
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into Rh size-dependent reactivity of CO2 methanation over Rh–Al2O3 catalysts†
Jinshi Dong *a,
Hongli Yanga,
Shengtong Lia,
Panpan Changa and
Jiaqiang Yang*b
*a,
Hongli Yanga,
Shengtong Lia,
Panpan Changa and
Jiaqiang Yang*b
aLaboratory of New Energy and Environmental Catalysis, School of Biological and Chemical Engineering, Guangxi University of Science and Technology, Liuzhou 545006, Guangxi, China. E-mail: jinshidong@gxust.edu.cn
bZhongyuan Critical Metals Laboratory, Zhengzhou University, Zhengzhou 450001, Henan, China. E-mail: jqyang@zzu.edu.cn
First published on 15th July 2025
Abstract
The hydrogenation of CO2 to methane at atmospheric pressure is a significant chemical approach to achieve carbon neutrality and gain renewable energy. However, developing catalysts with high selectivity and high methane yield remains challenging. In this study, a series of Rh–Al2O3 catalysts with varying Rh particle sizes were prepared by modulating the Rh loading amounts. Rh nanoparticles (Rh NPs) were found to exhibit superior performance compared to Rh single atoms (Rh SAs) under identical reaction conditions. The sharp decrease in CH4 selectivity at high temperature is dominantly attributed to the side reaction of dry reforming of methane instead of the limitation of reaction thermodynamics. It was found that there was the coexistence of formate and CO pathways in CO2 methanation on Rh–Al2O3 catalysts regardless of Rh loadings and formate pathway is dominate for CO2 methanation when the temperature high than 400 °C. Turnover frequency (TOF) calculations indicated that the theoretical CH4 generation frequency of Rh NP was three times higher than that of Rh SA. Kinetic experiments and DFT calculations revealed that the dissociation and activation of H2 is the key factor affecting the performance of Rh–Al2O3 catalyst. This study facilitates our understanding of Rh size-dependent chemistry for CO2 methanation reaction.
1. Introduction
As human society and industry continue to advance, the rising concentration of CO2 in the atmosphere exacerbates the global greenhouse effect, thereby emphasizing the urgency of eliminating CO2 waste gas and utilizing such resources.1–4 Carbon dioxide could be converted into high value-added chemical products, including methane (CH4), carbon monoxide (CO), methanol (CH3OH), and even hydrocarbons, depending on the catalysts employed and specific reaction conditions.5–11 The hydrogenation of CO2 to CH4 (CO2 methanation) not only achieves CO2 emission reduction, but also is one of the effective ways to make full use of hydrogen obtained from renewable energy sources, which is of great significance in solving the problems of energy shortage and environmental pollution.12–16The molecular CO2 is very stable and it is a great challenge to convert it to the target product methane with high selectivity. CO2 methanation (eqn (1)) is a strongly exothermic reaction, which means that it will be limited by thermodynamic equilibrium at high temperatures.17,18 The reaction is accompanied by other side reactions such as reverse water gas shift (eqn (2), RWGS) and dry reforming of methane (eqn (3), DRM).19–21 Therefore, a highly active catalyst is essential to overcome high kinetic limitations of CO2 methanation while promoting methanation selectivity. Compared to other supported metals, supported Rh catalysts exhibit excellent catalytic performance and methane selectivity in CO2 methanation reactions.22–27 Previous studies indicated that CeO2, TiO2, ZrO2, and Al2O3 have been used as the support to investigate the catalytic performance of metal oxide-supported Rh catalysts in CO2 hydrogenation, among which the highest CO2 conversion towards methane is obtained for the Rh–Al2O3, benefiting from the strong adsorption capacity of the Al2O3 support for CO2.19,28
| CO2 + 4H2 ↔ CH4 + 2H2O; ΔH298K = −165 kJ mol−1 | (1) |
| CO2 + H2 ↔ CO + H2O; ΔH298K = 41.2 kJ mol−1 | (2) |
| CO2 + CH4 ↔ 2CO + 2H2; ΔH298K = 247 kJ mol−1 | (3) |
It is widely accepted that the CO2 methanation reaction activity and selectivity can be tuned by controlling metal particle size, elemental doping, and metal–support interactions.3,6,23,24,29–36 Karelovic et al.29 reported that larger Rh particles are up to four times more active than smaller particles at low temperature (135–150 °C), whereas at higher temperatures (200 °C) the influence of Rh particle size on catalytic activity is insignificant. A similar phenomenon was found in the Rh–TiO2 catalyst system, but the catalytic properties did not change appreciably when the particle size was increased to about 7 nm.25 Bentrup et al.33 studied the modification of Rh–Al2O3 catalysts with Ni and K to vary their acidity/basicity and redox behavior and found that the Ni modification promotes the formation of CH4, whereas K modification enhances the CO formation. Siang et al.23 showed that the metal–support interaction and the degree of basicity are significantly enhanced with increasing Rh contents, which facilitates CO2 adsorption and reduces the activation barriers (from 110.2 to 19.7 kJ mol−1) during the methanation reaction, thereby promoting catalytic activity.
Despite so much progress up to now, the fundamental factors affecting CH4 product yield still remain highly controversial. In order to gain deeper insights into the influence of particle size on catalytic activity and selectivity, a series of Al2O3-supported Rh catalysts with varying Rh particle sizes were synthesized and subsequently analyzed to reveal the structure–performance relationship for CO2 methanation. Our findings indicate that the adsorption and activation of H2 are critical factors influencing product selectivity and larger Rh particle sizes would facilitate H2 activation, thereby enhancing CO2 methanation activity. The decreased CH4 selectivity at high temperatures is mainly due to the side reaction of dry reforming of methane. Furthermore, in situ infrared spectroscopy studies have demonstrated that both the formate and CO pathways are main reaction pathways for the CO2 methanation reaction catalyzed by the Rh–Al2O3 catalyst.
2. Experimental section
2.1. Synthesis of Rh–Al2O3 catalysts
Rh–Al2O3 catalysts were prepared by incipient wetness impregnation (IWI) using ammonium aquopentachlororhodate ((NH4)2RhCl5·H2O, Macklin) and γ-alumina (Macklin). The quantity of (NH4)2RhCl5·H2O necessary for each weight loading was dissolved in a small beaker with 500 μL of water. Four weight loads of rhodium (0.1%, 0.5%, 1% and 2%, weight fractions) were deposited on the Al2O3 powder in the crucible, to produce total sample masses of 300 mg. The samples were dried under an infrared lamp, ground up with a mortar and pestle, and then calcined at 400 °C in air for 4 h in a muffle furnace to obtain catalysts with different Rh loads, which were designated as 0.1–2Rh–Al2O3.2.2. Catalyst characterization
The samples were characterized using a JEM-ARM 200F transmission electron microscope (TEM) at an acceleration voltage of 200 kV. The sample powder was sonicated and suspended in ethanol solution and added drop by drop to the carbon-coated copper mesh sample rack. The particle diameter of the sample was measured by DigitalMicrograph software to determine the Rh particle size distribution. The Rh content was determined by inductively coupled plasma (ICP) spectrometry on an Agilent 5800 instrument. Prior to testing, approximately 30 mg of sample was dissolved in nitrohydrochloric acid and kept in a digester for 30 min.CO adsorption diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS) was conducted on a Nicolet iS50 FTIR spectrometer equipped with a ZnSe window sample tank in a pike high temperature reaction chamber. Approximately 20 mg of catalyst was loaded into a sample tank, with the bottom surface lined with quartz wool and a metal mesh positioned beneath to prevent the obstruction of the gas outlet. The samples were pretreated by reduction in 5% H2/Ar (100 mL min−1) at 300 °C for 15 min before testing. After cooling to room temperature (RT), the system was purged with Ar for 10 min, and then the spectrum was recorded as the background. Subsequently, a 10% CO/Ar (50 mL min−1) gas flow was introduced into the sample tank and held for 3 min and then the inlet flow was switched to Ar (100 mL min−1) and held for 60 s, afterwards the spectrum was recorded.
In a typical in situ DRIFTS measurement, ∼15 mg of catalyst was used, and the reduction pretreatment and background acquisition operations were the same as above. Afterwards, the reaction atmosphere was introduced. The raw gas of CO2 hydrogenation consists of 1% CO2 and 5% H2 in a balanced mixture with 94% Ar (sourcing from 10% CO2/Ar, 20% H2/Ar and 99.99% Ar, Liuzhou Huaao Gas Company Limited), with a total flow rate of 40 mL min−1. The sample was heated to the target temperature at a rate of 15 °C min−1, held for 2 min, and then the spectra were recorded. The CO-DRIFTS measurement of the spent catalyst after the 400 °C in situ reaction was the same as the method of fresh catalyst after reduction pretreatment.
X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo Fisher Scientific K-Alpha spectrometer equipped with a monochromatic Al Kα X-ray source (1486.6 eV) operating at 12 kV. All catalyst samples were promptly transferred to vacuum tubes within an Ar-filled glove box and maintained under specified atmospheric conditions, before XPS testing. The samples were also loaded onto the holder in the glove box to prevent oxidation of Rh states. For XPS characterization of samples before the reaction, the samples were reduced in 5% H2/Ar (100 mL min−1) at 300 °C for 15 min on a Beijing Builder PCA-1200 chemisorption analyzer and cooled down in the reduction atmosphere then kept in sealed centrifuge tube. The spectrum of spent catalyst after 400 °C in situ DRIFTS reaction was also recorded. After the measurements, all binding energies were charge-corrected using the dominant sp2-hybridized carbon component (C–C/C–H) −40 of the C 1s adventitious carbon peak fixed at 284.8 eV. The fitting residual is controlled within ±0.1 eV. XPS peak fitting was performed with XPSPEAK, the baseline used a Shirley non-linear sigmoid-type. Rhodium was analyzed on the 3d5/2 and 3d3/2 doublet separated from 4.84 eV. The ratio of peak areas for Rh 3d5/2 and Rh 3d3/2 is constrained to 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The fraction of rhodium in a metallic state was evaluated as the ratio between atomic surface concentration of Rh0 and the total Rh concentration (Rh0/Rhtot).
:2. The fraction of rhodium in a metallic state was evaluated as the ratio between atomic surface concentration of Rh0 and the total Rh concentration (Rh0/Rhtot).
2.3. Computational details
The density functional theory (DFT) calculations were present in supplementary Note 1 in the ESI.†2.4. Catalytic activity and kinetics measurements
A typical CO2 hydrogenation activity evaluation test was performed by mixing 50 mg of catalyst with 750 mg of quartz sand uniformly filled between quartz wool in a U-shaped quartz tube reactor. The tests were carried out at a heating rate of 15 °C min−1. The reaction atmosphere comprised 1% CO2 and 5% H2 in a balanced mixture with Ar (sourced from 10% CO2/Ar, 20% H2/Ar and 99.99% Ar, Liuzhou Huaao Gas Company Limited), with a total flow rate of 100 mL min−1. An on-line gas chromatograph was used to quantitatively analyse the reactants and product gases. FID1 was used to detect organics in this experiment and the CH4 converter equipped with FID2 was used to detect CO, CO2, CH4. Before the test, the catalysts were reduced in 5% H2/Ar at 300 °C for 15 min with at a flow rate of 100 mL min−1. CO2 conversion and product selectivity were calculated using the following equation: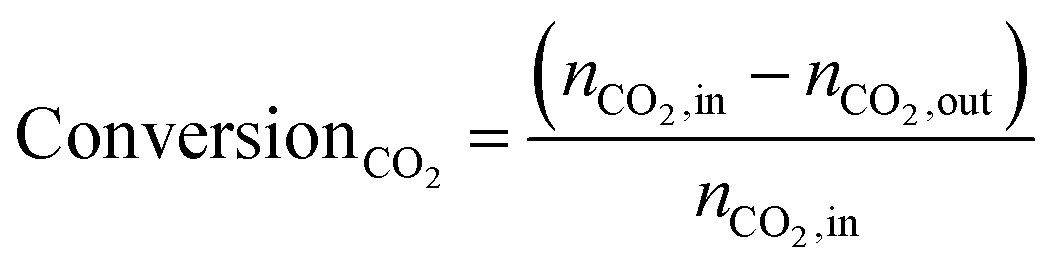

Temperature programmed surface reaction (TPSR) test pretreatment conditions and experimental conditions were consistent with the activity evaluation test. The concentrations of CO2, H2, CO and CH4 were monitored using the HPR-20 R&D online Mass Spectrometer (Hiden Co. Ltd). Prior to the test, the initial gas concentration was calibrated using the quantitative analysis software “QGA Professional” according to the exact flow rate of each gas measured by the mass flowmeter. The kinetic measurements of the CO2 hydrogenation reaction were performed under the condition that CO2 conversion was controlled at a low level (below 20%). In order to prove that our kinetic testing conditions are limited neither by mass transfer nor by heat transfer, we conducted the calculations of Weisz–Prater criteria (CWP) and Mears criteria (CM) for all catalysts with CO2 conversions higher than 20% (uniformly applying the values at 500 °C), and the calculation results show that the mass transfer and heat transfer effects can be entirely ignored in our system. Detailed calculation processes are shown in ESI Note 2.†
3. Results and discussion
3.1 Morphology of Rh–Al2O3
A series of Rh–Al2O3 catalysts with Rh loadings from 0.1 to 2 wt% were synthesized by the method of incipient wetness impregnation, subsequently calcined at 400 °C for 4 h, and reduced in 5% H2/Ar at 300 °C for 15 min. Fig. 1 shows the TEM images of Rh–Al2O3 with different loadings after reduction. The presence of Rh single atoms (Rh SA) was detected in all catalysts despite various loading, as indicated by the red circles (Fig. 1). To prove the presence of the single-atom catalysts, we took TEM images of the Al2O3 support without Rh loading and confirmed that there was no white dot on its surface (Fig. S1†). We classified the nanoparticle by two criterions: (1) the amount of aggregated Rh atoms is more than 4; (2) the distance between each atom is not more than 0.4 nm. It is notable that Rh NPs are present in all samples and marked by yellow circles (Fig. 1b–d), with the exception of the sample with a Rh loading of 0.1 wt%. The results of particle size statistics show that the particle sizes of Rh increase with the increase of Rh loading. The exclusive Rh SA was further verified by the HAADF-TEM image as shown in Fig. S2.† The TEM images of before reduction are shown in Fig. S3.† It is found that the mean particle size of the catalyst after reduction increases, indicating that sintering occurs during the reduction process.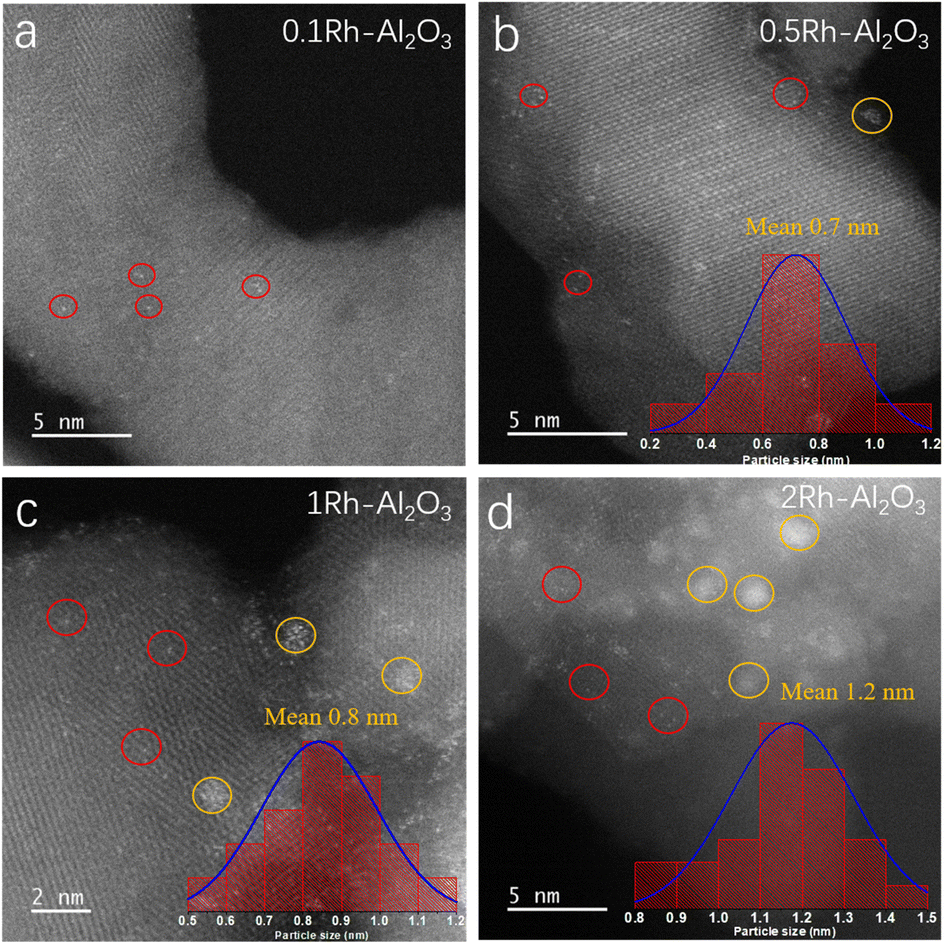 | ||
| Fig. 1 TEM images of reduced (a) 0.1Rh–Al2O3, (b) 0.5Rh–Al2O3, (c) 1Rh–Al2O3 and (d) 2Rh–Al2O3 catalysts. | ||
3.2 Catalytic performances of Rh–Al2O3 in CO2 hydrogenation
Variations in the catalytic environment and reaction conditions can lead to the formation of diverse products; the primary products of CO2 hydrogenation include methane, methanol, carbon monoxide, among others.3,8,34,37 In this study, the products of CO2 hydrogenation were identified as two carbon-containing substances, namely methane (CH4) and carbon monoxide (CO), through online gas chromatography for quantitative analysis of the gas (Fig. S4†). The catalytic performance of CO2 hydrogenation over Rh–Al2O3 catalysts with varying loadings was evaluated at atmospheric pressure and CO2/H2 = 1/5, and the findings are presented in Fig. 2. It can be seen that the catalytic activity and product selectivity changed significantly with the increasing of Rh loading. At the same reaction temperature, the CO2 reaction rate gradually increased with the increasing in Rh loading. Nevertheless, when the Rh loading was increased from 1% to 2%, the enhancement in CO2 reaction rate under identical conditions was not significant, indicating that the number of Rh active sites was sufficient to effectively convert CO2 under the experimental conditions with a Rh loading of 1%. In all Rh–Al2O3 catalyst samples, the reaction rate of CO2 exhibited a gradual increasing trend with the elevation of reaction temperature. However, the reaction rates of the 1Rh–Al2O3 and 2Rh–Al2O3 catalysts exhibited only marginal increases when the temperature was elevated from 400 °C to 500 °C. This observation indicates that the CO2 reaction rate is constrained by the thermodynamic equilibrium of the reaction; it is difficult to continue to increase up to ∼60% under the conditions of the present experiments. | ||
| Fig. 2 The CO2 reaction rate and product selectivity of (a) 0.1Rh–Al2O3, (b) 0.5Rh–Al2O3, (c) 1Rh–Al2O3 and (d) 2Rh–Al2O3 at different temperatures. Equilibrium conversions for CO2 methanation (pink line) as a function of temperature are plotted. The catalyst was reduced by 5% H2/Ar at 300 °C for 15 min before the reaction. Feed gas stream: 1% CO2 and 5% H2 balanced with Ar, flow rate: 100 mL min−1. 50 mg catalyst was used for each sample. | ||
It is also noteworthy that product selectivity showed a tendency towards regular change. At the same temperature, the CH4 selectivity gradually increased and the CO selectivity decreased with the increase of Rh loading, but when Rh loading was increased from 1% to 2%, the CH4 selectivity under the same conditions showed an insignificant increase, and the decrease in CO selectivity was also not obvious. Studies above have indicated that further increases in Rh loading do not result in a significant enhancement in CH4 selectivity and a loading of 1% Rh could represent the optimal condition for the CO2 methanation reaction. Furthermore, methane production was not observed for 0.1Rh–Al2O3 at 300 °C, indicating that Rh NPs exhibit superior CO2 methanation activity compared to Rh SAs at low temperatures.
In order to reveal the difference of intrinsic performance between catalysts with different Rh loadings, we used the temperature programmed surface reaction (TPSR) method to investigate the real-time concentration changes of the reactants CO2 and H2 and the products CH4 and CO. The partial pressure signals of CH4 (m/z = 16), CO (m/z = 28), CO2 (m/z = 44) and H2 (m/z = 2) were recorded by mass spectrometry (MS) in real time from 200 °C to 500 °C, as shown in Fig. 3. It can be observed that the reaction starting temperatures (288 °C, 240 °C, 234 °C and 206 °C, respectively) decreased with increasing of Rh loading, indicating that a higher loading of Rh favors higher reaction activity. Meanwhile, the concentration of CH4 and CO gradually increased with the Rh loading increasing at temperatures lower than 400 °C, which is consistent with the conclusion of Fig. 2. This is because the rising reaction temperatures accelerate both CO2 methanation and reverse water–gas shift (RWGS) reactions for CO2 conversion. It is noteworthy that when operated temperatures exceeded approximately 415 °C, the partial pressure of CO initially increased while that of CH4 posed a gradual decline. Concurrently, the partial pressure of CO2 fell at slower rate for 1Rh–Al2O3 and 2Rh–Al2O3 samples, whereas H2 partial pressure started rising. This indicated that a side reaction occurred and resulted in hydrogen production at elevated temperatures. Combined with higher temperature conditions for hydrogen production and concentration changes of reactants, we infer that dry reforming of methane (DRM) side reactions occurred at temperatures higher than 415 °C.38 To verify the existence of DRM reaction at high temperature, we carried out the temperature-programmed surface reaction (TPSR) of DRM on 1Rh–Al2O3 (Fig. S5†). The generation of H2 and CO begins at ∼400 °C because of the strongly endothermic characteristic (ΔH = +247 kJ mol−1), which is in well line with DRM reaction occurring at ∼415 °C in our system. Combined with the actual CO2 conversion curve in Fig. 2, it is proposed that the decrease of CH4 concentration at high temperatures is mainly ascribed to the consuming of methane in dry reforming, rather than being limited by thermodynamic equilibrium.
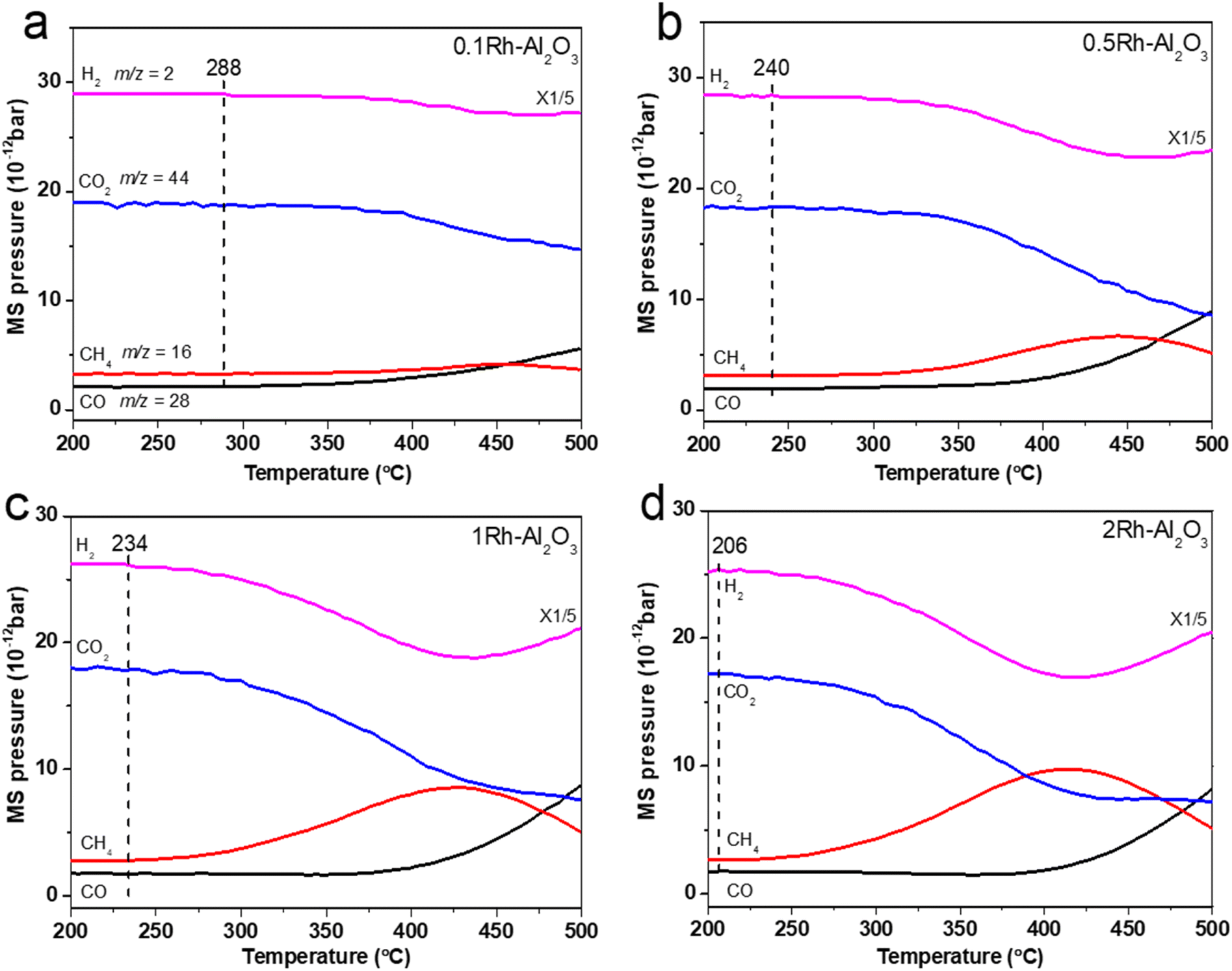 | ||
| Fig. 3 The TPSR profiles of (a) 0.1Rh–Al2O3, (b) 0.5Rh–Al2O3, (c) 1Rh–Al2O3 and (d) 2Rh–Al2O3. The TPSR experimental conditions were the same as those for activity evaluation. | ||
Fig. 4a showed the CO-DRIFTS spectra of Rh–Al2O3 before reaction. Both peaks at ∼2087 and ∼2016 cm−1 can be observed in all the catalysts, which are attributed to the symmetric and asymmetric stretching vibrations of Rh(CO)2 adsorbed on Rh SAs, respectively.30,39 However, when the Rh loading exceeds 0.1 wt%, two distinct adsorption peaks emerge at approximately 1867 cm−1 and 2053 cm−1, ascribed to bridge and linear CO adsorption vibrations on Rh NPs, respectively.30,39 As the Rh loading increases, the CO adsorption signal on Rh NPs progressively intensifies, corresponding to the rise in a quantity of Rh NPs, which aligns with TEM image presented in Fig. 1. Fig. 4b showed the CO-DRIFTS spectra falling to room temperature after reaction at 400 °C. It can be seen that the various peaks appear at almost the same positions as peaks in the samples before reaction, indicating that the form of Rh remains unchanged in all the Rh–Al2O3 catalysts after the reaction. The redshift of the CO adsorption peak after the reaction mainly originates from the change in the electronic state of Rh (Δν ≈ 7 cm−1) such as the aggregation of Rh single atoms and instrumental errors or minor environmental fluctuations (Δν ≈ 5 cm−1), rather than from changes in the particle morphology.
 | ||
| Fig. 4 Room temperature CO-DRIFT spectra of Rh–Al2O3 with different loadings. (a) Before reaction, (b) after reaction at 400 °C. The catalyst was treated with 5% H2/Ar at 300 °C for 15 minutes before the test. CO2 hydrogenation reaction conditions: 1% CO2 and 5% H2 balanced with Ar, flow rate: 40 mL min−1. 20 mg catalyst was used for each sample. | ||
Furthermore, the stability of four Rh–Al2O3 catalysts was studied through durability tests (Fig. S6†). The results show that the CO2 conversion rate of the 0.1Rh–Al2O3 catalyst drops to zero within one hour of the reaction, indicating that the isolated Rh single atom is unstable under the reaction conditions and deactivates rapidly. The CO2 conversion rate of the 0.5–2Rh–Al2O3 catalyst remained basically unchanged within 10 hours, demonstrating excellent long-term stability. The CO-DRIFTS spectra of the catalysts after the durability test showed that the 0.1Rh–Al2O3 catalyst exhibits bridging (1867 cm−1) and linear CO (2053 cm−1) adsorption vibrations on Rh NPs (Fig. S7†), which strongly indicates that isolated Rh single atoms agglomerate to form inactive aggregates under reaction conditions. This is consistent with the phenomenon reported in our previous work that single-atom catalysts are prone to sintering at high temperatures due to the weak stability of isolated metal sites.40,41 In contrast, the CO-DRIFTS spectra of the 0.5–2Rh–Al2O3 catalyst showed no significant changes compared with those before reaction, indicating that Rh nanoparticles in these samples were stable, which is in consistence with the durability test results.
The X-ray photoelectron spectra (XPS) of the Rh–Al2O3 catalyst before the reaction are shown in Fig. 5a–d. The negligible signal of Rh material observed on the 0.1Rh–Al2O3 catalyst in Fig. 5a can be attributed primarily to the low Rh content, which falls outside the detection range of XPS spectroscopy. The peaks with binding energies of ∼307.9 eV and ∼310.2 eV in the Rh 3d5/2 spectrum are attributed to Rh0 and Rh3+ species, respectively;42 with the Rh loading increases from 0.5% to 2%, the percentage of Rh0 also rises from 39% to 47%, as shown in Fig. 5b–d. It suggests a gradual increase of the particle size of Rh, which aligns with the findings from TEM analysis. Fig. 5e–h illustrates the Rh 3d XPS spectra after the 400 °C reaction, and we observe that the percentages of Rh0 in each catalyst after reaction have no obvious change compared to the corresponding catalysts before reaction, which is consistent with the findings in Fig. 4. The detailed results of XPS deconvolution of the Rh–Al2O3 catalyst are shown in Tables S1 and S2.†
 | ||
| Fig. 5 XPS spectra in the Rh 3d region of different loadings Rh–Al2O3. (a–d) Before reaction, (e–h) after reaction at 400 °C. All catalysts before reaction were pretreated by reduction in 5% H2/Ar at 300 °C for 15 min before testing. | ||
The actual Rh loadings were further measured by ICP and are shown in Table 1, and it is seen that the values consequently approached the nominal Rh loading. The significant discrepancies in the stoichiometric ratios of CO adsorbed onto the surfaces of Rh SA and Rh NP, along with the variations in the ratios of CO adsorbed onto Rh NP surfaces with differing particle sizes,33 render traditional CO chemisorption techniques inadequate for precisely determining the dispersion of Rh–Al2O3 samples. In this study, the Rh SA content in other Rh–Al2O3 samples is semi-quantified based on the adsorption strength of Rh SA in the CO-DRIFT spectrum of each Rh–Al2O3 sample, with the 0.1Rh–Al2O3 sample serving as a reference point, and corresponding calculation process is outlined in Table S3 and Fig. S8.† The dispersion of Rh NP is derived from the mean particle size of Rh NP measured by TEM particle size statistics. Fig. 1 and S9† show representative TEM images of Rh NP, and the statistical mean size of Rh NP is listed in Table 1. The Rh NP dispersion was determined by calculating the reciprocal of the average size of Rh NP (in nm) and subsequently multiplying this value by a correction factor, following the methodology outlined by Zhang et al..43 Fig. S10† shows that the calculated Rh NP dispersion is consistent with the actual Rh NP dispersion when the correction coefficient is 0.3. Accordingly, the turnover frequency of CO2 conversion (TOFCO2) and CH4 generation (TOFCH4) as well as the theoretical turnover frequency of Rh NP CH4 generation (TOFCH4 of Rh NP) were calculated for the Rh–Al2O3 catalysts at 400 °C and detailed calculations are shown in Tables S4 and S5† As observed in Table 1, the minimal variation observed in the turnover frequency of CO2 (TOFCO2) across different Rh–Al2O3 samples indicates a comparable CO2 conversion efficiency between Rh single atoms (Rh SA) and Rh nanoparticles (Rh NP). Furthermore, the turnover frequency of CH4 (TOFCH4) for samples containing Rh loadings exceeding 0.5% is notably higher than that of samples with a Rh loading of 0.1%. This suggests that Rh NPs exhibit a superior capacity for CH4 generation compared to Rh SA. Additionally, the theoretical TOFCH4 of Rh NP was calculated and approximately three times the value of Rh SA (Table 1), suggesting better CO2 methanation activity of Rh sites on Rh NP than Rh SA. In this study, we conducted a comparative analysis of the turnover frequency values of CH4 generation (TOFCH4) calculated in this work with those in CO2 methanation using various Rh-based catalysts (Table S6†). The TOF values reported here are superior than others.
| Sample | Rh actual contenta (wt%) | Rh SA contentb (wt%) | Rh NP content (wt%) | Mean size of Rhc NP (nm) | Rh NP dispersiond (%) | Rh total dispersione (%) | TOFCO2, netf (s−1) | gTOFCH4, net (s−1) | TOFCH4, net of Rhg NP (s−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured by ICP-AES.b Determined by the IR intensity of CO adsorption on Rh single atom (SA), and the detailed calculation is shown in Table S3.c Determined by TEM image counts.d Rh NP dispersion = 1/mean size of Rh NP × 0.3.e Total dispersion = (Rh SA content × 100 + Rh NP content × Rh NP di2persion)/Rh actual content.f Net reaction rate of CO2, measured at 400 °C, and the detailed calculation is shown in Table S4.g Net CH4 formation rate, measured at 400 °C, and the detailed calculation is shown in Table S5. | |||||||||
| 0.1Rh–Al2O3 | 0.09 | 0.09 | — | — | — | 100 | 0.11 | 0.06 | — |
| 0.5Rh–Al2O3 | 0.46 | 0.17 | 0.29 | 0.7 | 42 | 64 | 0.13 | 0.10 | 0.16 |
| 1Rh–Al2O3 | 1.16 | 0.19 | 0.97 | 0.8 | 36 | 46 | 0.15 | 0.14 | 0.18 |
| 2Rh–Al2O3 | 1.81 | 0.22 | 1.59 | 1.2 | 25 | 34 | 0.14 | 0.13 | 0.17 |
We performed kinetic experiments on the CO2 methanation reaction on Rh–Al2O3, as shown in Fig. 6. From Fig. 6a, the apparent activation energy (Ea) gradually decreased with increasing Rh loading, indicating that Rh NP is more prone to catalyze CO2 methanation reaction. In Fig. 6b, the negative measured CO2 apparent reaction orders (nCO2) indicated that increasing partial pressure of CO2 for 0.1% Rh-loaded catalyst would inhibit the CH4 formation and conversely, partial pressure of CO2 could achieve the different degree of enhancement in the formation rate of CH4 for other samples, due to non-negative apparent reaction orders. In addition, the reaction orders of CO2 (nCO2) are approximately zero (−0.21–0.23), also indicating that CO2 adsorption and activation is not a significant influencing factor in the CO2 methanation reaction.
 | ||
| Fig. 6 Arrhenius plots of (a) CO2 methanation reaction. Reaction orders of (b) CO2 and (c) H2 with the different loadings Rh–Al2O3 catalysts in the CO2 methanation reactions. (d) The adsorption energies of H2 adsorbing on Rh1–Al2O3, Rh4–Al2O3 and Rh13–Al2O3 by DFT calculations. | ||
On the other hand, as shown in Fig. 6c, an increase in the partial pressure of H2 could significantly increase CH4 formation rates for all Rh–Al2O3 catalyst samples, because the H2 reaction orders (nH2) are greater than 0.80 and much higher than nCO2 (Fig. 6c), suggesting that H2 dissociation and activation should be key factors affecting catalytic performance of Rh–Al2O3 catalysts. In addition, the nH2 gradually decreases with increasing sizes of Rh particles and it also indicates that H2 adsorption becomes more pronounced for Rh nanoparticles with larger size, thus contributing to deep hydrogenation for the product CO or other intermediates, thereby indicating that high Rh loading favors methane generation. Subsequent density functional theory (DFT) calculations were employed to ascertain H2 adsorption strength on different supported Rh sites. The structural models of Rh1–Al2O3, Rh4–Al2O3, and Rh13–Al2O3 were used to represent Rh single atoms, Rh clusters, and Rh nanoparticles loaded on the Al2O3 surface, respectively40 and the calculated results and H2 adsorption structures are shown in Fig. 6d and Tables S7–9,† respectively. It can be found that the H2 adsorption becomes stronger for supported Rh catalysts with larger size, which is consistent with the experimental results of the reaction orders. The reaction orders of the reverse water–gas shift (RWGS) process are not determined because of low CO2 conversion rates of Rh–Al2O3 samples, with the exception of the 0.1Rh–Al2O3 samples (in Table S10†).
Table 2 shows the calculation results of mass and heat transfer criteria for CO2 hydrogenation reaction with different catalysts at 773.15 K. The average particle size of catalyst was measured by laser particle size analyzer (Fig. S11†). The relevant parameters to calculate mass and heat transfer limits of CO2 hydrogenation reaction was given in Tables S11 and S12.† The results show that both the mass transfer and heat transfer effects can be ignored under our experimental conditions.
| Sample | T (K) | Deff (×10−6 m2 s−1) | kc (m s−1) | h (W m−2 K−1) | CWP (×10−3) | CM, mass (×10−3) | CM, heat (×10−6) |
|---|---|---|---|---|---|---|---|
| 0.1Rh–Al2O3 | 773.15 | 8.09 | 1.15 | 580.04 | 1.32 | 0.132 | 3.73 |
| 0.5Rh–Al2O3 | 773.15 | 8.09 | 0.86 | 436.54 | 6.12 | 0.612 | 289 |
| 1Rh–Al2O3 | 773.15 | 8.09 | 0.84 | 426.82 | 7.19 | 0.719 | 400 |
| 2Rh–Al2O3 | 773.15 | 8.09 | 1.27 | 644.35 | 3.15 | 0.315 | 145 |
3.3 Reaction mechanism of Rh–Al2O3 in CO2 hydrogenation
In order to deeply investigate the reaction mechanism and intermediates of Rh–Al2O3 catalyzed CO2 hydrogenation, the in situ DRIFTS spectra of Rh–Al2O3 with different loadings were measured and the results are presented in Fig. 7. The peaks at 1438–1433 cm−1, 1587 cm−1 and 1372 cm−1 are attributed to the absorption peaks of, CO32−*,32,34 the vibrations of monodentate formate (m-HCOO*) and bidentate formate (b-HCOO*), respectively.44–46 It can be observed that the intensity of CO32−* absorption peak decreased with increasing temperature due to CO32−* binding with H* to produce formates, which further caused the appearance of formate signals (1587 cm−1 and 1372 cm−1)44 and the appearance temperatures gradually decreased with the increasing Rh loading. The CO32−* adsorption signal finally disappeared at 400 °C, 300 °C, 250 °C and 200 °C for 0.1Rh–Al2O3, 0.5Rh–Al2O3, 1Rh–Al2O3, 2Rh–Al2O3, respectively. The temperature at which HCOO* species began to decrease gradually (from 300 °C to 100 °C) with the increase of Rh loading amount. The gaseous CH4 (2907 cm−1) is also generated along with the HCOO* species,33,47 suggesting that formate should be the key intermediate species for methane production.45,48,49 These results illustrated that high Rh loading would bring about the superior catalytic performance of CO2 hydrogenation, in accordance to the catalytic performance tests in Fig. 2.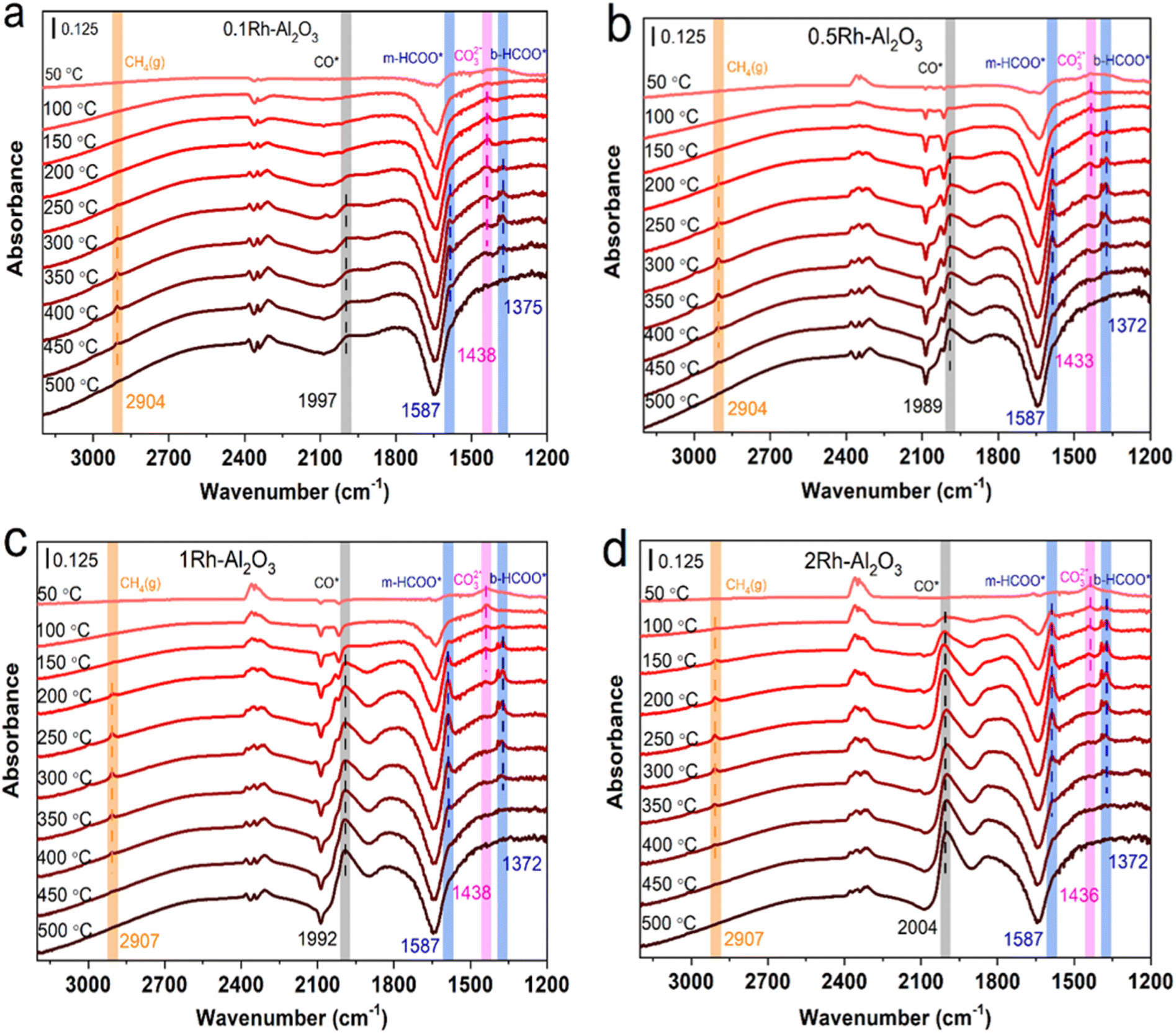 | ||
| Fig. 7 In situ DRIFTS spectra of (a) 0.1Rh–Al2O3, (b) 0.5Rh–Al2O3, (c) 1Rh–Al2O3 and (d) 2Rh–Al2O3 during CO2 hydrogenation at different temperatures. | ||
The peak appearing at ∼2000 cm−1 is attributed to the CO adsorption peak on Rh,20,34,50 and its appearance temperatures gradually fell with increasing Rh loadings, but the product temperature of CO gas increased with increasing Rh loadings, as shown in Fig. 3. The inconsistency suggested that the formed CO species should first be adsorbed on the surface sites and then released as gas CO via overcoming desorption barriers and the catalysts with higher Rh loading should pose the higher CO desorption barriers. Otherwise, it is found that the signal of CO (CO*) and gaseous CH4 appears at the same temperature and thus it is inferred that CO* is also a crucial intermediate species of methane generation.31,34,45,51 It is worth noting that at temperatures higher than 450 °C, the signals of gaseous CH4 and HCOO* disappeared, but the CO adsorption signal still existed, which further confirmed that DRM or RWGS reactions occur at high temperatures. The situation agrees with the conclusion that CO selectivity at high temperatures is enhanced as shown in Fig. 2.
To determine the change of each pathway to the formation of CH4, the intensity of the CO* (∼2000 cm−1) and m-HCOO* (1587 cm−1) bands were calculated by the adsorbing intensity of the highest point of the peak position minus the adsorbing intensity of the baseline in each IR spectrum. Intensity values were plotted in function of temperature for each sample, as shown in Fig. 8. It can be found that the intensities of both intermediates increase with the elevated temperatures until up to 250 °C or 300 °C for all samples. When the temperature exceeded that temperature, the intensity of HCOO* reversed to gradually decrease, illustrating the conversion of formate to the product CH4. However, the intensity of CO* started to increase again when the temperature high than 350 °C, attributing to the produced CO species of DRM reaction at high temperature. Accordingly, both pathways contributed to CO2 methanation reaction, the intensity of CO* increased at high temperature mainly attributed to DRM reaction, as evidenced by the TPSR results of mere DRM reaction (Fig. S5†).
 | ||
| Fig. 8 In situ DRIFTS normalized band intensity versus temperature of CO* (∼2000 cm−1) and m-HCOO* (1587 cm−1). (a) 0.1Rh–Al2O3. (b) 0.5Rh–Al2O3. (c) 1Rh–Al2O3. (d) 2Rh–Al2O3. | ||
4. Conclusions
In this study, a series of Rh–Al2O3 catalysts with varying Rh particle sizes were prepared by modulating Rh loading amounts. TEM and CO-DRIFTS showed that the 0.1Rh–Al2O3 sample existed in the form of single atoms and 0.5–2Rh–Al2O3 catalyst samples existed with a mixture of Rh single atoms and Rh nanoparticles. The activity tests showed that the CH4 selectivity gradually increased with increasing Rh loading at the same reaction temperature. The CH4 yield of each Rh–Al2O3 catalyst first increases and then decreases with increasing reaction temperature; CH4 selectivity decreases sharply at temperatures higher than 400 °C. The TPSR experiments revealed that when the temperature exceeds approximately 415 °C, the partial pressure of H2 begins to increase significantly reversing from its downward trend, attributed to the occurrence of DRM reaction at elevated temperatures. Therefore, the decreased CH4 selectivity at high temperatures is not entirely limited by the thermodynamics of CO2 methanation, but largely due to DRM side reaction consuming large amounts of methane.Experimental results from in situ infrared spectroscopy confirm that both formate and CO pathways coexist for Rh–Al2O3 catalyzing CO2 methanation regardless of Rh loading amounts and formate pathway is dominate for CO2 methanation when the temperature high than 400 °C. TOF calculation showed that TOFCO2 did not differ much between different Rh particle sizes, but TOFCH4 on Rh NP was three times higher than that of TOFCH4 on Rh SA, suggesting better CO2 methanation activity of Rh sites on Rh NP. Overall, the reaction orders of CO2 on Rh–Al2O3 catalyst are approximately 0, whereas nH2 approaches 1 and decreases with increasing Rh loading, suggesting that H2 dissociation and activation are critical factors influencing performance of Rh–Al2O3 catalysts. Additionally, H2 adsorbed more strongly on Rh catalysts with large particle size by DFT calculations. These results substantially enhance the systematic and thorough comprehension of the catalytic hydrogenation of CO2 over Rh–Al2O3 catalysts.
Data availability
Data will be made available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22162005) and the Innovation Project of Guangxi University of Science and Technology Graduate education (GKYC202333). We thank Pan from Shiyanjia Lab (http://https:\www.shiyanjia.com) for the ICP and XPS tests.References
- A. D. N. Kamkeng, M. Wang, J. Hu, W. Du and F. Qian, Chem. Eng. J., 2021, 409, 128138 CrossRef CAS.
- H. Sun, C. Wu, B. Shen, X. Zhang, Y. Zhang and J. Huang, Mater. Today Sustain., 2018, 1–2, 1–27 Search PubMed.
- H. Zheng, W. Liao, J. Ding, F. Xu, A. Jia, W. Huang and Z. Zhang, ACS Catal., 2022, 12, 15451–15462 CrossRef CAS.
- K. Li, X. Li, L. Li, X. Chang, S. Wu, C. Yang, X. Song, Z.-J. Zhao and J. Gong, JACS Au, 2023, 3, 508–515 CrossRef CAS PubMed.
- S. Sharma, Z. Hu, P. Zhang, E. W. McFarland and H. Metiu, J. Catal., 2011, 278, 297–309 CrossRef CAS.
- S. Li, Y. Xu, Y. Chen, W. Li, L. Lin, M. Li, Y. Deng, X. Wang, B. Ge, C. Yang, S. Yao, J. Xie, Y. Li, X. Liu and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 10761–10765 CrossRef CAS PubMed.
- O. Martin, A. J. Martín, C. Mondelli, S. Mitchell, T. F. Segawa, R. Hauert, C. Drouilly, D. Curulla-Ferré and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2016, 55, 6261–6265 CrossRef CAS PubMed.
- N. Rui, X. Wang, K. Deng, J. Moncada, R. Rosales, F. Zhang, W. Xu, I. Waluyo, A. Hunt, E. Stavitski, S. D. Senanayake, P. Liu and J. A. Rodriguez, ACS Catal., 2023, 13, 3187–3200 CrossRef CAS.
- J. Lu, B. Fu, M. C. Kung, G. Xiao, J. W. Elam, H. H. Kung and P. C. Stair, Science, 2012, 335, 1205–1208 CrossRef CAS PubMed.
- K. Nakagawa, C. Kajita, K. Okumura, N.-o. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Catal., 2001, 203, 87–93 CrossRef CAS.
- X. Li, J. Lin, L. Li, Y. Huang, X. Pan, S. E. Collins, Y. Ren, Y. Su, L. Kang, X. Liu, Y. Zhou, H. Wang, A. Wang, B. Qiao, X. Wang and T. Zhang, Angew. Chem., Int. Ed., 2020, 59, 19983–19989 CrossRef CAS PubMed.
- W. Zhen, F. Gao, B. Tian, P. Ding, Y. Deng, Z. Li, H. Gao and G. Lu, J. Catal., 2017, 348, 200–211 CrossRef CAS.
- Z.-W. Zhao, X. Zhou, Y.-N. Liu, C.-C. Shen, C.-Z. Yuan, Y.-F. Jiang, S.-J. Zhao, L.-B. Ma, T.-Y. Cheang and A.-W. Xu, Catal. Sci. Technol., 2018, 8, 3160–3165 RSC.
- F. Hu, R. Ye, C. Jin, D. Liu, X. Chen, C. Li, K. H. Lim, G. Song, T. Wang, G. Feng, R. Zhang and S. Kawi, Appl. Catal., B, 2022, 317, 121715 CrossRef CAS.
- Q. Li, Y. Gao, M. Zhang, H. Gao, J. Chen and H. Jia, Appl. Catal., B, 2022, 303, 120905 CrossRef CAS.
- J. Dong, D. Li, Y. Zhang, P. Chang and Q. Jin, J. Catal., 2022, 407, 174–185 CrossRef CAS.
- Y. Du, C. Qin, Y. Xu, D. Xu, J. Bai, G. Ma and M. Ding, Chem. Eng. J., 2021, 418, 129402 CrossRef CAS.
- C. Italiano, G. Drago Ferrante, L. Pino, M. Laganà, M. Ferraro, V. Antonucci and A. Vita, Chem. Eng. J., 2022, 434, 134685 CrossRef CAS.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 RSC.
- J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei and S. Kawi, Catal. Today, 2020, 356, 471–489 CrossRef CAS.
- C. He, Q. Li, Z. Ye, L. Wang, Y. Gong, S. Li, J. Wu, Z. Lu, S. Wu and J. Zhang, Angew. Chem., Int. Ed., 2024, e202412308 CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- T. J. Siang, A. A. Jalil, N. A. A. Fatah and M. E. Chung, J. Environ. Chem. Eng., 2021, 9, 104616 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2016, 9, 120–127 CrossRef PubMed.
- A. Karelovic and P. Ruiz, J. Catal., 2013, 301, 141–153 CrossRef CAS.
- H. Arandiyan, Y. Wang, J. Scott, S. Mesgari, H. Dai and R. Amal, ACS Appl. Mater. Interfaces, 2018, 10, 16352–16357 CrossRef CAS PubMed.
- R. Thalinger, T. Götsch, C. Zhuo, W. Hetaba, W. Wallisch, M. Stöger-Pollach, D. Schmidmair, B. Klötzer and S. Penner, ChemCatChem, 2016, 8, 2057–2067 CrossRef CAS.
- D. Pandey and G. Deo, J. Ind. Eng. Chem., 2016, 33, 99–107 CrossRef CAS.
- A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 237–249 CrossRef CAS.
- J. C. Matsubu, V. N. Yang and P. Christopher, J. Am. Chem. Soc., 2015, 137, 3076–3084 CrossRef CAS PubMed.
- B. Yang, Y. Wang, B. Gao, L. Zhang and L. Guo, ACS Catal., 2023, 13, 10364–10374 CrossRef CAS.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, ACS Catal., 2018, 8, 6203–6215 CrossRef CAS.
- D. Heyl, U. Rodemerck and U. Bentrup, ACS Catal., 2016, 6, 6275–6284 CrossRef CAS.
- S. Li, Y. Xu, H. Wang, B. Teng, Q. Liu, Q. Li, L. Xu, X. Liu and J. Lu, Angew. Chem., Int. Ed., 2023, 62, e202218167 CrossRef CAS PubMed.
- X. Xu, L. Liu, Y. Tong, X. Fang, J. Xu, D.-e. Jiang and X. Wang, ACS Catal., 2021, 11, 5762–5775 CrossRef CAS.
- H. Xin, L. Lin, R. Li, D. Li, T. Song, R. Mu, Q. Fu and X. Bao, J. Am. Chem. Soc., 2022, 144, 4874–4882 CrossRef CAS PubMed.
- H. Zhao, R. Yu, S. Ma, K. Xu, Y. Chen, K. Jiang, Y. Fang, C. Zhu, X. Liu, Y. Tang, L. Wu, Y. Wu, Q. Jiang, P. He, Z. Liu and L. Tan, Nature Catalysis, 2022, 5, 818–831 CrossRef CAS.
- Y. Wang, L. Li, G. Li, Q. Zhao, X. s. Wu, Y. Wang, Y. Sun and C. Hu, ACS Catal., 2023, 13, 6486–6496 CrossRef CAS.
- Y. Kwon, T. Y. Kim, G. Kwon, J. Yi and H. Lee, J. Am. Chem. Soc., 2017, 139, 17694–17699 CrossRef CAS PubMed.
- J. Dong, Y. Zhang, D. Li, A. Adogwa, S. Huang, M. Yang, J. Yang and Q. Jin, Appl. Catal., B, 2023, 330, 122662 CrossRef CAS.
- J. Dong, S. Huang, S. Li, P. Chang and J. Yang, Catal. Sci. Technol., 2024, 14, 5211–5217 RSC.
- Y. Feng, Y. Zhang, J. Wang, L. Ling, R. Zhang, M. Fan, B. Hou, D. Li and B. Wang, ACS Catal., 2024, 14, 1874–1881 CrossRef CAS.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- A. Cárdenas-Arenas, A. Quindimil, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, U. De-La-Torre, B. Pereda-Ayo, J. A. González-Marcos, J. R. González-Velasco and A. Bueno-López, Appl. Catal., B, 2020, 265, 118538 CrossRef.
- C. Wang, Y. Lu, Y. Zhang, H. Fu, S. Sun, F. Li, Z. Duan, Z. Liu, C. Wu, Y. Wang, H. Sun and Z. Yan, Nano Res., 2023, 16, 12153–12164 CrossRef CAS.
- A. Holmgren, B. Andersson and D. Duprez, Appl. Catal., B, 1999, 22, 215–230 CrossRef CAS.
- R.-P. Ye, Q. Li, W. Gong, T. Wang, J. J. Razink, L. Lin, Y.-Y. Qin, Z. Zhou, H. Adidharma, J. Tang, A. G. Russell, M. Fan and Y.-G. Yao, Appl. Catal., B, 2020, 268, 118474 CrossRef CAS.
- F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans and X. Duan, J. Am. Chem. Soc., 2016, 138, 6298–6305 CrossRef CAS PubMed.
- B. Miao, S. S. K. Ma, X. Wang, H. Su and S. H. Chan, Catal. Sci. Technol., 2016, 6, 4048–4058 RSC.
- Y. Tang, T. Zhao, H. Han, Z. Yang, J. Liu, X. Wen and F. Wang, Adv. Sci., 2023, 10, 2300122 CrossRef CAS PubMed.
- Y. Li, Z. Liu, Z. Rao, F. Yu, W. Bao, Y. Tang, H. Zhao, J. Zhang, Z. Wang, J. Li, Z. Huang, Y. Zhou, Y. Li and B. Dai, Appl. Catal., B, 2022, 319, 121903 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03953c |
| This journal is © The Royal Society of Chemistry 2025 |