
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, characterization, biomolecular interaction, potent aqueous singlet oxygen generation and NADH photo-oxidation of phenanthroline-based cyclometalated Ir(III) complexes
Bhumika Joshia,
Rishav Dasa,
Rinku Chakrabarty b,
Murugesh Shivashankar*a and
Priyankar Paira*a
b,
Murugesh Shivashankar*a and
Priyankar Paira*a
aDepartment of Chemistry, School of Advanced Sciences, Vellore Institute of Technology, Vellore 632014, Tamil Nadu, India. E-mail: priyankar.paira@vit.ac.in; mshivashankar@vit.ac.in
bDepartment of Chemistry, Alipurduar University, Alipurduar 736122, West Bengal, India
First published on 18th November 2025
Abstract
Cyclometalated iridium complexes have been a great choice for PDT agents over the last few decades. Herein, we endeavoured to introduce a newly synthesized Ir(III) complex [LIr], focusing on its photophysical properties, DNA interactions, and catalytic potential. DNA-binding studies indicated a combination of binding interactions with notable affinity (K = 1.04 × 105 M−1) and a high Stern–Volmer quenching constant (KSV = 7.35 × 109 M−1), suggesting strong DNA association, while UV-visible spectroscopy confirmed robust MLCT transitions. The complex efficiently generated singlet oxygen with a quantum yield of 0.0613, highlighting its potential for photodynamic therapy. Additionally, it demonstrated moderate catalytic activity in NADH oxidation, with a turnover number (TON) of 4.22 and a turnover frequency (TOF) of 8.46 h−1, both enhanced under visible light, thereby establishing complex [LIr] as a promising future candidate for DNA-targeted therapies and photoactivated catalysis.
Introduction
Cancer is considered as one of the most deleterious diseases for mankind. The breakthrough success of first platinum-based drug cisplatin revolutionized cancer treatment and paved the way for the development of metal-based therapies. Cisplatin is a fortuitous discovery by Barnett Rosenberg1 in 1965, which binds to DNA, forming crosslinks that disrupt cancer cell replication and ultimately lead to cell death. Its widespread clinical success, along with its derivatives carboplatin and oxaliplatin,2 highlighted the potential of metal complexes in oncology followed by the development of trinuclear platinum drugs like triplatin or BBR3464.3 However, significant drawbacks such as severe side effects, drug resistance, and limited effectiveness against certain cancers have restricted their use.4,5These challenges have driven researchers to explore alternative metal-based complexes with distinct mechanisms of action, reduced toxicity, and improved selectivity for cancer cells. Traditional treatments such as radiation therapy, chemotherapy, and surgery often suffer from invasiveness, systemic toxicity, and lack of selectivity. Photodynamic therapy (PDT) is considered as a promising alternative, exhibiting reduced invasiveness, high selectivity, and strong efficiency.6
Cyclometalated iridium complexes have garnered immense attention as PDT agents in recent years.7–9 d-Block noble metal Ir possesses atomic no 77 and belongs to group 9 of the periodic table.10 This rare element Ir is non-corrosive and inert in nature. It is used as a catalyst11,12 in various organic reactions like C–H activation13 and asymmetric hydrogenation reactions14 and in alloys.15
Iridium (Ir) has gained significant attention as a potential anticancer agent16 due to its unique physical and chemical properties. Unlike platinum, iridium complexes offer diverse structural and electronic configurations that can be fine-tuned through ligand design and choice of oxidation state (+1, +3, and +4). This flexibility makes Ir complexes highly stable, selective, and suitable for targeted therapies. Their stability in biological systems, combined with rational ligand design, enables the development of cancer-selective compounds with minimal off-target toxicity.17
One of the most exciting applications of iridium-based complexes is in photodynamic therapy (PDT),17 a minimally invasive approach in which light activates photosensitizers to generate reactive oxygen species (ROS) that selectively destroy cancer cells. Compared to conventional chemotherapy, PDT offers several advantages including localized treatment, reduced damage to healthy tissue, and lower systemic toxicity. Iridium complexes, with their strong phosphorescence, high quantum yields, long-lived excited states, and tunable emission wavelengths, have emerged as highly effective photosensitizers for PDT.18 The rational design of ligands to improve water solubility, biocompatibility, and cellular uptake is crucial in developing efficient iridium-based PDT agents. Structural modifications aim to maximize photophysical properties for efficient light absorption and ROS generation upon irradiation. To ensure safety and efficacy, thorough biological evaluations including phototoxicity under light and dark conditions, are essential.18
A considerable number of research groups are paying attention for the designing of Ir complexes for their successful application as the PDT agent in the field of cancer biology.19 Mitochondria-targeting triphenylphosphine-functionalized cyclometalated iridium(III) complexes have been introduced by Fu et al. for their application as anticancer agents.20 A series of carbene Ir complexes have been reported by Wang et al.,21 whereas a phenanthroline-based cyclometalated Ir complex has been reported by Castor et al.22 for the G-Quadruplex DNA binding; Negi et al. emphasized on the application of Ir complexes in combination therapy with PDT and PACT methodology.23
This study explores the potential of iridium complexes as photosensitizers for their potential application in photodynamic therapy (PDT), focusing on their stability in biological environments and interactions with key biomolecules. We have introduced a cyclometalated iridium complex having – Br as the electron-withdrawing group, which will help the attraction towards the cancer cells, and the planar molecule helps to enter into the base pairs and aids the intercalation process. A hydrophobic moiety present within the designed Ir complex will help in cellular accumulation, while the overall positive charge will increase the attraction of the complex towards the target (Fig. 1).
 | ||
| Fig. 1 Schematic representation for the designing of the complex [LIr]. | ||
An earlier investigation by our research team revealed GSH-resistant, luminescent Ru(II)/Ir(III)/Re(I) complexes containing 2-(pyren-1-yl)-1H-imidazo[4,5-f][1,10]phenanthroline ligands, demonstrating significant phototoxicity towards triple-negative breast cancer (MDA-MB-231) cells when exposed to light irradiation.24 Expanding on that research, this study presents a newly crafted bromine-substituted imidazophenanthroline-based cyclometalated Ir(III) complex that exhibits a stronger electron-withdrawing nature and superior DNA-binding capacity. In contrast to previous mixed-metal systems that primarily focused on phototoxicity and luminescence, this manuscript offers a comprehensive analysis of the photophysical characteristics, DNA/protein interactions, singlet-oxygen production, and NADH-oxidation properties of an individual Ir(III) species. This method provides a mechanistic understanding of its light-triggered reactivity and therapeutic capabilities, thus enhancing the current advancements in Ir(III)-based photodynamic agents.
Results and discussion
Synthesis and characterization
Scheme 1 delineates a concise summary of the synthesis of ligand L and the complex [LIr]. An imidazophenanthroline-based cyclometalated Ir(III) complex was designed, synthesized, and characterized in the present study. The ligand 2-(6-bromopyridin-2-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (L) was synthesized by treating 1,10-phenanthroline-5,6-dione (1 equiv.) and 6-bromopicolin aldehyde (1 equiv.) using glacial acetic acid and ammonium acetate. Refluxing ligand L with [(ppy)2IrCl]2 in a methanol–toluene mixture of solvent afforded complex [LIr]. Synthesized ligand L and the complex [LIr] were characterized by 1H, 13C, FT-IR, and high-resolution mass spectrometry (HRMS) (Fig. S1–S8). | ||
| Scheme 1 Synthesis of ligand L and complex [LIr]. | ||
In the 1H NMR spectrum of 2-(6-bromopyridin-2-yl)-1H-imidazo[4,5-f][1,10]phenanthroline (L), aromatic protons appeared between δ 7.76 and 9.12 ppm. In the 13C NMR spectrum, peaks were observed between δ 121.1 and 149.9 ppm. The FTIR spectrum confirms the presence of characteristic functional groups. HRMS spectra supports the formation of ligand L; the observed [M + H]+ peak at 376.0100 corresponds to the protonated ligand.
In the 1H NMR spectrum of complex [LIr], aromatic protons were found between δ 8.49 and 6.29 ppm, whereas in 13C NMR peaks appeared between δ 167.3 and 120.4. The HRMS spectra of complex [LIr] displayed a peak corresponding to [M + H]+ at 876.1322, proving the formation of the expected complex [LIr].
Photophysical studies
The UV-visible absorption spectra of the free ligand (L) and the synthesized iridium complex [LIr] were recorded at ambient temperature in 10% acetonitrile (ACN) solution. The ligand exhibited a prominent absorption band in the range of 250–350 nm, which is attributed to π–π* electronic transitions of the aromatic system.For the [LIr] complex, two distinct absorption maxima were observed at 262 nm and 378 nm, corresponding to the π–π* and metal-to-ligand charge transfer (MLCT) transitions, respectively. The peak at ∼340 nm indicates contributions from both MLCT and ligand-to-ligand charge transfer (LLCT) processes (Fig. 2a and b), which is consistent with previously reported Ir(III) complexes.
 | ||
| Fig. 2 Absorption spectra of (a) L and (b) complex [LIr]. (c) Emission spectra of L (black line) λex = 330 nm and [LIr] (red) λex = 340 nm. | ||
The photoluminescence emission spectra were recorded by exciting the ligand and complex at 330 nm and 340 nm, respectively (Fig. 2c). The free ligand (L) exhibited a strong emission band centered at 425 nm, characteristic of ligand-centered fluorescence. In contrast, the [LIr] complex showed a broader and red-shifted emission band centered at 586 nm, typical for cyclometalated Ir(III) complexes. This red shift and broadening indicate significant contribution from MLCT emission, which is associated with the efficient intersystem crossing to a triplet excited state, common in Ir(III)-based complexes. The weaker emission intensity of [LIr] compared to L is consistent with enhanced non-radiative decay pathways due to strong spin–orbit coupling in the heavy Ir center. The luminescence quantum yields were calculated using quinoline sulfate (Φ = 0.54) as a reference, resulting in values of 0.052 for the ligand and 0.025 for the [LIr] complex (eqn (i)). These photophysical properties suggest that the [LIr] complex is capable of efficient MLCT processes, which supports its proposed role as a potential photosensitizer in photodynamic therapy (PDT).
Solubility and lipophilicity
While evaluating metal complexes' potential for biological applications, especially in anti-tumor therapies, it is essential to comprehend their solubility and lipophilicity. Ligand L and its corresponding Ir complex, [LIr], are highly soluble in different organic solvents, such as methanol, acetone, toluene, dichloromethane (DCM), acetonitrile (ACN), dimethyl sulfoxide (DMSO), and dimethylformamide (DMF). The versatility of these compounds in a range of biological and experimental settings is guaranteed by their broad solubility.Finding the ideal balance between hydrophilicity and lipophilicity is crucial for metal complexes to demonstrate potent anti-tumor activity. Their capacity to dissolve in blood (hydrophilicity) and still pass through cellular membranes (lipophilicity) is determined by this balance. We used the popular shaking flask method to assess this, and the results are summarized in Table 1.
| Sample | λa (nm) | λb (nm) | Stokes shift | O.D.c | φd | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Po/we Po/we |
|
|---|---|---|---|---|---|---|---|
| π–π* | MLCT | ||||||
| a Absorption maxima.b Emission wavelength.c Optical density.d Luminescence quantum yield.e n-Octanol/water partition coefficient. | |||||||
| L | 262 | 425 | 163 | 1.6 | 0.052 | ||
| Complex [LIr] | 262 | 378 | 586 | 208 | 1.1 | 0.025 | 1.3 |
| Quinoline sulphate | 350 | 442 | 102 | 0.25 | 0.54 | ||
Stability study
Studying the stability of metal complexes in various biological environments is critical for practical applications. UV-visible spectroscopy was used to monitor the stability of the complex [LIr] over 72 hours in three relevant media: PBS (phosphate-buffered saline), GSH (glutathione), and 10% DMSO (aqueous solution). These habitats were selected to mimic the physiological conditions, intracellular reducing environments, and aqueous solubility conditions, respectively.In the 10% DMSO solution, which mimics aqueous conditions, the complex [LIr] remained highly stable, showing only a very slight decrease in absorbance over time. This indicates its potential suitability for applications requiring prolonged exposure to aqueous environments. In the GSH medium, representing the intracellular reductive environment, there was a notable decrease in absorbance at 24 h, suggesting an initial interaction with glutathione. Interestingly, after this point, the absorbance of the complex is stabilized, indicating that while there is an initial response to the reducing agent, the complex resists further degradation, which is essential for intracellular applications. In a PBS buffer medium, the complex demonstrated exceptional stability, with only a minimal and gradual decrease in absorbance throughout the 72 h period, reinforcing its robustness under physiological conditions.
All of these results highlight the exceptional stability profile of the complex [LIr] under a range of conditions, which is essential for both its biological compatibility and therapeutic potential. It is a potential option for use in drug administration, phototherapy, and other biomedical domains where long-term stability is a crucial necessity due to its capacity to preserve structural integrity under both aqueous and physiological conditions while also fending off glutathione-induced degradation.
Extensive research on DNA and protein binding reveals that these complexes can efficiently intercalate into DNA or attach to its grooves, disrupting critical processes such as transcription and replication in cancer cells. Protein-binding studies further suggest that iridium complexes can interfere with essential enzymatic functions needed for tumor growth and survival, enhancing their anticancer effects. Another crucial factor in developing effective PDT agents is their stability under physiological conditions. Our findings indicate that the synthesized iridium complex exhibits remarkable stability in biological environments, allowing for prolonged circulation and improved tumor targeting. This stability is vital for maintaining their structural integrity and therapeutic efficiency during treatment, ensuring their effectiveness in combating cancer.
DNA binding study by UV-vis method
The binding of DNA and the destruction of cancer cells are essential criteria for a drug molecule to meet, which diminish the uncontrolled proliferation, DNA replication, etc. DNA damage directly affects the phenomena of tumor growth, resulting in cell cycle arrest and cell death. Capability and the mechanism of binding of ligand L and complex [LIr] and calf thymus DNA (ct-DNA) were examined by UV-vis absorption spectroscopy in Tris–HCl buffer (pH 7.4) at 25 °C. Non-covalent interaction is very common while binding DNA and small molecules. Noncovalent interaction can be of three types: intercalative, partial intercalative and groove binding mode. This noncovalent binding includes hydrogen bonding, hydrophobic and electrostatic interaction. It is well known that the purine base pairs (adenine–guanine) and pyrimidine base pairs (thymine–cytosine) generally induce electronic transition. With the increase in the concentration of DNA, the pronounced change in absorbance is observed in the transition region of ligand L as well as in complex [LIr]. Table 2 displays the intrinsic binding constant (kb) (eqn (ii)) value of ligand L and complex [LIr]. The intrinsic binding constant (kb) value of complex [LIr] is found to be 1 × 106 M−1. Interestingly, hypochromic effects were observed during the binding process of ligand L and complex [LIr] (Fig. 3a and b) with DNA, maintaining the ligand and the complex concentration constant throughout the study. This decrease in absorption indicates the intercalative mode of binding. Such intercalative interaction highlights the versatility of these compounds in their interactions with DNA and provides valuable insights into potential applications in biological targeting and therapeutic development.| Sample | λ (nm) | Change in absorbance intensity | Kba × (108 M−1) | KSVb × (106 M−1) | Kappc × (106 M−1) |
|---|---|---|---|---|---|
| a Kb, DNA binding constant derived from UV-visible absorption spectra.b KSV, Stern–Volmer quenching constant.c Kapp, apparent coupling constant. | |||||
| L | 604 | Hypochromic | 5.89 | 1.8 | 0.32 |
| Complex [LIr] | 605 | Hypochromic | 0.01 | 3.5 | 0.34 |
 | ||
| Fig. 3 DNA binding study in UV-vis method of (a) L; (b) complex [LIr]; EtBr quenching study of (c) L and (d) complex [LIr] in the UV-vis method. BSA binding study of (e) L and (f) complex [LIr] in the UV-vis method. | ||
Ethidium bromide displacement assay
To identify the intensity of interaction between the synthesized complex and calf-thymus DNA (ct-DNA), ethidium bromide (EtBr) displacement assay was carried out. The interaction of EtBr (which is nonfluorescent in nature) between the base pairs of ct-DNA significantly improves the fluorescent intensity. EtBr does not exhibit any fluorescence in its free form as its fluorescence is quenched by the solvent molecules. However, its fluorescence intensity increases in the presence of ct-DNA, which suggests the intercalative mode of binding of EtBr with DNA. The fluorescence intensity was found to decrease with the further increase in the concentration of the complexes. Thus, the fluorescence intensity decreases when the complex [LIr] displaces EtBr from ct-DNA and it gets bound by itself to the DNA base pairs. During the current study, successful reciprocal agreement was confirmed during the titration process. The DNA–EtBr complex showed strong fluorescence with an emission peak at 604 nm when excited at 485 nm. Upon incremental addition of ligand L and complex [LIr] (0–50 µM), a gradual decrease in the fluorescence intensity is observed, indicating a hypochromic effect. This decrease in fluorescent intensity suggests that the complex [LIr] effectively competes with EtBr around the internal binding point of the DNA helix, thus replacing EtBr from the DNA–EtBr complex. The strength of this interaction was quantified by calculating the apparent binding constant (Kapp) 0.32 × 106 M−1 for L and 0.34 × 106 M−1 for the complex [LIr] (Table 2, eqn (iii), Fig. 3c and d).To further understand the quenching mechanism, the Stern–Volmer quenching constant (kSV) (eqn (iv)) was calculated. The complex [LIr] showed a rather high quenching constant (7.35 × 109 M−1) compared to ligand L (1.92 × 108 M−1), thereby proving a stronger interaction with DNA. Overall, these results show that complex [LIr] can effectively bind to DNA. Such interactions are important for the potential biological and therapeutic applications of these complexes.
BSA binding study
Bovine serum albumin (BSA) is a protein found in cow serum that closely resembles human serum albumin (HSA). This similarity makes BSA useful for studying how proteins interact with ligands in human biology. In this study, we studied how ligand L and its complex [LIr] interact with BSA by spectrofluorometric analysis. Fig. 3e and f reveals that with the increase in the concentration of BSA, the fluorescence intensity deceases. For ligand L, an isosbestic point at 382 nm indicates a clear balance between free and bound forms, showing good binding interaction. The binding constant for ligand L is 1.47 × 10−8 M−1, showing moderate affinity for BSA (Fig. 3e). The complex [LIr] has a slightly lower binding constant of 1.19 × 10−8 M−1 (eqn (v), (vi), Table 3 and Fig. 3e), indicating that metal coordination slightly affects binding strength. Ligand L has about 0.75 binding sites, while the complex [LIr] has 1.00 binding site, suggesting a more defined interaction. These results are important for understanding how drugs interact with proteins in the bloodstream, with the complex [LIr], showing potential for better drug delivery.Viscosity
To gain deeper insights into the binding mode between the complex [LIr] and calf-thymus DNA (ct-DNA), viscosity measurements are performed using an Ostwald viscometer under carefully controlled temperature conditions. Viscosity studies are a valuable tool for DNA binding studies, providing evidence of indirect but critical interactions. Changes in the viscosity of the DNA solution can indicate whether the connection is bound via interference. This generally causes minimal changes, either by increasing the viscosity due to the expansion of the DNA helix or by combining grooves or electrostatic interactions.In the experiments, there is a relative concentration of complex [LIr], but the ct-DNA concentration remains constant. The results indicated that the viscosity increased slightly with the increase in the concentrations of the complex [LIr]. This conservative increase in viscosity indicates that the interaction does not significantly distinguish or extend DNA double helix. This indicates that the classical inter-analysis, which leads to a significant increase in viscosity, is probably not the only binding mode. Instead, the data show a mixed coupling mechanism with slight or large coupling of grooves playing a major role, possibly with partial internality. This observation is adjusted with previously known ethidium bromide (EtBr) displacement assays. This assay has previously showed that DNA interactions are usually associated with strong intercalators, although without structural distortion. The consistent viscosity values increase the idea that the complex [LIr] forms a stable but non-invasive association with the DNA structure. Overall, viscosity studies complement spectroscopic findings and provide valuable information on sustained and stable DNA binding interactions by the complex [LIr] (Table 4). This is probably determined by the combination of groove connections and weak interactions.
| Sample condition | Observed viscosity change | Proposed binding mechanism | Remarks |
|---|---|---|---|
| ct-DNA alone | Baseline viscosity | — | Reference control; native B-DNA structure |
| ct-DNA + low [LIr] ratio | Negligible change | Weak groove binding/electrostatic association | Minimal structural alteration of DNA duplex |
| ct-DNA + moderate [LIr] ratio | Slight, gradual increase | Groove binding with partial intercalation | Minor base pair stacking interactions; no significant helix unwinding |
| ct-DNA + high [LIr] ratio | Conservative but consistent increase | Mixed binding mode; groove binding dominant | Stable, non-invasive complexation |
| EtBr (classical intercalator) control | Pronounced increase in viscosity | Strong intercalation | Helical lengthening and unwinding typical of planar aromatic intercalators |
Photobiology
The ability of the complex [LIr] to generate 1O2 under visible light (400–700 nm) was evaluated using 1,3-diphenylisobenzofuran (DPBF) as a chemical probe. DPBF reacts with 1O2, causing a decrease in its absorption at 410 nm, which allows quantitative assessment of 1O2 generation. Rose Bengal, a well-known photosensitizer, was used as a ref. 26.
Upon visible light irradiation, [LIr] exhibited a singlet oxygen quantum yield (ΦΔ) of 0.0613. This value is lower than that of typical PDT sensitizers such as porphyrins (ΦΔ ∼ 0.4–0.7) and Ru(II) complexes (ΦΔ ∼ 0.1–0.5). Despite this modest ΦΔ, [LIr] demonstrates good photostability and visible-light absorption, suggesting potential utility in photodynamic or photoactivated antimicrobial therapies. The quantum yield was calculated using the relative DPBF degradation rates of [LIr] and Rose Bengal, accounting for absorption differences (correction factor F = 1.020) and experimental parameters, ensuring accurate quantification (Fig. 4).27
 | ||
| Fig. 4 Change in the absorbance of DPBF with complex [LIr] under (a) dark condition and (b) light condition. (c) Change in the absorbance of DPBF with RB (Rose Bengal) under (c) dark condition and (d) light condition. (e) Plot of the relative change of absorbance (A/A0) vs. time (s) of RB (Rose Bengal) and complex [LIr]. | ||
NADH oxidation study
Nicotinamide adenine dinucleotide (NADH) plays a crucial role in maintaining cellular redox homeostasis by controlling electron transfer during metabolic reactions (Fig. 5). It acts as a key coenzyme in the mitochondrial electron transport chain (ETC), facilitating energy production in cells. The oxidation of NADH to NAD+ disrupts this chain and negatively impacts the metabolic process, which may ultimately lead to severe cellular damage (Table 5).28 | ||
| Fig. 5 Change in the absorbance of NADH at 340 nm in the presence of complex [LIr] under (a) dark conditions and (b) light irradiation, indicating enhanced NADH oxidation upon photoactivation. | ||
| Condition | Monitoring wavelength (nm) | TON | TOF (h−1) | Relative activity | Interpretation |
|---|---|---|---|---|---|
| Light irradiation | 340 | 4.22 | 8.46 | High | Significant oxidation of NADH; photoactivation enhances electron transfer efficiency |
| Dark | 340 | Low | Low | Baseline | Minimal catalytic activity; confirms light-dependent activation of the complex |
| Control (no [LIr]) | 340 | — | — | None | No detectable NADH oxidation; validates role of [LIr] as catalyst |
To gain further insights into the redox behavior and reactive oxygen species (ROS)-generating ability of the iridium complex, we conducted NADPH and DPBF assays. NADH depletion was used as a direct indicator of ROS generation and oxidative stress induction. A notable decrease in NADH concentration after treatment with [LIr] confirmed its effective ROS production and the resulting oxidative damage to cancer cells. The DPBF assay, which directly measures ROS generation, further validated the strong photodynamic activity of [LIr] under light irradiation.
Mechanistically, the process likely involves the photoexcitation of Ir(III) to its excited state upon visible light exposure. This is followed by electron transfer, oxidizing Ir(III) to an unstable Ir(IV) intermediate. The high oxidation state facilitates the abstraction of an electron from NADH, forming the radical intermediate NADH˙+, which is subsequently converted into NAD˙ and eventually NAD+ (Fig. 6).
 | ||
| Fig. 6 Probable mechanism for the oxidation process of NADH using complex [LIr] as catalyst in the presence of visible light. | ||
Overall, these results emphasize the strong photoactive capacity of the Iridium complex [LIr] to generate ROS, which is essential for the development of photoresponsive therapeutic agents. The complex's moderate quantum yield combined with advantageous absorption in the visible light region further supports its application in photocatalytic and medicinal settings.
Moreover, a broad analysis of ROS generation, stability, and biomolecular interactions highlights the significant therapeutic potential of Ir(III)-based complexes in photodynamic therapy (PDT). Their ability to selectively target cancer cells, combined with high photostability and strong interactions with DNA and proteins, makes them highly promising candidates for future clinical applications.29
Beyond PDT, there is growing interest in developing multifunctional iridium-based platforms capable of both therapeutic and diagnostic (theranostic) applications. These advanced systems provide the dual benefit of effective cancer treatment and real-time disease monitoring, potentially transforming personalized medicine.29
Despite these promising outcomes, certain challenges remain for the widespread adoption of Ir(III)-based therapies. The high cost and limited availability of iridium underscore the importance of developing sustainable metal recycling strategies and optimizing metal usage without compromising therapeutic efficacy.30
Ultimately, the full potential of iridium complexes in cancer therapy will be unlocked through cross-disciplinary collaboration involving chemistry, biology, medicine, and industrial research. Advances in medicinal chemistry, molecular biology, and nanotechnology are expected to accelerate the development of safer, efficient, and patient-friendly iridium-based therapeutics, contributing to more personalized and minimally invasive cancer treatment solutions in the future.
Conclusion
The evolution of iridium-based treatments showcases the remarkable progress in metal-based cancer therapies. This study focuses on the development of iridium complex [LIr] for photodynamic therapy, exploring its interactions with DNA, stability in biological environments, and ROS generation through NADH and DPBF assays. The promising results provide a strong foundation for further research aimed at refining these treatments for clinical use. With continued advancements, iridium-based therapies have the potential to drive precision medicine forward, ultimately improving patient care and outcomes.In summary, this study highlights the potential of the complex [LIr] as a versatile candidate for photochemical applications. Its strong photophysical properties, including metal-to-ligand charge transfer (MLCT) transitions and enhanced π–π* absorption, improve its interaction with light, making it suitable for photodynamic therapy (PDT). DNA-binding studies confirm intercalation, with a high binding constant (K = 1.04 × 105 M−1) and a significant Stern–Volmer quenching constant (KSV = 7.35 × 109 M−1), indicating a strong affinity for DNA. Additionally, its ability to generate reactive oxygen species (ROS) under visible light, with a singlet oxygen quantum yield (Φ) of 0.0613, underscores its effectiveness as a photosensitizer. The complex [LIr] also exhibits moderate catalytic activity in NADH oxidation, with improved performance when exposed to light, as reflected in its turnover number (TON) of 4.22 and turnover frequency (TOF) of 8.46 h−1. With balanced lipophilicity (logPo/w = 1.3), strong biomolecular interactions, and photo-enhanced catalytic efficiency, the complex [LIr] presents significant promise for future applications in PDT as both type I PDT agent and type II PDT agent to combat both normoxic tumors and hypoxic tumor cells, DNA-targeted therapies, and redox-based biomedical treatments. Its ability to harness light for selective therapeutic and catalytic effects makes it a compelling candidate for further exploration in cancer treatment, photochemical research, and advanced biomedical technologies.
Experimental section
Materials and methods
The reagents and solvents used were of high purity and market ratings. The analytical grade organic solvents and chemicals, e.g. 1,10-phenanthroline-5,6-dione and 6-bromopicolin aldehyde, used in chemical synthesis and chromatographic purposes, were obtained from trusted vendors of companies such as Sigma-Aldrich, Spectrochem, and E-Merck. Thin-layer chromatography was performed on pre-coated silica gel 60 F254 aluminum sheets (E. Merck, Germany). Sigma-Aldrich Chemical Limited supplied bovine serum albumin. The 1H and 13C NMR spectra were recorded using a 400 MHz Advance Bruker DPX spectrometer with tetramethyl silane (TMS) as the internal reference. The molecular changes were quantified using ppm units. The shortened words are: ‘d’ for doublet, ‘t’ for triplet, and ‘m’ for multiplet. An Ostwald viscometer was used to measure viscosity. A Shimadzu Affinity FT-IR spectrometer was used to record the IR spectra from 4000 to 400 cm−1. The mass spectra of the discovered compounds were recorded using a Shimadzu ESI-MS-4000 mass spectrometer with a 4000 triple quadrupole MS and methanol as the solvent. UV-visible spectra were recorded using a JASCO V-730 spectrophotometer with a 1 cm quartz cell, while fluorescence spectra were recorded using a JASCO FP-8440 fluorescence spectrophotometer with a xenon lamp.Chemistry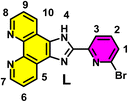

![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching (1669), CC stretching (1545), CN stretching (1411), C–Br stretching (630); ESI-MS (MeOH): observed m/z = 376.0100 [M + H]+, calcd m/z = 376.0178 [M + H]+.
N stretching (1669), CC stretching (1545), CN stretching (1411), C–Br stretching (630); ESI-MS (MeOH): observed m/z = 376.0100 [M + H]+, calcd m/z = 376.0178 [M + H]+. Complex [(ppy)2Ir(µ-Cl)]2 (obtained from the reaction of IrCl3 hydrate and 2-phenyl pyridine in a 2-methoxy ethanol–water mixture following the previously reported procedure) and ligand L at a molar ratio of 1/2 were dissolved in a solvent mixture of methanol (MeOH)/toluene at 1/4 (v/v), and the mixture was refluxed for 12 hours to obtain the complex [LIr] with a high yield. Yield: 82%; 1H NMR (DMSO-d6, 400 MHz): δ 8.49 (d, J = 7.7 Hz, 1H, H9, pyridine ArH), 8.28 (d, J = 8.2 Hz, 2H, H5, H14, phenanthroline ArH), 8.17 (d, 2H, J = 5.1 Hz, H7, H12, phenanthroline ArH), 8.12–7.93 (m, 2H, H4, H12), 8.04 (t, 1H, J = 7.4 Hz, H4, pyridine ArH), 7.53 (d, 2H, J = 5.9 Hz, H25, H18), 7.89–7.84 (m, 4H, H2, H16, H19, H24), 7.52 (d, 2H, J = 6.0 Hz, H1, H17), 7.07 (t, 2H, J = 7.2 Hz, H22, H26), 7.01–6.94 (m, 5H, H11, H23, H15, H6, H13), 6.29 (d, 2H, J = 7.6 Hz, H3, H20); 13C NMR (DMSO-d6, 100 MHz): δ 167.3, 150.8, 149.7, 145.1, 144.5, 139.2, 131.7, 130.7, 125.5, 124.3, 122.9, 120.4; IR (cm−1): ν NH stretching (3366), CH stretching (3064), CN stretching (1610), CC stretching (1572), CN stretching (1412), Ir–N stretching (490); ESI-MS (MeOH): observed m/z = 876.1322 [M + H]+, calcd m/z = 876.1062 [M + H]+.
Complex [(ppy)2Ir(µ-Cl)]2 (obtained from the reaction of IrCl3 hydrate and 2-phenyl pyridine in a 2-methoxy ethanol–water mixture following the previously reported procedure) and ligand L at a molar ratio of 1/2 were dissolved in a solvent mixture of methanol (MeOH)/toluene at 1/4 (v/v), and the mixture was refluxed for 12 hours to obtain the complex [LIr] with a high yield. Yield: 82%; 1H NMR (DMSO-d6, 400 MHz): δ 8.49 (d, J = 7.7 Hz, 1H, H9, pyridine ArH), 8.28 (d, J = 8.2 Hz, 2H, H5, H14, phenanthroline ArH), 8.17 (d, 2H, J = 5.1 Hz, H7, H12, phenanthroline ArH), 8.12–7.93 (m, 2H, H4, H12), 8.04 (t, 1H, J = 7.4 Hz, H4, pyridine ArH), 7.53 (d, 2H, J = 5.9 Hz, H25, H18), 7.89–7.84 (m, 4H, H2, H16, H19, H24), 7.52 (d, 2H, J = 6.0 Hz, H1, H17), 7.07 (t, 2H, J = 7.2 Hz, H22, H26), 7.01–6.94 (m, 5H, H11, H23, H15, H6, H13), 6.29 (d, 2H, J = 7.6 Hz, H3, H20); 13C NMR (DMSO-d6, 100 MHz): δ 167.3, 150.8, 149.7, 145.1, 144.5, 139.2, 131.7, 130.7, 125.5, 124.3, 122.9, 120.4; IR (cm−1): ν NH stretching (3366), CH stretching (3064), CN stretching (1610), CC stretching (1572), CN stretching (1412), Ir–N stretching (490); ESI-MS (MeOH): observed m/z = 876.1322 [M + H]+, calcd m/z = 876.1062 [M + H]+.Experimental procedure
 | (i) |
 | (ii) |
| Kapp × [complex]50 = KEtBr × [EtBr] | (iii) |
 | (iv) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) Po/w). The logPo/w value of the complex [LIr] was calculated by a shake flask method using the previously published procedure.31 A known amount of each complex was suspended in water (prostrated with n-octanol) and shaken for 48 h on an orbital shaker. To allow the phase separation, the solution was centrifuged for 10 min at 3000 rpm. After the separation of two layers, analysis was performed by UV-vis spectroscopic technique. The partition coefficient (logPo/w) values were calculated using the OD of the complex in n-octanol and water.
Po/w). The logPo/w value of the complex [LIr] was calculated by a shake flask method using the previously published procedure.31 A known amount of each complex was suspended in water (prostrated with n-octanol) and shaken for 48 h on an orbital shaker. To allow the phase separation, the solution was centrifuged for 10 min at 3000 rpm. After the separation of two layers, analysis was performed by UV-vis spectroscopic technique. The partition coefficient (logPo/w) values were calculated using the OD of the complex in n-octanol and water.
 | (v) |
 | (vi) |
 | (vii) |
| F = 1 − 10−OD | (viii) |
| [NAD+/NADP+] = [(Abs340nm)initial − (Abs340nm)final]/(Abs340 nm)initial × [NADH] | (ix) |
| Turnover number (TON) = [NAD+/NADP+]/[catalyst] | (x) |
| Turnover frequency (TOF) = TON/time (h) | (xi) |
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra06269a.Acknowledgements
The authors express gratitude to the Department of Science and Technology, Government of India, for their support of this research through the DST-SERB CRG project grant (CRG/2021/002267). Additionally, this work was financially supported by Vellore Institute of Technology (VIT), Vellore, under the Faculty Seed Grant (REGMS) (Sanction Order No. SG20250014). Recognition is also extended to DST, New Delhi, India, for the DST-FIST project.References
- J. D. Hoeschele, Dalton Trans., 2016, 45, 12966–12969 RSC.
- P. O'Sullivan, V. Previtali, B. Twamley, C. J. Marmion, A. R. McDonald and I. Rozas, RSC Adv., 2025, 15, 3427–3435 RSC.
- M. A. Tesoriero and N. J. Wheate, Dalton Trans., 2025, 54, 2199–2208 RSC.
- Q. Zhang and Q.-B. Lu, Sci. Rep., 2021, 11, 788 CrossRef PubMed.
- R. Oun, Y. E. Moussa and N. J. Wheate, Dalton Trans., 2018, 47, 6645–6653 RSC.
- D. E. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 5, 380–387 CrossRef PubMed.
- S. U and P. Paira, Chem.–Asian J., 2025, e202401047 CrossRef PubMed.
- G. Vigueras, G. Gasser and J. Ruiz, Dalton Trans., 2025, 54, 1320–1330 RSC.
- R. Das, U. Das, N. Roy, C. Mukherjee, S. U and P. Paira, Dyes Pigm., 2024, 226, 112134 CrossRef.
- C. Housecroft and A. Sharpe, Inorganic Chemistry, Pearson, London, U.K., 4th edn, 2012 Search PubMed.
- G. J. Sunley and D. J. Watson, Catal. Today, 2000, 58, 293–307 CrossRef CAS.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730–8731 CrossRef CAS.
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka Jr and M. R. Smith III, Science, 2002, 295, 305–308 CrossRef CAS.
- K. Murata, T. Ikariya and R. Noyori, J. Org. Chem., 1999, 64, 2186–2187 CrossRef CAS.
- P. Štarha, Inorg. Chem. Front., 2025, 12, 897–910 RSC.
- I. Kostova, Molecules, 2025, 30, 801 CrossRef CAS PubMed.
- S. U and P. Paira, Chem.–Asian J., 2025, e202401047 CrossRef CAS.
- S. Ghosh and P. Paira, Eur. J. Inorg. Chem., 2025, e202400769 CrossRef CAS.
- S. U, R. Chakrabarty and P. Paira, Dalton Trans., 2025, 54, 7602–7610 RSC.
- H. Fu, S. Wang, Y. Gong, H. Dong, K. Lai, Z. Yang, C. Fan, Z. Liu and L. Guo, Bioorg. Chem., 2025, 155, 108148 CrossRef CAS PubMed.
- X. Wang, C. Zhang, R. Madji, C. Voros, S. Mazères, C. Bijani, C. Deraeve, O. Cuvillier, H. Gornitzka, M. Maddelein and C. Hemmert, Molecules, 2023, 28, 691 CrossRef CAS PubMed.
- K. J. Castor, K. L. Metera, U. M. Tefashe, C. J. Serpell, J. Mauzeroll and H. F. Sleiman, Inorg. Chem., 2015, 54, 6958–6967 CrossRef CAS PubMed.
- M. Negi and V. Venkatesh, Chem. Sci., 2025, 16, 6376–6382 RSC.
- R. Das and P. Paira, Dalton Trans., 2023, 52, 15365–15376 RSC.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS.
- S. Monro, K. L. Colón, H. Yin, J. Roque, P. Konda, S. Gujar, R. P. Thummel, L. Lilge, C. G. Cameron and S. A. McFarland, Chem. Rev., 2019, 119, 797–828 CrossRef CAS.
- R. Kushwaha, A. Upadhyay, S. Saha, A. K. Yadav, A. Bera, A. Dutta and S. Banerjee, Dalton Trans., 2024, 53, 13591–13601 RSC.
- E. V. Potapova, E. A. Zherebtsov, V. V. Shupletsov, V. V. Dremin, K. Y. Kandurova, A. V. Mamoshin, A. Y. Abramov and A. V. Dunaev, FEBS J., 2024, 291, 2674–2682 CrossRef CAS PubMed.
- P. Szymaszek, M. Tyszka-Czochara and J. Ortyl, Eur. J. Med. Chem., 2024, 276, 116648 CrossRef CAS.
- E. M. Boreham, L. Jones, A. N. Swinburne, M. Blanchard-Desce, V. Hugues, C. Terryn, F. Miomandre, G. Lemercier and L. Natrajan, Dalton Trans., 2015, 44, 16127–16135 RSC.
- N. Roy, T. Dasgupta, S. Ghosh, M. Jayaprakash, M. Pal, S. Shanavas, S. K. Pal, V. Muthukumar, A. S. Kumar, R. Tamizhselvi, M. Roy, B. Bose, D. Panda, R. Chakrabarty and P. Paira, Langmuir, 2024, 40, 25390–25404 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |