DOI:
10.1039/D5RA05851A
(Paper)
RSC Adv., 2025,
15, 37888-37898
Metal-free access to 4-indolyl coumarin from coumarin 3-carboxylic acid and indole via a Michael addition-decarboxylation and dehydrogenation protocol: a photo-physical study and utility as an invisible ink
Received
9th August 2025
, Accepted 22nd September 2025
First published on 10th October 2025
Abstract
Herein, on water tandem Michael addition-decarboxylation of coumarin-3-carboxylic acids with indoles under catalysis of Amberlite IR120 afforded 4-indolyl 3,4-dihydrocoumarin. The process was extended to the synthesis of 4-indolyl coumarin derivatives upon oxidation using an organic oxidant. The wide substrate scope, high product yields, catalytic recyclability and large-scale preparation of intermediates are the noteworthy features of this methodology. This two-step process provides better yields of 4-indole-substituted coumarins as compared to the corresponding single-step palladium-catalyzed cross-coupling reactions. We also report herein for the first time that the indole-coumarin hybrid can be used as a thermal- and moisture-resistant invisible ink.
Introduction
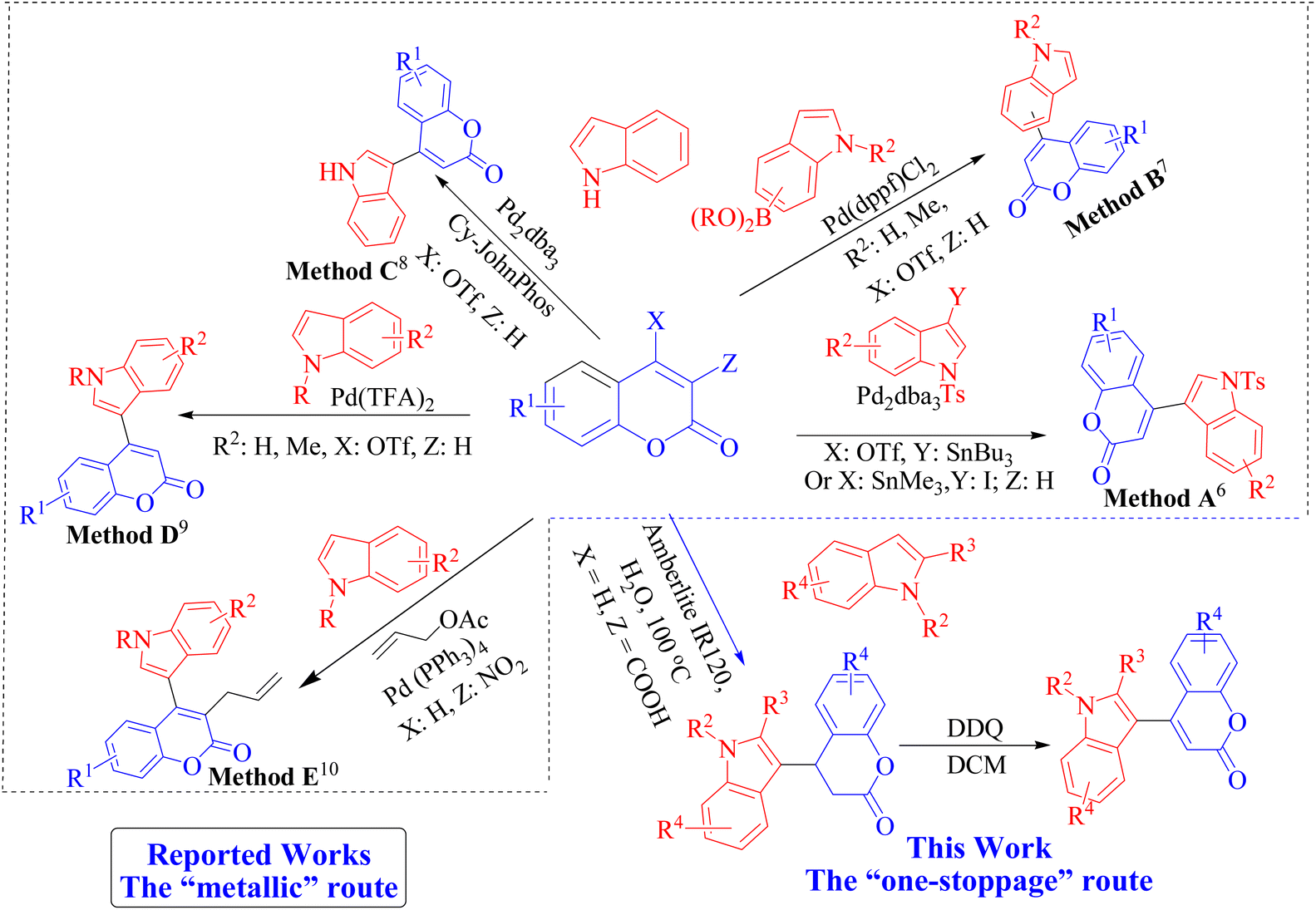
In recent decades, donor–acceptor substituted conjugated organic molecules have attracted much attention due to their wide range of applications in chemistry, biology and material sciences, where they are used in organic electronics, dye-sensitized solar cells (DSSCs), nonlinear optics (NLO), etc.1 Such organic molecules exhibit D–π–A type push–pull phenomena through their conjugated π-systems since one part of the molecule acts as an electron donor and another part acts as an electron acceptor. This structural feature enables intra-molecular charge transfer with increased polarizability and dipole moment, which ultimately impacts the nonlinear optical (NLO) and other properties.2 Coumarins (1) and indoles (2) are two molecular fragments of ample importance because of the frequent occurrence of these scaffolds in numerous natural products,3 pharmaceuticals,4 and materials science.5 Due to their overwhelming significance in drug discovery, the synthesis of compounds containing both these moieties is highly desirable. Thus, the head-to-head connection of these two immensely important molecular motifs into one framework leads to a molecule that exhibits different properties than the parent materials, and the resulting materials are expected to be propitious. However, a challenging task from the synthetic point of view is to gain green, environmentally benign, efficient access to indole-substituted coumarins by forming a new carbon–carbon bond between the 4-position of the coumarin and 3-position of the indole to provide 4-indolyl coumarins (3) (Fig. 1). For this reason, several research groups around the globe have long been engaged in developing high-yielding procedures for the synthesis of indole-coumarin hybrids (3). After the first attempt at the synthesis of 4-indolyl coumarins in 1994 by Ortar6 through a palladium-catalysed cross-coupling reaction of organostannanes with triflates or halides (Method A, Scheme 1), a Suzuki–Miyaura cross-coupling reaction between 4-coumarinyl triflates and indolylboronic acids and pinacolate esters was reported by Combs et al.7 (Method B, Scheme 1) in 2008. Another Pd-catalysed reaction between indoles and 4-coumarinyl triflates was reported by Beletskaya8 (Method C, Scheme 1) and Guo et al.9 (Method D, Scheme 1) for the synthesis of indole-coumarin hybrid molecules. Very recently, a Pd-catalyzed three-component cine, ipso-di-substitution of 3-nitro coumarin towards the synthesis of structurally diverse 4-indolyl coumarin was reported10 by Greening and co-workers (Method E, Scheme 1).
 |
| | Fig. 1 Structures of coumarin, indole and indole-coumarin hybrids. | |
 |
| | Scheme 1 Schematic of reported Pd-catalysed protocols and the present two-step protocol for the synthesis of 4-indolyl coumarin derivatives. | |
Apart from the difficulties associated with the handling of palladium-catalyzed reactions, the availability of starting materials such as triflate, indolylboronic acids, organostannanes, 3-iodoindoles or pinacolate esters in all the reported procedures is a central drawback, stemming from the implementation of the palladium-catalysed cross-coupling strategy. Although a capable catalyst, palladium presents intrinsic potential environmental health hazards, and the extraction of palladium from its ore emits huge amounts of CO2, which burdens our environment.11a–c These procedures also have other objectionable issues, such as the requirement for toxic organic solvents, special setups to create inert atmospheres, lengthy reaction times, expensive reagents and unsatisfactory yields. It is therefore highly desirable to synthesize 4-indolyl coumarin derivatives with reduced generation of metal waste, without compromising the efficiency, by redesigning this reaction using a sustainable and environmentally benign, large-scale-tolerant, and highly atom-economic protocol. Alternatively, 4-indole substituted 3,4-dihydrocoumarins (4) are privileged molecules that are considered biologically and pharmaceutically important molecules that show a wide spectrum of pharmacological and biological activities12 on their own, such as anti-hyperlipidemic, anti-leishmanial, cytotoxic, anti-metastatic and anti-cholinestearase activity. Unfortunately, not many methodologies are available that fulfil the environmental constraints as set by green chemistry principles and satisfy the concerns of the industrial community for the synthesis of 4. Recent reports indicated that 3-nitrocoumarin and coumarin-3-carboxylic acid can be used as Michael acceptors to generate dihydrocoumarin motifs under different reaction conditions.13 Srivastava et al. reported14a a saccharin-based functional ionic liquid mediated multicomponent reaction involving Knoevenagel condensation and Michael addition to yield the indole-3-substituted dihydrocoumarins, and, very recently, an uncatalyzed high-temperature version of the same reaction was reported by Xiao et al.14b However, these reactions have their own drawbacks, such as long reaction times, harsh conditions, high energy requirements, designer catalyst preparation, and tedious workup, which limit the scope of the reaction. Owing to our research activity on the manipulation of bis-heterocyclic compounds,15 we were particularly interested in the metal-free synthesis of 4-indolyl coumarins because their structural uniqueness as D–π–A systems could provide a significant motif for electronic, opto-electronic and spectroscopic studies. Thus, in the present research, we aimed to synthesize 4-indolyl 3,4-dihydrocomarin (4) by adopting a benign protocol as directed by green chemistry principles, followed by oxidative dehydrogenation to the 4-indole substituted coumarin (3) (Scheme 1). We also explored the photophysical properties of the synthesized indole-coumarin hybrid compounds and successfully utilized them as a component of invisible ink.
During our pursuit of sustainable alternative methodologies, we and others have explored the efficacy and catalytic potency of Amberlite IR120 and Amberlyst 15 in various organic transformations.15c,16 Thus, to attain the synthesis of 4-indole substituted 3,4-dihydrocoumarins using a domino Michael addition-decarboxylation reaction between indole and coumarin-3-carboxylic acids, while exploring the possibility of using much greener reaction conditions, guided by our previous experiences,17 we envisioned the use of water as a cheap, nontoxic alternative to organic solvents. However, to reap the benefits that water can offer, we had to search for a suitable water-compatible acid catalyst system that was cheap, stable, recyclable and reusable by simple filtration from the reaction mixture. Based on our recent experience and on literature reports, we were fascinated by Amberlite IR120 and Amberlyst 15 as both contain polymer-supported sulfonic acid (–SO3H) residues for the activation of functional groups. We decided to use these materials as catalysts for the present study under benign conditions, and subsequently advanced our study to access 4-indolyl coumarins by dehydrogenation of the synthesised 3,4-dihydrocoumarins using DDQ as oxidant.18
Results and discussion
To explore the eco-friendly synthetic setup for the 4-indole substituted 3,4-dihydrocoumarins, the reaction between coumarin-3-carboxylic acid (1 mmol) and indole (1.2 mmol), taking their respective roles as Michael acceptor and nucleophilic partner, was adopted as a model reaction (Table 1). At first, the possibilities with solvent- and catalyst-free conditions were explored at 80 °C, but a poor yield (20%) of 4a was obtained after 3 hours (Table 1, entry 1). Then, the scope of the reaction was studied using either pure solvent or a combination of solvents without any catalyst. It was clear that the use of ethanol or ethanol–water mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) did not give encouraging results, even at an elevated temperature of 80 °C (Table 1, entries 2–3). However, water medium, despite showing sluggish results at room temperature (Table 1, entry 4), gave a clear sign of hope by providing a spectacular jump to 60% yield of 4a (Table 1, entry 5) in comparison to ethanol at 80 °C, indicating that water could be a good choice of medium for this reaction. Motivated by this result, to improve the reaction further, the scope of heterogeneous solid acid catalysts was evaluated (Table 1, entries 6–14) at different reaction temperatures in aqueous medium. Heating is crucial for this reaction, given that the uncatalyzed aqueous reaction failed (Table 1, entry 4) and the solid acid catalysts such as Amberlite IR120 and Amberlyst 15 followed the same fate, generating 4a in only 42% (Table 1, entry 6) and 40% (Table 1, entry 7) yields, respectively, at room temperature. Surprisingly, Amberlite IR120 (∼16% wt/wt of coumarin-3-carboxylic acid, 30 mg) performed well at a higher temperature of 80 °C, in combination with water, to produce an encouraging 75% yield of the product 4a (Table 1, entry 8). At the same concentration, Amberlyst 15 struggled to produce any improvement over the previously obtained product yield by using Amberlite IR120 and thus brought about only 65% yield of product 4a at the same temperature (Table 1, entry 9). The most promising result of this ongoing study was found at an elevated temperature of 100 °C with Amberlite IR120, taken at the same concentration (∼16% wt/wt, 30 mg) in an aqueous medium, which gave an 89% yield of product 4a (Table 1, entry 10) and was considered to be the optimum conditions for this reaction. However, under identical conditions, Amberlyst 15 failed to perform consistently, providing only 70% yield of product 4a (Table 1, entry 11), which consolidated the selection of Amberlite IR120 over Amberlyst 15 for this particular reaction. Further, increasing the amount of Amberlite IR120 catalyst threefold (∼48% wt/wt, 90 mg) produced only a marginal improvement in the yield of 4a (90%; Table 1, entry 12) over the standard Amberlite IR120 conditions. Decreasing the Amberlite IR120 catalyst amount further, from ∼16% wt/wt to ∼8% wt/wt, led to a reduction in the yield of 4a from 89%, under the standard conditions, to 65% (Table 1, entry 13), while keeping the temperature constant at 100 °C. We also confirmed that with an increment in temperature from 100 °C to 120 °C (in a closed vessel), while maintaining 16% wt/wt concentration of Amberlite IR120, no noticeable improvement in reaction course was detected (Table 1, entry 14), thus it was considered unnecessary.
:2) did not give encouraging results, even at an elevated temperature of 80 °C (Table 1, entries 2–3). However, water medium, despite showing sluggish results at room temperature (Table 1, entry 4), gave a clear sign of hope by providing a spectacular jump to 60% yield of 4a (Table 1, entry 5) in comparison to ethanol at 80 °C, indicating that water could be a good choice of medium for this reaction. Motivated by this result, to improve the reaction further, the scope of heterogeneous solid acid catalysts was evaluated (Table 1, entries 6–14) at different reaction temperatures in aqueous medium. Heating is crucial for this reaction, given that the uncatalyzed aqueous reaction failed (Table 1, entry 4) and the solid acid catalysts such as Amberlite IR120 and Amberlyst 15 followed the same fate, generating 4a in only 42% (Table 1, entry 6) and 40% (Table 1, entry 7) yields, respectively, at room temperature. Surprisingly, Amberlite IR120 (∼16% wt/wt of coumarin-3-carboxylic acid, 30 mg) performed well at a higher temperature of 80 °C, in combination with water, to produce an encouraging 75% yield of the product 4a (Table 1, entry 8). At the same concentration, Amberlyst 15 struggled to produce any improvement over the previously obtained product yield by using Amberlite IR120 and thus brought about only 65% yield of product 4a at the same temperature (Table 1, entry 9). The most promising result of this ongoing study was found at an elevated temperature of 100 °C with Amberlite IR120, taken at the same concentration (∼16% wt/wt, 30 mg) in an aqueous medium, which gave an 89% yield of product 4a (Table 1, entry 10) and was considered to be the optimum conditions for this reaction. However, under identical conditions, Amberlyst 15 failed to perform consistently, providing only 70% yield of product 4a (Table 1, entry 11), which consolidated the selection of Amberlite IR120 over Amberlyst 15 for this particular reaction. Further, increasing the amount of Amberlite IR120 catalyst threefold (∼48% wt/wt, 90 mg) produced only a marginal improvement in the yield of 4a (90%; Table 1, entry 12) over the standard Amberlite IR120 conditions. Decreasing the Amberlite IR120 catalyst amount further, from ∼16% wt/wt to ∼8% wt/wt, led to a reduction in the yield of 4a from 89%, under the standard conditions, to 65% (Table 1, entry 13), while keeping the temperature constant at 100 °C. We also confirmed that with an increment in temperature from 100 °C to 120 °C (in a closed vessel), while maintaining 16% wt/wt concentration of Amberlite IR120, no noticeable improvement in reaction course was detected (Table 1, entry 14), thus it was considered unnecessary.
Table 1 Optimization studies of 4-indole substituted 3,4-dihydrocoumarin

|
| Entry |
Catalyst |
Solvent/medium |
Temp (°C) |
Time (hrs) |
Yielda (%) |
| Isolated yield. 10 mL. 10 mL (1:2). Coumarin-3-carboxylic acid (1 mmol), indole (1.2 mmol). |
| 1 |
No |
No |
80 |
3 |
20 |
| 2 |
No |
EtOHb |
80 |
3 |
32 |
| 3 |
No |
EtOH–H2Oc |
80 |
3 |
43 |
| 4 |
No |
H2Ob |
r.t |
12 |
Trace |
| 5 |
No |
H2Ob |
80 |
3 |
60 |
| 6 |
Amberlite IR120 (∼16% wt/wt, 30 mg) |
H2Ob |
r.t |
3 |
42 |
| 7 |
Amberlyst 15 (∼16% wt/wt, 30 mg) |
H2Ob |
r.t |
3 |
40 |
| 8 |
Amberlite IR120 (∼16% wt/wt, 30 mg) |
H2Ob |
80 |
3 |
75 |
| 9 |
Amberlyst 15 (∼16% wt/wt, 30 mg) |
H2Ob |
80 |
3 |
65 |
| 10 |
Amberlite IR120 (∼16% wt/wt, 30 mg) |
H2Ob |
100 |
3 |
89 |
| 11 |
Amberlyst 15 (∼16% wt/wt, 30 mg) |
H2Ob |
100 |
3 |
70 |
| 12 |
Amberlite IR120 (∼48% wt/wt, 90 mg) |
H2Ob |
100 |
3 |
90 |
| 13 |
Amberlite IR120 (∼8% wt/wt, 15 mg) |
H2Ob |
100 |
3 |
65 |
| 14 |
Amberlite IR120 (∼16% wt/wt, 30 mg) |
H2Ob |
120 |
3 |
89 |
After the initial success of the reaction between indole and coumarin-3-carboxylic acid under optimized reaction conditions (Table 1, entry 10) with the appropriate catalyst proportion, we explored the outcomes by varying indole substitutions with different coumarin-3-carboxylic acids (Fig. 2). Indole rings bearing electron-donating groups, such as –OCH3, or withdrawing groups, such as –Br, performed nicely, giving good yields of 4b (91%) and 4c (87%), respectively, when they were allowed to react with unsubstituted coumarin-3-carboxylic acids. However, of course, indoles containing an electron-donating group produced slightly better yields because of the enhanced nucleophilicity of the C-3 position of the indole moiety. Evidently, unsubstituted coumarin-3-carboxylic acid reacted swiftly with indole bearing methyl substitution at C-2, despite the steric hindrance, producing 89% yield of 4d. Again, N-substituted indoles, such as N-methyl and N-benzyl substituted substrates, performed well to produce 4e and 4f in 88% and 92% yield, respectively. The reaction scope was then extended by using coumarin-3-carboxylic acid derivatives bearing an electron-withdrawing –Cl group with differently substituted indoles, producing consistent 89–92% yields of the expected products 4g–k. Similarly, coumarin-3-carboxylic acids bearing –Br or sterically hindered tert-butyl groups fared encouragingly, generating products 4l and 4m in 91% and 90% yields, respectively. However, this reaction protocol failed for the reaction between coumarin-3-carboxylic acids with a heteroatom bearing electron-donating groups like –NEt2, –OMe or –OH attached at the 7-position, even after 12 h. A reason may be –NEt2, –OMe or –OH groups exhibit highly electron-pushing natures by exerting a +R effect, which can decrease the electrophilicity of the 4-position of the coumarin-3-carboxylic acid, thereby rendering the nucleophilic attack by the indole unfavorable. Thus, this protocol is evidently applicable to unsubstituted coumarin-3-carboxylic acid or derivatives bearing electron-withdrawing groups.
 |
| | Fig. 2 Substrate scope for the synthesis of 4-indolyl 3,4-dihydrocoumarins. | |
Mechanism
A plausible mechanism for the reaction between coumarin-3-carboxylic acid and indole in heterogeneous solid acid catalyst on water is presented in Fig. 3. It is reasonably believed that an initial stabilization of coumarin-3-carboxylic acid takes place by a sulfonic acid group of the solid acid via a transition state involving two six-membered rings (A) by the participation of multiple hydrogen bonds between the sulfonic acid proton of Amberlite IR120 and the lactone carbonyl of the coumarin unit as well as a carboxylic acid proton of the coumarin-3-carboxylic acid.
 |
| | Fig. 3 Proposed mechanism. | |
Further, simultaneous H-bond donation by the carboxylic acid group and H-bond acceptance by the lactone carbonyl of the coumarin-3-carboxylic acid leads to an H-bond donor–acceptor dual activation provided by Amberlite IR120 to coumarin-3-carboxylic acid, which concertedly increases the electrophilicity at the 4 C center of coumarin-3-carboxylic acid. The increased electrophilicity of the coumarin-3-carboxylic acid is fully exploited by the donor indole to undergo a Michael addition followed by a facile decarboxylation, which is assisted by the adjacent lactone carbonyl, to afford the 4-indole substituted 3,4-dihydrocoumarin derivatives (4). This type of H-bonding donor–acceptor dual activation is anticipated to become important for this reaction because the reactions with ethyl coumarin-3-carboxylate or 3-acetyl coumarin and indole were drastic failures. It may be reasoned that simple coumarin/ethyl coumarin-3-carboxylate/3-acetyl coumarin compounds bearing no carboxylic acid proton may prevent the formation of hydrogen bonding possibilities required for the H-bonded transition state (A), thus the H-bonding donor–acceptor dual activation mechanism is interrupted, and the reaction does not proceed any further. To substantiate the proposed mechanism, we conducted 1H NMR experiments with coumarin-3-carboxylic acid (1) and Amberlite IR120 in D2O, as suggested by one of the esteemed reviewers. We were pleased to observe a 0.03 ppm downfield shift of H-4 in the 1H NMR spectrum due to the interaction of Amberlite IR 120 with the carboxylic acid functional group (see the SI for supporting spectra). These observations confirm that the presence of a carboxylic acid group on the 3-position of the coumarin is absolutely indispensable.
Scale-up of the methodology and recyclability of the catalyst
To establish the industrial credibility of this reaction protocol, it is interesting to scale up the reaction based on the successful synthesis of 4. As the vessel becomes larger, the parameters controlling the ‘length scale’ and ‘time scale’ are difficult to maintain constantly, so there will be some expected deviation from the perspective of industrial scalability. For that, the initial reaction between coumarin-3-carboxylic acid and indole was up-scaled to 30 mmol, and we were pleased to isolate the product 4a in 87% yield (6.86 g), which is very much comparable to the yield of the 1 mmol scale (89%) reaction.
Also, with this newly developed protocol involving Amberlite IR120 catalyst, a consistent yield of product was observed with a very tolerable increase in reaction time (4 hours) in comparison to the initially performed reaction at 1 mmol scale (3 hours). The scale comparison results are shown in Fig. 4. To exploit the advantages of this heterogeneous solid acid catalyst, the recyclability aspects of the Amberlite IR120 were assessed on a 1 mmol scale. After completion of the initial reaction between coumarin-3-carboxylic acid and indole, the reaction mixture was then filtered from the water and dried. The product was then separated from the mixture by dissolving it using ethyl acetate. Pure product 4a was obtained from the ethyl acetate by crystallization or chromatographic techniques, while the dried resin was utilized for the next cycle. The process was repeated for five consecutive cycles following the initial run, and the recyclability results are shown in the Fig. 4. To our pleasure, no substantial reduction in yield was observed, with recurring yields losing no more than 1% of the initial yield over five consecutive cycles, which proves the enormous recyclable potential of the Amberlite IR120 solid acid catalyst and indicates that it can be used industrially as a promising “no waste” catalyst.
 |
| | Fig. 4 Methodology scale-up and recyclability of the catalyst. | |
Oxidation of 4-indolyl dihydrocoumarin (4) to 4-indolyl coumarin (3)
After the successful synthesis of the series of 4-indole substituted 3,4-dihydrocoumarins, the products were treated with DDQ as a versatile and widely accepted oxidant of choice because of its high reduction potential. In an initial trial, we considered dihydrocoumarin 4a (1 mmol) as a model substrate and the DDQ (1.3 mmol) in dichloromethane and THF at room temperature. However, this attempt produced only 42% and 35% yield of the oxidized product 3a after 60 minutes. Then, hoping to improve the reaction yield, we screened the reactions at a higher temperature (60 °C) while keeping the same amount of DDQ in different solvents, such as DCM, THF, MeOH and ACN. Astonishingly, at 60 °C, DCM (using a cold-bath circulator cooling system) was found to be the most suited solvent for this reaction, providing 95% yield of the oxidized product 3a after 60 minutes, while the other solvents (THF: 84%; MeOH: 66%; ACN: 60%) lagged behind. The reaction was also conducted with a 1:1 ratio of DDQ to 3a, but only 83% of product 3a was noted, which justifies the use of a slight excess of DDQ (standard 1.3 equivalent) over the starting dihydrocoumarin. After the optimization of the synthesis of 3a from 4a, a series of 4-indolyl 3,4-dihydrocoumarins (4b, 4d–e, 4g–h and 4j; Fig. 2) were utilized to synthesize the corresponding 4-indolyl coumarins. We observed a clean conversion of 4 into the oxidised products 3b–g within 60 min in yields of 94–98% (Fig. 5).
 |
| | Fig. 5 DDQ oxidation of 4-indolyl 3,4-dihydrocoumarin. | |
It is also worth mentioning that the overall yields calculated for the two-step reaction, consisting of Amberlite IR120 and water-accelerated Michael addition-decarboxylation and the subsequent oxidation reaction, were in the range of 84–91%. These overall yields are encouragingly higher than the yields found in the reported single-step protocol involving palladium cross-coupling between 4-coumarinyltriflate and substituted indole7,9 to produce comparable 4-indolylcoumarins. So, this work provides a new sustainable, green approach towards the synthesis of 4-indolylcoumarins without compromising the efficiency, while avoiding the metal cross-coupling route (Scheme 2).
 |
| | Scheme 2 Comparison of the present methodology with the reported9 methods. | |
Structural features
The structures and identities of all the synthesised compounds were determined by 1H, 13C NMR, IR and mass spectral analyses. We also tried to have suitable crystals for single crystal XRD studies since the crystal structures of organic molecules are often reported to stabilize peculiar supramolecular architecture supported by non-classical interactions.19,20 The non-classical interactions are very important for understanding and designing advanced functional materials with specific properties and in drug design and delivery. Therefore, after the characterization of all the synthesised compounds, single crystals of 4-indolyl 3,4-dihydrocoumarin (4g) and 4-indolyl coumarin (3g) were grown in methanol–water (3:1) and DCM–hexane (2:1) solvent systems, respectively, for X-ray diffraction study. The ORTEP diagrams at 50% thermal probability are presented in Fig. 6(a) and (c). Compound 4g is found to have a nonplanar structure in which the indole and dihydrocoumarin rings lie in different planes. Furthermore, the lattice stacking of compound 4g clearly suggested the presence of intermolecular H-bonding between the indole N–H of one molecule and the lactone carbonyl oxygen of another molecule, with a bond length of 2.42 Å [Fig. 6(b); green dotted lines]. Similarly, the crystal structure of 3g revealed that the two heterocyclic rings are located in different planes, with a dihedral angle of ∼41.7° [Fig. 6(d)]. The distortion of the internal planes makes the molecule 3g ‘V-shaped’, which is responsible for a unique stacking of molecules within its crystal lattice, whereby the molecules are stacked upside-down alternatively over one another as shown in Fig. 6(e). There is also extensive intermolecular hydrogen bonding between the N–H proton of the indole moiety of one molecule and the lactone carbonyl oxygen of the coumarin moiety of another molecule, with the two molecules residing in two different planes [Fig. 6(f)]. The lengths of these interlayer and intermolecular H-bonds were found to be 2.008 Å (green dotted lines) and 1.985 Å (orange dotted lines), respectively. The solved crystal structures of compounds 4g and 3g were deposited in the Cambridge Crystallographic Centre (CCDC 2326700, CCDC 2326704), and all the crystallographic parameters of compounds 4g and 3g are provided as SI (Table S1). In the crystal structure of 3g, containing Cl and OMe substituents, a rare pentamer of water molecules was observed; these water molecules are stabilized by hydrogen-bonding interaction with the carbonyl units of the two coumarin molecules at the two extreme ends of the molecule. An outstanding architecture was revealed upon expanding the hydrogen-bonding contacts in the crystal structure [Fig. 7(B)], where five hydrogen-bonded water molecules could be clearly seen forming a zig–zag open pentameric structure, supported by two units of 3g at the distal ends involving the carbonyl groups by hydrogen-bonding in the same type of interaction. Apart from the aforementioned interactions, 3g forms a symmetric polymeric sheet by a C–H⋯Cl type hydrogen-bonding interaction [Fig. 7(C)].
 |
| | Fig. 6 (a) ORTEP diagram of 4g; (b) H-bonded network in the solid state of 4g; (c) ORTEP diagram of 3g; (d) dihedral angle between the two heterocyclic rings in 3g; (e) stacking patterns of 3g in the crystal lattice and (f) H-bonded network in the solid state of 3g. | |
 |
| | Fig. 7 (A) ORTEP diagram of 3g showing the presence of water molecules in the crystal; (B) pentamer of water stabilized by hydrogen-bonding interactions and (C) The C–H⋯Cl hydrogen-bonded 1D network formed in 3g. | |
Photo-physical studies
The photophysical properties of coumarin derivatives, particularly those bearing an electron-releasing group at the 7-position, have been extensively studied for a long time.21 However, no previous report existed on the photophysical dissection of coumarins substituted with an indole ring, a towering lacuna because indole derivatives are also reported to be efficient design-specific fluorophores.22 For this reason, the photophysical study of the synthesized coumarin-indole derivatives was deemed important. To understand the effect of the substituents on the optical properties of the coumarin-indole derivatives, the fully unsubstituted 4-indolyl coumarin (3a) and 4-indolyl coumarin, containing Cl and OMe groups in different locations (3g), were chosen. To begin with, the photophysical aspects of 3a and 3g were investigated in the solution phase. The steady-state and time-resolved data are presented in Fig. 8. Both of the compounds showed a broad absorption maximum at ∼350 nm in chloroform and acetonitrile, indicative of an S0–S1 type transition. The almost non-variant position of the absorption maximum in solvents of varying polarity (εchloroform = 4.1 and εacetonitrile = 5.8) signified little dependence on the polarity of the ground state absorption of the molecules [Fig. 8(A)]. Upon excitation at 330 nm [Fig. 8(B)], 3a did not show any significant change in the emission maximum upon changing the solvent (λmaxem in chloroform ∼465 nm and λmaxem in acetonitrile ∼475 nm). However, the notable Stokes Shift (ν = 8798 cm−1 in chloroform and 9250 cm−1 in acetonitrile) signified considerable charge separation in the excited state of 3a, an outcome of the innate push–pull nature of the molecular architecture of 3a itself (indole being the donor and coumarin being the acceptor). The shift in emission maximum was remarkable in 3g (λemmax in chloroform ∼470 nm and λemmax in acetonitrile ∼570 nm), indicating the added advantage of introducing the Cl and OMe groups in the core of 3a. The relative quantum yield (Φf) in the solution phase was calculated by taking anthracene as a standard, the quantum yield of which is known (0.36 in n-hexane), using the standard equation,  where the subscripts R and S refer to the reference anthracene and samples (i.e. 3a and 3g), respectively. Ai denotes the absorbance values at the wavelength monitored (i.e., 330 nm), Areai refers to the integrated area of the fluorescence spectra obtained for the samples and the reference, and ηi refers to the refractive index of the medium. The quantum yield values for 3a in hexane and acetonitrile were calculated to be 0.07 and 0.054, respectively. The quantum yields for 3g in hexane and acetonitrile were calculated to be 0.018 and 0.012, respectively. A perusal of the quantum yields leads to the following observations: (i) The quantum yield values decreased for each of 3a and 3g upon increment in the polarity of the solvents; (ii) the overall quantum yield of 3g was appreciably lower than that of 3a, irrespective of solvent. These observations strongly support an ICT process operative in 3a and 3g and indicate that it is more predominant in 3g. Our assignment finds justification by considering the established result that ICT is essentially a non-radiative process in the molecule that leads to a reduction in the quantum yield of fluorescence in organic fluorophores.23 However, we could not calculate the absolute quantum yield in the solid-state due to the limited experimental setup; nevertheless, the red-shift in the steady-state emission spectrum of 3g (λmax ∼500 nm) as compared to 3a (λmax ∼450 nm) in the solid-state strongly indicates the existence of ICT in the solid-state as well.24
where the subscripts R and S refer to the reference anthracene and samples (i.e. 3a and 3g), respectively. Ai denotes the absorbance values at the wavelength monitored (i.e., 330 nm), Areai refers to the integrated area of the fluorescence spectra obtained for the samples and the reference, and ηi refers to the refractive index of the medium. The quantum yield values for 3a in hexane and acetonitrile were calculated to be 0.07 and 0.054, respectively. The quantum yields for 3g in hexane and acetonitrile were calculated to be 0.018 and 0.012, respectively. A perusal of the quantum yields leads to the following observations: (i) The quantum yield values decreased for each of 3a and 3g upon increment in the polarity of the solvents; (ii) the overall quantum yield of 3g was appreciably lower than that of 3a, irrespective of solvent. These observations strongly support an ICT process operative in 3a and 3g and indicate that it is more predominant in 3g. Our assignment finds justification by considering the established result that ICT is essentially a non-radiative process in the molecule that leads to a reduction in the quantum yield of fluorescence in organic fluorophores.23 However, we could not calculate the absolute quantum yield in the solid-state due to the limited experimental setup; nevertheless, the red-shift in the steady-state emission spectrum of 3g (λmax ∼500 nm) as compared to 3a (λmax ∼450 nm) in the solid-state strongly indicates the existence of ICT in the solid-state as well.24
 |
| | Fig. 8 (A) Absorption spectra of 3a and 3g in the solution phase. (B) Emission spectra of 3a and 3g in the solution phase. (inset) digital photographs of 3a and 3g in chloroform and acetonitrile taken using a handheld UV torch of 365 nm. (C) Emission lifetime decays of 3a and 3g in chloroform upon excitation at 366 nm and monitoring the emission at 500 nm. | |
The lifetime emission studies were confined to 3a due to very poor emission from 3g in solution [Fig. 8(C)]. It was observed that 3a relaxes to the ground state following a mono-exponential decay with a lifetime of 5.4 ns, indicative of intramolecular charge transfer (ICT) occurring in the same.20 The poor emission from 3g could be explained by considering the enhanced ICT, which is often associated with the loss of emission aptitude in organic fluorophores.25 In the solid state (λmax = 330 nm), the trend in the emission maxima (λemmax for 3a ∼450 nm and λemmax for 3g ∼500 nm) was the same as in the solution phase, albeit with a considerably reduced Stokes shift (ν = 8100 cm−1 in 3a and 10303 cm−1 in 3g) (Fig. 9). The decrement in the Stokes shift bore further testament to the occurrence of excited-state ICT in the molecular scaffolds, which requires dielectric stabilization from the solvent molecules, the absence of which reduced the extent of ICT, as reflected in the lower value of the Stokes shift in the solid phase as compared to the solution phase. However, the greater extent of ICT in 3g could be perceived by observing its color under UV light (365 nm), where the sample emitted a yellowish green color (Fig. 9 inset). The emission lifetime decays in the solid state were observed at various wavelengths across the emission spectrum for both compounds, but showed a bi-exponential nature of decay with lifetime values of τ1 = 0.6–1.0 ns and τ2 = 2.0–2.5 ns. The lower lifetime values compared to the solution phase are indicative of collisional quenching in the solid state. At this stage, Scheme 3 was introduced to illustrate the photophysical events26 occurring in 3a and 3g. The influence of the conjugation was vital for the emission properties of 3a and 3g. To validate this, we recorded the steady-state spectra of the corresponding dihydro analogues 4a and 4j. To our satisfaction, neither of the two compounds showed any shift in emission maxima upon changing the polarity of the solvents, thereby ruling out the possibility of an ICT operating in the dihydro analogue. Hence, the nature of the ICT in 3a and 3g was a “through-bond” type, as noted in other reports.25
 |
| | Fig. 9 (A) Emission spectra of 3a and 3g in the solid state [inset – color of 3g under ambient light (left) and UV-light of wavelength 365 nm (right)]; (B) emission lifetime decays of 3a in the solid state and (C) emission lifetime decays of 3g in the solid state upon excitation at 366 nm and monitoring the emission at the indicated wavelength values. | |
 |
| | Scheme 3 Schematic representation of photo-physical events in 3a and 3g (not to scale). | |
DFT treatment of the ICT phenomenon in 3a and 3g
Steady-state and time-resolved spectroscopic experiments established the ICT process operative in 3a and 3g. To further visualize the ICT, we performed DFT calculations on the two compounds using Gaussian 09W software and B3LYP 6-311 g(d, p) functional.15a,b Upon optimization, the Mulliken charge densities were visualized (Fig. 10) from the optimized HOMO and LUMO diagrams of 3a and 3g. For both 3a and 3g, the charge densities underwent a complete shift moving from the HOMO to the LUMO, indicating a push–pull charge transfer is innately operative in molecules 3a and 3g. Compared to 3a, the extent of ICT in 3g was even more pronounced, as evidenced by the complete delocalisation of charge from the indole moiety to the coumarin moiety. Hence, DFT calculations further supported the ICT mechanism.
 |
| | Fig. 10 HOMO and LUMO diagrams of 3a and 3g showing the Mulliken charge distribution. The green lobes represent the positive charge, and the red lobes represent the negative charge. | |
Invisible ink
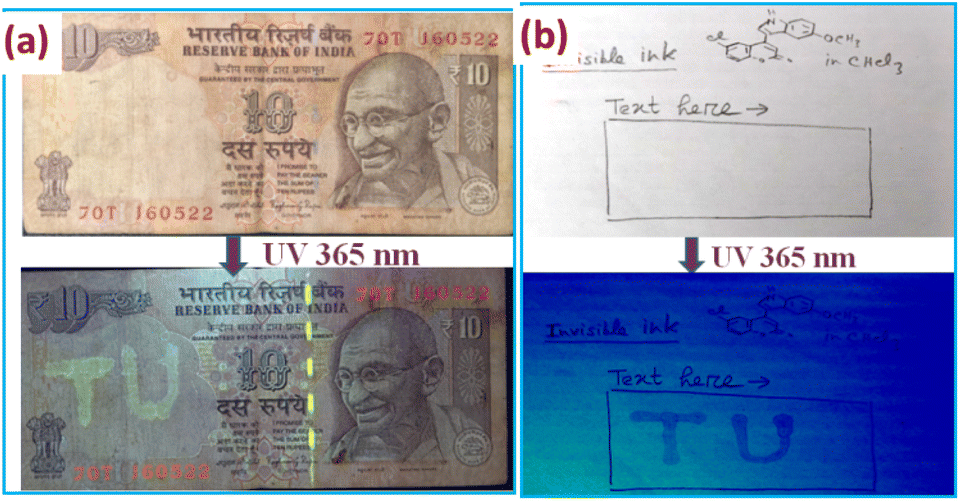
Encouraged by the green emission of 3g in the solid state, its potential to be used as an invisible ink was explored on two substrates: an Indian currency note and writing paper (Fig. 11). Currency notes with TU (abbreviation for Tripura University) written on them with a chloroform solution of 3g was exposed under UV light, revealing the letters having a yellowish green emission [Fig. 11(a)]. The stability of the ink was tested by wetting the currency notes with tap water, which led to minimal change in the intensity of the ink as observed under UV light. The longevity of the ink was also tested by leaving the note under ambient conditions for six months, whereupon the intensity of the ink remained the same. The utility of 3g as an invisible ink on white paper as a substrate is also commendable, although the colour of the ink was observed to be bluish [(Fig. 11(b))] instead of the usual yellowish green, a consequence of strong reflectance of the excitation UV light by the surface of paper, thereby masking the true emission colour of 3g.
 |
| | Fig. 11 (a) Indian currency notes with TU written on it, as observed under ambient and UV light. (b) Writing paper with TU written on it as observed under ambient (top) and UV light (bottom). | |
Conclusion
In conclusion, we have developed a metal-free, eco-friendly, two-step approach to the synthesis of 4-indolyl coumarins starting from structurally diverse coumarin-3-carboxylic acids and substituted indoles, resulting in the synthesis of 4-indolyl 3,4-dihydrocoumarins, which are then oxidized using an organic oxidant in a two-step sequence. The primary highlights of the current approach over the traditional cross-coupling route are the use of water as a green medium, instead of harmful organic solvents, together with the recoverable and reusable heterogeneous solid acid Amberlite IR120, while providing the same level of efficiency and easy-to-handle protocol. The structures of the synthesized compounds of both classes were fully characterized by NMR and IR spectroscopy, and the newly synthesized compounds were also characterized by high-resolution mass spectral analysis. For structural assessment, X-ray diffraction studies were carried out for two compounds that were highly crystalline in nature, namely 4g and 3g, as representatives of the 4-indole substituted 3,4-dihydrocoumains and 4-indolyl coumarins, respectively, to establish the spatial structures within their single crystals. From steady-state and time-resolved spectroscopic studies, it was established that the donation of the push–pull groups Cl and OMe leads to brightening of the emission of the molecular core from blue (3a) to yellowish green (3g) in the solution and solid-phase (powdered form). Finally, the yellowish-green emission of 3g was successfully exploited as a moisture-resistant invisible ink that is revealed only under UV light, an extremely important utility that has been observed for other classes of fluorophores for anti-counterfeiting purposes.27,28 To our knowledge, this is the first report of the use of a coumarin-indole hybrid as an invisible ink.
Experimental section
General procedure for the synthesis of dihydrocoumarins (4)
A mixture of coumarin-3-carboxylic acid (1.0 mmol, 0.190 g), indole (1.2 mmol, 0.140 g), Amberlite IR120 (10 mol%, 30 mg) and water (10 mL) was refluxed with constant stirring (Table 1, entry 10). After completion of the reaction, as indicated by TLC, the Amberlite resin was filtered, the reaction mixture was dissolved in ethyl acetate to recover the resin, and the organic layer was dried over anhydrous sodium sulphate. Finally, the product was purified over column chromatography using a silica gel pad (100–200 mesh) and EtOAc:hexane (1:4) as mobile phase, to give the analytically pure sample.
General procedure for the synthesis of 4-indolylcoumarins (3)
A suitable indole-substituted 3,4-dihydrocoumarin (4, 1 mmol) was dissolved in dichloromethane (5 mL) taken in a 25 mL round-bottomed flask, then DDQ (1.3 equiv.) was added to the stirring solution, and the mixture was heated at 60 °C for 60 min using a cold bath circulator. After the disappearance of the starting material, as indicated by TLC, the mixture was filtered to remove the formed DDQ-H2 and washed thoroughly with dichloromethane. The combined filtrate was evaporated to a residue, and the product was purified over silica gel (100–200 mesh) column using silica EtOAc:hexane (2:3) as eluant, to obtain the analytically pure product (3).
Author contributions
S. M. designed the work. S. C. and B. D. performed all experiments. A. B. contributed to the photo-physical and lifetime measurement studies. B. P. and R. N. performed XRD studies and the processing of the X-ray data. S. M. and S. C. analyzed the spectral data and wrote the manuscript with input from A. B. All authors reviewed and approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare. The authors also declare that no funds, grants, or other support were received during the preparation of this manuscript except for the fellowships.
Data availability
CCDC 2326700 and 2326704 contain the supplementary crystallographic data for this paper.29
The data supporting this article are available as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ra05851a.
Acknowledgements
We are grateful to the Central Instrumentation Center (CIC), Tripura University, for the instrumental facility. S. C. is thankful to the University Grant Commission (UGC), Govt. Of India, for providing a UGC-Senior Research Fellowship [No. 16–6(DEC. 2018)/2019(NET/CSIR)]. B. D. sincerely acknowledges Tripura University for providing the Non-NET fellowship.
Notes and references
-
(a) S. Nizamuddin, S. Nousheen, M. Navadharsana, B. Yadagiri, K. Narayanaswamy, S. P. Singh and G. D. Sharma, J. Mater. Chem. A, 2025, 13, 11114 RSC;
(b) K. E. Dorfman, F. Schlawin and S. Mukamel, Rev. Mod. Phys., 2016, 88, 045008 CrossRef;
(c) X. Wan, C. Li, M. Zhang and Y. Chen, Chem. Soc. Rev., 2020, 49, 2828 RSC.
-
(a) M. Hao, W. Chi, C. Wang, Z. Xu, Z. Li and X. Liu, Phys. Chem. C, 2020, 124, 16820 CrossRef CAS;
(b) Y. Li, P. Han, X. Zhang, J. Zhou, X. Q. D. Yang, A. Qin, B. Z. Tang, J. Peng and D. Ma, J. Mater. Chem. C, 2023, 11, 3284 RSC.
-
(a) M. I. Hussain, Q. A. Syed, M. N. K. Khattak, B. Hafez, M. J. Reigosa and A. El-Keblawy, Biol., 2019, 74, 863 Search PubMed;
(b) V. M. Norwood and R. W. Huigens, Chem. Bio. Chem., 2019, 20, 2273 CrossRef CAS PubMed;
(c) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecule, 2013, 18, 6620 CrossRef CAS PubMed.
-
(a) L. Zhang and Z. Xu, Eur. J. Med. Chem., 2019, 181, 111587 CrossRef CAS PubMed;
(b) M. Yildirim, S. Poyraz and M. Ersatir, Med. Chem. Res., 2023, 32, 617 CrossRef CAS.
-
(a) O. Younis, M. Sayed, A. A. K. Mohammed, M. S. Tolba, R. Hassanien, A. M. Kamal El-Dean, O. Tsutsumi and M. Ahmed, ACS Omega, 2022, 7, 15016 CrossRef CAS PubMed;
(b) J. M. Cuevas, R. Seoane-Rivero, R. Navarro and Á. Marcos-Fernández, Polym. J., 2020, 12, 630 CAS.
-
(a) P. G. Ciattini, E. Morera and G. Ortar, Synth. Commun., 1995, 25, 2883 CrossRef CAS;
(b) P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1994, 35, 2405 CrossRef CAS.
- O. G. Ganina, E. Daras, V. Bourgarel-Rey, V. Peyrot, A. N. Andresyuk, J. P. Finet, A. Y. Fedorov, I. P. Beletskaya and S. Combes, Bioorg. Med. Chem., 2008, 16, 8806 CrossRef CAS PubMed.
- O. G. Ganina, A. Y. Fedorov and I. P. Beletskaya, Synthesis, 2009, 21, 3689 Search PubMed.
- T. Guo, Y. Liu, Y. H. Zhao, P. K. Zhang, S. L. Han and H. M. Liu, Tetrahedron Lett., 2016, 57, 4629 CrossRef CAS.
- V. Vedovato, A. J. Gangano, I. Ghiviriga and A. J. Grenning, Org. Lett., 2024, 26, 647 CrossRef CAS PubMed.
-
(a) J. Kielhorn, C. Melber, D. Keller and I. Mangelsdorf, Int. J. Hyg. Environ. Health., 2002, 205, 417 CrossRef CAS PubMed;
(b) S. Mccarthy, A. Lee, W. Jie, D. C. Braddock, A. Serpe and J. D. E. T. Wilton-Ely, Molecules, 2021, 26, 5217 CrossRef CAS PubMed;
(c) T. Perić, V. L. Jakovljević, V. Zivkovic, J. Krkeljic, Z. D. Petrović, D. Simijonović, S. Novokmet, D. M. Djuric and S. M. Janković, Med. Chem., 2012, 8, 9 CrossRef PubMed.
-
(a) J. N. Sangshetti, F. A. Kalam Khan, A. A. Kulkarni, R. H. Patil, A. M. Pachpinde, K. S. Lohar and D. B. Shinde, Bioorg. Med. Chem. Lett., 2016, 26, 829 CrossRef CAS PubMed;
(b) A. H. Halawa, S. M. Abd El-Gilil, A. H. Bedair, E. M. Eliwa, M. Frese, N. Sewald, M. Shaaban and A. M. El-Agrody, Med. Chem. Res., 2018, 27, 796 CrossRef CAS;
(c) P. R. Kamath, D. Sunil, M. M. Joseph, A. A. Abdul Salam and T. T. Sreelekha, Eur. J. Med. Chem., 2017, 136, 442 CrossRef CAS PubMed;
(d) S. Ghanei-Nasab, M. Khoobi, F. Hadizadeh, A. Marjani, A. Moradi, H. Nadri, S. Emami, A. Foroumadi and A. Shafiee, Eur. J. Med. Chem., 2016, 121, 40 CrossRef CAS PubMed.
- M. C. Ye, Y. Y. Yang, Y. Tang, X. L. Sun, Z. Ma and W. M. Qin, Synlett, 2006, 8, 1240 Search PubMed.
-
(a) A. Kumar, P. Kumar, V. D. Tripathi and S. Srivastava, RSC Adv., 2012, 2, 11641 RSC;
(b) Z. Shao, L. Xu, L. Wang, H. Wei and J. Xiao, Org. Biomol. Chem., 2014, 12, 2185 RSC.
-
(a) B. Das, A. Bhattacharyya, B. Paul, R. Natarajan and S. Majumdar, RSC Adv., 2024, 14, 33512 RSC;
(b) B. Das, K. Das, U. C. De and S. Majumdar, RSC Adv., 2025, 15, 5523 RSC;
(c) B. Das, A. Rudra Paul, U. C. De, B. Paul, R. Natarajan, S. Chakraborty and S. Majumdar, ChemistrySelect, 2023, 8, e20230323 Search PubMed.
-
(a) M. Chakraborty, B. Deb, B. Dey, S. A. Hussain, D. K. Maiti and S. Majumdar, ChemistrySelect, 2017, 2, 241 CrossRef CAS;
(b) A. Rudra Paul, S. Sarkar, J. Hossain, S. A. Hussain and S. Majumdar, Res. Chem. Intermed., 2022, 48, 4963 CrossRef CAS;
(c) K. Niknam, H. Hashemi, M. Karimzadeh and D. Saberi, J. Iran. Chem. Soc., 2020, 17, 3095 CrossRef CAS;
(d) M. Chakraborty, B. Deb, B. Dey, S. A. Hussain, D. K. Maiti and S. Majumdar, ChemistrySelect, 2017, 2, 241 CrossRef CAS.
-
(a) S. Chakraborty, A. Rudra Paul and S. Majumdar, Results Chem., 2022, 4, e100294 CrossRef;
(b) S. Chakraborty, B. Paul, U. C. De, R. Natarajan and S. Majumdar, RSC Adv., 2023, 13, 6747 RSC;
(c) S. Chakraborty, B. Paul, R. Natarajan, J. Hossain and S. Majumdar, ChemistrySelect, 2023, 8, e202204128 CrossRef CAS.
- M. A. Alsharif, Q. A. Raja, N. A. Majeed, R. S. Jassas, A. A. Alsimaree, A. Sadiq, N. Naeem, E. U. Mughal, R. I. Alsantali, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 29826 Search PubMed.
- S. Dalapati, M. A. Alam, R. Saha, S. Jana and N. Guchhait, Cryst. Eng. Comm., 2012, 14, 1527 RSC.
- V. Bhakta, S. Chatterjee, A. Bhattacharyya and N. Guchhait, J. Phys. Chem. A, 2024, 128, 8914 CrossRef CAS PubMed.
- A. Rudra Paul, B. Dey, S. Suklabaidya, S. A. Hussain and S. Majumdar, RSC Adv., 2021, 11, 10212 RSC.
- H. L. Liu, K. Zhan, K. L. Zhong, X. L. Chen and X. H. Xia, Int. J. Mol. Sci., 2023, 24, 1711 CrossRef CAS PubMed.
-
(a) R. Ghosh and B. Manna, Phys. Chem. Chem. Phys., 2017, 19, 23078 RSC;
(b) H. Zhu, M. Li, J. Hu, X. Wang, J. Jie, Q. Guo, C. Chen and A. Xia, Sci. Rep., 2016, 6, 24313 CrossRef CAS PubMed;
(c) J. Hu, Y. Li, H. Zhu, S. Qiu, G. He, X. Zhu and A. Xia, ChemPhysChem, 2015, 16, 2357 CrossRef CAS PubMed.
-
(a) M. K. Bera, P. Pal and S. Malik, J. Mater. Chem. C, 2020, 8, 788 RSC;
(b) P. K. Megha and K. Singh, Spectrochem. Acta A, 2024, 307, 123649 CrossRef PubMed.
- S. C. Makhal, A. Bhattacharyya, S. Ghosh and N. Guchhait, J. Photochem. Photobiol A: Chem., 2018, 365, 67 CrossRef CAS.
- V. Nidhisha, R. Gopal, C. Anjali, T. P. Amrutha, K. K. Arunima, V. K. Praveen and R. N. Kizhakayil, Nanoscale Adv, 2024, 6, 1535 RSC.
- K. Muthamma, S. Acharya, D. Sunil, P. Shetty, A. A. Abdul Salam, S. D. Kulkarni and P. J. Anand, J. Colloid Interface Sci., 2024, 653, 209 CrossRef CAS PubMed.
- M. Yousaf and M. Lazzouni, Dyes Pigm., 1995, 27, 297 CrossRef CAS.
- CCDC 2326700: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2j33vm; CCDC 2326704: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2j33zr.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Arghyadeep Bhattachrjyya
a,
Arghyadeep Bhattachrjyya








 where the subscripts R and S refer to the reference anthracene and samples (i.e. 3a and 3g), respectively. Ai denotes the absorbance values at the wavelength monitored (i.e., 330 nm), Areai refers to the integrated area of the fluorescence spectra obtained for the samples and the reference, and ηi refers to the refractive index of the medium. The quantum yield values for 3a in hexane and acetonitrile were calculated to be 0.07 and 0.054, respectively. The quantum yields for 3g in hexane and acetonitrile were calculated to be 0.018 and 0.012, respectively. A perusal of the quantum yields leads to the following observations: (i) The quantum yield values decreased for each of 3a and 3g upon increment in the polarity of the solvents; (ii) the overall quantum yield of 3g was appreciably lower than that of 3a, irrespective of solvent. These observations strongly support an ICT process operative in 3a and 3g and indicate that it is more predominant in 3g. Our assignment finds justification by considering the established result that ICT is essentially a non-radiative process in the molecule that leads to a reduction in the quantum yield of fluorescence in organic fluorophores.23 However, we could not calculate the absolute quantum yield in the solid-state due to the limited experimental setup; nevertheless, the red-shift in the steady-state emission spectrum of 3g (λmax ∼500 nm) as compared to 3a (λmax ∼450 nm) in the solid-state strongly indicates the existence of ICT in the solid-state as well.24
where the subscripts R and S refer to the reference anthracene and samples (i.e. 3a and 3g), respectively. Ai denotes the absorbance values at the wavelength monitored (i.e., 330 nm), Areai refers to the integrated area of the fluorescence spectra obtained for the samples and the reference, and ηi refers to the refractive index of the medium. The quantum yield values for 3a in hexane and acetonitrile were calculated to be 0.07 and 0.054, respectively. The quantum yields for 3g in hexane and acetonitrile were calculated to be 0.018 and 0.012, respectively. A perusal of the quantum yields leads to the following observations: (i) The quantum yield values decreased for each of 3a and 3g upon increment in the polarity of the solvents; (ii) the overall quantum yield of 3g was appreciably lower than that of 3a, irrespective of solvent. These observations strongly support an ICT process operative in 3a and 3g and indicate that it is more predominant in 3g. Our assignment finds justification by considering the established result that ICT is essentially a non-radiative process in the molecule that leads to a reduction in the quantum yield of fluorescence in organic fluorophores.23 However, we could not calculate the absolute quantum yield in the solid-state due to the limited experimental setup; nevertheless, the red-shift in the steady-state emission spectrum of 3g (λmax ∼500 nm) as compared to 3a (λmax ∼450 nm) in the solid-state strongly indicates the existence of ICT in the solid-state as well.24