product distribution shifted toward higher oligomer derived from condensation. These findings establish that precise control of acid concentration and operation window is critical for maximizing monomer yields while minimizing side reactions.
The structural evolution of fractionated lignin under varying acid concentrations was further characterized by 2D HSQC NMR (Fig. 4). Pronounced β-O-4 motif signals (δC/δH = 65–85/3.3–4.5 ppm) in lignin from acid-free RCF conditions indicate effective retention of native β-ether linkages. Concurrently, dominant guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H) units in the aromatic region (δC/δH = 100–140/6.0–7.5 ppm) confirm structural preservation.41,42 As 0.3‰ acid was added to the RCF system, the peaks of β-ether linkage remain dominant, and the presence of phenylcoumaran signal indicates lignin fraction was more effectively extracted. Conversely, the introduction of 10‰ acid fundamentally altered lignin architecture, with β-ether signals almost disappeared while resinol (Cγ) emerged in the aliphatic region, demonstrating extensive acid-driven condensation at such condition.43,44
 | ||
| Fig. 4 HSQC spectra of RCF samples collected at 0 h varied H3PO4 concentration of (a and b) none (c and d) 0.3‰ and (e and f) 10‰. | ||
3.4 Composition analysis of solid residual from acid-assisted RCF
Compositional tracking of solid residues sampled from varied reaction times provides critical insights into the conversion dynamics under varied acid-catalyzed RCF conditions (Table 2). The acid-free system exhibited progressive delignification from 37% to 76% with extended reaction time from 0 h to 3 h, accompanied by moderate cellulose enrichment to 80.7% purity and good cellulose retention (90%). This gradual lignin removal aligns with the previously observed sluggish monomer evolution, confirming that thermal solvolysis cleavage proceeds slowly through rate-limited, stepwise depolymerization without significant carbohydrate degradation.45 In contrast, the optimized 0.3‰ H3PO4 condition shows a fundamentally altered scenario. Even at the initial reaction stage (0 h), delignification reached 58.0% that exceeds the 0.5 h performance of the acid-free system, demonstrating rapid acid-facilitated lignin solvolysis during heating. This acceleration was especially intensified during the early stage of the reaction, with a high delignification (81.6%) and high purity (82.8%) cellulose was achieved within 0.5 h reaction time. Crucially, cellulose retention remained high (>75.9%) after a 3 h reaction, reinforcing that the optimal operation window through precisely controlled low acid to rapidly extract lignin into soluble forms conducive to depolymerization while robustly preserving polysaccharide integrity.| Acid-time | Delignification (%) | Composition (%) | Cellulose | |||
|---|---|---|---|---|---|---|
| Glucan | Xylan | Lignin | Purity (%) | Retention (%) | ||
| Raw poplar | — | 43.05 | 16.43 | 20.15 | — | — |
| 0‰–0 h | 37.18 | 60.03 | 14.75 | 17.86 | 64.80 | 98.84 |
| 0‰–0.5 h | 57.06 | 61.32 | 10.23 | 13.08 | 72.46 | 94.22 |
| 0‰–1.5 h | 67.04 | 64.59 | 9.53 | 10.70 | 76.15 | 93.13 |
| 0‰–3 h | 76.01 | 67.64 | 7.77 | 8.43 | 80.68 | 90.12 |
| 0.3‰–0 h | 57.98 | 62.86 | 12.56 | 12.69 | 71.34 | 97.44 |
| 0.3‰–0.5 h | 81.51 | 74.34 | 8.53 | 6.96 | 81.64 | 92.45 |
| 0.3‰–1.5 h | 85.05 | 75.36 | 6.01 | 6.47 | 85.79 | 81.52 |
| 0.3‰–3 h | 88.03 | 78.70 | 4.63 | 5.81 | 88.29 | 75.87 |
| 10‰–0 h | 60.07 | 66.54 | 5.19 | 16.65 | 75.29 | 74.69 |
| 10‰–0.5 h | 50.06 | 58.74 | 4.21 | 23.26 | 68.14 | 59.01 |
| 10‰–1.5 h | 43.59 | 51.11 | 0.00 | 27.02 | 67.50 | 49.95 |
| 10‰–3 h | 39.12 | 44.63 | 0.00 | 31.08 | 63.72 | 40.92 |
Conversely, high acid loading (10‰ H3PO4) induced severe condensation and degradation despite high initial delignification efficacy (60% at 0 h). At 0.5 h, lignin content increased to 23% in the residue and delignification efficiency declined to 50%, indicating that acid-driven recombination of lignin fragments into condensed, insoluble structures. The post-reaction woodmeal formed dark brown, densely textured particles with high insoluble matter content, consistent with pseudo-lignin formation.46 Supporting this hypothesis, subjecting commercial cellulose to the acid-assisted RCF conditions generated acid-insoluble material (Table S6), suggesting potential contributions to the measured lignin content. Concurrent severe cellulose loss (retention plummeted to 41%) and complete xylan degradation confirm excessive acid promotes hydrolytic fragmentation of carbohydrates, likely through dehydration pathways.31 Analysis on the liquid product derived from varied acid concentrations show that dominant degraded products include xylose, glycerol, glycol and propanediol (Fig. S7).47 Interestingly, the portion of xylose accounts high as acid concentration increase above 3‰ H3PO4, which might be jointly attributed to the enhanced xylan hydrolysis and deteriorating catalyst under such high acidity.
Overall these compositional shifts reveal a critical temporal dimension to acid-assisted RCF process. While 10‰ acid initially solubilizes lignin rapidly (<0.5 h), prolonged exposure favors condensation over extraction, generating insoluble component and resulting in a declined delignification degree. Conversely, 0.3‰ acid maintains lignin in reactive soluble forms for depolymerization over extended periods. This outcome reinforces that optimal acid concentration balances extraction kinetics against recombination thermodynamics, enabling maximal monomer production from extracted lignin while preserving cellulose for downstream valorization, a dual objective essential to the economic viability of integrated biorefineries.
3.5 Solid residual conversion and catalyst recovery
The carbohydrate-rich solid residue obtained under optimized RCF condition (210 °C, 0.3‰ acid, 0.5 h) were subjected to hydrogenolysis, using the Ni/C catalyst from RCF stage that mixed with solid residue. To accelerate cellulose conversion, sulfuric acid was introduced to via one-pot tandem acid hydrolysis and catalytic hydrogenation (Fig. 5a). Control reactions without acid (0‰ H2SO4) confirmed minimal cellulose conversion (10%), yielding only 3.3% glucose and no detectable alcohol products. Introducing 0.1‰ H2SO4 causes rapid protonation of glycosidic linkages48 and liberated glucose monomers which underwent sequential hydrogenation over Ni sites, achieving 61% carbohydrate conversion. Sorbitol became a major product at such condition, implying preferential glucose hydrogenation over C–C cleavage.49 At 0.5‰ acid addition, a significant shift in product distribution, characterized by a sharp decline in sorbitol yield (to only 0.5%) and the concurrent emergence of ethylene glycol (EG) production (reaching 22.6%). Crucially, increasing the H2SO4 concentration further to 1‰ H2SO4 drove carbohydrate conversion to 86.3% and EG yield to its maximum value of 34.7%. Higher acid concentrations (2.5‰ to 10‰) marginally increased overall conversion (up to 93.7%) but failed to further elevate EG yield further, which plateaued at 32.4–34.7%. | ||
| Fig. 5 Hydrogenolysis of the solid residue from acid-assisted RCF process, impact of (a) acid ratio, (b) temperature and pressure, (c) solid loading. Reaction condition: 0.2 g residue in 10 mL pure water, 1‰ H2SO4 content, 2 MPa initial H2 pressure, 160 °C, 5 h, 700 rpm. | ||
Reaction temperature and hydrogen pressure was optimized (Fig. 5b). Elevated temperatures universally enhanced overall carbohydrate conversion, rising from 66.4% at 180 °C to 91.4% at 220 °C. Crucially, EG yield maximized at 34.7% at 200 °C. While conversion further increased at 220 °C, EG yield slightly declined to 30.65%. This pattern signifies intensified competitive reactions, likely retro-aldol condensation fragmentation leading to C1–C2 products and/or dehydration/hydrogenation cascades forming heavier byproducts at higher severity,50 reducing EG selectivity despite enhanced depolymerization. Hydrogen pressure significantly modulated reaction pathways. Low pressure (1 MPa) severely suppressed both overall conversion (78.6%) and critically, EG yield (15.5%), indicating insufficient hydrogen availability impedes key hydrogenation steps required for EG formation from glucose retro-aldol fragments (like glycolaldehyde).51 Elevating H2 pressure to 2 MPa dramatically boosted conversion to 86.3% and maximized EG yield, while higher pressure (4 MPa) plateaued, suggesting saturation of beneficial catalytic hydrogenation effects.
Notably, the solid loading investigation demonstrated that this one-pot conversion system can maintain stable performance at high solid/liquid ratios (Fig. 5c). While conversion and product yields showed only minor reductions below 60 g L−1 (e.g., conversion 80.1% and EG yield 31.8% at 60 g L−1), a pronounced downturn occurred above this threshold, with the sharpest decreases correlating with solid loadings of 80 g L−1 and higher, due to transfer limitation. Such findings highlight a critical balance for scale-up processing, where high solid loadings reduce reactor volumes and operational costs by minimizing solvent use, yet require enhanced mixing to overcome mass-transfer constraints and preserve conversion economics in industrially relevant contexts.
The Ni/C catalyst recovered from solid residue hydrogenolysis was directly used in the RCF process. Morphological inspect of recovered catalyst showed no obvious change compared with the original catalyst (Fig. S8), and analysis on the recovered catalysts through ICP-OES showed that only marginal (10.07 wt% vs. 10.86 wt%) leaching was observed for recovered catalyst. As shown in Fig. 6, the recovered catalyst showed good stability in the first reuse cycle, retaining approximately 90% of the initial monomer yield during the second run. However, a significant increase in the proportion of propenyl monomers was observed, potentially attributable to the loss of reduction-active Ni sites caused by acid exposure during RCF and hydrogenolysis processing.39 A further yield decrease occurred in the third cycle. Nevertheless, the catalytic performance could be fully regained following catalyst regeneration, with monomer composition similar to those from original catalyst.
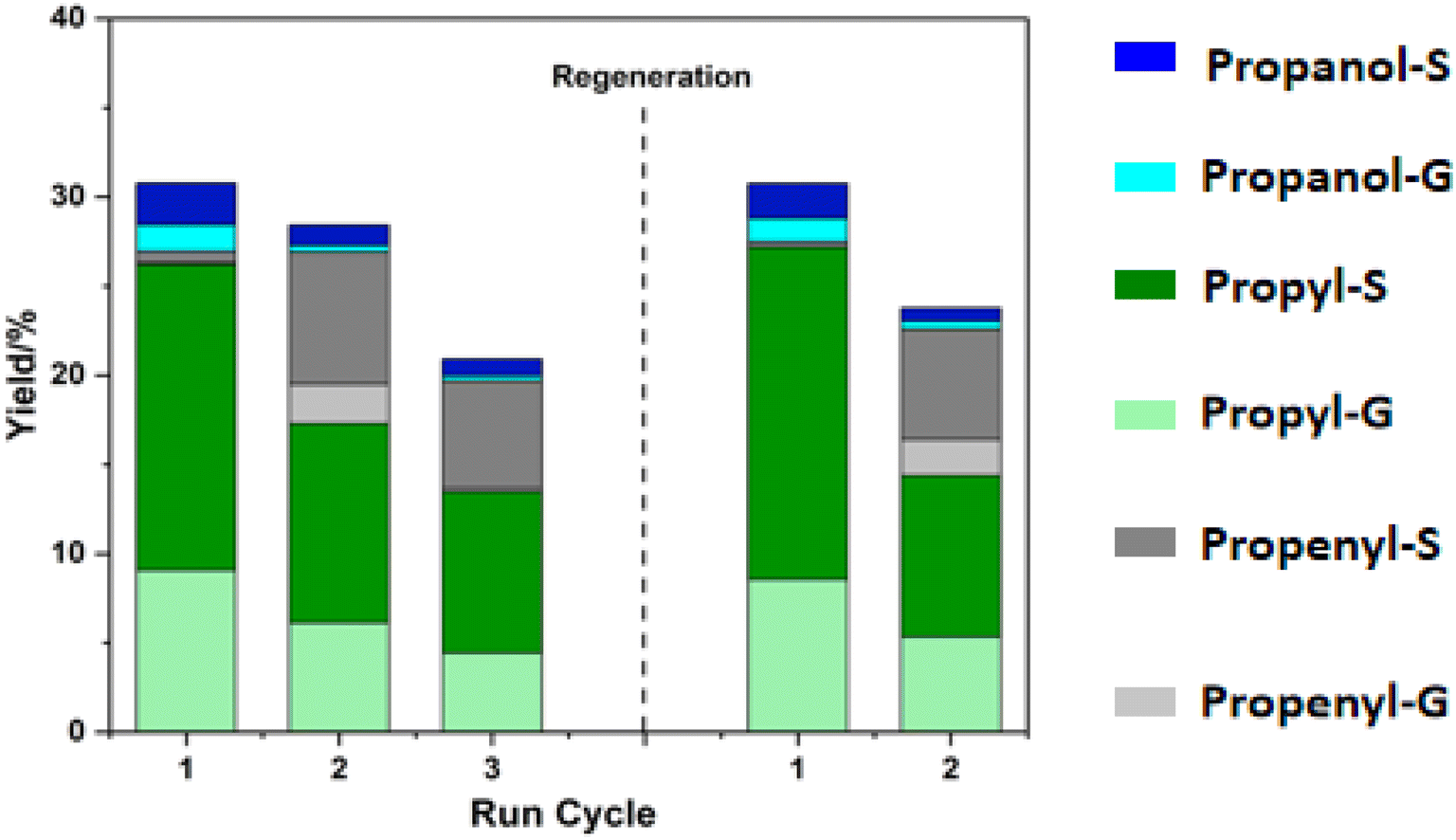 | ||
| Fig. 6 Catalyst recycle test after recovering from solid residue hydrogenolysis. Reaction conditions: 10 mL ethanol:H2O = 1:1, 0.4 g of poplar wood 0.08 g of recycled Ni/C, and 2 MPa H2, 0 h. | ||
4 Conclusion
This work establishes an efficient approach for valorizing lignin and cellulose component in biomass through integrated RCF fractionation and upgrading. The synergy of ethanol–water solvent and acid addition considerably accelerated lignin oil extraction and aromatic monomer formation. Crucially, 31% monomer yield was obtained at 0 h under optimized low phosphoric acid loading (0.3‰ H3PO4, 210 °C), achieving a production efficiency of 3.44 mg monomers per g lignin per min. In contrast, high acid concentration stimulated both propenyl monomers and phenol generation via dehydration and dealkylation pathways. Time course study demonstrated that precision acid concentration tuning and temporal operation window is critical, with 0.3‰ phosphoric acid addition unlocks dramatically accelerated monomer generation (43% yield) within 30 min, while preserving cellulose integrity (92.5% retention, 81.6% purity). The resulting cellulose-rich residue serves as optimal feedstock for EG synthesis via tandem hydrolysis and hydrogenolysis, where a specific sulfuric acid concentration threshold (1‰) is essential to steer retro-aldol fragmentation and maximize EG yield. The collected catalyst maintained good stability and can be effectively regenerated for recycle use. Overall, this cascaded, acid-modulated RCF biorefinery approach establishes an efficient and scalable paradigm for biomass fractionation and upgrading.Author contributions
Peidong Li: conceptualization, writing – original draft, visualization, data curation; Wangyue Xie: edit & review, data curation, formal analysis; Muhammad Fayyaz: validation, investigation, data curation; Tianyu Ren: software, formal analysis; Cheng Tung Chong: formal analysis, review and editing; Zhuo He: validation, visualization; Yuhe Liao: resources; resources; Yanbin Cui: writing – review & editing, supervision, funding acquisition. Chenguang Wang: review, resources, supervision, funding acquisition.Conflicts of interest
The authors declare that they have no competing interests.Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request. Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5ra06710c.Acknowledgements
This work was financially supported by Guangdong Basic and Applied Basic Research Foundation of China (2023A1515110745), and National natural Science Foundation of China (52276220, 22408365).References
- J. Wang, J. Xi and Y. Wang, Recent Advances in the Catalytic Production of Glucose from Lignocellulosic Biomass, Green Chem., 2015, 17(2), 737–751, 10.1039/C4GC02034K.
- L. Hu, L. Lin, Z. Wu, S. Zhou and S. Liu, Chemocatalytic Hydrolysis of Cellulose into Glucose over Solid Acid Catalysts, Appl. Catal. B Environ. Energy, 2015, 174–175, 225–243, DOI:10.1016/j.apcatb.2015.03.003.
- J. S. Luterbacher, J. M. Rand, D. M. Alonso, J. Han, J. T. Youngquist, C. T. Maravelias, B. F. Pfleger and J. A. Dumesic, Nonenzymatic Sugar Production from Biomass Using Biomass-Derived γ-Valerolactone, Science, 2014, 343(6168), 277–280, DOI:10.1126/science.1246748.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Metal Chlorides in Ionic Liquid Solvents Convert Sugars to 5-Hydroxymethylfurfural, Science, 2007, 316(5831), 1597–1600, DOI:10.1126/science.1141199.
- C. S. Lancefield, I. Panovic, P. J. Deuss, K. Barta and N. J. Westwood, Pre-Treatment of Lignocellulosic Feedstocks Using Biorenewable Alcohols: Towards Complete Biomass Valorisation, Green Chem., 2017, 19(1), 202–214, 10.1039/C6GC02739C.
- S. Van den Bosch, S.-F. Koelewijn, T. Renders, G. Van den Bossche, T. Vangeel, W. Schutyser and B. F. Sels, Catalytic Strategies Towards Lignin-Derived Chemicals, Top. Curr. Chem., 2018, 376(5), 36, DOI:10.1007/s41061-018-0214-3.
- P. J. Deuss, C. S. Lancefield, A. Narani, J. G. de Vries, N. J. Westwood and K. Barta, Phenolic Acetals from Lignins of Varying Compositions via Iron(III) Triflate Catalysed Depolymerisation, Green Chem., 2017, 19(12), 2774–2782, 10.1039/C7GC00195A.
- S. V. den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. D. Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Reductive Lignocellulose Fractionation into Soluble Lignin-Derived Phenolic Monomers and Dimers and Processable Carbohydrate Pulps, Energy Environ. Sci., 2015, 8(6), 1748–1763, 10.1039/C5EE00204D.
- S. V. den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. V. den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. M. Courtin, J. M. Thevelein, W. Schutyser and B. F. Sels, Integrating Lignin Valorization and Bio-Ethanol Production: On the Role of Ni-Al2O3 Catalyst Pellets during Lignin-First Fractionation, Green Chem., 2017, 19(14), 3313–3326, 10.1039/C7GC01324H.
- S. Qiu, X. Guo, Y. Huang, Y. Fang and T. Tan, Task-Specific Catalyst Development for Lignin-First Biorefinery toward Hemicellulose Retention or Feedstock Extension, ChemSusChem, 2019, 12(4), 944–954, DOI:10.1002/cssc.201802130.
- J. Sun, H. Li, L.-P. Xiao, X. Guo, Y. Fang, R.-C. Sun and G. Song, Fragmentation of Woody Lignocellulose into Primary Monolignols and Their Derivatives, ACS Sustainable Chem. Eng., 2019, 7(5), 4666–4674, DOI:10.1021/acssuschemeng.8b04032.
- J. H. Jang, A. R. C. Morais, M. Browning, D. G. Brandner, J. K. Kenny, L. M. Stanley, R. M. Happs, A. S. Kovvali, J. I. Cutler, Y. Román-Leshkov, J. R. Bielenberg and G. T. Beckham, Feedstock-Agnostic Reductive Catalytic Fractionation in Alcohol and Alcohol–Water Mixtures, Green Chem., 2023, 25(9), 3660–3670, 10.1039/D2GC04464A.
- X. Liu, H. Li, L.-P. Xiao, R.-C. Sun and G. Song, Chemodivergent Hydrogenolysis of Eucalyptus Lignin with Ni@ZIF-8 Catalyst, Green Chem., 2019, 21(6), 1498–1504, 10.1039/C8GC03511C.
- Y. Cheng, Y. Qu, S. Yang, K. Zhuang and J. Wang, Staged Biorefinery of Moso Bamboo by Integrating Polysaccharide Hydrolysis and Lignin Reductive Catalytic Fractionation (RCF) for the Sequential Production of Sugars and Aromatics, Ind. Crops Prod., 2021, 164, 113358, DOI:10.1016/j.indcrop.2021.113358.
- Y.-L. Loow, T. Y. Wu, J. Md. Jahim, A. W. Mohammad and W. H. Teoh, Typical Conversion of Lignocellulosic Biomass into Reducing Sugars Using Dilute Acid Hydrolysis and Alkaline Pretreatment, Cellulose, 2016, 23(3), 1491–1520, DOI:10.1007/s10570-016-0936-8.
- D. Hebbale and T. V. Ramachandra, Optimal Sugar Release from Macroalgal Feedstock with Dilute Acid Pretreatment and Enzymatic Hydrolysis, Biomass Convers. Biorefin., 2023, 13(9), 8287–8300, DOI:10.1007/s13399-021-01845-8.
- D. M. Miles-Barrett, A. R. Neal, C. Hand, J. R. D. Montgomery, I. Panovic, O. S. Ojo, C. S. Lancefield, D. B. Cordes, A. M. Z. Slawin, T. Lebl and N. J. Westwood, The Synthesis and Analysis of Lignin-Bound Hibbert Ketone Structures in Technical Lignins, Org. Biomol. Chem., 2016, 14(42), 10023–10030, 10.1039/C6OB01915C.
- L. Shuai, M. T. Amiri, Y. M. Questell-Santiago, F. Héroguel, Y. Li, H. Kim, R. Meilan, C. Chapple, J. Ralph and J. S. Luterbacher, Formaldehyde Stabilization Facilitates Lignin Monomer Production during Biomass Depolymerization, Science, 2016, 354(6310), 329–333, DOI:10.1126/science.aaf7810.
- P. J. Deuss, M. Scott, F. Tran, N. J. Westwood, J. G. de Vries and K. Barta, Aromatic Monomers by in Situ Conversion of Reactive Intermediates in the Acid-Catalyzed Depolymerization of Lignin, J. Am. Chem. Soc., 2015, 137(23), 7456–7467, DOI:10.1021/jacs.5b03693.
- P. J. Deuss, C. W. Lahive, C. S. Lancefield, N. J. Westwood, P. C. J. Kamer, K. Barta and J. G. de Vries, Metal Triflates for the Production of Aromatics from Lignin, ChemSusChem, 2016, 9(20), 2974–2981, DOI:10.1002/cssc.201600831.
- X. Sun, J. Ni, Y. Lou, P. Zhao, Y. Yu, Y. Li, Q. Tang, H. Yu and Y. Liu, Solvent-Triggered Directional Lignin Valorization towards Monomeric Acetals or Lignin Polyols, Green Chem., 2024, 26(7), 4151–4160, 10.1039/D3GC05133A.
- X. Zeng, Y. Xu, Q. Dai, J. Li, Q. Lin, J. Ye, C. Liu and W. Lan, Low-Temperature Degradation of Lignin in Aprotic Solvent System for Preparation of Monophenolic Platform Chemicals, Chem. Eng. J., 2023, 476, 146466, DOI:10.1016/j.cej.2023.146466.
- T. Renders, E. Cooreman, S. V. den Bosch, W. Schutyser, S.-F. Koelewijn, T. Vangeel, A. Deneyer, G. V. den Bossche, C. M. Courtin and B. F. Sels, Catalytic Lignocellulose Biorefining in N-Butanol/Water: A One-Pot Approach toward Phenolics, Polyols, and Cellulose, Green Chem., 2018, 20(20), 4607–4619, 10.1039/C8GC01031E.
- F. Brienza, K. Van Aelst, F. Devred, D. Magnin, B. F. Sels, P. Gerin, I. Cybulska and D. P. Debecker, Toward a Hydrogen-Free Reductive Catalytic Fractionation of Wheat Straw Biomass, ChemSusChem, 2023, 16(13), e202300103, DOI:10.1002/cssc.202300103.
- T. Renders, W. Schutyser, S. Van den Bosch, S.-F. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, Influence of Acidic (H3PO4) and Alkaline (NaOH) Additives on the Catalytic Reductive Fractionation of Lignocellulose, ACS Catal., 2016, 6(3), 2055–2066, DOI:10.1021/acscatal.5b02906.
- A. D. Sluiter, B. Hames, R. Ruiz, C. J. Scarlata, J. Sluiter, D. Templeton and D. P. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass, Technical Report NREL/TP-510−42618, National Renewable Energy Laboratory, Golden, CO, 2010 Search PubMed.
- J. H. Jang, D. G. Brandner, R. J. Dreiling, A. J. Ringsby, J. R. Bussard, L. M. Stanley, R. M. Happs, A. S. Kovvali, J. I. Cutler, T. Renders, J. R. Bielenberg, Y. Román-Leshkov and G. T. Beckham, Multi-Pass Flow-through Reductive Catalytic Fractionation, Joule, 2022, 6(8), 1859–1875, DOI:10.1016/j.joule.2022.06.016.
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S.-F. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, Synergetic Effects of Alcohol/Water Mixing on the Catalytic Reductive Fractionation of Poplar Wood, ACS Sustainable Chem. Eng., 2016, 4(12), 6894–6904, DOI:10.1021/acssuschemeng.6b01844.
- G. Li, B. Wang and D. E. Resasco, Solvent Effects on Catalytic Reactions and Related Phenomena at Liquid-Solid Interfaces, Surf. Sci. Rep., 2021, 76(4), 100541, DOI:10.1016/j.surfrep.2021.100541.
- H. Huang, X. Zhang, L. Ma and Y. Liao, Reductive Catalytic Fractionation of Lignocellulose Toward Propyl- or Propenyl-Substituted Monomers and Mechanistic Understanding, Angew. Chem., Int. Ed., 2025, 64(24), e202502545, DOI:10.1002/anie.202502545.
- A. Corti, E. Torrens and D. Montané, Acid-Catalyzed Fractionation of Almond Shells in γ-Valerolactone/Water, Biomass Convers. Biorefin., 2023, 13(4), 2729–2743, DOI:10.1007/s13399-020-01261-4.
- Y. Liao, S.-F. Koelewijn, G. Van den Bossche, J. Van Aelst, S. Van den Bosch, T. Renders, K. Navare, T. Nicolaï, K. Van Aelst, M. Maesen, H. Matsushima, J. M. Thevelein, K. Van Acker, B. Lagrain, D. Verboekend and B. F. Sels, A Sustainable Wood Biorefinery for Low–Carbon Footprint Chemicals Production, Science, 2020, 367(6484), 1385–1390, DOI:10.1126/science.aau1567.
- J.-P. Lange, Renewable Feedstocks: The Problem of Catalyst Deactivation and Its Mitigation, Angew. Chem., Int. Ed., 2015, 54(45), 13186–13197, DOI:10.1002/anie.201503595.
- X. Yu, Z. Wei, Z. Lu, H. Pei and H. Wang, Activation of Lignin by Selective Oxidation: An Emerging Strategy for Boosting Lignin Depolymerization to Aromatics, Bioresour. Technol., 2019, 291, 121885, DOI:10.1016/j.biortech.2019.121885.
- A. W. Bartling, M. L. Stone, R. J. Hanes, A. Bhatt, Y. Zhang, M. J. Biddy, R. Davis, J. S. Kruger, N. E. Thornburg, J. S. Luterbacher, R. Rinaldi, J. S. M. Samec, B. F. Sels, Y. Román-Leshkov and G. T. Beckham, Techno-Economic Analysis and Life Cycle Assessment of a Biorefinery Utilizing Reductive Catalytic Fractionation, Energy Environ. Sci., 2021, 14(8), 4147–4168, 10.1039/D1EE01642C.
- E. M. Anderson, R. Katahira, M. Reed, M. G. Resch, E. M. Karp, G. T. Beckham and Y. Román-Leshkov, Reductive Catalytic Fractionation of Corn Stover Lignin, ACS Sustainable Chem. Eng., 2016, 4(12), 6940–6950, DOI:10.1021/acssuschemeng.6b01858.
- I. Klein, B. Saha and M. M. Abu-Omar, Lignin Depolymerization over Ni/C Catalyst in Methanol, a Continuation: Effect of Substrate and Catalyst Loading, Catal. Sci. Technol., 2015, 5(6), 3242–3245, 10.1039/C5CY00490J.
- H. Luo and M. M. Abu-Omar, Lignin Extraction and Catalytic Upgrading from Genetically Modified Poplar, Green Chem., 2018, 20(3), 745–753, 10.1039/C7GC03417B.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Liu, H. I. Kenttämaa and M. M. Abu-Omar, Total Utilization of Miscanthus Biomass, Lignin and Carbohydrates, Using Earth Abundant Nickel Catalyst, ACS Sustainable Chem. Eng., 2016, 4(4), 2316–2322, DOI:10.1021/acssuschemeng.5b01776.
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Lignin Depolymerization (LDP) in Alcohol over Nickel-Based Catalysts via a Fragmentation–Hydrogenolysis Process, Energy Environ. Sci., 2013, 6(3), 994–1007, 10.1039/C2EE23741E.
- H.-M. Wang, C.-Y. Ma, H.-Y. Li, T.-Y. Chen, J.-L. Wen, X.-F. Cao, X.-L. Wang, T.-Q. Yuan and R.-C. Sun, Structural Variations of Lignin Macromolecules from Early Growth Stages of Poplar Cell Walls, ACS Sustainable Chem. Eng., 2020, 8(4), 1813–1822, DOI:10.1021/acssuschemeng.9b05845.
- F. Unda, L. de Vries, S. D. Karlen, J. Rainbow, C. Zhang, L. E. Bartley, H. Kim, J. Ralph and S. D. Mansfield, Enhancing Monolignol Ferulate Conjugate Levels in Poplar Lignin via OsFMT1, Biotechnol. Biofuels Bioprod., 2024, 17(1), 97, DOI:10.1186/s13068-024-02544-y.
- E. Jasiukaitytė-Grojzdek, M. Huš, M. Grilc and B. Likozar, Acid-Catalyzed α-O-4 Aryl-Ether Cleavage Mechanisms in (Aqueous) γ-Valerolactone: Catalytic Depolymerization Reactions of Lignin Model Compound During Organosolv Pretreatment, ACS Sustainable Chem. Eng., 2020, 8(47), 17475–17486, DOI:10.1021/acssuschemeng.0c06099.
- S. N. Obame, I. Ziegler-Devin, R. Safou-Tchima and N. Brosse, Homolytic and Heterolytic Cleavage of β-Ether Linkages in Hardwood Lignin by Steam Explosion, J. Agric. Food Chem., 2019, 67(21), 5989–5996, DOI:10.1021/acs.jafc.9b01744.
- J. Kim, C. Kim, C. Jeong, S. Won, S.-G. Kim, H. Lim, S. Kim and H. W. Kwak, Integrated Process for Lignin Depolymerization and Nanoparticle Production Using Deep Eutectic Solvent, Sci. Rep., 2025, 15(1), 11770, DOI:10.1038/s41598-025-96237-7.
- F. Hu, S. Jung and A. Ragauskas, Pseudo-Lignin Formation and Its Impact on Enzymatic Hydrolysis, Bioresour. Technol., 2012, 117, 7–12, DOI:10.1016/j.biortech.2012.04.037.
- L. Alves de Souza, P. Magalhães de Souza, G. Tozzi Wurzler, V. Teixeira da Silva, D. A. Azevedo, R. Wojcieszak and F. Bellot Noronha, Reductive Catalytic Fractionation of Lignocellulosic Biomass: Unveiling of the Reaction Mechanism, ACS Sustainable Chem. Eng., 2023, 11(1), 67–77, DOI:10.1021/acssuschemeng.2c03788.
- H. Zhao, Y. Jia, X. Liang, J. Hao, G. Xu, B. Chen, C. He, Y. Jiao and C. Chang, Theoretical and Experimental Study of 5-Ethoxymethylfurfural and Ethyl Levulinate Production from Cellulose, Chem. Eng. J., 2024, 480, 148093, DOI:10.1016/j.cej.2023.148093.
- C. Liu, C. Zhang, K. Liu, Y. Wang, G. Fan, S. Sun, J. Xu, Y. Zhu and Y. Li, Aqueous-Phase Hydrogenolysis of Glucose to Value-Added Chemicals and Biofuels: A Comparative Study of Active Metals, Biomass Bioenergy, 2015, 72, 189–199, DOI:10.1016/j.biombioe.2014.11.005.
- M. Zheng, J. Pang, R. Sun, A. Wang and T. Zhang, Selectivity Control for Cellulose to Diols: Dancing on Eggs, ACS Catal., 2017, 7(3), 1939–1954, DOI:10.1021/acscatal.6b03469.
- H. Wang, X. Hu, S. Liu, C. Cai, C. Zhu, H. Xin, Z. Xiu, C. Wang, Q. Liu, Q. Zhang, X. Zhang and L. Ma, Selective (Ligno) Cellulose Hydrogenolysis to Ethylene Glycol and Propyl Monophenolics over Ni–W@C Catalysts, Cellulose, 2020, 27(13), 7591–7605, DOI:10.1007/s10570-020-03340-1.
| This journal is © The Royal Society of Chemistry 2025 |