
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA plug and play approach to structural variations of bio-inspired anticancer lipidic phenyl dialkynylcarbinols
Margaux Bossuata,
Nadège Preuilhb,
Patrick Seigneura,
Isabelle Fabing a,
Christian Pradelc,
Antonio Peixotob,
Valérie Maravald,
Vania Bernardes-Génissond,
Stéphanie Ballereaua,
Sébastien Britton*b and
Yves Génisson*a
a,
Christian Pradelc,
Antonio Peixotob,
Valérie Maravald,
Vania Bernardes-Génissond,
Stéphanie Ballereaua,
Sébastien Britton*b and
Yves Génisson*a
aToulouse University, Laboratoire de Synthèse et de Physico-Chimie de Molécules d’Intérêt Biologie (SPCMIB), CNRS (UMR 5068), Toulouse, France. E-mail: yves.genisson@utoulouse.fr
bInstitut de Pharmacologie et de Biologie Structurale (IPBS), Université de Toulouse, CNRS, Toulouse, France. E-mail: sebastien.britton@ipbs.fr
cUniversité de Toulouse, CNRS, LHFA – UMR 5069, 118 Route de Narbonne, 31062, France
dToulouse University, Laboratoire de Chimie de Coordination du CNRS (UPR 8241), 205 Route de Narbonne, 31077, Toulouse, Cedex 4, France
First published on 14th October 2025
Abstract
Phenyl dialkynylcarbinols (PACs) are analogues of natural acetylenic lipids acting on human cells as cytotoxic prodrugs enantiospecifically bioactivated by HSD17B11. Here, we report a highly convergent and modular synthetic strategy to accelerate the exploration of their anticancer structure–activity relationships. Late-stage PAC assembly was achieved by Pd/Cu-catalysed coupling of three chiral alkynylcarbinol warheads, racemic or enantioenriched, with various functionalised lipidic aryl iodides. The added value of this methodology to directly access enantioenriched PAC analogues from an enzymatically resolved alkynylcarbinol precursor was also demonstrated. A total of 22 new compounds were prepared, including two butadiynylcarbinol congeners, with IC50 values as low as 0.13 μM in HCT116 cancer cells. Enantiomeric comparisons confirmed strong eudismic ratios in this series. Genetic inactivation of HSD17B11 in U2OS cells demonstrated its key role in the cytotoxicity of most compounds, while 1,2,3-triazolyl allenyl alkynylcarbinols emerged as particularly promising, combining a novel chemotype, a potent activity, and an alternative mechanism of action.
Introduction
Acetylenic lipids represent a fascinating family of natural products1 found both in marine2,3 and terrestrial organisms.4–6 Due to the combination of their unique structure and potent bioactivity, these so-called polyyne natural products have been the subject of numerous structural,7–9 synthetic10,11 and biological reports.12–14 Extensive mechanism of action-driven and structure–activity relationship (SAR)-oriented chemical and biological studies remain however scarce in this series. We focused our attention on a sub-class of anticancer acetylenic lipids designated as lipidic alkynylcarbinols. Encompassing several emblematic representatives such as petrocortyne,3,7 duryne,3,7 lembehyne2,3 and falcarindiol,6,9,13 these natural compounds all share the presence of one or several chiral alkynylcarbinol pharmacophoric units along their lipidic backbone (Fig. 1). | ||
| Fig. 1 Representative examples of natural lipids embedding alkynylcarbinol moieties. | ||
An in-depth SAR exploration initiated by our groups,15,16 originally inspired by the natural product (S,E)-eicos-4-en-3-ol (Fig. 2), was ultimately enlightened by the discovery of the cellular mechanism of action accounting for the cytotoxicity of representative synthetic analogues.17 The pro-apoptotic behaviour of these prodrugs results from the enantiospecific oxidation of the pharmacophoric dialkynylcarbinol (DAC) unit into a highly protein-reactive dialkynylketone warhead. The lipidic portion of the molecule is considered to contribute both to the substrate recognition by Short-chain Dehydrogenase/Reductase (SDR) bioactivating enzymes and to the cellular toxicity of the resulting protein lipoxidation process. On these bases, we recently designed a new generation of lipidic phenyl dialkynylcarbinol (PAC) anticancer prodrugs (Fig. 2).18 These compounds were conceived by conjugating the key DAC moiety to an (hetero)aromatic ring, such as a phenyl group, bearing the lipophilic appendage. Not only this modification led to a gain in stability and selectivity towards the main bioactivation enzyme, as compared to the earlier generation of synthetic DAC compounds, but it also afforded new opportunities for geometric and electronic structural optimization.18
 | ||
| Fig. 2 Molecular evolution of an archetypical natural acetylenic lipid towards the last generation of synthetic phenyl dialkynylcarbinol (PAC) analogues. | ||
A first set of 25 PAC derivatives was reported.18 The SAR data allowed identification of the prototypical molecule (S)-1 embedding a benzene ring para-substituted with an octyl chain. This compound exhibits a more than 100-fold increase in cytotoxicity against HCT116 colon cancer cells as compared to the natural product of reference (Fig. 2). A mechanistic study was carried out, using notably an enantioenriched ω-alkyne-tagged clickable probe. It confirmed the capacity of these new prodrugs, once bioactivated, to modify cellular proteins and to induce endoplasmic reticulum stress, ubiquitin-proteasome system inhibition and apoptosis, as previously reported for DACs.17 Lipidic backbone functionalisation led to the gem-difluorinated analogue (S)-2, which displays an IC50 down to 0.042 μM. Additional structural diversification in racemic series showed that decoration of the benzene ring, or its replacement by a heteroaromatic nucleus, allowed increasing the levels of cytotoxicity and/or selectivity towards the main bioactivation enzyme HSD17B11 vs. its paralogue HSD17B13. Prompted by these promising results, we pursued the development of a versatile synthetic approach to further explore the molecular diversity in the PAC series. Our first results along this line are reported herein.
One of the assets of the PAC series lies in its synthetic accessibility, which arises from the smooth reactivity of terminal alkynes under transition metal-catalysis. Their retrosynthetic analysis thus privileged disconnection of the key Csp–Csp2 bond between the alkyne moiety and the aromatic ring. Following this logic, our groups previously implemented two synthetic sequences relying on a late-stage formation of the dialkynylcarbinol unit.18 The present work focuses on the retrosynthesis depicted in Scheme 1 as an alternative pathway offering increased convergence and modularity.
 | ||
| Scheme 1 The retrosynthetic analysis at the basis of the plug and play approach. | ||
Thanks to the robust Sonogashira reaction, this so-called plug and play approach was expected to warrant direct access to a collection of analogues through the cross-coupling of a set of halogenated (hetero)aromatic intermediates with different alkynylcarbinol precursors. Previous structural exploration indicated that replacing the typical dialkynylcarbinol pharmacophoric unit for an alkenyl-19 or allenyl-alkynylcarbinol20 moiety offers opportunities for the identification of new preferential bioactivation enzymes, while preserving significant levels of cytotoxicity.17
A similar trend was also observed in the PAC series, as shown by the relative activity of racemic 6, 7 and 8 (Fig. 3) (see SI). We thus considered the three corresponding alkynylcarbinol precursors 3, 4 and 5 as relevant building blocks for the PAC series diversification (Scheme 1). In addition, not only the early-stage assembly of the chiral alkynylcarbinol precursors 3–5 was planned to facilitate modulation of the lipophilic core, but their use in enantioenriched form could also afford direct access to the targeted PAC compounds in non-racemic series.
 | ||
| Fig. 3 Series of PAC analogues carrying three different alkynylcarbinol warheads. | ||
The alkenyl alkynylcarbinol precursor 3 has been used, under racemic or enantioenriched form, in several syntheses of falcarindiol,21–23 panaxytriol24,25 and other related polyacetylenic natural products.26–28 The reactivity of the terminal alkyne moiety under smooth Cadiot–Chodkiewicz reaction conditions is indeed well-suited for the elaboration of the characteristic 1,3-diyne unit. In contrast, the C-silylated dialkynylcarbinol precursor 4 has only been scarcely used for the synthesis of acetylenic lipids. Elegant pioneering studies have taken advantage of the resolved enantiomers of the TBS-protected form of compound 4 for the concise preparation of the natural (S)-eicos-(4E)-en-1-yn-3-ol29 and the synthetic (15E)-isomer of (3R,28R)-duryne.30 Our groups applied the Crabbé-Ma asymmetric allenylation reaction to access stereoisomerically enriched samples of lipidic allenyl alkynylcarbinols from both enantiomers of the TIPS-protected precursor 4.6 Finally, the preparation and lipase-mediated kinetic resolution of a TMS derivative of the allenyl alkynylcarbinol precursor 5 have been described, but no further use of this compound has been reported.31
Results and discussion
Preparation of the alkynylcarbinol precursors
The alkenyl alkynylcarbinol precursor 3 was prepared from acrolein and ethynyl magnesium bromide as previously described (see SI).32 The dialkynylcarbinol precursor 4 was obtained by addition of ethynyl magnesium bromide onto TIPS-protected propynal resulting from the formylation of TIPS-acetylene with DMF (see SI).33 Finally, the allenyl alkynylcarbinol precursor 5 was synthesized from 4 thanks to the Crabbé-Ma procedure34 with paraformaldehyde followed by desilylation with TBAF (see SI).Preparation of racemic samples
|
|||
|---|---|---|---|
| Alkyne | Iodide | Product | Yield |
| 3 | PhI |  |
52% |
| 4 | PhI |  |
45% |
| 5 | PhI |  |
39% |
| 4 | 12 |  |
36% |
| R2 = R3 = H | |||
| X = CH2 | |||
| R4 = n-heptyl | |||
| 4 | 13 |  |
36% |
| R2 = R3 = H | |||
| X = CF2 | |||
| R4 = n-heptyl | |||
| 3 | 14 |  |
50% |
| R2 = 3-F, R3 = H | |||
| X = O | |||
| R4 = n-heptyl | |||
| 5 | 15 |  |
67% |
| R2 = 3-F, R3 = H | |||
| X = O | |||
| R4 = n-butyl | |||
| 3 | 16 |  |
46% |
| R2 = 3-F, R3 = 5-F | |||
| X = O | |||
| R4 = n-heptyl | |||
| 3 | 16 |  |
73% |
| R2 = 3-F, R3 = 5-F | |||
| X = O | |||
| R4 = n-heptyl | |||
| 3 | 17 |  |
40% |
| R2 = 3-F, R3 = H | |||
| X = O | |||
| R4 = geranyl | |||
| 3 | 17 |  |
58% |
| R2 = 3-F, R3 = H | |||
| X = O | |||
| R4 = geranyl | |||

|
|||
|---|---|---|---|
| Alkyne | Bromide | Product | Yield |
| 3 |  |
 |
58% |
| 5 |  |
 |
57% |

At this stage, the Cadiot–Chodkiewicz reaction with a trialkylsilyl-protected acetylene was considered (Table 3). The plan was to install an alkylated heterocyclic nucleus next to the alkynylcarbinol unit by means of a click-type 1,3-dipolar cycloaddition. Several examples of CuAAC reactions on the terminal position of the polyyne appendage of complex molecules being recently reported,41–43 we investigated this versatile transformation. The three different alkynylcarbinol precursors 3–5 were alternatively tested. Due to the expected volatility and sensitivity of the intermediate diynes, the three consecutive steps, i.e., the Cadiot–Chodkiewicz coupling, the C-desilylation and the CuAAC reaction, were carried out without isolation of the intermediates to ensure optimal global yields. Attempts to run the first two steps of this sequence using the dialkynylcarbinol precursor 4 invariably gave rise to decomposition of the starting material, potentially because of the instability of the butadiynyl alkynylcarbinol intermediate. In contrast, the approach proved viable with the precursors 3 and 5 (Table 3).
|
|||
|---|---|---|---|
| Alkyne | Azide | Product | Yield |
| 3 |  |
 |
34% |
| 3 |  |
 |
21% |
| 3 |  |
 |
2% |
| 5 |  |
 |
50% |
| 5 |  |
 |
34% |
| 5 |  |
 |
4% |

A set of three representative aliphatic, either linear or branched, and aromatic azide reagents was selected. The transformation proved efficient with 1-azidononane, giving overall yields of 34–50% over 3 steps. The bulkier 5-azidononane led to a less productive transformation while the aromatic reagent 1-azido-4-butylbenzene gave only traces of expected cycloadduct, possibly due to its reduced stability associated to the longer needed reaction time. Overall, allenyl alkynylcarbinol precursor 5 gave better yields than its vinyl alkynylcarbinol counterpart 3.
Encouraged by these first results, we envisioned to generalize this approach to isoxazole PAC analogues obtained by 1,3-dipolar cycloaddition with a nitrile oxide reagent. Base-promoted in situ generation of the latter from the chlorooxime precursor 35 derived from decanal was selected as an appropriate option to explore the feasibility of this pathway (Scheme 2).
 | ||
| Scheme 2 Attempted preparation of isoxazole-containing PAC analogues via Cadiot–Chodkiewicz coupling reaction-1,3-dipolar cycloaddition sequence. | ||
Several standard conditions were attempted with the more accessible alkynylcarbinol precursor 3. Neither the use of Et3N in CH2Cl2,44 nor NaHCO3 in AcOEt,45,46 nor CuSO4 and NaAsc in the presence of KHCO3 in t-BuOH/THF47,48 gave the expected cycloadduct 36, despite the complete consumption of the starting alkynylcarbinol 3. Since model studies with the chlorooxime 35 and phenyl acetylene had shown that some of these procedures gave fair yields in the corresponding isoxazole (see SI), this failure may be explained by the sensitivity of the dipolarophile intermediate 34 combined to the reduced reactivity of the long chain nitrile oxide. The retrosynthetic approach presented in Scheme 1 was thus considered.
|
|||
|---|---|---|---|
| Alkyne | Iodide | Product | Yield |
| 3 |  |
 |
38% |
| 4 |  |
 |
30% |

|
|||
|---|---|---|---|
| Alkyne | Iodide | Product | Yield |
| 3 |  |
 |
39% |
| 4 |  |
 |
52% |
| 5 |  |
 |
20% |

In order to diversify the panel of analogues, the approach was challenged to generate new furan derivatives. The high level of cytotoxicity previously demonstrated for this heterocyclic series, potentially resulting from the favourable −I/+M effect of the conjugated oxygen atom, suggested an appropriate tuning of their reactivity towards the SDR-catalysed dehydrogenation.4 The allenyl alkynylcarbinol derivative 40 was thus obtained in 18% yield through the coupling of precursor 5 with 2-hexyl-4-iodofuran (39), prepared following the methodology reported by Gevorgyan51 (see SI) (Scheme 3).
 | ||
| Scheme 3 Sonogashira coupling reaction to prepare a furan-containing PAC analogue. | ||
With the aim of fully exploring the influence of nitrogenated heterocyclic nucleus, pyrazoles were selected as readily accessible analogues (Table 5). The intermediates 41 and 42 were prepared by simple N-alkylation of commercially available 4-iodo-1H-pyrazole (see SI).52 Coupling reactions with alkynylcarbinol precursors 3–5 proceeded smoothly to deliver the targeted pyrazole-containing PAC analogues 43–45 in yields ranging from 20% to 52% (Table 5).
|
|||
|---|---|---|---|
| Substrate | Conditions | Product | Yield |
| 23 | Dry THF + 3% H2O, 1.5 eq. TBAF, 0 °C to RT, 3 h |  |
60% |
| 25 | Dry THF, 1.3 eq. TBAF, 0 °C to RT, 0.5 h |  |
40% |
| 38 | Dry THF + 2.5% H2O, 4 eq. TBAF, 0 °C to RT, 24 h |  |
54% |
| 44 | Dry THF, 1.5 eq. TBAF, −20 °C to 10 °C, 2.5 h |  |
42% |

Preparation of enantioenriched samples
One of the potential advantages of the present strategy is to offer a direct access to enantioenriched PAC analogues starting from enantiomerically resolved alkynylcarbinol precursors. We previously separated and individually used both enantiomers of 4 to access original dialkynylcarbinol derivatives.20 This precursor, otherwise scarcely exploited in the literature, was selected to implement the strategy. The racemic alkynylcarbinol 4 was thus subjected to enzymatic kinetic resolution by means of Candida antartica lipase B (Novozyme 435)-catalysed acetylation (Scheme 4). Treatment of rac-4 with acrylic resin-immobilized enzyme and vinyl acetate in heptane at 40 °C for 6 h allowed isolation of the corresponding (R)-acetate in 41% yield. The latter was hydrolysed by use of the same enzyme in the presence of a pH 6 phosphate buffer solution for 120 h to give (R)-4. The unreacted alcohol (S)-4 was subjected again to the enzymatic acetylation procedure for 20 h to upgrade its enantiomeric enrichment. (R)- and (S)-4 were typically obtained with e.e. of 98 and >99%, respectively, according to chiral gas chromatography analysis of a re-acetylated sample (see SI). | ||
| Scheme 4 Lipase-catalysed enzymatic kinetic resolution of the precursor 4. | ||
The model coupling reaction with phenyl iodide was first explored with (S)-4. Chiral analytical supercritical fluid chromatography (SFC) analysis on Chiralpak IC column evidenced the formation of (S)-10 with an e.e. >99%. Prompted by this result, assembly of the prototypical PAC framework was studied. Coupling of (S)- and (R)-4 with iodide 12 delivered (S)- and (R)-18, respectively. Both compounds were isolated with e.e. >99% according to SFC analysis on Chiralpak IB column, confirming preservation of the enantiomeric enrichment of 4 during the Pd-catalysed Sonogashira coupling. Finally, the efficiency of this approach was illustrated with one of the most active PAC analogues. Enantioenriched (R)- and (S)-46 were thus secured from coupling of the functionalised iodide 16 with (S)- or (R)-4, respectively, in a straightforward two-step sequence.
Biological evaluations
The cytotoxicity of all compounds was assessed in HCT116 colon cancer cells, a cell line of reference that was so far used to evaluate all analogues (Table 7). Comparing the relative potency of 50, 26 and 27 allowed to directly assess the contribution of the pharmacophoric alkynylcarbinol moiety on cytotoxicity in the most active BAC series. The activity was gradually reduced when moving from the dialkynylcarbinol to the alkenyl alkynylcarbinol and the allenyl alkynylcarbinol warhead. The same trend was observed in the PAC series with IC50 increasing from 0.17 to 1.47 μM for 6, 7 and 8. Of note, the PAC derivatives revealed to be only 3 to 6 times less active than their BAC congeners. Since the unique potency of BAC 50 was previously shown to take place at the expense of both its stability and its selectivity towards the main bioactivation enzyme HSD17B11,17 this observation confirmed the relevance of the PAC series.| Compound | IC50 | Compound | IC50 |
|---|---|---|---|
 |
0.17 |  |
0.53 |
 |
1.47 |  |
0.48 |
 |
6.37 |  |
0.19 |
 |
0.35 |  |
0.09 |
 |
0.32 |  |
0.49 |
 |
6.50 |  |
2.51 |
 |
0.40 |  |
4.71 |
 |
1.49 |  |
0.76 |
 |
17.1 |  |
1.35 |
 |
21.1 |  |
0.26 |
 |
5.71 |  |
0.14 |
 |
0.13 |  |
2.21 |
 |
1.37 |  |
0.06 |
Five out of the six representatives of para-alkoxy phenyl series displayed a submicromolar level of IC50 with an average value of 0.26 μM. This result is in line with previously reported data for the PAC derivatives, indicating the combined favourable effect on cytotoxicity of the presence of fluorine and oxygen atoms at the positions 3 and 5 or 4 of the benzene ring, respectively.17 Of note, comparison of compounds 46 and 22 evidenced that the alkenyl alkynylcarbinol pharmacophore can confer potent cytotoxicity with an IC50 0.19 μM. This is also the case for 36, which is three times more efficient than its dialkynylcarbinol analogue 48. In agreement with our previous data, a significant eudismic ratio was found between (S)- and (R)-46, the dextrogyre (S)-eutomer being 40 times more potent than the levogyre (R)-distomer, with and IC50 of 0.14 μM. On the other hand, the dialkynylcarbinol derivative 47 was found to possess an IC50 of 0.13 μM, illustrating the favourable impact the bioinspired O-geranyl tail in comparison with its previously described O-heptyl congener displaying an IC50 of 0.36 μM.18
Regarding the 1,2,3-triazole-containing derivatives, cytotoxicity data were more contrasted. The influence of the lipophilic appendage was prominent, the branched non-5-yl chain in compounds 29 and 32 being unfavourable (IC50 of 6.5 and 4.7 μM, respectively), whereas the aromatic moiety in compounds 30 and 33 gave appreciable levels of cytotoxicity (IC50 of 2.5 and 1.5 μM, respectively). Interestingly, both linear N-nonyl compounds 28 and 31 showed equivalent potency, with the allenyl alkynylcarbinol 31 presenting an IC50 of 0.40 μM. This result illustrates the promise of this synthetic pharmacophore, which has been only scarcely exploited so far.
As far as the other five-membered ring heterocyclic derivatives were concerned, furan 40 and pyrazole 45 displayed only marginal cytotoxicity (IC50 of 17 and 21 μM, respectively), indicating the detrimental influence of the allenyl alkynylcarbinol combined with a shorter alkyl chain, whether with a pyrazole or a furan nucleus. On the other hand, isoxazole and pyrazole derivatives 43, 48 and 49 displayed comparable IC50 values in the low μM, whereas isoxazole 36, having an alkenyl alkynylcarbinol warhead, was the most potent compound of this group with an IC50 of 0.76 μM. When compared to that of the dialkynylcarbinol 48, its three-time higher potency highlighted once again the favourable role of the alkenyl alkynylcarbinol pharmacophoric moiety on cytotoxicity.
Having previously identified several alkynylcarbinol-containing compounds as prodrugs enantiospecifically bioactivated by the human SDR HSD17B11 into reactive, cytotoxic alkynylketones,17 we sought to determine whether our new series also depend on HSD17B11 for their cytotoxic activities. For this, racemic compounds with an IC50 below 2 μM on HCT116 cells were selected and their cytotoxic activity evaluated on the human osteosarcoma cell line U2OS, either wild-type (WT) or inactivated for HSD17B11 via CRISPR/Cas9 (KO HSD17B11).17 The ratio between the IC50 on KO HSD17B11 versus WT U2OS was calculated as an indication of HSD17B11-dependent cytotoxicity (Table 8). The data showed that compounds with a terminal dialkynylcarbinol motif are strongly dependent on HSD17B11 for their cytotoxicity (ratio greater than 62 for 3 out of 4 compounds), in agreement with our previous observations.17 Compounds with a terminal alkenyl alkynylcarbinol motif had lower dependency on HSD17B11 for their activity (ratio between 2 and 57, eight compounds tested), which may result from their activation by RDH11, another human SDR for which we reported an ability to oxidize several alkenyl alkynylcarbinols.17 Finally, allenyl alkynylcarbinols displayed the lowest dependency on HSD17B11 (ratio between 1 and 8.9, four compounds tested) and among those, two related 1,2,3-triazolyl allenyl alkynylcarbinol compounds, 31 and 33, had a ratio close to 1, suggesting that their cytotoxicity is independent of HSD17B11 and mediated by another SDR or via an alternative mechanism.
| Compound | WT | KO HSD17B11 | Ratio IC50 KO/WT |
|---|---|---|---|
 |
0.08 | >5 | >62.5 |
 |
0.33 | 12.0 | 36.3 |
 |
1.18 | 8.61 | 7.3 |
 |
0.48 | 11.1 | 23.2 |
 |
0.24 | 13.9 | 57.8 |
 |
1.74 | 7.38 | 4.2 |
 |
0.06 | 1.68 | 28.0 |
 |
0.15 | 1.34 | 8.9 |
 |
0.34 | 1.52 | 4.5 |
 |
0.14 | 0.21 | 1.5 |
 |
0.97 | 1.08 | 1.1 |
 |
0.22 | 0.49 | 2.2 |
 |
0.91 | 4.22 | 4.6 |
 |
0.21 | >20 | >95.2 |
 |
0.07 | 9.18 | 131.1 |
 |
0.58 | 4.20 | 7.2 |
Conclusions
Discovery of the latest generation analogues of natural acetylenic lipids, namely phenyl dialkynylcarbinol derivatives, has prompted the development of a highly convergent synthetic route to facilitate their structural exploration. Cadiot–Chodkiewicz or Sonogashira Pd/Cu-catalysed coupling of chiral alkynylcarbinol precursors with various functionalised acetylenic or aromatic halides was developed. Whereas the CuAAC click reaction of a 1,3-butadiyne intermediate led to a 1,2,3-triazole-containing series in a sequential process, phenyl, furyl, pyrazolyl and isoxazolyl congeners were produced in a single Sonogashira coupling step. The added-value of this modular approach for the direct access to enantioenriched compounds from a chemoenzymatically-resolved alkynylcarbinol precursor was demonstrated in the benzenic series. Cell viability data highlighted that the alkynylcarbinol warhead and the aromatic fragment display complementary contributions, giving rise to analogues with submicromolar IC50 of cytotoxicity against cancer cells when combined in an optimal fashion. Among these series, the 1,2,3-triazolyl allenyl alkynylcarbinols were identified as particularly interesting as their cytotoxic activity (IC50 of 0.14 μM for 31 on U2OS) is independent of HSD17B11, a finding that opens new prospects for further chemical and biological studies to decipher and exploit their mechanism of action.Experimental section
General methods
All reagents were obtained from commercial suppliers and used without any further purification. If not specified, reactions were run under nitrogen atmosphere in oven-dried glassware. Standard inert atmosphere techniques were used in handling all air and moisture sensitive reagents. Toluene, dichloromethane (DCM), tetrahydrofuran (THF), dimethylformamide (DMF) and diethyl ether (Et2O) were obtained by filtration through a drying column on a filtration system. Thin-layer chromatography (TLC) analyses were performed on precoated, aluminum-backed silica gel (Merck 60 F254). Visualization of the developed chromatogram was performed by UV light (254 nm) and using 10% phosphomolybdic acid in EtOH, or aqueous potassium permanganate (KMnO4) stain. Flash chromatography columns were performed using flash silica gel (SDS 35–70 μm). Nuclear magnetic resonance spectra were recorded on a Bruker Advance 300 or 400 or 600 MHz spectrometer. Chemical shifts for 1H NMR spectra are given in parts per million (ppm) with the residual solvent resonance as the reference CHCl3 (δ = 7.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet and br = broad), coupling constant in Hz and integration. Chemical shifts for 13C NMR spectra are given in ppm using the central peak of CDCl3 (δ = 77.16 ppm) as the reference. All 13C NMR spectra were obtained with complete proton decoupling. High-resolution mass spectrometry (HRMS) was performed on a Thermo-Finnigan MAT 95 XL instrument. Mass spectrometry m/z values are given in Dalton units.General procedures
5-(4-Octylphenyl)pent-1-en-4-yn-3-ol (7). To a solution of 3-(4-octylphenyl)propiolaldehyde18 (38 mg, 0.157 mmol, 1.0 equiv.) in dry THF (2 mL) under N2 at 0 °C was added vinyl magnesium bromide (190 μL, 0.19 mmol, 1.0 M solution in THF, 1.2 equiv.). The resulting solution was stirred at 0 °C for 1 h and allowed to warm to RT. After completion of the reaction, monitored by TLC, a saturated aqueous NH4Cl solution was added to quench the reaction. The resulting mixture was extracted with diethyl ether. Combined organic layers were washed with brine and dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by flash chromatography on silica gel using a gradient of up to 20% of diethyl ether in pentane. The alkenyl alkynylcarbinol 7 (11 mg, 26%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.41–7.31 (m, 2H), 7.17–7.07 (m, 2H), 6.06 (ddd, J = 17.0, 10.1, 5.3 Hz, 1H), 5.54 (dt,J = 17.0, 1.5, 1.2 Hz, 1H), 5.27 (dt, J = 10.1, 1.3 Hz, 1H), 5.10 (ddt, J = 6.7, 5.3, 1.3 Hz, 1H), 2.61 (t, J = 7.4 Hz 2H), 1.96 (s, J = 6.4 Hz, 1H), 1.65–1.57 (m, 2H), 1.33–1.16 (m, 10H), 0.94–0.82 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 143.8, 137.1, 131.7, 128.4, 119.5, 116.5, 87.0, 86.6, 63.8, 35.9, 31.9, 31.2, 29.5, 29.3 (*2), 22.7, 14.1. HRMS (DCI-CH4): calcd for C19H27O [MH]+: 271.2062 m/z, found: 271.2055 m/z.
1-(4-Octylphenyl)hexa-4,5-dien-1-yn-3-ol (8). In a sealed tube, 1-(4-octylphenyl)penta-1,4-diyn-3-ol (6)18 (50 mg, 0.186 mmol), CuI (2.6 mg, 7.5 mol%) and paraformaldehyde (8.9 mg, 0.014 mmol) were dissolved in dioxane. Then DIPA (37 μL, 0.261 mmol) was added. The reaction mixture was evacuated and backfilled with argon three times, then heated to 130 °C overnight. After completion of the reaction, the reaction mixture was cooled down to RT and directly concentrated under reduced pressure. The crude mixture was then purified by flash chromatography on silica gel using a gradient of up to 20% of diethyl ether in pentane. The allenyl alkynylcarbinol 8 (42 mg, 81%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.32 (d, J = 8.2 Hz, 2H), 7.12 (d, J = 8.0 Hz, 2H), 5.48 (q, J = 6.4 Hz, 1H), 5.14 (br s, 1H), 5.01 (dd, J = 6.4, 2.5 Hz, 2H), 2.59 (t, J = 7.8 Hz, 2H), 2.09 (br s, 1H), 1.68–1.52 (m, 2H), 1.34–1.25 (m, 10H), 0.94–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.8, 144.0, 131.8, 128.5, 119.5, 93.4, 87.5, 86.0, 79.1, 61.2, 36.0, 32.0, 31.4, 29.6, 29.4, 22.8, 14.2. HRMS (DCI-CH4): calcd for C20H27O [MH]+: 283.2062 m/z, found: 283.2064 m/z.
5-(3-Fluoro-4-(heptyloxy)phenyl)pent-1-en-4-yn-3-ol (20). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of diethyl ether in pentane. The alkenyl alkynylcarbinol 20 (35 mg, 50%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.23–7.13 (m, 2H), 6.89 (t, J = 8.9 Hz, 1H), 6.07 (ddd, J = 17.0, 10.1, 5.4 Hz, 1H), 5.55 (dt, J = 17.0, 1.3 Hz, 1H), 5.30 (dt, J = 10.1, 1.3 Hz, 1H), 5.11 (ddt, J = 6.7, 5.4, 1.3 Hz, 1H), 4.05 (t, J = 6.5 Hz, 2H) 1.99 (d, J = 6.5 Hz, 1H), 1.92–1.76 (m, 2H), 1.54–1.25 (m, 8H), 0.96–0.87 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 151.9 (d, J = 246.8 Hz), 148.0 (d, J = 7.5 Hz), 136.9, 128.3 (d, J = 3.5 Hz), 119.4 (d, J = 19.8 Hz), 116.6, 114.6 (d, J = 8.5 Hz), 114.3 (d, J = 2.6 Hz), 86.9, 85.3 (d, J = 7.5 Hz), 69.4, 63.7, 31.7, 29.1, 29.0, 25.9, 22.6, 14.1. 19F NMR (282 MHz, CDCl3) δ (ppm) −134.2. HRMS (DCI-CH4): calcd for C18H24O2F [MH]+: 291.1760 m/z, found: 291.1750 m/z.
1-(4-Butoxy-3-fluorophenyl)hexa-4,5-dien-1-yn-3-ol (21). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of diethyl ether in pentane. The allenyl alkynylcarbinol 21 (37 mg, 67%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.23–7.12 (m, 2H), 6.89 (t, J = 8.9 Hz, 1H), 5.49 (q, J = 6.4 Hz, 1H), 5.15 (s, 1H), 5.04 (dd, J = 6.4, 2.5 Hz, 2H), 4.06 (t, J = 6.5 Hz, 2H) 2.09 (d, J = 6.5 Hz, 1H), 1.90–1.75 (m, 2H), 1.55–1.46 (m, 2H), 0.94–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.9, 168.1, 151.8 (d, J = 246.7 Hz), 128.4 (d, J = 3.5 Hz), 119.4 (d, J = 19.8 Hz), 114.5 (d, J = 8.5 Hz), 114.2 (d, J = 2.6 Hz), 93.1, 87.3, 84.6 (d, J = 3.6 Hz), 79.0, 69.1, 61.0, 31.2, 19.1, 13.8. 19F NMR (282 MHz, CDCl3) δ (ppm) −134.2. HRMS (DCI-CH4): calcd for C16H18O2F [MH]+: 261.1291 m/z, found: 261.1285 m/z.
5-(3,5-Difluoro-4-(heptyloxy)phenyl)pent-1-en-4-yn-3-ol (22). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of diethyl ether in pentane. The alkenyl alkynylcarbinol 22 (87 mg, 46%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.08–6.93 (m, 2H), 6.06 (ddd, J = 17.0, 10.1, 5.4 Hz, 1H), 5.54 (dt, J = 17.0, 1.2 Hz, 1H), 5.31 (dt, J = 10.1, 1.2 Hz, 1H), 5.10 (ddt, J = 6.7, 5.4, 1.2 Hz, 1H), 4.17 (tt, J = 6.7, 0.9 Hz, 2H) 2.00 (d, J = 6.7 Hz, 1H), 1.83–1.71 (m, 2H), 1.54–1.18 (m, 8H), 0.96–0.86 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 155.5 (dd, J = 248.6, 6.9 Hz), 136.8 (t, J = 14.1 Hz), 136.5, 116.9, 116.6 (t, J = 11.2 Hz), 116.1–115.3 (m), 88.5, 77.2, 74.9 (t, J = 3.2 Hz), 63.6, 29.9, 29.0, 25.6, 22.6, 14.1. 19F NMR (282 MHz, CDCl3) δ (ppm) −127.7. HRMS (DCI-CH4): calcd for C18H23O2F2 [MH]+: 309.1666 m/z, found: 309.1656 m/z.
(E)-5-(4-((3,6-Dimethylhepta-2,5-dien-1-yl)oxy)-3-fluorophenyl)-pent-1-en-4-yn-3-ol (24). Following the general procedure A, the obtained crude mixture was purified on preparative reversed phase HPLC Waters (XSelect C18, 5 μm (19 × 150 mm) column 80
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 to 70:30 then to 60:40H2O/ACN, 20 mL min−1). The geranyl alkenyl alkynylcarbinol 24 (10 mg, 40%) was isolated as a colorless oil. 1H NMR (600 MHz, CDCl3) δ (ppm) 7.19–7.13 (m, 2H), 6.91–6.85 (m, 1H), 6.05 (ddd, J = 17.0, 10.1, 5.4 Hz, 1H), 5.53 (dt, J = 17.0, 1.3 Hz, 1H), 5.47 (tq, J = 6.6, 1.3 Hz, 1H), 5.27 (dt, J = 10.1, 1.3 Hz, 1H), 5.12–5.07 (m, 2H), 4.64 (d, J = 6.6 Hz, 2H), 2.17–2.06 (m, 4H), 1.96 (d, J = 6.5 Hz, 1H), 1.73 (br s, 3H), 1.67 (d, J = 1.3 Hz, 3H), 1.60 (d, J = 1.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.1 (d, J = 246.8 Hz), 147.6 (d, J = 10.6 Hz), 142.2, 136.9, 131.9, 128.2 (d, J = 3.4 Hz), 123.7, 119.4 (d, J = 20.1 Hz), 118.8, 116.6, 114.8 (d, J = 13.3 Hz), 109.9, 87.0, 85.2 (d, J = 2.8 Hz), 66.2, 63.7, 39.5, 26.2, 25.7, 17.7, 16.7. 19F NMR (282 MHz, CDCl3) δ (ppm) −133.7. HRMS (DCI-CH4): calcd for C21H24O2F [MH]+: 327.1760 m/z, found: 327.1758 m/z.
:20 to 70:30 then to 60:40H2O/ACN, 20 mL min−1). The geranyl alkenyl alkynylcarbinol 24 (10 mg, 40%) was isolated as a colorless oil. 1H NMR (600 MHz, CDCl3) δ (ppm) 7.19–7.13 (m, 2H), 6.91–6.85 (m, 1H), 6.05 (ddd, J = 17.0, 10.1, 5.4 Hz, 1H), 5.53 (dt, J = 17.0, 1.3 Hz, 1H), 5.47 (tq, J = 6.6, 1.3 Hz, 1H), 5.27 (dt, J = 10.1, 1.3 Hz, 1H), 5.12–5.07 (m, 2H), 4.64 (d, J = 6.6 Hz, 2H), 2.17–2.06 (m, 4H), 1.96 (d, J = 6.5 Hz, 1H), 1.73 (br s, 3H), 1.67 (d, J = 1.3 Hz, 3H), 1.60 (d, J = 1.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.1 (d, J = 246.8 Hz), 147.6 (d, J = 10.6 Hz), 142.2, 136.9, 131.9, 128.2 (d, J = 3.4 Hz), 123.7, 119.4 (d, J = 20.1 Hz), 118.8, 116.6, 114.8 (d, J = 13.3 Hz), 109.9, 87.0, 85.2 (d, J = 2.8 Hz), 66.2, 63.7, 39.5, 26.2, 25.7, 17.7, 16.7. 19F NMR (282 MHz, CDCl3) δ (ppm) −133.7. HRMS (DCI-CH4): calcd for C21H24O2F [MH]+: 327.1760 m/z, found: 327.1758 m/z.
Heptadeca-1-en-4,6-diyn-3-ol (26). Following the general procedure B, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 20% of diethyl ether in pentane. The butadiynyl alkenyl alkynylcarbinol 26 (87 mg, 58%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 5.95 (ddd, J = 17.0, 10.1, 5.3 Hz, 1H), 5.47 (ddd, J = 17.0, 1.4, 1.0 Hz, 1H), 5.24 (ddd, J = 10.1, 1.4, 1.0 Hz, 1H), 4.92 (ddt, J = 6.7, 5.3, 1.4 Hz, 1H), 2.28 (td, J = 7.0, 1.0 Hz, 2H), 1.91–1.82 (m, 1H), 1.61–1.45 (m, 2H), 1.43–1.23 (m, 14H), 0.93–0.83 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 136.4, 117.2, 82.7, 73.9, 71.6, 64.4, 63.7, 32.0, 29.7, 29.6, 29.4, 29.2, 29.0, 28.2, 22.8, 19.4, 14.3. HRMS (DCI-CH4): calcd for C17H27O [MH]+: 247.2062 m/z, found: 247.2064 m/z.
Octadeca-1-en-4,6-diyn-3-ol (27). Following the general procedure B, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 20% of diethyl ether in pentane. The butadiynyl allenyl alkynylcarbinol 27 (156 mg, 57%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 5.44–5.31 (m, 1H), 5.04–4.90 (m, 3H), 2.28 (td, J = 7.0, 0.9 Hz, 2H), 1.97 (dd, J = 6.2, 1.5 Hz, 1H), 1.63–1.42 (m, 2H), 1.44–1.20 (m, 14H), 0.94–0.81 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.8, 92.7, 82.8, 79.3, 74.2, 70.9, 64.4, 61.1, 32.0, 29.7, 29.6, 29.4, 29.2, 29.0, 28.2, 22.8, 19.4, 14.3. HRMS (DCI-CH4): calcd for C18H27O [MH]+: 259.2062 m/z, found: 259.2068 m/z.
5-(1-Nonyl-1H-1,2,3-triazol-4-yl)pent-1-en-4-yn-3-ol (28). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 28 (41 mg, 34%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.66 (s, 1H), 6.08 (ddd, J = 17.1, 10.2, 5.4 Hz, 1H), 5.58 (dt, J = 17.1, 1.3 Hz, 1H), 5.32 (dt, J = 10.2, 1.3 Hz, 1H), 5.16 (dt, J = 5.4, 1.5 Hz, 1H), 4.38 (t, J = 7.2 Hz, 2H), 2.07–1.82 (m, 3H), 1.39–1.25 (m, 12H), 0.96–0.86 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 136.3, 130.6, 126.0, 117.0, 91.3, 75.7, 63.6, 50.6, 31.8, 30.2, 29.3, 29.2, 20.0, 26.4, 22.7, 14.1. HRMS (DCI-CH4): calcd for C16H26N3O [MH]+: 276.2076 m/z, found: 276.2089 m/z.
5-((1-Nonan-5-yl)-1H-1,2,3-triazol-4-yl)pent-1-en-4-yn-3-ol (29). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 29 (22 mg, 21%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.62 (s, 1H), 6.08 (ddd, J = 17.1, 10.2, 5.4 Hz, 1H), 5.57 (ddd, J = 17.1, 1.6, 1.0 Hz, 1H), 5.32 (dt, J = 10.2, 1.6 Hz, 1H), 5.16 (t, J = 5.4 Hz, 1H), 4.48 (quint, J = 7.2 Hz, 1H), 2.10 (d, J = 6.4 Hz, 1H), 1.97–1.78 (m, 4H), 1.43–0.97 (m, 8H), 0.87 (t, J = 7.2 Hz, 6H). 13C NMR (75 MHz, CDCl3): δ (ppm) 136.4, 130.0, 124.6, 116.9, 91.5, 75.9, 63.5, 62.8, 35.4, 28.0, 22.2, 13.8. HRMS (DCI-CH4): calcd for C16H26N3O [MH]+: 276.2076 m/z, found: 276.2084 m/z.
5-(1-(4-Butylphenyl)-1H-1,2,3-triazol-4-yl)pent-1-en-4-yn-3-ol (30). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 30 (3 mg, 2%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.03 (s, 1H), 7.50 (d, J = 8.33 Hz, 2H), 7.33 (d, J = 8.33 Hz, 2H), 6.07 (ddd, J = 17.1, 10.2, 5.4 Hz, 1H), 5.57 (ddd, J = 17.1, 1.6, 1.0 Hz, 1H), 5.31 (dt, J = 10.2, 1.6 Hz, 1H), 5.16 (dt, J = 5.4, 1.5 Hz, 1H), 2.68 (t, J = 7.6 Hz, 2H), 1.86 (br s, 1H), 1.71–1.55 (m, 2H), 1.46–1.28 (m, 2H), 0.94 (t, J = 7.6 Hz, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 144.5, 136.2, 134.3, 130.8, 129.8, 124.2, 120.6, 117.1, 92.0, 75.5, 63.6, 35.2, 33.5, 22.3, 13.9. HRMS (DCI-CH4): calcd for C17H19N3O [MH]+: 281.1506 m/z, found: 281.1509 m/z.
1-(1-Nonyl-1H-1,2,3-triazol-4-yl)hexa-4,5-dien-1-yn-3-ol (31). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 31 (58 mg, 51%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.63 (s, 1H), 5.49 (q, J = 6.4 Hz, 1H), 5.17 (dt, J = 6.4, 2.4 Hz, 1H), 5.03 (dd, J = 6.4, 2.4 Hz, 2H), 4.36 (t, J = 7.2 Hz, 2H), 2.06 (br s, 1H), 1.97–1.84 (m, 2H), 1.35–1.22 (m, 12H), 0.94–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.6, 126.0, 92.7, 91.6, 79.2, 75.2, 60.9, 50.6, 31.8, 30.2, 29.3,29.2, 28.9, 26.4, 22.7, 14.1. HRMS (DCI-CH4): calcd for C17H26N3O [MH]+: 288.2076 m/z, found: 288.2072 m/z.
1-(1-(Nonan-5-yl)-1H-1,2,3-triazol-4-yl)hexa-4,5-dien-1-yn-3-ol (32). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 32 (24 mg, 34%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.61 (s, 1H), 5.49 (q, J = 6.4 Hz, 1H), 5.17 (dt, J = 6.4, 2.4 Hz, 1H), 5.03 (dd, J = 6.4, 2.4 Hz, 2H), 4.54–4.39 (m, 1H), 1.95–1.76 (m, 4H), 1.41–0.94 (m, 8H), 0.85 (t, J = 7.5 Hz, 6H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.7, 129.9, 124.7, 92.7, 91.8, 79.0, 75.2, 62.8, 60.9, 35.4, 28.0, 22.2, 13.8. HRMS (DCI-CH4): calcd for C17H26N3O [MH]+: 288.2076 m/z, found: 288.2072 m/z.
1-(1-(Nonan-5-yl)-1H-1,2,3-triazol-4-yl)hexa-4,5-dien-1-yn-3-ol (33). Following the general procedure C, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of a mixture of DCM/AcOEt (8/2) in pentane. The triazole 33 (4 mg, 4%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.03 (s, 1H), 7.63–7.57 (m, 2H), 7.36–7.28 (m, 2H), 5.50 (q, J = 6.3 Hz, 1H), 5.19 (dt, J = 6.3, 2.4 Hz, 1H), 5.02 (dd, J = 6.3, 2.4 Hz, 2H), 2.68 (t, J = 7.6 Hz, 2H), 1.68–1.58 (m, 2H), 1.41–1.33 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 207.7, 144.4, 134.3, 130.8, 129.8, 124.2, 120.6, 92.6, 92.3, 79.2, 74.8, 61.0, 35.2, 33.3, 22.2, 13.8. HRMS (DCI-CH4): calcd for C18H20N3O [MH]+: 294.1606 m/z, found: 294.1609 m/z.
5-(3-Nonylisoxazol-5-yl)pent-1-en-4-yn-3-ol (36). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of diethyl ether in pentane. The isoxazole 36 (26 mg, 38%) was isolated as a brown oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 6.31 (s, 1H), 6.04 (ddd, J = 17.0, 10.2, 5.4 Hz, 1H), 5.55 (dt, J = 17.0, 1.3 Hz, 1H), 5.33 (dt, J = 10.1, 1.3 Hz, 1H), 5.20–5.10 (m, 1H), 2.72 (d, J = 5.4 Hz, 1H), 2.67 (t, J = 7.6 Hz, 2H), 1.72–1.58 (m, 2H), 1.42–1.20 (m, 12H), 0.96–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3) δ (ppm) 164.2, 152.3, 135.5, 117.5, 108.0, 97.1, 72.9, 63.3, 31.9, 29.4, 29.3, 29.2, 29.1, 28.2, 25.9, 22.7, 14.1. HRMS (DCI-CH4): calcd for C17H26NO2 [MH]+: 276.1964 m/z, found: 276.1968 m/z.
1-(6-Hexylfuran-2-yl) hexa-1,2-dien-5-yn-4-ol (40). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 20% diethyl ether in pentane. The allenyl alkynylcarbinol 40 (14 mg, 18%) was isolated as a brown oil. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.51 (d, J = 0.9 Hz, 1H), 6.04 (m, 1H), 5.48 (q, J = 6.4 Hz, 1H), 5.17–5.09 (m, 1H), 5.08–4.97 (m, 2H), 2.60 (td, J = 7.5, 0.9 Hz, 2H), 2.12 (br s, 1H), 1.71–1.55 (m, 2H), 1.50–1.21 (m, 6H), 0.99–0.84 (m, 3H). 13C NMR (100 MHz, CDCl3): δ (ppm) 207.6, 157.1, 144.4, 107.5, 106.9, 93.1, 89.4, 78.9, 77.7, 61.1, 31.5, 28.7, 27.8, 27.7, 22.6, 14.1. HRMS (DCI-CH4): calcd for C16H21O2 [MH]+: 245.1542 m/z, found: 245.1547 m/z.
5-(1-Nonyl-1H-pyrazol-4-yl)pent-1-en-4-yn-3-ol (43). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 40% of diethyl ether in pentane. The alkenyl alkynylcarbinol 43 (91 mg, 39%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.60 (d, J = 0.7 Hz, 1H), 7.52 (d, J = 0.7 Hz, 1H), 6.06 (ddd, J = 17.0, 10.1, 5.5 Hz, 1H), 5.52 (dt, J = 17.0, 1.5 Hz, 1H), 5.27 (dt, J = 10.1, 1.5 Hz, 1H), 5.09 (ddt, J = 6.7, 5.5, 1.5 Hz, 1H), 4.09 (t, J = 7.1 Hz, 2H), 2.08 (d, J = 6.4 Hz, 1H), 1.85 (quint, J = 7.3 Hz, 2H), 1.37–1.22 (m, 12H), 0.93–0.83 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 141.9, 137.1, 131.9, 116.4, 101.9, 88.5, 78.0, 63.8, 52.5, 31.8, 30.2, 29.4, 29.2, 26.5, 22.6, 14.1. HRMS (DCI-CH4): calcd for C17H27N2O [MH]+: 275.2123 m/z, found: 275.2114 m/z.
1-(1-Hexyl-1H-pyrazol-4-yl)hexa-4,5-dien-1-yn-3-ol (45). Following the general procedure A, the obtained crude mixture was purified by flash chromatography on silica gel using a gradient of up to 30% of diethyl ether in pentane. The allenyl alkynylcarbinol 45 (26 mg, 20%) was isolated as a colourless oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.60 (d, J = 0.6 Hz, 1H), 7.52 (d, J = 0.6 Hz, 1H), 5.48 (q, J = 6.4 Hz, 1H), 5.14, (br d, J = 6.5 Hz, 1H), 5.07–4.97 (m, 2H), 4.10 (t, J = 7.1 Hz, 2H), 2.22 (s, 1H), 1.85 (quint, J = 7.2 Hz, 2H), 1.38–1.22 (m, 6H), 0.97–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 207.6, 141.9, 132.0, 93.2, 88.8, 79.4, 78.9, 77.3, 61.1, 52.5, 31.2, 30.1, 26.1, 22.5, 14.0. HRMS (DCI-CH4): calcd for C15H21N2O [M]+: 245.1654 m/z, found: 245.1654 m/z.
1-(3,5-Difluoro-4-(heptyloxy)phenyl)penta-1,4-diyn-3-ol (46). Compound 23 (45.5 mg, 98.3 μmol) was dissolved in wet THF (1 mL, 3% v/v H2O). At 0 °C under an atmosphere of N2, TBAF (1 M in THF, 147 μL, 0.147 mmol, 1.5 equiv.) was added dropwise. After 3 h, a saturated aqueous solution of NH4Cl was added to quench the reaction. The resulting mixture was extracted with diethyl ether. The combined organic layers were washed with brine and dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified by flash chromatography on silica gel using a gradient of up to 10% of diethyl ether in pentane. The dialkynylcarbinol 46 (21 mg, 60%) was isolated as an orange oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.08–6.93 (m, 2H), 5.31 (dd, J = 7.6, 2.3 Hz, 1H), 4.15 (tt, J = 6.6, 0.9 Hz, 2H), 2.63 (d, J = 2.3 Hz, 1H), 2.32 (d, J = 7.6 Hz, 1H), 1.88–1.64 (m, 2H), 1.50–1.24 (m, 8H), 0.94–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 155.5 (dd, J = 248.9, 6.8 Hz), 137.2 (t, J = 14.1 Hz), 116.2–115.9 (m), 115.9–115.7 (m), 86.1, 82.5 (t, J = 3.4 Hz), 80.4, 74.9 (t, J = 3.4 Hz), 73.3, 52.5 (d, J = 6.6 Hz), 31.7, 29.9, 28.9, 25.6, 22.6, 14.1. 19F NMR (282 MHz, CDCl3) δ (ppm) −127.5. HRMS (DCI-CH4): calcd for C18H21O2F2 [MH]+: 307.1510 m/z, found: 307.1511 m/z.
(E)-1-(4-((3,6-Dimethylhepta-2,5-dien-1-yl)oxy)-3-fluorophenyl)-penta-1,4-diyn-3-ol (47). Compound 25 (66 mg, 0.140 mmol, 1.0 equiv.) was dissolved in THF (1.5 mL). At 0 °C under N2 atmosphere, TBAF (1 M in THF, 183 μL, 0.183 mmol, 1.3 equiv.) was added dropwise. After 30 minutes, a saturated aqueous solution of NH4Cl was added to quench the reaction mixture. The resulting mixture was extracted with diethyl ether. Combined organic layers were washed with brine and dried over MgSO4, filtered and concentrated under reduced pressure. The crude mixture was purified on preparative reversed phase HPLC Waters (XSelect C18, 5 μm (19 × 150 mm) column 70
:30 to 30:70 in 15 minutes H2O/ACN, 0.6 mL min−1). The dialkynylcarbinol 47 (14 mg, 40%) was isolated as a colorless oil. 1H NMR (600 MHz, CDCl3) δ (ppm) 7.25–7.17 (m, 2H), 6.91 (t, J = 8.4 Hz, 2H), 5.47 (td, J = 6.6, 1.4 Hz, 1H), 5.35 (dd, J = 7.3, 2.3 Hz, 1H), 5.09 (tt, J = 6.8, 1.5 Hz, 1H), 4.65 (d, J = 6.6 Hz, 2H), 2.64 (d, J = 2.3 Hz, 1H), 2.36 (d, J = 7.3 Hz, 1H), 2.18–2.07 (m, 4H), 1.73 (br s, 3H), 1.67 (br s, 3H), 1.60 (br s, 3H). 13C NMR (125 MHz, CDCl3): δ (ppm) 152.0 (d, J = 246.9 Hz), 148.0 (d, J = 10.7 Hz), 142.2, 131.9, 128.5 (d, J = 3.4 Hz), 123.6, 119.5 (d, J = 20.2 Hz), 118.7, 114.7 (d, J = 2.7 Hz), 114.0 (d, J = 8.5 Hz), 84.7, 83.7 (d, J = 2.8 Hz), 80.8, 73.0, 66.2, 52.5, 39.5, 26.2, 25.7, 17.7, 16.7. 19F NMR (282 MHz, CDCl3) δ (ppm) −133.7. HRMS (DCI-CH4): calcd for C21H22O2F [MH]−: 325.1604 m/z, found: 325.1605 m/z.
1-(3-Nonylisoxazol-5-yl)pent-1,4-diyn-3-ol (48). At 0 °C, under N2 atmosphere the isoxazole 38 (58 mg, 0.135 mmol, 1.0 equiv.) was dissolved into wet THF (1 mL, 2.5% v/v H2O). TBAF (1 M in THF, 540 μL, 0.540 mmol, 4.0 equiv.) was added dropwise in three portions. After completion of the reaction, a saturated aqueous solution of NH4Cl was added to quench the reaction. The resulting mixture was extracted with diethyl ether. Combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of up to 20% of diethyl ether in pentane. The isoxazole 48 (12 mg, 54%) was isolated as a brown oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 6.35 (s, 1H), 5.36 (dd, J = 7.8, 2.4 Hz, 1H), 2.75–2.60 (m, 4H, containing at 2.69 (t, J = 7.7 Hz, 2H) and at 2.65 (d, J = 2.4 Hz, 1H)), 1.72–1.58 (m, 2H), 1.42–1.20 (m, 12H), 0.96–0.84 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 164.2, 151.8, 108.7, 94.4, 79.3, 74.1, 71.5, 52.1, 31.8, 29.4, 29.2 (*2), 29.1, 28.1, 25.9, 22.7, 14. HRMS (DCI-CH4): calcd for C17H24NO2 [MH]+: 274.1807 m/z, found: 274.1796 m/z.
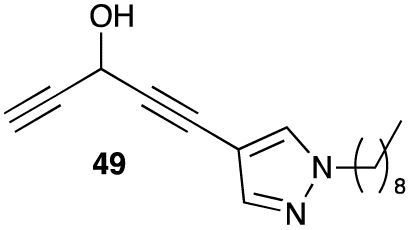
1-(1-Nonyl-1H-pyrazol-4-yl)penta-1,4-diyn-3-ol (49). Compound 44 was dissolved in THF (1.5 mL). Under N2 atmosphere, TBAF (1 M in THF, 573 μL, 0.573 mmol, 1.3 equiv.) was added dropwise at −20 °C. The mixture was warmed to 10 °C over 2.5 h. After completion of the reaction, a saturated aqueous solution of NH4Cl was added to quench the reaction. The resulting mixture was extracted with diethyl ether. Combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel using a gradient of up to 40% of diethyl ether in pentane. The dialkynylcarbinol 49 (51 mg, 42%) was isolated as a yellow oil. 1H NMR (300 MHz, CDCl3) δ (ppm) 7.61 (d, J = 0.7 Hz, 1H), 7.52 (d, J = 0.7 Hz, 1H), 5.34 (dd, J = 6.4, 2.3 Hz, 1H), 4.10 (t, J = 7.1 Hz, 2H), 3.74 (d, J = 6.4 Hz, 1H), 2.59 (d, J = 2.3 Hz, 1H), 1.83 (quint, J = 7.4 Hz, 2H), 1.37–1.16 (m, 12H), 0.96–0.85 (m, 3H). 13C NMR (75 MHz, CDCl3): δ (ppm) 142.0, 132.3, 101.5, 86.7, 81.2, 76.3, 72.5, 52.6, 52.4, 31.8, 30.1, 29.4, 29.2, 29.1, 26.5, 22.6, 14.1. HRMS (DCI-CH4): calcd for C17H25N2O [MH]+: 273.1967 m/z, found: 273.1970 m/z.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general synthetic experimental details, characterization data, UPC2 chromatograms and NMR spectra. See DOI: https://doi.org/10.1039/d5ra06473b.Acknowledgements
The analysis by supercritical CO2 were recorded on Acquity UPC2, from Waters, which is part of the “Integrated Screening Platform of Toulouse” (PICT, GenoToul, IBiSA). We are grateful to the Université Fédérale Toulouse Midi-Pyrénées (UFTMiP) and Région Occitanie for a PhD fellowship to MB. Part of this work was funded by the prematuration program Prostacure from Région Occitanie and by the TIRIS “Scaling-Up Science” program (PhD fellowship to PS) supported by the Région Occitanie, the European Regional Development Fund (ERDF), and the French government, through the France 2030 project managed by the National Research Agency (ANR) with the reference number “ANR-22-EXES-0015”.Notes and references
- R. E. Minto and B. J. Blacklock, Prog. Lipid Res., 2008, 47, 233–306 CrossRef PubMed.
- V. M. Dembitsky, D. O. Levitsky, T. A. Gloriozova and V. V. Poroikov, Nat. Prod. Commun., 2006, 1, 773–811 Search PubMed.
- Z. F. Zhou, M. Menna, Y. S. Cai and Y. W. Guo, Chem. Rev., 2015, 115, 1543–1596 CrossRef PubMed.
- V. M. Dembitsky and D. O. Levitsky, Nat. Prod. Commun., 2006, 1, 405–429 Search PubMed.
- Q. Xie and C. H. Wang, Phytochemistry, 2022, 201, 113288 CrossRef.
- J. S. Qi, Y. C. Duan, Z. C. Li, J. M. Gao, J. Z. Qi and C. W. Liu, Nat. Prod. Bioprospect., 2023, 13, 50 CrossRef.
- Y. J. Lee, Y. Cho and H. N. K. Tran, Mar. Drugs, 2021, 19, 122 CrossRef PubMed.
- L. H. N. de Sousa, R. D. de Araujo, D. Sousa-Fontoura, F. G. Menezes and R. M. Araujo, Mar. Drugs, 2021, 19, 663 CrossRef PubMed.
- P. Santos, L. Busta, W. C. Yim, E. B. Cahoon and D. K. Kosma, J. Exp. Bot., 2022, 73, 2889–2904 CrossRef PubMed.
- B. W. Gung, C. R. Chim., 2009, 12, 489–505 CrossRef.
- A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 CrossRef PubMed.
- A. Siddiq and V. Dembitsky, Anti-Cancer Agents Med. Chem., 2008, 8, 132–170 CrossRef PubMed.
- C. Dawid, F. Dunemann, W. Schwab, T. Nothnagel and T. Hofmann, J. Agric. Food Chem., 2015, 63, 9211–9222 CAS.
- J. Bouvet, V. Maraval, S. Ballereau, V. Bernardes-Génisson and Y. Génisson, J. Nat. Prod., 2024, 87, 2550–2566 CAS.
- D. Listunov, V. Maraval, R. Chauvin and Y. Génisson, Nat. Prod. Rep., 2015, 32, 49–75 CAS.
- D. Listunov, E. Joly, P. Rullière, H. Gaspard, V. Bernardes-Génisson, Y. Génisson, V. Maraval and R. Chauvin, Synthesis, 2018, 50, 3114–3130 CAS.
- P. Demange, E. Joly, J. Marcoux, P. R. A. Zanon, D. Listunov, P. Rullière, C. Barthes, C. Noirot, J. B. Izquierdo, A. Rozie, K. Pradines, R. Hee, M. V. de Brito, M. Marcellin, R. F. Serre, O. Bouchez, O. Burlet-Schiltz, M. C. F. Oliveira, S. Ballereau, V. Bernardes-Génisson, V. Maraval, P. Calsou, S. M. Hacker, Y. Génisson, R. Chauvin and S. Britton, eLife, 2022, 11, e73913 CrossRef CAS.
- M. Bossuat, P. Rullière, N. Preuilh, A. Peixoto, E. Joly, J.-G. Gomez, M. Bourkhis, F. Rodriguez, F. Gonçalves, I. Fabing, H. Gaspard, V. Bernardes-Génisson, V. Maraval, S. Ballereau, R. Chauvin, S. Britton and Y. Génisson, J. Med. Chem., 2023, 66, 13918–13945 CrossRef CAS.
- D. El Arfaoui, D. Listunov, I. Fabing, M. Oukessou, C. Frongia, V. Lobjois, A. Samson, F. Ausseil, A. Ben-Tama, E. El Hadrami, R. Chauvin and Y. Génisson, ChemMedChem, 2013, 8, 1779–1786 CrossRef CAS PubMed.
- D. Listunov, E. Joly, C. Duhayon, N. Saffon-Merceron, I. Fabing, Y. Génisson, V. Maraval and R. Chauvin, ChemMedChem, 2018, 13, 1711–1722 CrossRef CAS PubMed.
- G. R. Zheng, W. Lu and J. C. Cai, J. Nat. Prod., 1999, 62, 626–628 CrossRef CAS.
- S. Tamura, T. Ohno, Y. Hattori and N. Murakami, Tetrahedron Lett., 2010, 51, 1523–1525 CrossRef CAS.
- T. J. Mann, A. W. H. Speed, R. R. Schrock and A. H. Hoveyda, Angew. Chem. Int. Ed., 2013, 52, 8395–8400 CAS.
- W. Lu, G. R. Zheng, D. X. Gao and J. C. Cai, Tetrahedron, 1999, 55, 7157–7168 CAS.
- M. K. Gurjar, V. S. Kumar and B. V. Rao, Tetrahedron, 1999, 55, 12563–12576 Search PubMed.
- K. R. Prasad and B. Swain, J. Org. Chem., 2011, 76, 2029–2039 Search PubMed.
- G. Kumaraswamy and K. Sadaiah, Tetrahedron, 2012, 68, 262–271 CrossRef.
- L. Rycek, V. Ticli, J. Pyszkowski, S. Latkolik, X. Liu, A. G. Atanasov, T. Steinacher, R. Bauer, D. Schuster, V. M. Dirsch, M. Schnurch, M. Ernst and M. D. Mihovilovic, J. Nat. Prod., 2018, 81, 2419–2428 CrossRef.
- A. Sharma and S. Chattopadhyay, Tetrahedron Asymmetry, 1998, 9, 2635–2639 CrossRef.
- A. Sharma and S. Chattopadhyay, J. Org. Chem., 1998, 63, 6128–6131 CAS.
- W. H. Li, Z. M. Lin, L. Chen, X. C. Tian, Y. Wang, S. H. Huang and R. Hong, Tetrahedron Lett., 2016, 57, 603–606 CrossRef.
- C. Elgindy, J. S. Ward and M. S. Sherburn, Angew. Chem. Int. Ed., 2019, 58, 14573–14577 CrossRef PubMed.
- K. Cocq, N. Saffon-Merceron, Y. Coppel, C. Poidevin, V. Maraval and R. Chauvin, Angew. Chem. Int. Ed., 2016, 55, 15133–15136 CrossRef PubMed.
- H. W. Luo and S. M. Ma, Eur. J. Org Chem., 2013, 2013, 3041–3048 Search PubMed.
- D. W. Xu, Z. Y. Li and S. M. Ma, Tetrahedron Asymmetry, 2003, 14, 3657–3666 CrossRef.
- For a preliminary disclosure by our groups of the conversion of rac-4 into rac-10, see: M. Vieira de Brito, F.-X. Toublet, M. Bossuat, D. M. Barbosa Davi, D. K. Almeida, T. de Sousa Fonseca, F. Miranda Nunes, M. Carlos de Mattos, M. Caroff, S. Britton, S. Ballereau, V. Maraval, M. C. Ferreira Oliveira, Y. Génisson and V. Bernardes-Génisson, Eur. J. Org. Chem., 2025, 00, e202500762 CrossRef.
- M. Schilz and H. Plenio, J. Org. Chem., 2012, 77, 2798–2807 Search PubMed.
- M. Bourkhis, H. Gaspard, P. Rulliere, D. K. C. de Almeida, D. Listunov, E. Joly, R. Abderrahim, M. C. de Mattos, M. C. F. de Oliveira, V. Maraval, R. Chauvin and Y. Génisson, ChemMedChem, 2018, 13, 1124–1130 CrossRef PubMed.
- D. Listunov, N. Saffon-Merceron, E. Joly, I. Fabing, Y. Génisson, V. Maraval and R. Chauvin, Tetrahedron, 2016, 72, 6697–6704 CrossRef.
- M. A. M. Subbaiah and N. A. Meanwell, J. Med. Chem., 2021, 64, 14046–14128 CrossRef PubMed.
- E. S. Hems, B. A. Wagstaff, G. Saalbach and R. A. Field, Chem. Commun., 2018, 54, 12234–12237 RSC.
- K. Kai, M. Sogame, F. Sakurai, N. Nasu and M. Fujita, Org. Lett., 2018, 20, 3536–3540 Search PubMed.
- G. S. Chen, X. X. Yan, S. J. Chen, X. Y. Mao, Z. D. Li and Y. L. Liu, J. Org. Chem., 2020, 85, 6252–6260 CAS.
- J. M. Guan, C. Spry, E. T. Tjhin, P. H. Yang, T. Kittikool, V. M. Howieson, H. Ling, L. Starrs, D. Duncan, G. Burgio, K. J. Saliba and K. Auclair, J. Med. Chem., 2021, 64, 4478–4497 CAS.
- J. S. Poh, C. García-Ruiz, A. Zúñiga, F. Meroni, D. C. Blakemore, D. L. Browne and S. V. Ley, Org. Biomol. Chem., 2016, 14, 5983–5991 CAS.
- M. De Amici, P. Conti, C. Dallanoce, L. Kassi, S. Castellano, G. Stefancich and C. De Micheli, Med. Chem. Res., 2000, 10, 69–80 CAS.
- X. Z. Wang, J. Jia, Y. Zhang, W. R. Xu, W. Liu, F. N. Shi and J. W. Wang, J. Chin. Chem. Soc., 2007, 54, 643–652 CAS.
- R. da Rosa, M. H. de Moraes, L. A. Zimmermann, E. P. Schenkel, M. Steindel and L. S. C. Bernardes, Eur. J. Med. Chem., 2017, 128, 25–35 CAS.
- J. S. Lee, Y. S. Cho, M. H. Chang, H. Y. Koh, B. Y. Chung and A. N. Pae, Bioorg. Med. Chem. Lett., 2003, 13, 4117–4120 CAS.
- F. Ushiyama, H. Takashima, Y. Matsuda, Y. Ogata, N. Sasamoto, R. Kurimoto-Tsuruta, K. Ueki, N. Tanaka-Yamamoto, M. Endo, M. Mima, K. Fujita, I. Takata, S. Tsuji, H. Yamashita, H. Okumura, K. Otake and H. Sugiyama, Bioorg. Med. Chem., 2021, 30, 115964 CrossRef CAS.
- A. W. Sromek, M. Rubina and V. Gevorgyan, J. Am. Chem. Soc., 2005, 127, 10500–10501 CrossRef CAS.
- J. D. Katz, J. P. Jewell, D. J. Guerin, J. Lim, C. J. Dinsmore, S. V. Deshmukh, B. S. Pan, C. G. Marshall, W. Lu, M. D. Altman, W. K. Dahlberg, L. Davis, D. Falcone, A. E. Gabarda, G. Z. Hang, H. Hatch, R. Holmes, K. Kunii, K. J. Lumb, B. Lutterbach, R. Mathvink, N. Nazef, S. B. Patel, X. L. Qu, J. F. Reilly, K. W. Rickert, C. Rosenstein, S. M. Soisson, K. B. Spencer, A. A. Szewczak, D. Walker, W. X. Wang, J. Young and Q. W. Zeng, J. Med. Chem., 2011, 54, 4092–4108 CrossRef CAS PubMed.
- P. García-Domínguez, I. Lepore, C. Erb, H. Gronemeyer, L. Altucci, R. Alvarez and A. R. de Lera, Org. Biomol. Chem., 2011, 9, 6979–6987 Search PubMed.
- D. Listunov, I. Fabing, N. Saffon-Merceron, H. Gaspard, Y. Volovenko, V. Maraval, R. Chauvin and Y. Génisson, J. Org. Chem., 2015, 80, 5386–5394 CAS.
- M. de Lèsèleuc, É. Godin, S. Parisien-Collette, A. Lévesque and S. K. Collins, J. Org. Chem., 2016, 81, 6750–6756 Search PubMed.
- B. M. Trost, M. R. Machacek and B. D. Faulk, J. Am. Chem. Soc., 2006, 128, 6745–6754 CAS.
- P. Manini, W. Amrein, V. Gramlich and F. Diederich, Angew. Chem. Int. Ed., 2002, 41, 4339–4343 CAS.
- O. Klein, H. Hopf and J. Grunenberg, Eur. J. Org Chem., 2009, 2009, 2141–2148 Search PubMed.
- D. Prenzel, T. Sander, J. Gebhardt, H. Soni, F. Hampel, A. Görling, S. Maier and R. R. Tykwinski, Chem.–Eur. J., 2017, 23, 1846–1852 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |