
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO, H2S, and NH3 gas sensing on Mn-doped WSe2 monolayers: a DFT and machine learning study
N. V. Hoang *ab and
Tr. Q. Trieuc
*ab and
Tr. Q. Trieuc
aAtomic Molecular and Optical Physics Research Group, Institute for Advanced Study in Technology, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: hoangvanngoc@tdtu.edu.vn
bFaculty of Electrical and Electronics Engineering, Ton Duc Thang University, Ho Chi Minh City, Vietnam
cNam Dinh Teacher Training's College, Nam Dinh City, Nam Dinh Province, Vietnam
First published on 14th July 2025
Abstract
The structural, electronic, magnetic, optical, and thermo-mechanical properties of Mn-doped WSe2 (WSe2Mn) monolayers and their CO-, H2S-, and NH3-adsorbed counterparts were systematically investigated using density functional theory and machine learning techniques. All systems exhibited spin-resolved semimetallic behavior, characterized by metallic spin-up and semiconducting spin-down channels. Magnetic ordering was maintained upon gas adsorption, with a slight enhancement in magnetic moment observed for the H2S-adsorbed configuration. Thermo-mechanical analysis revealed that gas adsorption increases the bulk modulus and heat capacities while preserving the shear modulus, suggesting enhanced compressive stiffness without compromising shear resistance. The thermal expansion coefficient and Debye temperature showed gas-dependent variations, particularly under NH3 exposure, indicating strong vibrational coupling. Optical calculations demonstrated strong absorption in the ultraviolet and partial visible range. These findings underscore the potential of WSe2Mn monolayers for multifunctional applications in nanoelectronics, gas sensing, and thermally responsive devices.
1. Introduction
Nanostructures play a crucial role in modern science and technology. Among them, two-dimensional (2D),1–3 one-dimensional (1D),4–6 and zero-dimensional (0D)7–9 materials have been extensively investigated due to their distinct quantum confinement effects. Graphene, a single-layer 2D material,10–12 is the basic structural unit of graphite,13,14 carbon nanotubes,15,16 and fullerenes.17,18 Monolayer WSe2, a prominent 2D semiconductor, exhibits a direct bandgap of ∼1.566 eV, while its bulk form shows an indirect bandgap.19–22 Its electronic properties are highly strain-sensitive, with a semiconductor-to-metal transition observed under ∼−10% biaxial compressive strain. WSe2 also demonstrates excellent performance in sub-5 nm field-effect transistors (FETs), where DFT studies emphasize the critical influence of metal–WSe2 contacts on carrier mobility and ON-current characteristics.23The chemical vapor deposition (CVD) growth of WSe2 monolayers on silica substrates was systematically investigated by Bilu Liu et al.,24 who demonstrated that the electrical behavior of WSe2 (p-type or bipolar) strongly depends on the choice of metal contacts. In a separate work, Yang Gao et al. reported the rapid CVD growth of high-quality WSe2 monolayers on Au substrates.25 These monolayers exhibited excellent crystallinity and electrical performance, comparable to mechanically exfoliated samples. Density functional theory (DFT) calculations attributed this to the exothermic diffusion and low energy barriers for W and Se atom incorporation on Au surfaces.
Efforts to tailor the properties of WSe2 through chemical doping have also been widely reported.26–28 Eleonora Pavoni et al. studied vanadium (V) doping at various concentrations (1.4–11.2%) and found that V incorporation reduced the bandgap and significantly altered the electronic and optical properties, including shifts in absorption spectra relevant to optoelectronic applications.29 Another study explored the effects of Mn, Fe, and V doping, identifying Mn and Fe as n-type and V as p-type dopants. The formation of impurity clusters was shown to weaken magnetic exchange interactions, leading to more dynamic magnetic behavior and underscoring the importance of controlling dopant distribution.30
Additionally, Antonia Kagkoura et al. investigated Co- and Ni-doped WSe2 for electrocatalytic applications in sustainable energy technologies.31 Both doped systems exhibited efficient catalytic activity toward the oxygen evolution reaction (OER), with overpotentials of 370 mV (Co) and 400 mV (Ni) at 10 mA cm−2, and toward the hydrogen evolution reaction (HER), with potentials of −0.22 V (Co) and −0.20 V (Ni) at −10 mA cm−2. The catalysts also demonstrated high stability and performance as cathodes in polymer electrolyte membrane water electrolyzers.
Recent studies have explored doping strategies to enhance the gas sensing performance of WSe2-based materials.32–34 For instance, Pt-doped WSe2 has shown improved adsorption and sensitivity toward NO2, CO2, SO2, and H2, with significant bandgap reduction and strong binding energies.33 Mo-doping has also been demonstrated to enhance CO2, CH4, and N2O adsorption, with favorable charge transfer characteristics and efficient gas desorption at ambient conditions.35 Additionally, Re-doped WSe2 has exhibited selective adsorption toward C2H4, C2H2, and CO, as revealed by DFT combined with nonequilibrium Green's function analysis.36 In addition to the aforementioned studies, many other research efforts have explored gas sensing based on materials, leading to promising applications in sensor technology.37–41
Despite the growing interest in doped WSe2 for gas sensing, the specific potential of Mn-doped WSe2 (WSe2Mn) monolayers has received limited attention in the literature. The incorporation of Mn atoms into the WSe2 lattice is expected to induce notable modifications in the electronic structure, magnetic properties, and chemical reactivity, which may enhance the interaction between the material surface and gas molecules. In this work, we present a detailed theoretical investigation of the electronic, magnetic, and gas adsorption properties of WSe2Mn monolayers with a particular focus on the detection of CO, H2S, and NH3 gases-molecules of high environmental and industrial relevance. Our approach combines density functional theory (DFT) with crystal graph convolutional neural networks (CGCNN), a state-of-the-art machine learning technique, to both characterize the structural and electronic response of WSe2Mn to gas adsorption and predict adsorption-related properties with enhanced efficiency and accuracy. This integrated DFT-ML framework enables not only high-fidelity simulations but also improved generalization in evaluating structure–property relationships, offering a robust platform for rapid materials screening. These findings provide new insights into high-performance 2D gas sensors and broaden the application scope of WSe2-based materials in nanoscale sensing technologies.
2. Computational methods
In this study, a combination of density functional theory (DFT) and machine learning techniques was employed to investigate the structural, electronic, and thermo-mechanical properties of WSe2Mn monolayers and their interaction with gas molecules (CO, H2S, and NH3).All DFT calculations were performed using the Quantum ESPRESSO package, which implements plane-wave-based pseudopotential methods. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was used to describe the exchange–correlation interaction. The projector augmented-wave (PAW) method was employed, and a kinetic energy cutoff of 50 Ry was chosen for the plane-wave basis set. To account for van der Waals interactions during gas adsorption, the Grimme-D2 dispersion correction scheme was applied. A vacuum layer of 20 Å was applied along the out-of-plane direction to prevent interaction between periodic images. Brillouin zone integration was carried out using a 11 × 11 × 1 Monkhorst–Pack k-point mesh for geometry optimizations and force converged below thresholds of 10−7 Ry and 10−4 Ry Bohr−1, respectively.
To investigate thermo-mechanical properties such as bulk modulus, shear modulus, heat capacities, thermal expansion coefficient, and Debye temperature, we employed the Crystal Graph Convolutional Neural Network (CGCNN) framework. The combination of DFT and CGCNN allows for comprehensive analysis of both the energetics and thermo-mechanical responses of WSe2Mn monolayers under gas adsorption.
Adsorption energy is calculated by the formula:42
| EAE = ET − EP − EM | (1) |
3. Results and discussion
3.1. Electromagnetic properties
The monolayer structure of WSe2Mn and WSe2Mn structures adsorbing CO/H2S/NH3 gas are shown in Fig. 1, WSe2Mn has three atomic layers, the projection on the x0y plane is a hexagonal structure. The structural parameters of WSe2Mn and WSe2Mn adsorbing CO/H2S/NH3 gas are shown in Table 1. The bond length between W–Se is d1, d1 has a value from 2.464 Å to 2.468 Å, these values are not significantly different, this result proves that the presence of CO/H2S/NH3 does not affect the W–Se distance much. Similar to d1, d2 and d3 also vary in a very small range, d2 has a value from 3.439 Å to 3.441 Å, while d3 has a value from 3.011 Å to 3.021 Å, this result once again confirms that WSe2Mn structure is less distorted when adsorbed by gas. When compared with structures similar to WSe2Mn, such as monolayer MoS2 with a bond length between Mo–S of 2.41 Å,43 we see that the bond length between W–Se is about 0.05 Å larger, this result shows that there is not much difference, so the bonding force between W–Se in WSe2Mn is almost similar to that between Mo–S in MoS2. To see more clearly the strong or weak bond between atoms, we compare the bond length between W–Se with the bond length in the graphene structure, the bond length between C atoms in graphene is 1.42 Å,44 which shows that the bond between C–C is stronger than the bond between W–Se. | ||
| Fig. 1 Top view and side view of structures: (a and b) pristine WSe2Mn monolayer, (c and d) CO/H2S/NH3-adsorbed WSe2Mn monolayer (the atoms W, Se, Mn correspond to the colors purple, green and gray, respectively). | ||
| Configurations | d1 (Å) | d2 (Å) | d3 (Å) | Ea (eV) | μ (μB) |
|---|---|---|---|---|---|
| WSe2Mn monolayer | 2.465 | 3.439 | 3.011 | — | 2.175 |
| CO adsorption | 2.464 | 3.441 | 3.013 | −0.91 | 2.174 |
| H2S adsorption | 2.468 | 3.441 | 3.021 | −0.86 | 2.670 |
| NH3 adsorption | 2.465 | 3.439 | 3.013 | −0.96 | 2.175 |
The adsorption energy values for CO, H2S, and NH3 on WSe2Mn monolayer are calculated to be −0.91 eV, −0.86 eV, and −0.96 eV, respectively. The negative adsorption energies indicate that all three gas adsorption processes are energetically favorable and exothermic. Among them, the NH3 adsorption configuration exhibits the lowest adsorption energy, suggesting that NH3 adsorption is the most thermodynamically favorable, making NH3 more readily adsorbed onto WSe2Mn surface compared to CO and H2S.
Regarding the magnetic properties, both the pristine WSe2Mn monolayer and its gas-adsorbed configurations exhibit magnetism, with total magnetic moments ranging from 2.670μB to 2.175μB. Notably, the pristine WSe2Mn and the NH3-adsorbed WSe2Mn configurations share the highest magnetic moment value. This result suggests that the adsorption of CO and H2S leads to a slight reduction in the system's magnetic moment, whereas NH3 adsorption does not induce any significant change in the magnetic properties.
To investigate the impact of gas adsorption on the thermo-mechanical properties of the material, key parameters including the bulk modulus (B), shear modulus (G), constant-pressure heat capacity (Cp), constant-volume heat capacity (CV), thermal expansion coefficient (α), and Debye temperature (TD) were computed for WSe2Mn system in both its pristine state and after the adsorption of CO, H2S, and NH3 gas molecules, employing the CGCNN method (Table 2). The results reveal that gas adsorption leads to an enhancement in the material's compressive modulus. Specifically, the bulk modulus increased from 17.36 GPa in the pristine system to 22.60 GPa (CO), 22.41 GPa (H2S), and 21.28 GPa (NH3), respectively. This increase indicates a trend toward structural stiffening induced by the interaction between gas molecules and WSe2Mn monolayer, thereby improving the system's compressive strength. In contrast, the shear modulus (G) exhibits negligible variation, remaining in the range of 25.8–26.3 GPa, suggesting that the resistance to shear deformation is largely preserved following gas adsorption.
| Configurations | B (GPa) | G (GPa) | Cp (kB per atom) | CV (kB per atom) | α (K−1) | TD (K) |
|---|---|---|---|---|---|---|
| WSe2Mn monolayer | 17.36 | 26.38 | 2.93 | 2.84 | 4.75 × 10−5 | 272.28 |
| CO adsorption | 22.60 | 26.22 | 2.96 | 2.84 | 4.72 × 10−5 | 272.87 |
| H2S adsorption | 22.41 | 25.80 | 2.99 | 2.85 | 4.96 × 10−5 | 261.33 |
| NH3 adsorption | 21.28 | 26.30 | 2.97 | 2.85 | 5.16 × 10−5 | 262.51 |
Regarding thermal properties, both the constant-pressure heat capacity (Cp) and constant-volume heat capacity (CV) exhibited a slight increase following gas adsorption. Specifically, Cp increased from 2.93 kB per atom in the pristine system to 2.96–2.99 kB per atom in the adsorption configurations, while CV reached a value of 2.85 kB per atom. This enhancement is likely attributed to the vibrational contributions of the adsorbed gas molecules, which increase the total number of accessible vibrational modes in the system. Notably, the thermal expansion coefficient (α) showed a more significant variation. The pristine configuration exhibited α = 4.75 × 10−5 K−1, which increased to 5.16 × 10−5 K−1 upon NH3 adsorption-higher than the corresponding values for CO (4.72 × 10−5 K−1) and H2S (4.96 × 10−5 K−1). This pronounced change indicates a strong interaction between NH3 molecules and WSe2Mn lattice, highlighting both the influence of NH3 on lattice vibrations and the high sensitivity of the material system to this particular gas.
From a dynamical perspective, the Debye temperature (TD) serves as an indicator of the overall stiffness of the crystal lattice and is directly associated with the maximum vibrational frequency of the system. For the pristine WSe2Mn monolayer, TD was calculated to be 272.28 K-significantly higher than that of bulk WSe2 (160 K (ref. 45)). This enhancement can be attributed to the two-dimensional nature of the material and the local lattice stiffening induced by Mn doping. Upon adsorption of H2S and NH3 molecules, TD exhibited a slight reduction to 261.33 K and 262.51 K, respectively, suggesting a softening of lattice vibrations due to gas–surface interactions.
In comparison with bulk WSe2 (B = 63.7 GPa, G = 52.5 GPa,46 α = 5.7 × 10−5 K−1,47 TD = 160 K (ref. 45)), the Mn-doped monolayer clearly possesses lower mechanical stiffness but exhibits a Debye temperature approximately 70% higher. This behavior is characteristic of two-dimensional materials, where weak interlayer interactions along the out-of-plane (z) direction reduce resistance to compressive and shear forces, yet the restricted dimensionality leads to elevated vibrational frequencies due to limited degrees of freedom. These findings suggest that gas adsorption exerts a measurable influence on the thermo-mechanical behavior of WSe2Mn monolayer. In particular, the observed sensitivity of the thermal expansion coefficient and Debye temperature to different adsorbed gas species highlights the potential of this material for application in gas sensing technologies, where detection may be achieved through mechanical–thermodynamic response signatures.
The electronic and magnetic properties of pristine and gas-adsorbed WSe2Mn systems are examined via band structure and density of states (DOS) analyses (Fig. 2). All configurations exhibit spin-polarized semi-metallic behavior, with metallic characteristics in the spin-up channel and semiconducting features in the spin-down channel, confirming their magnetic nature. For the CO-adsorbed system, the band structure remains largely similar to that of pristine WSe2Mn in the valence band region, but shows increased DOS above 2 eV in the conduction band. The DOS peak heights are ∼11.5 states per eV for both pristine and CO-adsorbed systems, while slightly higher values are observed for H2S (∼12 states per eV) and NH3 (∼16 states per eV), indicating stronger electronic perturbations induced by these gases.
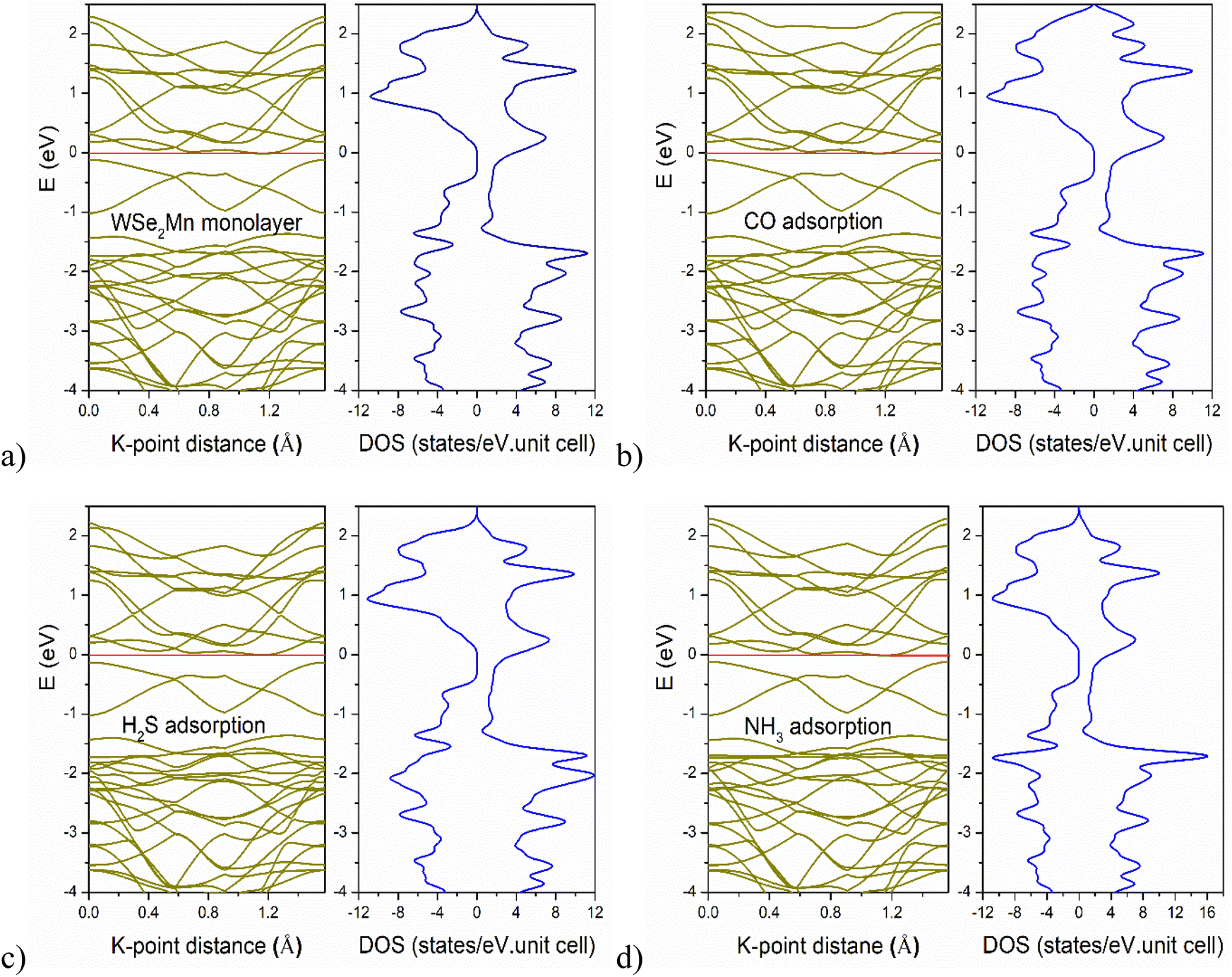 | ||
| Fig. 2 Band structure and density of states of configurations: (a) pristine WSe2Mn monolayer; (b) CO-adsorbed WSe2Mn monolayer; (c) H2S-adsorbed WSe2Mn monolayer; (d) NH3-adsorbed WSe2Mn monolayer. | ||
Partial DOS (PDOS) analysis (Fig. 3) reveals that Se orbitals (particularly px) dominate the valence band in the pristine structure, with Se(s) peaking at −4.5 eV and W/Mn(s) contributions localized deeper (∼−6 eV). Upon CO adsorption, C and O states appear mainly at −4.5 eV and 2.5 eV, showing localized hybridization with Se. H2S adsorption introduces a broader energy distribution of S states (−6.5 eV to 4 eV) and clear spin asymmetry, indicating stronger and more complex hybridization compared to CO and NH3. In contrast, N states in the NH3-adsorbed system are confined near −6.5 eV and −1.6 eV, suggesting limited interaction with Se.
 | ||
| Fig. 3 Partial DOS (PDOS) of configurations: (a) pristine WSe2Mn monolayer; (b) CO-adsorbed WSe2Mn monolayer; (c) H2S-adsorbed WSe2Mn monolayer; (d) NH3-adsorbed WSe2Mn monolayer. | ||
Overall, the results highlight that gas adsorption not only modifies the electronic structure of WSe2Mn but also induces distinct spin-dependent hybridization patterns, particularly pronounced in the case of H2S. These features may be leveraged to achieve gas-specific electronic responses in spintronic or sensing applications.
The charge density difference for the CO, H2S, and NH3 adsorption configurations is illustrated in Fig. 4. It is evident that gas adsorption induces a redistribution of charge around both the adsorbed molecules and WSe2Mn surface in proximity to these molecules. The interaction between the gas molecules and the substrate is visualized through charge accumulation and depletion regions, represented by blue and yellow areas in the interfacial space. The adsorption interaction primarily occurs between the Se atoms of WSe2Mn monolayer and the CO, H2S, and NH3 molecules. In contrast, the inner W and Mn atoms exhibit negligible interaction with the adsorbed species, as indicated by the minimal charge redistribution, appearing as small, dispersed yellow and blue regions around these atoms.
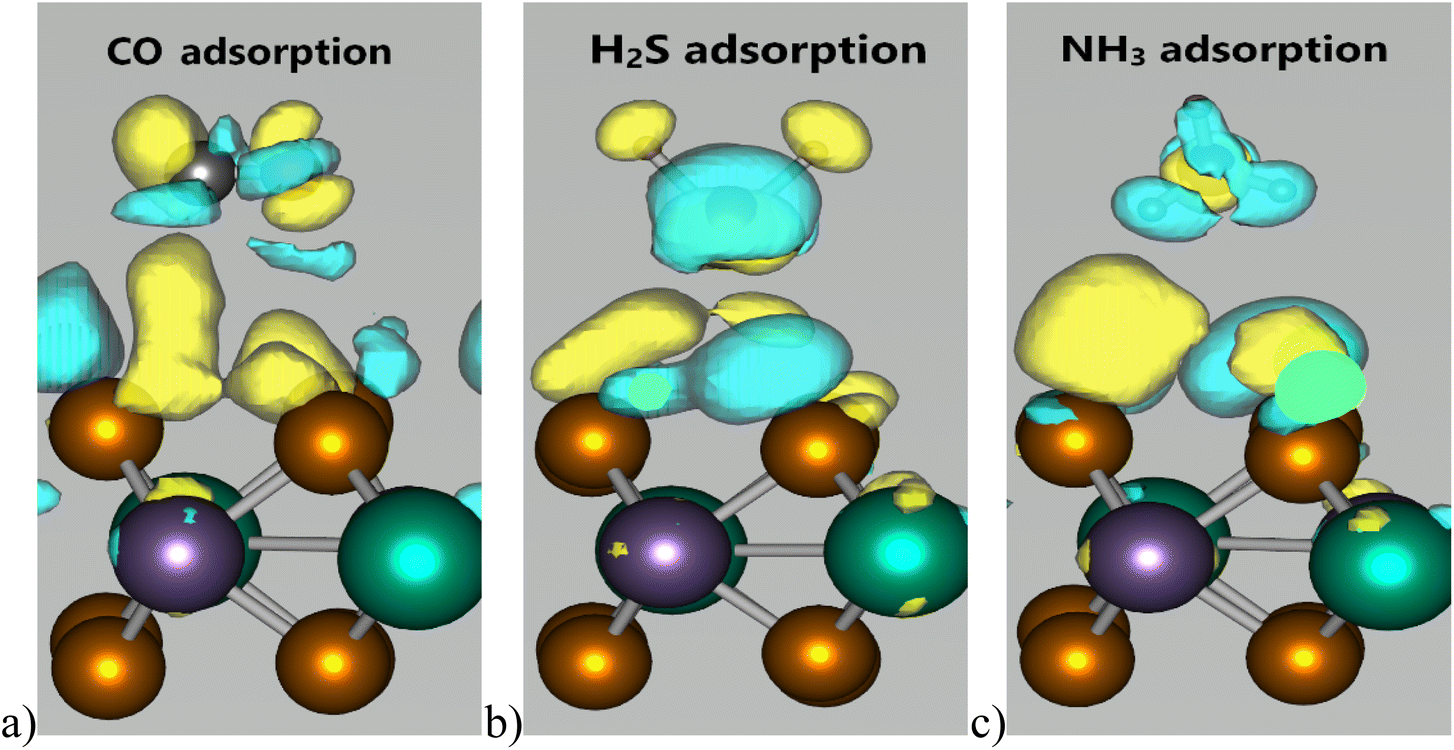 | ||
| Fig. 4 The charge density difference in the configurations: (a) CO-adsorbed WSe2Mn monolayer; (b) H2S-adsorbed WSe2Mn monolayer; (c) NH3-adsorbed WSe2Mn monolayer (the yellow region is where the charge is enhanced, the blue region is where the charge is reduced). | ||
3.2. Optical properties
The optical properties of WSe2Mn and its CO, H2S, and NH3 adsorption configurations are analyzed through the dielectric function (Fig. 5), absorption coefficient (Fig. 6), and electron–hole density distribution (Fig. 7). The dielectric function is presented in terms of its real and imaginary components. | ||
| Fig. 5 Dielectric function of the configurations: (a) pristine WSe2Mn monolayer; (b) CO-adsorbed WSe2Mn monolayer; (c) H2S-adsorbed WSe2Mn monolayer; (d) NH3-adsorbed WSe2Mn monolayer. | ||
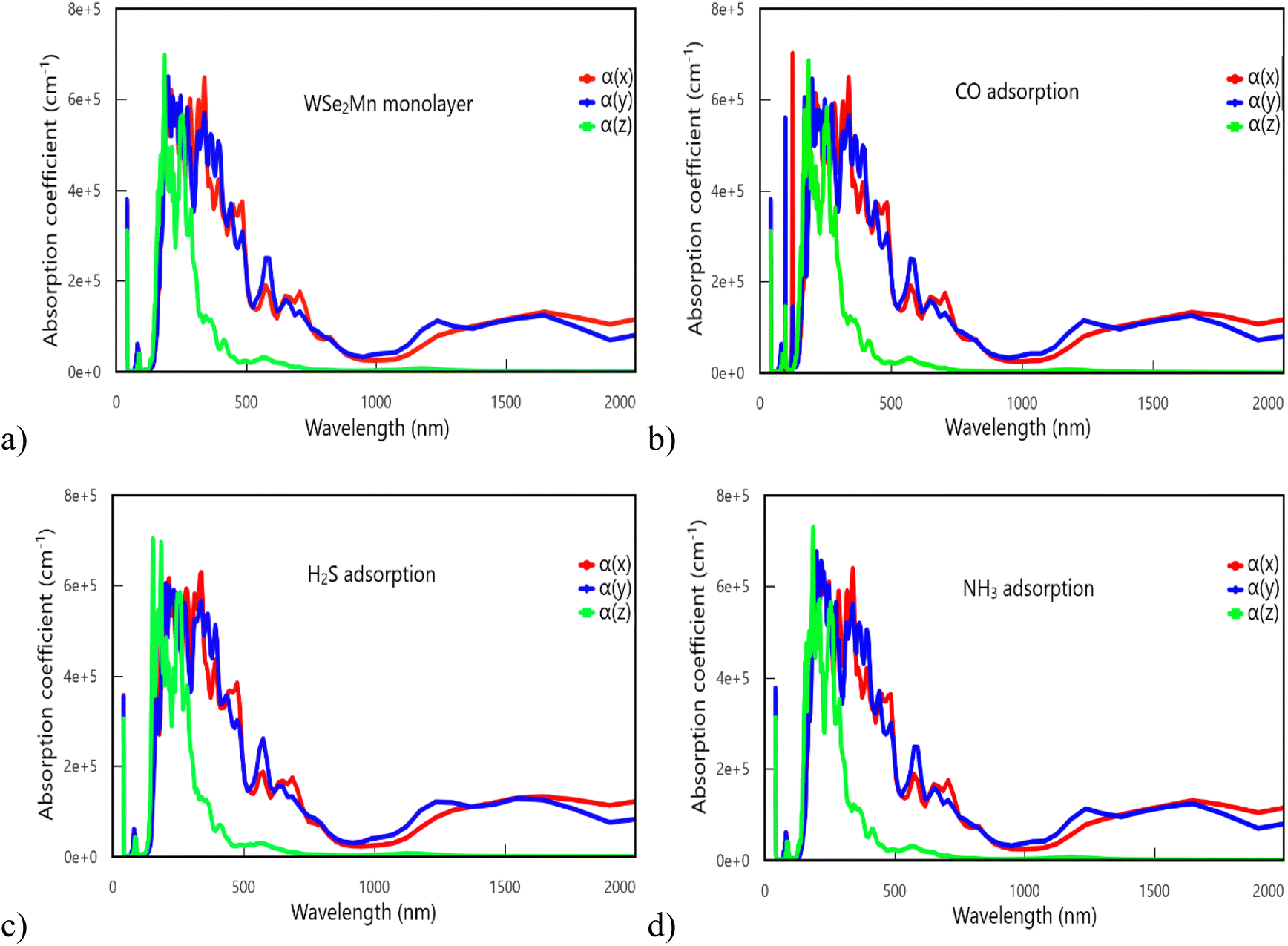 | ||
| Fig. 6 Absorption coefficient of the configurations: (a) pristine WSe2Mn monolayer; (b) CO-adsorbed WSe2Mn monolayer; (c) H2S-adsorbed WSe2Mn monolayer; (d) NH3-adsorbed WSe2Mn monolayer. | ||
 | ||
| Fig. 7 The joint density of states (jDOS) of the configurations. | ||
As illustrated in Fig. 5, the pristine and CO-adsorbed WSe2Mn configurations exhibit strong optical activity within the 0–8 eV energy range. In comparison, H2S- and NH3-adsorbed systems extend this activity slightly further, up to ∼8.5 eV and ∼8.3 eV, respectively, indicating enhanced optical responsiveness due to gas adsorption.
All systems show negative values in the real part of the dielectric function (ε1(ω)) along the x and y directions within certain energy ranges, implying a low-loss propagation of electromagnetic waves-an essential characteristic for optoelectronic and photonic applications. Along the z-direction, ε1(ω) exhibits lower intensity due to the limited thickness of the monolayer (three atomic layers), as opposed to the infinite extent in the x–y plane.
Prominent peaks in ε1(ω) appear near 2 eV for both the x and y directions, corresponding to the visible light region, where the material shows maximum attenuation. At higher photon energies (above ∼8 eV), attenuation rapidly diminishes, indicating high transparency in the ultraviolet range. These results suggest that WSe2Mn, particularly in gas-adsorbed forms, could be a promising candidate for tunable optoelectronic devices and optical communication components.
The imaginary part of the dielectric function ε2(ω) reveals a pronounced anisotropy. The most intense peak is observed along the x-direction at approximately 0.3 eV, within the infrared region, suggesting significant energy dissipation due to electromagnetic wave interaction at this energy. This peak is followed by a less intense response along the y-direction, while the z-direction exhibits minimal response, indicating negligible absorption along the out-of-plane axis.
In the energy range of 0–4 eV, dominant absorption features are retained in the x- and y-directions, confirming strong in-plane interaction with incident radiation. Beyond 4 eV, ε2(ω) gradually decreases for all directions, with convergence observed near 8 eV for the pristine and CO-adsorbed systems. For H2S- and NH3-adsorbed configurations, this convergence shifts slightly to ∼8.5 eV. These convergence points signify a transition beyond which the material exhibits minimal electromagnetic loss, indicating transparency to higher-energy photons.
Analysis of the absorption coefficient (α) further corroborates the material's strong light–matter interaction (Fig. 6). All configurations show intense absorption in the 160–480 nm range, corresponding to ultraviolet and visible light (particularly violet and blue regions). In the investigated configurations, the maximum absorption coefficient reaches approximately 7.1 × 105 cm−1 at an energy of about 6.2 eV. Compared to pristine monolayer WSe2, which was previously reported to exhibit a maximum absorption coefficient of around 1.6 × 106 cm−1 at 5.6 eV, the present results indicate a shift of the main absorption peak toward higher energy (blue-shift).48 This shift can be attributed to the effects of doping and gas adsorption, which modify the electronic band structure and the density of states, thereby altering the resonance conditions for optical transitions. Notably, distinct anisotropic features are observed: CO adsorption induces dual peaks in the x- and y-directions, whereas H2S adsorption leads to enhanced absorption along the z-axis.
At wavelengths exceeding 700 nm, absorption in the z-direction diminishes to nearly zero, while the x- and y-components persist at low but finite values beyond 800 nm. These findings underscore the directional dependence of light absorption in WSe2Mn and its modified states. Overall, the pronounced anisotropic optical response and strong absorption in the UV-visible region suggest that WSe2Mn is a promising candidate for applications in optoelectronics, sensing, photocatalysis, and biomedical technologies.
The joint density of states (jDOS) spectrum of the pristine system exhibits a prominent peak in the low-energy region (approximately 3–6 eV), with its intensity gradually diminishing as the photon energy increases (Fig. 7). This behavior suggests that the pristine material possesses a strong light absorption capability in the ultraviolet (UV) and partially visible regions, facilitating the generation of electron–hole pairs and thereby enhancing the system's conductivity. Upon CO adsorption, significant modifications in the jDOS spectrum are observed, characterized by the emergence of new peaks in the higher-energy regions (around 9–12 eV and 26–28 eV). In the case of H2S adsorption, the jDOS intensity is notably enhanced in the mid-energy range (approximately 9–12 eV) compared to the pristine state. Conversely, NH3 adsorption introduces subtle jDOS peaks in the high-energy region (around 18–21 eV). These alterations underscore the substantial influence of CO, H2S, and NH3 adsorption on the electronic structure of the material, leading to an expanded jDOS profile. This expansion in jDOS highlights the potential for utilizing these gas-adsorbed configurations in optical devices operating within high-energy regimes.
4. Conclusions
The structural, electronic, magnetic, optical, and thermo-mechanical properties of Mn-doped WSe2 (WSe2Mn) monolayers, both pristine and adsorbed with CO, H2S, and NH3 gas molecules, were systematically investigated using density functional theory (DFT) and machine learning (CGCNN) approaches. Electronic structure analysis revealed spin-resolved semimetallic behavior, characterized by a metallic spin-up channel and a semiconducting spin-down channel, which remained largely unchanged upon gas adsorption. The material's intrinsic magnetic moment was also preserved, indicating that adsorption induces minimal perturbation to its magnetic characteristics. All adsorption processes were exothermic with adsorption energies below 1 eV, confirming weak chemisorption as the dominant interaction mechanism. In terms of thermo-mechanical behavior, gas adsorption led to an increase in bulk modulus and heat capacities, while the shear modulus remained nearly constant, indicating selective stiffening under compressive strain. The thermal expansion coefficient (α) and Debye temperature (TD) exhibited notable gas-specific variations, particularly under NH3 exposure, suggesting enhanced vibrational coupling and mechanical–thermal sensitivity to gas adsorption. Optical analyses showed strong absorption in the ultraviolet and partial visible ranges for all configurations, supporting potential optoelectronic applications. These findings point to the potential of WSe2Mn monolayers as multifunctional materials for use in nanoelectronic devices, gas sensors, and thermomechanical-responsive systems.Data availability
Data supporting the findings of this study are available within the article.Author contributions
N. V. Hoang was responsible for designing and performing the computational research, including the preparation of input files. Tr. Q. Trieu handled the organization and presentation of the results in tables and figures. Both N. V. Hoang and Tr. Q. Trieu contributed equally to drafting and editing the main manuscript. All authors participated in the review and approval of the final version.Conflicts of interest
We declare we have no competing interests.Acknowledgements
The authors gratefully acknowledge the support of the HPC system at the Institute for Advanced Study in Technology (IAST), Ton Duc Thang University.References
- G. Fiori, et al., Electronics based on two-dimensional materials, Nat. Nanotechnol., 2014, 9(10), 768–779 CrossRef CAS PubMed.
- P. Miró, M. Audiffred and T. Heine, An atlas of two-dimensional materials, Chem. Soc. Rev., 2014, 43(18), 6537–6554 RSC.
- M. Xu, et al., Graphene-like two-dimensional materials, Chem. Rev., 2013, 113(5), 3766–3798 CrossRef CAS PubMed.
- S. Barth, et al., Synthesis and applications of one-dimensional semiconductors, Prog. Mater. Sci., 2010, 55(6), 563–627 CrossRef CAS.
- S. V. Kuchibhatla, et al., One dimensional nanostructured materials, Prog. Mater. Sci., 2007, 52(5), 699–913 CrossRef CAS.
- L. Wang, et al., One-dimensional electrical contact to a two-dimensional material, Science, 2013, 342(6158), 614–617 CrossRef CAS PubMed.
- Y. Ma, et al., Zero-dimensional to three-dimensional nanojoining: current status and potential applications, RSC Adv., 2016, 6(79), 75916–75936 RSC.
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Zero-dimensional, one-dimensional, two-dimensional and three-dimensional nanostructured materials for advanced electrochemical energy devices, Prog. Mater. Sci., 2012, 57(4), 724–803 CrossRef CAS.
- J. Zheng, et al., Advanced anode materials of potassium ion batteries: from zero dimension to three dimensions, Nano-Micro Lett., 2021, 13, 1–37 CrossRef CAS PubMed.
- D. Abergel, et al., Properties of graphene: a theoretical perspective, Adv. Phys., 2010, 59(4), 261–482 CrossRef CAS.
- L. Falkovsky and S. Pershoguba, Optical far-infrared properties of a graphene monolayer and multilayer, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76(15), 153410 CrossRef.
- Q. Lu, M. Arroyo and R. Huang, Elastic bending modulus of monolayer graphene, J. Phys. D: Appl. Phys., 2009, 42(10), 102002 CrossRef.
- D. Chung, Review graphite, J. Mater. Sci., 2002, 37, 1475–1489 CrossRef CAS.
- D. Chung, A review of exfoliated graphite, J. Mater. Sci., 2016, 51, 554–568 CrossRef CAS.
- H. Dai, Carbon nanotubes: opportunities and challenges, Surf. Sci., 2002, 500(1–3), 218–241 CrossRef CAS.
- V. N. Popov, Carbon nanotubes: properties and application, Mater. Sci. Eng., R, 2004, 43(3), 61–102 CrossRef.
- F. Giacalone and N. Martin, Fullerene polymers: synthesis and properties, Chem. Rev., 2006, 106(12), 5136–5190 CrossRef CAS PubMed.
- F. Wudl, Fullerene materials, J. Mater. Chem., 2002, 12(7), 1959–1963 RSC.
- W.-T. Hsu, et al., Evidence of indirect gap in monolayer WSe2, Nat. Commun., 2017, 8(1), 929 CrossRef PubMed.
- J.-K. Huang, et al., Large-area synthesis of highly crystalline WSe2 monolayers and device applications, ACS Nano, 2014, 8(1), 923–930 CrossRef CAS PubMed.
- R. Zhao, et al., First-Principle Study of Bandgap Engineering and Optical Properties of Monolayer WSe2 in Second Near-Infrared Windows, Adv. Mater. Interfaces, 2023, 10(23), 2300277 CrossRef CAS.
- D. Muoi, et al., Electronic properties of WS2 and WSe2 monolayers with biaxial strain: a first-principles study, Chem. Phys., 2019, 519, 69–73 CrossRef CAS.
- W. Liu, et al., High-performance field-effect-transistors on monolayer-WSe2, ECS Trans., 2013, 58(7), 281 CrossRef.
- B. Liu, et al., Chemical vapor deposition growth of monolayer WSe2 with tunable device characteristics and growth mechanism study, ACS Nano, 2015, 9(6), 6119–6127 CrossRef CAS PubMed.
- Y. Gao, et al., Ultrafast growth of high-quality monolayer WSe2 on Au, Adv. Mater., 2017, 29(29), 1700990 CrossRef PubMed.
- H.-Y. Chen, et al., Locally Doped Transferred Contacts for WSe2 Transistors, ACS Appl. Electron. Mater., 2024, 6(11), 8319–8327 CrossRef CAS.
- S. Chen, et al., Extension Doping with Low-Resistance Contacts for P-Type Monolayer WSe2 Field-Effect Transistors, Adv. Electron. Mater., 2024, 2400843 Search PubMed.
- Y.-R. Liu, et al., Sn-doped induced stable 1T-WSe2 nanosheets entrenched on N-doped carbon with extraordinary half/full sodium/potassium storage performance, Rare Met., 2023, 42(5), 1557–1569 CrossRef CAS.
- E. Pavoni, et al., First principles study of WSe2 and the effect of V doping on the optical and electronic properties, Mater. Adv., 2024, 5(6), 2230–2237 RSC.
- S. Tiwari, et al., Reduction of Magnetic Interaction Due to Clustering in Doped Transition-Metal Dichalcogenides: A Case Study of Mn-, V-, and Fe-Doped WSe2, ACS Appl. Mater. Interfaces, 2024, 16(4), 4991–4998 CrossRef CAS PubMed.
- A. Kagkoura, et al., Cobalt-and Nickel-Doped WSe2 as Efficient Electrocatalysts for Water Splitting and as Cathodes in Hydrogen Evolution Reaction Proton Exchange Membrane Water Electrolysis, J. Phys. Chem. C, 2025, 129, 2893–2903 CrossRef CAS.
- S. Cheng, et al., The adsorption and sensing mechanism of toxic gases HCN, NO2, NH3 and Cl2 on Mo, Ag-modified WSe2 monolayer: Insights from the first-principles computations, Mater. Today Commun., 2023, 35, 105906 CrossRef CAS.
- A. Kushwaha and N. Goel, A DFT study of superior adsorbate-surface bonding at Pt-WSe2 vertically aligned heterostructures upon NO2, SO2, CO2, and H2 interactions, Sci. Rep., 2024, 14(1), 15708 CrossRef CAS PubMed.
- L. Lin, et al., Adsorption of NO2, SO2, H2S, and NH3 on Os-Doped WSe2 Monolayers: A First-Principles Study, Langmuir, 2023, 39(42), 15142–15151 CrossRef CAS PubMed.
- M. Dong, et al., A Study Based on the First-Principle Study of the Adsorption and Sensing Properties of Mo-Doped WSe2 for N2O, CO2, and CH4, Chemosensors, 2024, 12(9), 192 CrossRef CAS.
- S. Sarkar, et al., DFT analysis of Re-modified WSe2 monolayers for adsorption of CO, C2H2, and C2H4, Modell. Simul. Mater. Sci. Eng., 2024, 32(7), 075003 CrossRef CAS.
- Z. Cui, et al., Highly sensitive and selective defect WS2 chemical sensor for detecting HCHO toxic gases, Sensors, 2024, 24(3), 762 CrossRef CAS PubMed.
- Z. Cui, et al., Adsorption of CO, NH3, NO, and NO2 on pristine and defective g-GaN: Improved gas sensing and functionalization, Appl. Surf. Sci., 2020, 530, 147275 CrossRef CAS.
- Z. Cui, et al., Adsorption of gas molecules on intrinsic and defective MoSi2N4 monolayer: Gas sensing and functionalization, Sens. Actuators, A, 2024, 366, 114954 CrossRef CAS.
- Z. Cui, et al., Toxic gas molecules adsorbed on intrinsic and defective WS2: gas sensing and detection, Appl. Surf. Sci., 2023, 613, 155978 CrossRef CAS.
- Y. Qin, T. Zhang and Z. Cui, Core-shell structure of polypyrrole grown on W18O49 nanorods for high performance gas sensor operating at room temperature, Org. Electron., 2017, 48, 254–261 CrossRef CAS.
- M. Monshi, S. Aghaei and I. Calizo, Edge functionalized germanene nanoribbons: impact on electronic and magnetic properties, RSC Adv., 2017, 7(31), 18900–18908 RSC.
- C. Li, et al., Bandgap engineering of monolayer MoS2 under strain: A DFT study, J. Korean Phys. Soc., 2015, 66, 1789–1793 CrossRef CAS.
- M. Pumera and C. H. A. Wong, Graphane and hydrogenated graphene, Chem. Soc. Rev., 2013, 42(14), 5987–5995 RSC.
- S. Mathew, et al., Temperature dependent structural evolution of WSe2: A synchrotron X-ray diffraction study, Condens. Matter, 2020, 5(4), 76 CrossRef CAS.
- L.-p. Feng, et al., Effect of pressure on elastic, mechanical and electronic properties of WSe2: A first-principles study, Mater. Res. Bull., 2014, 50, 503–508 CrossRef CAS.
- T. M. Kucinski, et al., Direct Measurement of the Thermal Expansion Coefficient of Epitaxial WSe2 by Four-Dimensional Scanning Transmission Electron Microscopy, ACS Nano, 2024, 18(27), 17725–17734 CrossRef CAS PubMed.
- H.-L. Liu, et al., Temperature-dependent optical constants of monolayer MoS2, MoSe2, WS2, and WSe2: spectroscopic ellipsometry and first-principles calculations, Sci. Rep., 2020, 10(1), 15282 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |