
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotochemical [2 + 2] cycloadditions of naphthalene acrylic acids: templated and untemplated photoreactivity, selective homo/heterodimerizations, and conformational analysis†
Merve Temel a,
Gizem Yildiza,
Kaan Berkay Ceyhana,
Fahri Alkana,
Onur Şahinb and
Yunus E. Türkmen*ac
a,
Gizem Yildiza,
Kaan Berkay Ceyhana,
Fahri Alkana,
Onur Şahinb and
Yunus E. Türkmen*ac
aDepartment of Chemistry, Faculty of Science, Bilkent University, Ankara, 06800, Turkiye. E-mail: yeturkmen@bilkent.edu.tr
bDepartment of Occupational Health & Safety, Faculty of Health Sciences, Sinop University, Sinop 57000, Turkiye
cUNAM, National Nanotechnology Research Center, Institute of Materials Science and Nanotechnology, Bilkent University, Ankara, 06800, Turkiye
First published on 14th July 2025
Abstract
Selective homo- and heterodimerization reactions of naphthalene acrylic acids have been achieved in a diastereocontrolled manner with the use of 1,8-dihydroxynaphthalene as a covalent template in photochemical [2 + 2] cycloadditions. When the reactions were run in solution, cycloaddition products were obtained in good yields (64–88%), which were subsequently detached from the template via a transesterification reaction affording the symmetrical and unsymmetrical cyclobutane products in 75–91% yields. A careful examination of the crystal structures of diester 6a and cycloadduct 15a along with detailed powder XRD and ATR-IR spectroscopic studies revealed an intriguing case of conformational isomerism in 15a arising from the different possible orientations of its two ester carbonyls. The rotational barriers between the different conformers of 15a were calculated to be within the range of 4.19–7.10 kcal mol−1, indicative of rapid interconversion between these conformers in solution at room temperature.
Introduction
Photodimerization reactions of cinnamic acids and related analogues to access multisubstituted cyclobutanes comprise an important subclass of photochemical [2 + 2] cycloadditions of olefins.1 Dimers of cinnamic acid derivatives, namely truxillic and truxinic acids, esters and amides, attracted significant attention from the synthetic community due to their presence in many cyclobutane-containing natural products, their interesting biological activities, and applications in polymer chemistry and materials science.2 A variety of template-directed strategies3 have been developed over the years in order to achieve [2 + 2] photodimerization of olefins in a regio- and stereoselective manner in both solid state4 and solution phase.5 In this respect, the use of diol- and bis-aniline-based covalent templates proved to be especially useful for the selective photochemical [2 + 2] cycloadditions of cinnamic acids.6 Moreover, Benaglia and co-workers developed in 2023 a chiral auxiliary-based approach to access δ-truxinate products with high enantiomeric excess values.7 Recently, in their elegant work, Yoon and co-workers reported enantioselective synthesis of β- and δ-truxinates as well as selective synthesis of α-truxillates.8Despite these advances in the photodimerization reactions of cinnamic acid derivatives, photocycloadditions of acrylic acids substituted with polycyclic aromatic groups have been considerably less explored.9 In one such study, Sawaki and co-workers reported in 1993 the photochemical [2 + 2] cycloaddition of sodium 1-naphthyl acrylate (1) in the presence of hydrotalcite clays, which gave the β-truxinate cycloadduct 2 in 75% yield along with the formation of cis-olefin 3 (25%, Scheme 1a).9b More recently, Mutlu, Barner-Kowollik and co-workers utilized the reversible [2 + 2] cycloaddition of 1-pyrenyl acrylic acid esters (4) giving cyclobutanes 5 in the design of systems that exhibit orthogonal photochemical reactivity in a single molecule (Scheme 1b).9d In 2020, Lu and co-workers investigated the photomechanical effects of [2 + 2] cycloaddition reactions of halogen-substituted 2-naphthalene acrylic acids, where the crystals of F–, Cl–, and Br-substituted 2-naphthalene acrylic acid derivatives were found to undergo a variety of motions such as bending, rotating, bursting, etc. upon irradiation with UV light.9e In 2021, we utilized 1,8-dihydroxynaphthalene (1,8-DHN) as a diol-based covalent template to provide a general solution to the selective homo- and heterodimerization of cinnamic acids (Scheme 1c).10 Recently, we extended our methodology to the photodimerization reactions of vinylogous cinnamic acids to access symmetrical and unsymmetrical dialkenyl cyclobutanes.11 In the present work, we aimed to apply our template-directed strategy to the photodimerization of naphthalene acrylic acids (Scheme 1d).
 | ||
| Scheme 1 Examples of photodimerization of β-aryl acrylic acids with aryl groups larger than a phenyl ring, and our work. | ||
There are a number of reasons for our interest in the photocycloadditions of this class of compounds. First, modification of a phenyl to a naphthyl ring, while seemingly simple, was shown to lead to superior results in various research areas possibly due to the larger size of the naphthyl ring in addition to its higher capability to participate in CH–π and π–π interactions compared to the phenyl ring. For instance, the presence of the naphthyl groups in 2-methoxy-2-naphthylpropanoic acids was shown by the Harada and Ichikawa groups to be highly beneficial in chiral recognition,12 and determination of absolute configuration of alcohols.13 Moreover, it was shown by Rawal and co-workers that much higher catalytic activity and enantioselectivity levels were obtained with the use of a 1-naphthyl-substituted taddol derivative as hydrogen bonding catalyst in enantioselective Diels–Alder reactions compared to the phenyl-substituted taddol catalyst.14 Second, the higher conjugation of the π system present in naphthalene acrylic acids was expected to change their photophysical and photochemical properties,15 which might affect their photoreactivity in [2 + 2] cycloadditions as well as their stabilities due to cis–trans isomerization under UV light or daylight. Finally, the larger size of the naphthyl ring has the potential to affect the crystal structures of template-bound naphthalene acrylic acids, which may lead to altered photochemical reactivity in solid state. Due to these reasons, we investigated in this work the photodimerization of naphthalene acrylic acids in solid state and in solution with the use of 1,8-DHN as a covalent template (Scheme 1d). In this regard, we achieved highly selective homo- and heterodimerizations of naphthalene acrylic acids, and compared the efficiencies and outcomes of the templated and untemplated photocycloadditions. Moreover, we performed a careful conformational analysis of the template-bound cycloadducts, which led to the identification of a highly interesting example of conformational isomerism. This phenomenon emerges as a result of the parallel and anti-parallel orientations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups with respect to each other in the template-bound, photochemical [2 + 2] cycloaddition products. Such instances of conformational isomerism are attractive from a stereochemical viewpoint, as they have the potential to lead to atropisomerism depending on the energy barrier for the interconversion of the specific conformers.
O groups with respect to each other in the template-bound, photochemical [2 + 2] cycloaddition products. Such instances of conformational isomerism are attractive from a stereochemical viewpoint, as they have the potential to lead to atropisomerism depending on the energy barrier for the interconversion of the specific conformers.
Results and discussion
Photochemical [2 + 2] cycloadditions of template-bound naphthalene acrylic acids
Our studies commenced with the synthesis of the template-bound cycloaddition precursors 6a–c (Scheme 2). For this purpose, we first prepared (E)-3-(naphthalen-1-yl)acrylic acid (10) in three steps starting from the commercially available 1-naphthol. Triflation of 1-naphthol with Tf2O in the presence of Et3N gave naphthyl triflate 8 in 95% yield. A subsequent Pd-catalyzed Heck coupling of 8 with methyl acrylate afforded α,β-unsaturated ester 9 in excellent yield (98%). The targeted 1-naphthalene acrylic acid 10 was obtained in 90% yield upon a final basic hydrolysis of ester 9. The 3J coupling constants of the olefinic protons of 9 and 10 were determined to be 15.8 and 15.7 Hz, respectively, which indicate that both compounds have trans alkene configurations. | ||
| Scheme 2 Synthesis of the [2 + 2] cycloaddition precursors 6a–c. | ||
With the naphthalene acrylic acid 10 in hand, we next synthesized the symmetrical diester 6a via the esterification of 1,8-DHN (12) with 10 using DCC (N,N′-dicyclohexylcarbodiimide) and DMAP (4-dimethylaminopyridine) in 48% yield (Scheme 2). Preparation of the desired unsymmetrical cycloaddition precursors 6b and 6c required the sequential introduction of two different α,β-unsaturated acyl groups to 1,8-DHN. To this end, first the previously reported mono-ester 13,10a was prepared by the mono-acylation of 1,8-DHN (12) with trans-cinnamoyl chloride in 78% yield. A second acylation of mono-ester 13 with the use of 1-naphthalene acryloyl chloride 11, which in turn was prepared from 10 via its reaction with oxalyl chloride, and NaH as base afforded the unsymmetrical diester 6b in 50% yield. A similar strategy was undertaken for the synthesis of diester 6c. Initially, the known mono-ester 14,10a was prepared in 76% yield from 1,8-DHN. The second acylation of mono-ester 14 with acyl chloride 11 in the presence of NaH resulted in the formation of the unsymmetrical cycloaddition precursor 6c in 51% yield.
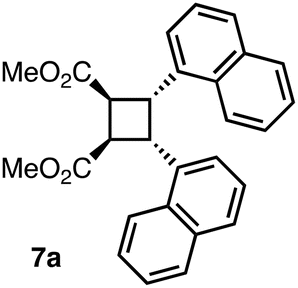
Following the successful synthesis of the cycloaddition precursors 6, we next investigated their [2 + 2] photodimerization reactions. To our delight, the irradiation of a powder sample of 6a with UV-A light (365 nm, 4 × 9 W fluorescent lamps) afforded [2 + 2] cycloadduct 15a in 88% yield as a single diastereomer (Table 1, entry 1). Moreover, the photochemical cycloaddition of the same compound in CHCl3 solution was found to work equally well (88% yield, entry 1). Single crystals of diester 6a were grown by the vapor diffusion of n-pentane to its solution in CH2Cl2. Single-crystal X-ray analysis of 6a (CCDC 2426760) revealed an almost parallel orientation of the reacting olefins with a distance of 3.71 Å between their centroids (Fig. 1a and S1†), which is in accordance with the Schmidt criteria,1c and hence provides a rationale for the successful cycloaddition result that we obtained in solid state. The two naphthalene rings are also close to being parallel to each other with a dihedral angle of 4° between the two naphthalene planes. In addition, the shortest distance of the centroid of one of the phenyl rings of the top naphthalene to the plane formed by the bottom naphthalene in Fig. 1a was observed to be 3.54 Å, which is indicative of a strong π–π stacking interaction between the two naphthalene rings. Furthermore, the crystal structure of 6a exhibited several intermolecular CH–π interactions with distances ranging between 2.74 and 2.94 Å (Fig. S2†).16 The structure of cycloadduct 15a was also analyzed by single-crystal X-ray crystallography (CCDC 2426761) confirming its β-truxinate nature and thus, the stereochemistry of the cycloaddition process (Fig. 1b, S3 and S4†). We should also add that we observed small amounts of product formation when compound 6a was kept in solution under ambient laboratory conditions. In order to check this in a controlled manner, a solution of 6a in CDCl3 in an NMR tube was exposed to daylight, and the conversion of 6a to 15a was regularly checked by 1H NMR spectroscopy (Fig. S5 and S6†). The conversion values were determined to be 25 and 38% after 48 and 72 h, respectively, whereas cycloadduct 15a was observed to form with 96% conversion at the end of 15 days. These results show that for the template-bound diester 6a, the major pathway in solution under daylight is the photochemical [2 + 2] cycloaddition without any significant cis–trans isomerization of the olefins.
|
|||||
|---|---|---|---|---|---|
| a Yields refer to isolated product yields after purification by column chromatography.b Photochemical solution reactions were conducted in CHCl3 in quartz test tubes.c Diastereomeric ratio (dr) values of the crude reaction mixtures were determined by 1H NMR spectroscopy. The products were obtained as single diastereomers after purification. | |||||
| Entry | Cycloaddition product | Yield of 15 (%)a | Final product | Yield of 7 (%)a | |
| Powder | solutionb | ||||
| 1 |  |
88 | 88 (dr = 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3)c :3)c |
 |
75 |
| 2 |  |
11 | 64 (dr = 93:7)c |
 |
91 |
| 3 | 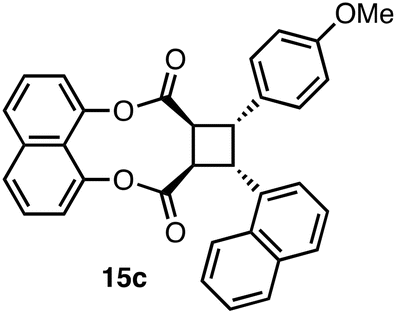 |
35 | 85 (dr = 96:4)c |
 |
87 |

 | ||
| Fig. 1 X-ray crystal structures of (a) 6a, and (b) 15a. | ||
With the successful results that we obtained on the homodimerization of 6a, we turned our attention to the photochemical heterodimerization reactions of diesters 6b and 6c with the aim of constructing unsymmetrical β-truxinic acid analogues. Whereas the photochemical cycloaddition of diester 6b worked poorly in solid state (11% yield), the heterodimerization product 15b was isolated in 64% yield upon irradiation of 6b in solution (Table 1, entry 2). Similarly, the heterodimerization of 6c bearing the 1-naphthyl and 4-methoxyphenyl substituents proceeded smoothly in solution with an isolated product yield of 85% compared to 35% yield in solid state (entry 3). The inferior results obtained for the cycloaddition of diesters 6b and 6c in the solid state may be due to a less suitable geometric orientation of their reacting olefins in their solid-state structures. The cycloadducts 15a–c were all detached from the template via a transesterification reaction using NaOMe in a 1:1 mixture of methanol and THF. At the end of these reactions, the cyclobutane dicarboxylic acid methyl esters 7a–c were isolated in 75–91% yields as single diastereomers (Table 1, entries 1–3).
Irradiation of crystals of diester 6a
Single-crystal-to-single-crystal (SCSC) transformations, where crystals retain their crystallinity at the end of chemical reactions, attracted considerable attention from the synthetic and crystallography communities.17 Following the success of the cycloaddition of diester 6a in powder form, we were curious to check if crystals of 6a would undergo photochemical [2 + 2] cycloaddition in an SCSC manner. When we irradiated crystals of diester 6a with UV light (365 nm), crystals did not crumble, and retained their integrity in terms of their shapes (Fig. 2 and S7†). We were also pleased to see that the cycloaddition reactions proceeded with full conversion within 20 h, as confirmed by 1H-NMR spectroscopy (Fig. 3). | ||
| Fig. 2 Appearance of four crystals of 6a after irradiation for (a) 1 h, (b) 5 h, (c) 10 h, and (d) 20 h. | ||
 | ||
| Fig. 3 Stacked 1H-NMR spectra for the conversion of different crystals of diester 6a to cycloadduct 15a at different irradiation times. | ||
However, disappointingly, during the course of the irradiation, crystals were observed to turn orange from colourless, lose their transparency, and become opaque (Fig. 2 and S7†).‡ As a consequence, despite our extensive efforts, single-crystal X-ray analysis of the crystals after irradiation with UV light did not give any diffraction.
A closer look at the crystal structures of 6a and 15a revealed the possibility of an interesting conformational isomerism for cycloadduct 15a, which arises from the different orientations of its carbonyl groups (Fig. 1 and Scheme 3). The three possible conformers of 15a are shown in Scheme 3a, where the stereochemical descriptors syn and anti are used to indicate the relative relationship between the corresponding CO group and the cyclobutane C–H, while the descriptor cis is used to describe the relative arrangement of the relevant C–H groups on the cyclobutane ring. It should be noted that the fourth possible conformation, namely the syn-cis-anti conformer, is the enantiomer of the already shown anti-cis-syn conformer, and therefore it was not included in Scheme 3a. In the X-ray structure of diester 6a, the two carbonyls have an anti-parallel orientation, whereas its two olefins are almost parallel to each other (Fig. 1a). The [2 + 2] cycloaddition of crystals of 6a is expected to proceed with retention of the orientations of the carbonyls affording the anti-cis-syn conformer of cycloadduct 15a (15a-1, Scheme 3b). However, as explained in the previous section, when cycloadduct 15a was purified by column chromatography and recrystallized, its X-ray analysis indicated the anti-cis-anti conformer (15a-4, Scheme 3b) with the CO groups having a parallel orientation (Fig. 1b). In order to check if 15a-1 is different from 15a-4, we performed a series of powder XRD and ATR-IR studies. Firstly, we wanted to confirm that the bulk structure of 15a-3 in powder form is the same as 15a-4, which was secured by X-ray crystallography. The powder XRD spectrum of 15a-3 is in nearly perfect agreement with the simulated powder XRD pattern of 15a-4 generated based on its single-crystal XRD data, indicating that they both have the same solid-state structure as the anti-cis-anti conformer (Fig. S8†). Next, we sought to compare the structures of 15a-2 and 15a-3. To this end, 15a-1 crystals, which were obtained via the irradiation of the crystals of 6a, were gently ground and analyzed by powder XRD. The powder XRD spectrum of 15a-2 showed differences compared to the spectrum of 15a-3, supporting our hypothesis that 15a-2 and thus 15a-1 might be a conformational isomer of 15a-3 (Fig. S8† and Scheme 3b).
 | ||
| Scheme 3 (a) Stereochemical description of the possible conformers of cycloadduct 15a; (b) different solid forms of 15a, and their conformational relationships (Ar = 1-naphthyl). | ||
The proposed conformational isomerism of the different solid forms of the cycloadduct 15a was further investigated by ATR-IR spectroscopy (Fig. 4 and S9–S12†). Whereas the ATR-IR spectrum of 15a-1 displays a strong CO stretching band at 1760 cm−1, the spectrum of 15a-3 surprisingly shows two separate signals in this region at 1740 and 1766 cm−1, indicating a clear difference in the structures of these two solid forms of 15a (Fig. 4, S10 and S11†). The reason for observing two CO stretching bands for 15a-3 is not clear, but in its X-ray structure, the two carbonyl oxygens of 15a-4 participate in different CH–O interactions with varying CH⋯O distances (Fig. S4†).18 These CH–O interactions with different strengths may cause the two carbonyls to display two separate IR bands. Finally, we should note that the ATR-IR spectra of 15a-3 and 15a-4 were found to be similar confirming that they are both anti-cis-anti conformers (Fig. S12†).
 | ||
| Fig. 4 Overlay of the ATR-IR spectra of 15a-1 and 15a-3. | ||
Overall, based on the above considerations, we propose that irradiation of crystals of 6a with the two carbonyls having anti-parallel orientation affords 15a-1, which is the anti-cis-syn conformer of cycloadduct 15a. This conformation is proposed to be locked in the solid state, presumably due to the lack of free rotation of the carbonyl groups inside the crystals. However, when any sample of 15a is dissolved in a solvent and concentrated, the resulting powder sample (15a-3) has anti-cis-anti conformation. The recrystallization of 15a-3 results in the formation of 15a-4 crystals, which are also anti-cis-anti conformers of 15a (Scheme 3). This may be due to the higher thermodynamic stability of the lattice structure of the anti-cis-anti conformer of 15a compared to that of the anti-cis-syn conformer.
Computational investigation of the conformational isomers of 15a
Despite the differences mentioned above in the various solid forms of 15a regarding their conformations, its 1H and 13C NMR spectra in CDCl3 always displayed a single set of signals indicating either only one major conformation in solution or a rapid interconversion between different conformers. This prompted us to investigate computationally the energies of the different conformers of 15a and the kinetic rotational barriers between these conformers.The calculations were performed using the B3LYP functional,19 with Grimmes D4 empirical dispersion corrections,20 and Ahlrichs' def2-SVP basis set.21 To minimize computational cost, the RIJCOSX approximation22 was utilized alongside the def2/J auxiliary basis set23 throughout the geometry optimizations of the initial and final states. The conductor-like polarizable continuum model (CPCM) was utilized to account for solvent effects using dichloromethane as the solvent in all computations.24 This level of theory will be referred to as RIJCOSX-B3LYP-D4/def2-SVP as implemented in ORCA 6.0.1.25 The minimum energy pathway (MEP) and the transition state between the different conformers were investigated with the nudged elastic band transition state (NEB-TS) procedure26 as implemented in ORCA 6.0.1, employing 16 images between minima. All NEB-TS calculations and the optimization of the conformers of 15a were carried out at the RIJCOSX-B3LYP-D4/def2-SVP level of theory. All calculated structures were generated and visualized using Gabedit27 and Mercury28 programs.
We first focused on the comparison of the 15a-anti-cis-syn conformer, which corresponds to the solid-state 15a-1 structure in Scheme 3, and the 15a-anti-cis-anti conformer, corresponding to the solid-state structure 15a-4 (Fig. 5). Please note that 15a-anti-cis-syn conformer is chiral, whereas 15a-anti-cis-anti conformer is achiral, increasing the importance of this analysis from a stereochemistry perspective. The energy difference between the 15a-anti-cis-syn and 15a-anti-cis-anti conformers was calculated to be 1.89 kcal mol−1, with the anti-cis-syn conformer being more stable. Employing the NEB-TS method, the calculated activation barriers in both rotation directions appeared to be 4.56 and 6.45 kcal mol−1, both of which point to a facile, rapid interconversion between the two conformational isomers in solution at room temperature. It should also be noted that in the transition state structure of this rotation (16a), the planes of two carbonyl groups are almost perpendicular to each other with a dihedral angle of 86° (Fig. 5). Next, we considered the third possible conformer of 15a, labeled as 15a-syn-cis-syn in Fig. 5. This conformer was calculated to be slightly less stable than the 15a-anti-cis-anti conformer being 1.02 kcal mol−1 higher in energy. The activation barriers for the interconversion between 15a-anti-cis-syn and 15a-syn-cis-syn were found to be 4.19 and 7.10 kcal mol−1 in the two rotation directions. This second transition state (16b) also has an almost perpendicular orientation of its two carbonyl groups. Overall, these computational studies indicate that the three conformational isomers of 15a have close energies in solution (less than 3 kcal mol−1) with activation barriers within the range of 4.19–7.10 kcal mol−1. Therefore, it can be concluded that all three conformers of 15a may coexist in solution with rapid interconversion at room temperature supporting its observed NMR spectroscopic features.
 | ||
| Fig. 5 The RIJCOSX-B3LYP-D4/def2-SVP reaction coordinates of 15a-anti-cis-anti (A) to 15a-anti-cis-syn (C) (blue), and 15a-anti-cis-syn (C) to 15a-syn-cis-syn (E) (orange). The relative energies of 15a-anti-cis-anti (A) is 1.89 kcal mol−1, 16a (B) is 6.45 kcal mol−1, 15a-anti-cis-syn (C) is 0 kcal mol−1, 16b (D) is 7.10 kcal mol−1, and 15a-syn-cis-syn (E) is 2.91 kcal mol−1. The potential energy diagram was generated by employing 16 images between two minima (32 images in total) with NEB-TS formalism. | ||
Photochemical reactivity of untemplated naphthalene acrylic acid derivatives
In the final stage of our studies, we examined the photochemical reactivities of the untemplated 1-naphthalene acrylic acid and ester derivatives. During our studies on the synthesis of the template-bound cycloaddition precursors, we observed that both naphthalene acrylic acid 10 and ester 9 were sensitive to daylight undergoing a cis–trans isomerization. In order to check this behaviour in a controlled manner, solutions of 9 (E:Z = 98:2) in CDCl3, and 10 (pure E isomer) in DMSO-d6 were kept under ambient daylight for 11 days, and the cis–trans isomerization was analyzed regularly by 1H-NMR spectroscopy. Whereas the formation of a [2 + 2] cycloadduct was not observed under these conditions, the E:Z (trans:cis) ratio reached a constant value of 25:75 for ester 9, and 15:85 for acid 10 after 9 days. The UV-vis absorption spectra of these compounds provide a rationale for their photochemical reactivities (Fig. S13 and S14†). The UV-vis spectrum of 9 (1 × 10−4 M in methanol) exhibited λmax value at 323 nm with an absorption tail up to ca. 378 nm. Likewise, a solution of 10 (1 × 10−4 M in methanol) displays λmax value at 322 nm in its UV-vis spectrum again with an absorption tail up to ca. 390 nm. The molar absorption coefficients (ε) of compounds 9 and 10 were determined to be 1.46 × 104 and 1.30 × 104 M−1 cm−1, respectively, at their λmax values. It should also be noted that the UV-vis spectrum of the cycloaddition precursor 6a is very similar to those of 9 and 10 with a λmax value of 326 nm (ε = 3.04 × 104 M−1 cm−1) and an absorption tail up to ca. 400 nm in CH2Cl2 (Fig. S15†). This provides a rationale for the observed photoreactivity of 6a both with UV light at 365 nm and ambient daylight. Moreover, the diffuse reflectance UV-vis spectrum of 6a recorded in solid powder form displayed absorbance below 398 nm (Fig. S16†). Finally, UV-vis absorption spectrum of cycloadduct 15a revealed a λmax value of 284 nm (ε = 5.90 × 103 M−1 cm−1) in CH2Cl2 (Fig. S17†). This decrease in the λmax value for cycloadduct 15a compared to compounds 6a, 9 and 10, which all have α,β-unsaturated ester and acid moieties, can be attributed to the reduced degree of conjugation in 15a after cyclobutane ring formation.
Next, we investigated the irradiation of ester 9 in neat form as oil and in solution by UV-A light (Scheme 4a). The reaction of neat 9 afforded a mixture of β-truxinate 7a and δ-truxinate 17 with 3:1 diastereomeric ratio (dr) and in 64% yield. The same reaction in a CDCl3 solution proceeded without any diastereoselectivity, and a mixture of 7a and 17 was observed to form in 1:1 ratio. On the other hand, treatment of ester 9 with the Ir(ppy)3 (tris(2-phenylpyridine)iridium) as a photocatalyst with visible light irradiation (450 nm) afforded δ-truxinate 17 as the major product with an unoptimized yield of 39% (Scheme 4b).29 This result is not surprising as it was shown independently by the Wu and Reiser groups that visible-light catalysis of cinnamates proceeded via a [2 + 2] cycloaddition in an anti-head-to-head fashion giving δ-truxinic acid derivatives as the major products.29,30 We then turned our attention to the photochemical behaviour of 1-naphthalene acrylic acid 10. When a powder sample of 10 was irradiated with UV-A light (365 nm) for 20 h, β-truxinic acid product 18 was isolated in 78% yield (Scheme 4c). Despite all our attempts, we could not manage to get single crystals of compound 10 for X-ray analysis.
 | ||
| Scheme 4 Photochemical reactions of untemplated naphthalene acrylate 9 and acrylic acid 10. | ||
Conclusions
Our template-directed photochemical [2 + 2] cycloaddition strategy using 1,8-dihydroxynaphthalene as a covalent template has been employed for the selective homo- and heterodimerization of naphthalene acrylic acids. While the cycloaddition reactions work successfully both in solid state and solution, the reactions in solution were observed to give equal or higher yields (64–88%). After the final detachment of the template from cycloadducts, symmetrical and unsymmetrical β-truxinic acid dimethyl ester analogues bearing one or two naphthyl rings were isolated in good yields (75–91%). Single-crystal XRD analyses not only provided a geometrical rationale for the success of the cycloaddition of diester 6a in solid state, but also confirmed the stereochemistry of the cycloadduct 15a. Conformational isomers of 15a in various solid forms, which are caused by the different possible orientations of the two carbonyl groups in powder and crystal forms, were carefully investigated by powder XRD and ATR-IR studies. Furthermore, computational analysis of the different conformers of 15a revealed low barriers of rotation (up to 7.10 kcal mol−1) in solution phase, which indicate fast interconversion between these conformational isomers of 15a at room temperature. Finally, the untemplated photoreactivity of the naphthalene acrylic acid 10 and ester 9 was examined. Whereas the irradiation of ester 9 in neat oil form and in solution gave a mixture of the β and δ truxinate products, the δ truxinate product 17 and β truxinic acid product 18 could be obtained selectively, upon photocatalytic reaction of 9 and UV-A irradiation of 10, respectively.Experimental
General information
Air or water sensitive reactions were carried out under an inert atmosphere of nitrogen using oven-dried glassware. Reactions were monitored by thin-layer chromatography (TLC) using aluminium-backed plates pre-coated with silica gel (Silicycle, 60 Å, F254). TLC visualization was done either by UV light (254 nm) or KMnO4 staining solution. Silicycle 40–63 μm (200–400 mesh) silica gel was used for purification by flash column chromatography. NMR spectra were recorded using a Bruker spectrometer at 400 MHz for 1H-NMR spectra and 100 MHz for 13C{1H} spectra, and calibrated from internal standard (TMS, 0 ppm) or residual solvent signals (chloroform at 7.26, DMSO at 2.50, and acetone at 2.05 ppm for 1H-NMR spectra; chloroform at 77.16, DMSO at 39.52, and acetone at 29.84 ppm for 13C{1H}-NMR spectra). 1H-NMR data are reported as follows: chemical shift (ppm, parts per million), integration, multiplicity (s = singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet, br s = broad signal, app = apparent), coupling constant (Hz). ATR-IR spectra were recorded using a Bruker Alpha-Platinum-ATR spectrometer, and selected peaks are reported. HRMS (high resolution mass spectrometry) analyses were conducted at UNAM-National Nanotechnology Research Center and Institute of Materials Science and Nanotechnology, Bilkent University, using Agilent Technologies 6224 TOF LC/MS instrument. Single-crystal XRD analysis was performed at the Scientific and Technological Research Application and Research Center, Sinop University, Türkiye. Powder XRD experiments were carried out using Malvern PANalytical (X'Pert PRO) X-ray diffractometer equipped with a Cu Kα X-ray source. An Agilent Cary 300 spectrophotometer was utilized for recording the UV-vis absorption spectra. Melting points are uncorrected. Photochemical reactions were conducted using a commercial UV gel nail dryer (Beurer, MP38) equipped with four 9W UV-A (365 nm) fluorescent lamps (Philips PL-S; Fig. S18†). Photographs of the crystals were acquired with a Ninyoon 4 K wifi digital microscope. Anhydrous CH2Cl2 and THF were purchased from Acros Organics (AcroSeal®). 1,8-dihydroxynaphthalene was purchased from abcr, and used as received. All commercially available reagents were used without further purification, unless stated otherwise.Naphthalen-1-yl trifluoromethanesulfonate (8)
This compound was prepared following the procedure reported in the literature.31 An oven-dried, 100 mL round-bottomed flask was subjected to vacuum–nitrogen cycle three times, and then, 1-naphthol (500 mg, 3.47 mmol) was mixed with 5.0 mL of anhydrous CH2Cl2. When the solid dissolved completely, the reaction flask was inserted in an ice bath, and Et3N (422 mg, 580 μL, 4.16 mmol) was added slowly. Afterwards, trifluoromethane sulfonic anhydride (1.08 g, 640 μL, 1.67 mmol) was added dropwise over 10 min. After ca. 10 min, the ice bath was removed, and the reaction mixture was stirred for 3 h at 23 °C. Then it was quenched with 5 mL of deionized water, and the aqueous phase was extracted three times with CH2Cl2. Anhydrous Na2SO4 was used to dry the combined organic phase. After the removal of the salt by filtration, the solvent was evaporated under reduced pressure. Purification by flash column chromatography (SiO2; EtOAc:hexanes = 1:2) afforded compound 8 as a colourless oil (906 mg, 95% yield). 1H NMR (400 MHz; CDCl3) δ: 8.13 (1H, d, J = 8.3 Hz), 7.91 (1H, d, J = 8.0 Hz), 7.86 (1H, d, J = 6.8 Hz), 7.66 (1H, t, J = 7.6 Hz), 7.60 (1H, t, J = 7.5 Hz), 7.50–7.45 (2H, m). The 1H NMR spectral data are in agreement with the data reported in the literature.32
Methyl (E)-3-(naphthalen-1-yl)acrylate (9)
An oven-dried, 25 mL Schlenk flask was subjected to vacuum–nitrogen cycle three times. Compound 8 (470 mg, 1.70 mmol) was dissolved in 10 mL of deoxygenated DMF, and methyl acrylate (732 mg, 792 μL, 8.50 mmol) was added to the reaction medium. Afterwards, Et3N (860 mg, 1.18 mL, 8.50 mmol) and PdCl2(PPh3)2 (59.6 mg, 0.085 mmol) were added sequentially. The Schlenk flask was then sealed with a glass stopper, and was heated gradually above the oil bath. After 10 min, it was dipped into a pre-heated oil bath at 80 °C. The reaction mixture was stirred at this temperature for 24 h. Then, the reaction mixture was cooled down to ambient temperature, and 5 mL of deionized water was added to quench the reaction. The mixture was transferred into a separatory funnel, and deionized water (5 mL) and brine (5 mL) were added. The product was extracted with EtOAc (3 × 10 mL). Anhydrous Na2SO4 was used to dry the combined organic phase. After the filtration of the salt, the solvent was evaporated under reduced pressure. Purification by column chromatography (SiO2; EtOAc:hexanes = 1:9) afforded compound 9 as a colourless oil (354 mg, 98% yield). 1H NMR (400 MHz; CDCl3) δ: 8.55 (1H, d, J = 15.8 Hz), 8.20 (1H, d, J = 8.4 Hz), 7.88 (2H, app t, J = 7.0 Hz), 7.75 (1H, d, J = 7.2 Hz), 7.58 (1H, ddd, J = 8.5, 6.9 and 1.4 Hz), 7.53 (1H, ddd, J = 8.1, 6.8 and 1.4 Hz), 7.48 (1H, t, J = 7.7 Hz), 6.54 (1H, d, J = 15.7 Hz), 3.87 (3H, s). The 1H NMR spectral data are in agreement with the data reported in the literature.33
(E)-3-(naphthalen-1-yl)acrylic acid (10)
In a 25 mL round-bottomed flask, compound 9 (77 mg, 0.36 mmol) was dissolved in 6 mL of a 1:2 mixture of MeOH:THF at 23 °C. An excess of 5 M aqueous solution of KOH (2.5 mL) was gradually added to the round-bottomed flask. After ca. 2 h, 0.5 mL additional KOH solution (5 M) was added. At the end of 4 h, TLC analysis indicated full consumption of the starting material. The reaction flask was placed in an ice bath, and HCl (ca. 1.2 M) was added into the flask to make the pH around 1–2. After the pH adjustment, the product was extracted by CH2Cl2 (3 × 10 mL). Anhydrous Na2SO4 was used to dry the combined organic phase. After the filtration of the salt, the solvent was evaporated under reduced pressure. Purification by flash column chromatography (SiO2; 1% acetic acid in EtOAc:hexanes = 1:1) afforded compound 10 as a white solid (64.8 mg, 90% yield). M.P. 211.8–212.4 °C (lit. 210–213 °C).34 1H NMR (400 MHz; DMSO-d6) δ: 12.54 (1H, bs), 8.39 (1H, d, J = 15.7 Hz), 8.20 (1H, d, J = 8.3 Hz), 8.01 (2H, t, J = 9.0 Hz), 7.95 (1H, d, J = 7.1 Hz), 7.66–7.55 (3H, m), 6.60 (1H, d, J = 15.7 Hz). The 1H NMR spectral data are in agreement with the data reported in the literature.34
Naphthalene-1,8-diyl (2E,2′E)-bis(3-(naphthalen-1-yl)acrylate) (6a)
An oven-dried, 25 mL round-bottomed flask was subjected to vacuum–nitrogen cycle three times. 1,8-DHN (12, 50.0 mg, 0.31 mmol) was dissolved in 4.0 mL of anhydrous CH2Cl2 under an inert atmosphere of N2. Afterwards, compound 10 (136 mg, 0.68 mmol) was added to the flask. To completely dissolve compound 10, additional 4.5 mL of anhydrous CH2Cl2 was added to the reaction medium. Sequentially, DCC (142 mg, 0.69 mmol) and DMAP (7.6 mg, 0.062 mmol) were added to the flask. The reaction mixture was stirred for 24 h under N2 at 23 °C. It was then quenched with 10 mL of deionized water. The aqueous phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic phase was dried over anhydrous Na2SO4. After the filtration of the salt, the solvent was evaporated under reduced pressure. Purification by flash column chromatography (SiO2; hexanes: DCM = 1:1) afforded diester 6a as a light yellow solid (77 mg, 48% yield). Rf = 0.23 (1:1 DCM: hexanes). M.P. 223.5–225.2 °C (CH2Cl2/pentane). 1H NMR (400 MHz; CDCl3) δ: 8.61 (2H, d, J = 15.8 Hz), 8.01–7.98 (2H, m), 7.79 (2H, d, J = 8.4 Hz), 7.65–7.63 (2H, m), 7.56 (2H, d, J = 8.2 Hz), 7.47 (2H, t, J = 7.9 Hz), 7.36–7.31 (4H, m), 7.26 (2H, d, J = 7.2 Hz), 7.19 (2H, d, J = 7.7 Hz), 6.75 (2H, t, J = 7.7 Hz), 6.66 (2H, d, J = 15.8 Hz). 13C{1H} NMR (100 MHz; CDCl3) δ: 165.9, 145.5, 143.7, 137.0, 133.6, 131.3, 130.9, 130.7, 128.7, 127.1, 126.3, 126.2, 125.2, 125.0, 123.0, 121.6, 120.8, 119.6. FTIR νmax (ATR, solid) per cm 3058, 1717, 1628, 1602, 1575, 1507, 1370, 1346. HRMS (APCI+) calcd for C36H25O4 [M + H]+: 521.1747, found: 521.1740.
Crystallization of compound 6a
In a 2.0 mL vial, 15.0 mg (0.029 mmol) of compound 6a was dissolved in 1.0 mL of CH2Cl2. When the solution became clear, it was placed inside a 20 mL scintillation vial that contained ca. 7.5 mL of n-pentane. The lid of the outer vial was sealed with parafilm. To keep it under dark, it was stored inside a cabinet. Crystal formation was started to be observed within 24 h, and crystals were collected after two additional days. Single crystals had transparent-white appearance.8-(Cinnamoyloxy)naphthalen-1-yl (E)-3-(naphthalen-1-yl)acrylate (6b)
Initially, acyl chloride 11 was prepared by dissolving 1-naphthalene acrylic acid 10 (51.8 mg, 0.26 mmol) in 0.7 mL of oxalyl chloride under N2 at 23 °C. After 10 min, the round-bottomed flask was inserted into a pre-heated oil bath at 60 °C, and was stirred for 2 h. At the end of this time, the reaction flask was cooled to ambient temperature, and the unreacted oxalyl chloride was evaporated carefully using a rotary evaporator. The resulting acyl chloride 11 (56.6 mg, 0.26 mmol) was obtained as a yellow solid. For the synthesis of diester 6b, an oven-dried 50 mL round-bottomed flask was subjected to vacuum–nitrogen cycle three times. Compound 13,10a (65.9 mg, 0.22 mmol) was dissolved in 3.0 mL of anhydrous THF. When the solution became clear after 10 min, an ice bath was placed under the round-bottomed flask. Then, a solution of acyl chloride 11 (56.6 mg, 0.26 mmol) in 3.0 mL of anydrous THF was added to the reaction mixture. Then, NaH (10.0 mg, 0.25 mmol, 60% dispersion in mineral oil) was added in small portions. After 5 min, the ice bath was removed, and the reaction mixture was stirred overnight. It was then quenched with 10 mL of saturated aqueous NH4Cl solution. The aqueous phase was extracted with EtOAc (3 × 10 mL). Combined organic phase was dried by over anhydrous Na2SO4. After the filtration of the salt, the solvent was removed under reduced pressure. Purification by flash column chromatography (SiO2; hexanes: CH2Cl2:MeOH = 8:1:1 to 7:1:1) afforded diester 6b as a yellow solid (53.9 mg, 50% yield). Rf = 0.21 (hexanes: CH2Cl2:MeOH = 7:1:1). M.P. 147.8–148.8 °C. 1H NMR (400 MHz; CDCl3) δ: 8.72 (1H, d, J = 15.8 Hz), 8.17 (1H, d, J = 8.2 Hz), 7.82–7.78 (4H, m), 7.74 (1H, d, J = 8.2 Hz), 7.54–7.45 (5H, m), 7.24 (1H, d, J = 7.5 Hz), 7.21 (1H, d, J = 7.5 Hz), 7.14–7.10 (3H, m), 7.05 (1H, t, J = 7.7 Hz), 6.90 (2H, t, J = 7.7 Hz), 6.72 (1H, d, J = 15.8 Hz), 6.60 (1H, d, J = 16.0 Hz). 13C{1H} NMR (100 MHz; CDCl3) δ: 166.0, 165.9, 147.1, 145.4, 143.6, 137.0, 133.8, 133.7, 131.5, 131.0, 130.9, 130.5, 128.8, 128.7, 128.0, 127.1, 127.0, 126.3, 126.2, 125.4, 125.1, 123.2, 121.5, 120.8, 119.6, 117.3. (26 signals were observed instead of the expected 30 signals in total, possibly due to the overlap of certain signals.) FTIR νmax (ATR, solid) per cm 3059, 2925, 1729, 1633, 1604, 1574, 1432, 1366. HRMS (APCI+) calcd for C32H23O4 [M + H]+: 471.1591, found: 471.1596.
8-(((E)-3-(4-methoxyphenyl)acryloyl)oxy)naphthalen-1-yl (E)-3-(naphthalen-1-yl)acrylate (6c)
Initially, acyl chloride 11 was prepared by dissolving 1-naphthalene acrylic acid 10 (69.6 mg, 0.35 mmol) in 1.0 mL of oxalyl chloride under N2 at 23 °C. After 10 min, the round-bottomed flask was dipped into pre-heated oil bath at 60 °C, and was stirred for 2 h. At the end of this time, the reaction flask was cooled to ambient temperature, and the unreacted oxalyl chloride was evaporated carefully using a rotary evaporator. The resulting acyl chloride 11 (76.1 mg, 0.35 mmol) was obtained as a yellow solid. However, 51.2 mg of compound 11 was used in the next step where diester 6c was synthesized. An oven-dried 50 mL round-bottomed flask was subjected to vacuum–nitrogen cycle three times. Compound 14,10a (52.7 mg, 0.216 mmol) was dissolved in 3.0 mL of anhydrous THF under nitrogen. When the solution became clear after 10 min, the flask was inserted into an ice bath. Then, acyl chloride 11 (51.2 mg, 0.23 mmol) was dissolved in 2.0 mL of anhydrous THF and was added to the reaction mixture. Then, NaH (6.5 mg, 0.164 mmol, 60% dispersion in mineral oil) was added in small portions. After 5 min, the ice bath was removed, and the reaction mixture was stirred overnight. It was then quenched with 10 mL of saturated aqueous solution of NH4Cl. The aqueous phase was extracted with EtOAc (3 × 15 mL). The combined organic phase was dried by over anhydrous Na2SO4. After the filtration of the salt, the solvent was removed under reduced pressure. Purification by flash column chromatography (SiO2; hexanes: CH2Cl2:MeOH = 11:1:1 to 7:1:1) gave diester 6c as a yellow solid (41.6 mg, 51% yield). Rf = 0.16 (hexanes: CH2Cl2:MeOH = 7:1:1). M.P. 156.7–159.9 °C. 1H NMR (400 MHz; CDCl3) δ: 8.63 (1H, d, J = 15.8 Hz), 8.11 (1H, d, J = 8.1 Hz), 7.76–7.65 (5H, m), 7.49–7.41 (5H, m), 7.17 (1H, d, J = 7.3 Hz), 7.14 (1H, d, J = 7.5 Hz), 7.06 (1H, t, J = 7.7 Hz), 6.97 (2H, d, J = 8.7 Hz), 6.66 (1H, d, J = 15.8 Hz), 6.38 (1H, d, J = 15.9 Hz), 6.28 (2H, d, J = 8.6 Hz), 3.62 (3H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 166.3, 165.9, 161.4, 146.8, 145.4, 143.3, 137.0, 133.8, 131.5, 131.0, 130.9, 129.7, 128.8, 127.1, 127.0, 126.9, 126.5, 126.3, 126.22, 126.17, 125.4, 125.23, 125.16, 123.2, 121.6, 120.8, 120.7, 119.8, 114.7, 114.0, 55.3. FTIR νmax (ATR, solid) per cm 3061, 2933, 2911, 2835, 1714, 1629, 1601, 1573, 1510, 1463. HRMS (APCI+) calcd for C33H25O5 [M + H]+: 501.1697, found: 501.1693.
(8 aR,9S,10R,10 aS)-9,10-di(naphthalen-1-yl)-8a,9,10,10a-tetrahydrocyclobuta[g]naphtho [1,8-bc][1,5]dioxonine-8,11-dione (15a)
Irradiation in solution: diester 6a (15.8 mg, 0.030 mmol) was dissolved in 3.5 mL of CHCl3 in a quartz test tube. The tube was equipped with a stir bar, and placed inside the nail dryer (365 nm, 4 × 9 W fluorescent bulbs). A fan was used to control the temperature, and the reaction was monitored by TLC. At the end of 9 h, the solvent was removed under reduced pressure. Purification by flash column chromatography (SiO2; DCM:hexanes = 1:2 to 1:1.2) afforded pure cycloadduct 15a as a yellow solid (13.9 mg, 88% yield). Irradiation in solid state: diester 6a (8.2 mg, 0.015 mmol) in powder form was placed between two quartz microscope slides. With the help of paper clips, the slides were fixed. They were then placed under UV lamps in a nail dryer (365 nm, 4 × 9 W). Every 4 h, the solid residue was mixed with a spatula. In addition to the mixing process to ensure homogeneous light distribution, the side of the slides facing the lamp was switched every 4 h. At the end of 16 h of irradiation, the conversion was determined as 98% by 1H NMR analysis. Purification by flash column chromatography (SiO2; DCM:hexanes = 1:1) afforded cycloadduct 15a as a yellow solid (7.2 mg, 88% yield). Rf = 0.62 (1:1 DCM: hexanes). 1H NMR (400 MHz; CDCl3) δ: 7.99–7.96 (2H, m), 7.75 (2H, d, J = 7.9 Hz), 7.58–7.55 (2H, m), 7.48 (2H, d, J = 8.2 Hz), 7.44 (2H, t, J = 8.0 Hz),7.26–7.19 (8H, m), 7.17–7.13 (2H, m), 5.68 (2H, app d, J = 6.2 Hz), 4.35 (2H, app d, J = 6.1 Hz). 13C{1H} NMR (100 MHz; CDCl3) δ: 170.0, 145.6, 137.2, 134.5, 133.6, 131.7, 128.7, 127.7, 127.1, 126.5, 126.0, 125.7, 124.9, 123.8, 123.7, 121.2, 119.8, 45.4, 41.1. FTIR νmax (ATR, solid, 15a-3) per cm 3052, 2958, 2922, 2851, 1766, 1740, 1603, 1512. FTIR νmax (ATR, solid, 15a-1) per cm 3050, 1760, 1606, 1577, 1510. HRMS (APCI+) calcd for C36H25O4 [M + H]+: 521.1747, found 521.1748.
Crystallization of compound 15a
In a 2.0 mL vial, 6.8 mg (0.013 mmol) of compound 15a was dissolved in 1.0 mL of CH2Cl2. When the solution became clear, it was placed inside a 20 mL scintillation vial that contained ca. 7.5 mL of n-pentane. The lid of the outer vial was sealed with parafilm. To keep it under dark, it was stored inside a cabinet. Crystal formation was started to be observed within 24 h, and crystals were collected after two additional days. Single crystals had needle-like, yellowish-white appearance.(8 aR,9S,10R,10 aS)-9-(naphthalen-1-yl)-10-phenyl-8a,9,10,10a-tetrahydrocyclobuta[g] naphtho[1,8-bc][1,5]dioxonine-8,11-dione (rac-15b)
Diester 6b (23.6 mg, 0.050 mmol) was dissolved in 2.0 mL of CHCl3 in a quartz test tube. The tube was equipped with a stir bar, and placed inside the nail dryer (365 nm, 4 × 9 W fluorescent bulbs). A fan was used to control the temperature, and the reaction was monitored by TLC. At the end of 20 h, the solvent was removed under reduced pressure. Purification by flash column chromatography (SiO2; EtOAc:hexanes = 1:10 to 1:9) afforded cycloadduct 15b as an amorphous foam (15.1 mg, 64% yield). Rf = 0.42 (hexanes: DCM:MeOH = 7:1:1). 1H NMR (400 MHz; CDCl3) δ: 7.98 (1H, d, J = 8.4 Hz), 7.82 (1H, dd, J = 8.3, 0.8 Hz), 7.81 (1H, dd, J = 8.3, 0.7 Hz), 7.71 (1H, d, J = 7.8 Hz), 7.62 (1H, dd, J = 7.5, 1.5 Hz), 7.55–7.44 (3H, m), 7.41–7.30 (4H, m), 7.25–7.23 (1H, m), 7.00–6.97 (2H, m), 6.95–6.90 (3H, m), 5.50 (1H, t, J = 9.5 Hz), 4.87 (1H, dd, J = 10.2, 4.9 Hz), 4.66 (1H, t, J = 9.8 Hz), 4.16 (1H, ddd, J = 10.8, 4.8, 1.1 Hz). 13C{1H} NMR (100 MHz; CDCl3) δ: 170.4, 169.7, 145.54, 145.49, 138.1, 137.1, 133.7, 133.6, 131.5, 128.7, 128.0, 127.7, 127.10, 127.09, 126.9, 126.6, 126.0, 125.9, 125.0, 124.1, 123.9, 121.2, 121.1, 119.7, 45.3, 43.8, 42.2. (27 signals were observed instead of the expected 30 signals in total, possibly due to the overlap of certain signals.) FTIR νmax (ATR, solid) per cm 2959, 2922, 2852, 1762, 1607, 1455, 1365. HRMS (APCI+) calcd for C32H23O4 [M + H]+: 471.1591, found: 471.1596.
(8 aS,9R,10S,10 aR)-9-(4-methoxyphenyl)-10-(naphthalen-1-yl)-8a,9,10,10a-tetrahydrocyclo buta[g]naphtho[1,8-bc][1,5]dioxonine-8,11-dione (rac-15c)
Diester 6c (14.9 mg, 0.030 mmol) was dissolved in 2.0 mL of CHCl3 in a quartz test tube. The tube was equipped with a stir bar, and placed inside the nail dryer (365 nm, 4 × 9 W fluorescent bulbs). A fan was used to control the temperature, and the reaction was monitored by TLC. At the end of 9 h, the solvent was removed under reduced pressure. Purification by flash column chromatography (SiO2; EtOAc:hexanes = 1:10 to 1:7) afforded cycloadduct 15c as yellowish-orange oil (12.6 mg, 85% yield). Rf = 0.34 (1:2 EtOAc:hexanes). 1H NMR (400 MHz; CDCl3) δ: 7.90 (1H, d, J = 8.4 Hz), 7.75 (1H, d, J = 8.3 Hz), 7.74 (1H, d, J = 8.3 Hz), 7.66 (1H, d, J = 8.0 Hz), 7.57 (1H, dd, J = 6.9, 2.1 Hz), 7.49–7.29 (6H, m), 7.24 (1H, d, J = 7.5 Hz), 7.17 (1H, d, J = 8.4 Hz), 6.84 (2H, d, J = 8.6 Hz), 6.40 (2H, d, J = 8.6 Hz), 5.40 (1H, t, J = 9.4 Hz), 4.76 (1H, dd, J = 10.1, 4.9 Hz), 4.58 (1H, t, J = 9.8 Hz), 4.04 (1H, dd, J = 10.6, 4.9 Hz), 3.51 (3H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 170.5, 169.8, 158.3, 145.54, 145.48, 137.1, 133.9, 133.7, 131.5, 130.3, 129.0, 128.7, 127.7, 127.1, 126.5, 126.0, 125.9, 125.1, 124.2, 123.8, 121.2, 121.1, 119.7, 113.4, 55.2, 45.8, 44.7, 43.6, 42.4. (29 signals were observed instead of the expected 31 signals in total, possibly due to the overlap of certain signals.) FTIR νmax (ATR, film) per cm 3058, 2954, 2922, 2850, 1760, 1732, 1629, 1607, 1577, 1513, 1364. HRMS (APCI+) calcd for C33H25O5 [M + H]+: 501.1697, found: 501.1699.
Dimethyl (1R,2S,3R,4S)-3,4-di(naphthalen-1-yl)cyclobutane-1,2-dicarboxylate (7a)
In a 20 mL scintillation vial, compound 15a (18.2 mg, 0.035 mmol) was dissolved in 6.0 mL of 1:1 MeOH:THF mixture. To this clear solution, NaOMe (4.7 mg, 0.087 mmol) was then added. The resulting cloudy mixture was stirred at 23 °C for 8 h. At the end of this time, all volatiles were removed under reduced pressure. Purification by flash column chromatography (SiO2; 1:1 = hexanes:CH2Cl2) afforded cyclobutane 7a as a pale yellow solid (11.2 mg, 75% yield). Rf = 0.39 (1:1 hexanes:CH2Cl2). M.P. 182.3–184.2 °C. 1H NMR (400 MHz; CDCl3) δ: 7.90 (2H, d, J = 8.2 Hz), 7.51 (2H, d, J = 8.4 Hz), 7.40 (2H, dd, J = 7.2, 2.0 Hz), 7.20–7.13 (4H, m), 7.10–7.05 (4H, m), 5.32 (2H, app d, J = 6.6 Hz), 4.00 (2H, app d, J = 6.3 Hz), 3.72 (6H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 173.1, 134.7, 133.5, 131.9, 128.5, 127.4, 125.7, 125.5, 124.8, 123.8, 123.6, 52.4, 43.9, 41.6. FTIR νmax (ATR, solid) per cm 2951, 2919, 1720, 1596, 1510, 1430, 1363. HRMS (APCI+) calcd for C28H25O4 [M + H]+: 425.1747, found: 425.1745.
Dimethyl (1S,2R,3S,4R)-3-(naphthalen-1-yl)-4-phenylcyclobutane-1,2-dicarboxylate (rac-7b)
Compound 15b (11.9 mg, 0.025 mmol) was dissolved in 3.0 mL of a 1:1 MeOH:THF mixture in a 20 mL scintillation vial. To this clear solution, NaOMe (6.8 mg, 0.13 mmol) was then added. The resulting mixture was stirred at 45 °C for 3.5 h. At the end of this time, all volatiles were removed under reduced pressure. Purification by flash column chromatography (SiO2; EtOAc:hexanes = 1:11 to 1:5) afforded cyclobutane 7b as orange oil (8.6 mg, 91% yield). Rf = 0.30 (1:5 EtOAc:hexanes). 1H NMR (400 MHz; CDCl3) δ: 7.96 (1H, d, J = 8.4 Hz), 7.69 (1H, d, J = 8.0 Hz), 7.57 (1H, d, J = 8.2 Hz), 7.45 (1H, t, J = 7.6 Hz), 7.38 (1H, t, J = 7.4 Hz), 7.29–7.26 (1H, m), 7.18 (1H, d, J = 7.0 Hz), 6.87 (5H, app s), 5.22 (1H, t, J = 9.6 Hz), 4.46 (1H, dd, J = 9.9, 4.7 Hz), 4.28 (1H, t, J = 9.7 Hz), 3.81 (3H, s), 3.80–3.76 (1H, m), 3.75 (3H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 173.5, 172.8, 138.4, 134.3, 133.5, 131.7, 128.6, 127.8 (overlapping of two signals), 127.4, 126.6, 125.8, 125.7, 125.0, 124.2, 123.6, 52.5, 52.3, 46.1, 44.5, 42.7, 41.7. FTIR νmax (ATR, film) per cm 2951, 2922, 2850, 1730, 1599, 1435. HRMS (APCI+) calcd for C24H23O4 [M + H]+: 375.1591, found: 375.1592.
Dimethyl (1R,2S,3R,4S)-3-(4-methoxyphenyl)-4-(naphthalen-1-yl)cyclobutane-1,2-dicarboxylate (rac-7c)
Compound 15c (11.7 mg, 0.023 mmol) was dissolved in 3.0 mL of a 1:1 MeOH:THF mixture in a 20 mL scintillation vial. To this clear solution, NaOMe (6.3 mg, 0.12 mmol) was then added. The resulting mixture was stirred at 45 °C for 5 h. Purification by flash column chromatography (SiO2; 1.25:1 = hexanes:EtOAc) afforded cyclobutane 7c as yellowish oil (8.2 mg, 87% yield). Rf = 0.22 (1:5 EtOAc:hexanes). 1H NMR (400 MHz; CDCl3) δ: 7.93 (1H, d, J = 8.4 Hz), 7.70 (1H, d, J = 8.1 Hz), 7.58 (1H, d, J = 8.2 Hz), 7.44 (1H, t, J = 7.1 Hz), 7.38 (1H, t, J = 7.3 Hz), 7.29 (1H, t, J = 7.7 Hz), 7.19 (1H, d, J = 7.1 Hz), 6.78 (2H, d, J = 8.6 Hz), 6.41 (2H, d, J = 8.6 Hz), 5.17 (1H, t, J = 9.7 Hz), 4.41 (1H, dd, J = 9.9, 4.7 Hz), 4.26 (1H, t, J = 9.7 Hz), 3.80 (3H, s), 3.74 (3H, s), 3.71 (1H, dd, J = 10.2, 4.8 Hz), 3.56 (3H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 173.5, 172.8, 158.1, 134.5, 133.6, 131.7, 130.7, 128.8, 128.6, 127.4, 125.8, 125.6, 125.0, 124.2, 123.5, 113.3, 55.1, 52.4, 52.3, 45.4, 45.0, 42.9, 41.6. FTIR νmax (ATR, film) per cm 2951, 2924, 2850, 1727, 1610, 1513, 1435. HRMS (APCI+) calcd for C25H25O5 [M + H]+: 405.1697, found: 405.1693.
Dimethyl (1R,2R,3S,4S)-3,4-di(naphthalen-1-yl)cyclobutane-1,2-dicarboxylate (rac-17)
In this reaction, the procedure developed by Wu and co-workers has been applied.29 In a quartz test tube, compound 9 (20.1 mg, 0.094 mmol) was dissolved in 0.94 mL of anhydrous 1,4-dioxane under nitrogen. Afterwards, tris(2-phenylpyridine)iridium(III) (Ir(ppy)3, 1.2 mg, 0.0018 mmol) was added. The test tube was irradiated using 450 nm light for 8 h. At the end of this time, all volatiles were removed under reduced pressure. The 1H NMR spectrum of the crude mixture indicated that the ratio of β truxinate isomer 7a to the δ truxinate isomer 17 was 0.16:1.00. Purification by flash column chromatography (SiO2; EtOAc:hexanes = 1:8 to 1:7) afforded cyclobutane 17 as a yellowish-white solid (7.9 mg, 39% yield). Rf = 0.32 (1:5 EtOAc:hexanes). M.P. 197.9–199.0 °C. 1H NMR (400 MHz; CDCl3) δ: 7.79 (2H, d, J = 8.2 Hz), 7.77–7.73 (6H, m), 7.50 (2H, t, J = 7.7 Hz), 7.36 (2H, ddd, J = 8.0, 6.8, 1.1 Hz), 7.21 (2H, ddd, J = 8.5, 6.8, 1.4 Hz), 4.75 (2H, app d, J = 9.5 Hz), 3.73 (2H, app d, J = 9.6 Hz), 3.71 (6H, s). 13C{1H} NMR (100 MHz; CDCl3) δ: 173.4, 136.7, 133.9, 132.0, 128.7, 128.0, 126.0, 125.8, 125.7, 124.1, 123.6, 52.4, 45.3, 43.9. FTIR νmax (ATR, solid) per cm 3050, 2954, 2923, 2850, 1723, 1597, 1510, 1445, 1435, 1397. HRMS (APCI+) calcd for C28H25O4 [M + H]+: 425.1747, found: 425.1742.
(1R,2S,3R,4S)-3,4-di(naphthalen-1-yl)cyclobutane-1,2-dicarboxylic acid (18)
Compound 10 (20.4 mg, 0.10 mmol) was placed between two quartz microscope slides. The slides were fixed with the help of paper clips. The solid was then irradiated by 365 nm UV light (4 × 9 W) using the nail dryer. Every 4 h, solid residue was mixed with a spatula. In addition to the mixing process to ensure homogenous light distribution, the side of the slides facing the lamp was turned every 4 h. The irradiation was continued for 20 h. Purification by flash column chromatography (SiO2; 1% acetic acid in EtOAc) afforded cyclobutane 18 as a white solid (15.9 mg, 78% yield). Rf = 0.64 (1% acetic acid in EtOAc). M.P. 195 °C (decomp). 1H NMR 400 MHz; (acetone-d6) δ: 8.17 (2H, d, J = 7.7 Hz), 7.64–7.61 (2H, m), 7.52 (2H, d, J = 8.1 Hz), 7.46 (2H, d, J = 7.2 Hz), 7.30–7.21 (6H, m), 5.45–5.44 (2H, m), 4.22–4.21 (2H, m). 13C{1H} NMR (100 MHz; (acetone-d6) δ: 174.1, 136.2, 134.4, 132.8, 129.2, 127.8, 126.3, 126.1, 125.7, 125.0, 124.7, 44.4, 42.2. FTIR νmax (ATR, solid) per cm 3041, 2956, 2921, 2851, 1698, 1597, 1509, 1417, 1397. HRMS (APCI-) calcd for C26H19O4 [M–H]−: 395.1289, found: 395.1290.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The numerical calculations reported in this article were fully performed at TUBITAK ULAKBIM, High Performance and Grid Computing Center (TRUBA resources). Y. E. T. acknowledges financial support by the GEBIP Award of the Turkish Academy of Sciences. The authors also acknowledge the Scientific and Technological Research Application and Research Center, Sinop University, Türkiye, for the use of the Bruker D8 Quest diffractometer.References
- (a) S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748 CrossRef CAS PubMed; (b) D. M. Bassani, The Dimerization of Cinnamic Acid Derivatives, in CRC Handbook of Photochemistry and Photobiology, ed. W. M. Horspool and F. Lenci, 2nd edn, CRC Press, Boca Raton, 2004; p. p. 20 Search PubMed; (c) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS.
- (a) P. Yang, Q. Jia, S. Song and X. Huang, Nat. Prod. Rep., 2023, 40, 1094 RSC; (b) X. Yuan, L. Men, Y. Liu, Y. Qiu, C. He and W. Huang, J. Holistic Integr. Pharm., 2020, 1, 48 CrossRef; (c) S. El-Arid, J. M. Lenihan, A. B. Beeler and M. W. Grinstaff, Polym. Chem., 2024, 15, 3935 RSC.
- Selected examples: (a) L. R. MacGillivray, J. L. Reid and J. A. Ripmeester, J. Am. Chem. Soc., 2000, 122, 7817 CrossRef CAS; (b) D. M. Bassani, V. Darcos, S. Mahony and J.-P. Desvergne, J. Am. Chem. Soc., 2000, 122, 8795 CrossRef CAS; (c) G. S. Papaefstathiou, A. J. Kipp and L. R. MacGillivray, Chem. Commun., 2001, 2462 RSC; (d) T. Caronna, R. Liantonio, T. A. Logothetis, P. Metrangolo, T. Pilati and G. Resnati, J. Am. Chem. Soc., 2004, 126, 4500 CrossRef CAS PubMed; (e) X. Mei, S. Liu and C. Wolf, Org. Lett., 2007, 9, 2729 CrossRef CAS PubMed; (f) B. R. Bhogala, B. Captain, A. Parthasarathy and V. Ramamurthy, J. Am. Chem. Soc., 2010, 132, 13434 CrossRef CAS PubMed; (g) T. P. Martyanov, A. P. Vorozhtsov, N. A. Aleksandrova, I. V. Sulimenkov, E. N. Ushakov and S. P. Gromov, ACS Omega, 2022, 7, 42370 CrossRef CAS PubMed; (h) J. Alfuth, O. Jeannin and M. Fourmigué, Angew. Chem., Int. Ed., 2022, 61, e202206249 CrossRef CAS.
- (a) L. R. MacGillivray, J. Org. Chem., 2008, 73, 3311 CrossRef CAS PubMed; (b) L. R. MacGillivray, G. S. Papaefstathiou, T. Friščić, T. D. Hamilton, D.-K. Bučar, Q. Chu, D. B. Varshney and I. G. Georgiev, Acc. Chem. Res., 2008, 41, 280 CrossRef CAS PubMed; (c) M. Nagarathinam, A. M. P. Peedikakkal and J. J. Vittal, Chem. Commun., 2008, 5277 RSC; (d) K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950 RSC; (e) M.-M. Gan, J.-G. Yu, Y.-Y. Wang and Y.-F. Han, Cryst. Growth Des., 2018, 18, 553 CrossRef CAS.
- (a) J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413 CrossRef CAS PubMed; (b) B. Bibal, C. Mongin and D. M. Bassani, Chem. Soc. Rev., 2014, 43, 4179 RSC.
- (a) D. Haag and H.-D. Scharf, J. Org. Chem., 1996, 61, 6127 CrossRef CAS; (b) B. König, S. Leue, C. Horn, A. Caudan, J.-P. Desvergne and H. Bouas-Laurent, Liebigs Ann., 1996, 1231 CrossRef; (c) H. Zitt, I. Dix, H. Hopf and P. G. Jones, Eur. J. Org Chem., 2002, 2298 CrossRef CAS; (d) H. Yuasa, M. Nakatani and H. Hashimoto, Org. Biomol. Chem., 2006, 4, 3694 RSC; (e) M. W. Ghosn and C. Wolf, J. Org. Chem., 2010, 75, 6653 CrossRef CAS PubMed; (f) J. M. Lenihan, M. J. Mailloux and A. B. Beeler, Org. Process Res. Dev., 2022, 26, 1812 CrossRef CAS; (g) S. El-Arid, J. M. Lenihan, A. Jacobsen, A. B. Beeler and M. W. Grinstaff, ACS Macro Lett., 2024, 13, 607 CrossRef CAS PubMed.
- F. Medici, A. Puglisi, S. Rossi, L. Raimondi and M. Benaglia, Org. Biomol. Chem., 2023, 21, 2899 RSC.
- (a) M. J. Genzink, M. D. Rossler, H. Recendiz and T. P. Yoon, J. Am. Chem. Soc., 2023, 145, 19182 CrossRef CAS; (b) E. F. Plachinski, H. J. Kim, M. J. Genzink, K. M. Sanders, R. M. Kelch, I. A. Guzei and T. P. Yoon, J. Am. Chem. Soc., 2024, 146, 14948 CrossRef CAS.
- (a) K. Takagi, H. Fukaya, N. Miyake and Y. Sawaki, Chem. Lett., 1988, 1053 CrossRef CAS; (b) K. Takagi, T. Shichi, H. Usami and Y. Sawaki, J. Am. Chem. Soc., 1993, 115, 4339 CrossRef CAS; (c) T. B. Nguyen and A. Al-Mourabit, Photochem. Photobiol. Sci., 2016, 15, 1115 CrossRef CAS; (d) D. E. Marschner, C. O. Franck, D. Abt, H. Mutlu and C. Barner-Kowollik, Chem. Commun., 2019, 55, 9877 RSC; (e) J. Liu, Y. Shen, J. Peng, J. Sun and R. Lu, J. Mater. Chem. C, 2020, 8, 3165 RSC; (f) T.-Y. Li, Y.-Z. Du, T.-Y. Xu, T.-L. Zhang and F. Tong, Crystals, 2024, 14, 492 CrossRef CAS.
- (a) B. B. Yagci, Y. Zorlu and Y. E. Türkmen, J. Org. Chem., 2021, 86, 13118 CrossRef CAS PubMed; (b) B. B. Yagci, B. Munir, Y. Zorlu and Y. E. Türkmen, Synthesis, 2023, 55, 3777 CrossRef CAS.
- B. Munir, B. B. Yagci, Y. Zorlu and Y. E. Türkmen, J. Org. Chem., 2024, 89, 10409 CrossRef CAS.
- (a) A. Ichikawa, H. Ono and Y. Mikata, Chem.–Asian J., 2012, 7, 2294 CrossRef CAS PubMed; (b) A. Ichikawa, H. Ono, T. Echigo and Y. Mikata, CrystEngComm, 2011, 13, 4536 RSC.
- (a) N. Harada, M. Watanabe, S. Kuwahara, A. Sugio, Y. Kasai and A. Ichikawa, Tetrahedron: Asymmetry, 2000, 11, 1249 CrossRef CAS; (b) N. Harada, Chirality, 2008, 20, 691 CrossRef CAS.
- (a) A. N. Thadani, A. R. Stankovic and V. H. Rawal, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5846 CrossRef CAS PubMed; (b) Y. Huang, A. K. Unni, A. N. Thadani and V. H. Rawal, Nature, 2003, 424, 146 CrossRef CAS; (c) C. D. Anderson, T. Dudding, R. Gordillo and K. N. Houk, Org. Lett., 2008, 10, 2749 CrossRef CAS.
- S. M. Sartor, Y. M. Lattke, B. G. McCarthy, G. M. Miyake and N. M. Damrauer, J. Phys. Chem. A, 2019, 123, 4727 CrossRef CAS.
- M. Nishio, CrystEngComm, 2004, 6, 130 RSC.
- (a) V. Enkelmann and G. Wegner, J. Am. Chem. Soc., 1993, 115, 10390 CrossRef CAS; (b) T. Friščić and L. R. MacGillivray, Z. Kristallogr., 2005, 220, 351 CrossRef; (c) A. Chaudhary, A. Mohammad and S. M. Mobin, Cryst. Growth Des., 2017, 17, 2893 CrossRef CAS.
- Y. Gu, T. Kar and S. Scheiner, J. Am. Chem. Soc., 1999, 121, 9411 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Kossmann and F. Neese, J. Chem. Theory Comput., 2010, 6, 2325 CrossRef CAS PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057 RSC.
- (a) S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS; (b) M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2022, e1606 Search PubMed.
- V. Asgeirsson, B. Orri Birgisson, R. Björnsson, U. Becker, F. Neese, C. Riplinger and H. Jonsson, J. Chem. Theory Comput., 2021, 17, 4929 CrossRef CAS PubMed.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174 CrossRef CAS PubMed.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS PubMed.
- T. Lei, C. Zhou, M.-Y. Huang, L.-M. Zhao, B. Yang, C. Ye, H. Xiao, Q.-Y. Meng, V. Ramamurthy, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2017, 56, 15407 CrossRef CAS PubMed.
- S. K. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072 RSC.
- B. Dogga, C. S. A. Kumar and J. T. Joseph, Eur. J. Org Chem., 2021, 309 CrossRef CAS.
- P. Daley-Dee, J. Clarke, S. Monfette and R. B. Bedford, Org. Lett., 2025, 27, 197 CrossRef CAS PubMed.
- A. M. Martínez, A. Puet, G. Domínguez, I. Alonso, R. Castro-Biondo and J. Pérez-Castells, Org. Lett., 2023, 25, 5923 CrossRef.
- A. A. Upare, P. K. Gadekar, H. Sivaramakrishnan, N. Naik, V. M. Khedkar, D. Sarkar, A. Choudhari and S. M. Roopan, Bioorg. Chem., 2019, 86, 507 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2426760 and 2426761. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra03698d |
| ‡ The source of this orange color may be an oxidation process occurring on the surface of the crystals of 6a during irradiation. |
| This journal is © The Royal Society of Chemistry 2025 |