
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the halogenated spirooxindoles as novel anticancer scaffolds: chemistry and bioactivity approach
Mohamed S. Nafie *ab,
Ihab Shawishc,
Sherif Ashraf Fahmy*d,
Mohamed K. Diabe,
Mariam M. Abdelfattahf,
Bassel M. Hassenf,
Khaled M. Darwishgh,
Ayman El-Fahamij and
Assem Barakat*k
*ab,
Ihab Shawishc,
Sherif Ashraf Fahmy*d,
Mohamed K. Diabe,
Mariam M. Abdelfattahf,
Bassel M. Hassenf,
Khaled M. Darwishgh,
Ayman El-Fahamij and
Assem Barakat*k
aDepartment of Chemistry, College of Sciences, University of Sharjah, Sharjah (27272), United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae; mohamed_nafie@science.suez.edu.eg
bDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia (41522), Egypt
cDepartment of Math and Sciences, College of Humanities and Sciences, Prince Sultan University, Riyadh 11586, Saudi Arabia
dDepartment of Pharmaceutics and Biopharmaceutics, University of Marburg, Robert-Koch-Str. 4, Marburg, (35037), Germany. E-mail: sheriffahmy@aucegypt.edu
ePest Physiology Department, Plant Protection Research Institute, Agricultural Research Center, Giza, (12311), Egypt
fCollege of Biotechnology, Misr University for Science and Technology (MUST), 6th of October City, Egypt
gDepartment of Medicinal Chemistry, Faculty of Pharmacy, Galala University, New Galala (P.O. 43713), Egypt
hMedicinal Chemistry Department, Faculty of Pharmacy, Suez Canal University, Ismailia (P.O. 41522), Egypt
iDepartment of Basic Medical Sciences, College of Medicine, Dar Al Uloom University, P. O. Box 45142, Riyadh 11512, Saudi Arabia
jDepartment of Chemistry, Faculty of Science, Alexandria University, P.O. Box 426, Ibrahimia, Alexandria 21321, Egypt
kDepartment of Chemistry, College of Science, King Saud University, Riyadh 11451, Saudi Arabia. E-mail: ambarakat@ksu.edu.sa
First published on 1st July 2025
Abstract
Halogenated spirooxindoles have garnered increasing attention as promising targeted anticancer therapy agents, owing to their improved binding affinity and ability to engage diverse molecular targets. These compounds demonstrate notable anticancer activity across various human malignancies, attributed to their selectivity targeting and reduced toxicity profiles. Functioning as multitarget agents, halogenated spirooxindoles exert biological effects through mechanisms including kinase inhibition, disruption of the MDM2–p53 interaction, DNA-binding modulation, and activation of apoptosis pathways. Their multifaceted activity profile supports their potential as next-generation anticancer agents, suggesting that further studies will be required in this direction. Areas covered: the recent developments of synthetic routes of derivatization and anticancer activities of halogenated spirooxindoles scaffolds during 2020–2025 are thoroughly covered. Expert opinion: halogenated spirooxindoles represent a strategic advance in the development of targeted cancer therapy, combining structural ingenuity with mechanistic precision. These findings encourage medicinal chemists to optimize further halogen placement, scaffold rigidity, and hybrid designs that could lead to new target-oriented chemotherapeutics.
Introduction
Cancer remains a leading cause of death in both developed and developing nations, with a global increase in burden expected due to population growth and aging demographics.1 In 2020, an estimated 19.3 million new cancer cases and 10 million cancer-related deaths were reported worldwide, according to GLOBOCAN data.2 Cancer is characterized by the abnormal growth of cells that typically proliferate uncontrollably. Meanwhile, the structural and functional heterogeneity of cancer cells makes it difficult to achieve high therapeutic selectivity.3 Increasing knowledge in understanding the molecular changes that control the cell cycle and the uncontrolled growth of cancer cells provides valuable guidance for designing cancer-specific drugs.4Spiro compounds have long held significance in organic synthesis due to their notable biological activities.5 Specifically, spirooxindole derivatives have emerged as attractive synthetic targets, mainly due to their occurrence in numerous natural products and pharmacologically active molecules.6 A defining structural feature of these compounds is the spiro-fused ring system at the C3 position of the oxindole core, which incorporates diverse heterocyclic motifs. Halogenated spirooxindoles, in particular, exhibit potent anticancer activity across multiple cancer types, owing to their ability to modulate a range of molecular targets.7,8
Halogenated spirooxindole alkaloids often exhibit marked biological activities strongly influenced by halogen substituents within their structures.9 Spiro compounds remain a subject of interest in research into synthetic chemistry and, indeed, the focus of attention in the synthesis of naturally occurring compounds, where reaction conditions are often tailored to expand the scope of applicable transformations or to favor the formation of specific diastereomers.10 In recent years, multicomponent reactions, recognized as efficient strategies for constructing complex heterocyclic systems, have been increasingly employed in the synthesis of spiro compounds.11
Halogenated spirooxindoles have garnered increasing attention for their ability to effectively suppress the growth and proliferation of various cancer cell types.12 Recent studies have indicated that the anticancer activities involve several pathways or processes via oxidative stress, autophagy, apoptosis induction,13 caspase activation, cyclooxygenase-2 (COX-2) downregulation, death receptor agonism and trafficking, mTOR pathway modulation and inhibition of survivin expression, designing and cellular anti-angiogenesis, p53 expression and activation,14 reactive oxygen species (ROS) generation, sphingomyelinases modulation,9 telomerase inhibition, topoisomerase I/IIα inhibition or tubulin interaction, mitochondrial membrane depolarization, mitochondrial respiratory chain complex.15 Moreover, halogenated spirooxindoles have been reported to interfere with caspase inhibition, intracellular substance accumulation, and STAT activation in combination with them.16 Their efficacy can be further enhanced through synergistic combinations, such as biphenyl carbamate arsenoxide and various organosilicon compounds, or by sensitizing cancer cells to tumor necrosis factor (TNF) cytokines, resulting in amplified antitumor effects.17 In these signaling pathways and antitumor effects, kinase inhibition, including both mono-target and multi-target pathways, plays a key role in exploring therapeutic mechanisms.8 Additionally, they have also shown some other effects of exerting their antitumor activity, such as HsPPIg-mediated suppression of pancreatic tumor growth by blocking paracrine signals within the tumor microenvironment.18 ROS and human serum albumin (HAS), combined with a few, can react with DNA to induce depurination of the guanine base, leading to a further cytotoxic effect.7
Kinases represent a significant family of proteins attracting attention both as therapeutic targets and experimental tools.19 Serine/threonine and tyrosine kinases can regulate a wide range of cellular functions, including growth, survival, and metabolism.20 The oncogenic potential of kinases has been demonstrated in various clinical cases, like BCR-ABL in chronic myeloid leukemia and ABL in gastrointestinal stromal tumors.21 Several studies have reported a direct correlation between enzymatic overexpression or mutational activation and cancer insurgence.22 For instance, approximately 50% of casein kinase isoforms are overexpressed in lung medium adenocarcinoma, an event associated with early tumor progression.23 Overexpression of cyclin-dependent kinase 2 (CDK2) was detected in 49% of invasive cancer cells in Asians, and it was demonstrated that the hepatocellular carcinoma with S/T somatic mutations chi-square equals 7.90.24 Furthermore, a less regulated C-terminus has been linked to the presence of Ala, whereas residues such as Glu, Asp, and Phe are related to protein delocalization.25,26 Kinases involved in critical signaling pathways include glycogen synthase kinase 3, Janus kinase, Ca2+-dependent protein kinase, Polo-like kinase, lipid kinase, casein kinase, cyclin-dependent kinases, mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK), protein kinase B (AKt), ERK, and PKC isoforms such as PKC-α, and PKC-δ.27–31
Over the past few decades, various kinase inhibitors have significantly impacted the clinical landscape, revolutionizing cancer treatments.28 Several decades of drug approval have drastically improved the methodological development and advancements in kinase-targeted therapies.32 Rational drug design and medicinal chemistry efforts, particularly in the later stages of the drug design process, have led to the discovery of several promising kinase-targeted drugs.33,34 Drug discovery was reanalyzed to identify new strategies for enhancing the therapeutic efficacy of kinase inhibitors.32 Most kinase inhibitors act as active site-directed, ATP-competitive inhibitors, and unfortunately, they are limited due to a lack of selectivity over other typical cellular kinases.35 The focus of current drug development is to overcome the therapeutic limitations of first-generation kinase inhibitors.36 The discovery of spirooxindole as a molecular core integrated into multikinase inhibitors has been extensively studied at the fundamental level, demonstrating both hit and lead compounds characterized by strong anticancer properties.9,37 Specifically, halogenated spirooxindole has shown promising anticancer activity by targeting kinases involved in breast cancer.18 Recent studies have demonstrated that halogenated spirooxindole derivatives target Polo-like kinase, cyclin-dependent kinase 2, phosphoinositide 3-kinase, and receptor tyrosine kinase families in breast cancer.38 This makes halogenated spirooxindole very promising as a potential anticancer agent, either as a multitarget or selective kinase inhibitor.7 In conclusion, this research provides the first scientific evidence supporting the development and synthesis of spirooxindole-derived molecules for the selective targeting of anticancer kinases in breast cancer cells.8
In addition to their direct cytostatic activity, which remains to be fully elucidated, halogenated spirooxindoles appear to influence several other cellular pathways and processes, including apoptosis, cellular senescence, and autophagy.14 They have been shown to promote increased oxidative stress within cancer cells, potentially releasing reactive oxygen accumulations to trigger cell cycle arrest.13 Furthermore, it is feasible that halogenated spirooxindoles interact with the tumor microenvironment, possibly influencing immune modulation.9 These multifaceted effects suggest that halogenated spirooxindoles possess a promising therapeutic potential extending beyond the traditional scope of kinase inhibitor research.18
Bioactivity of halogenated spirooxindoles as anticancer agents
In vitro studies have been conducted to determine the anticancer efficacy of halogenated spirooxindoles, comparing their cytotoxic and apoptotic effects to those of reference compounds or adjuvant treatments on several cancer cell lines and normal cells.39 These pharmacological studies specifically evaluate cell viability and/or cytotoxicity, apoptosis induction, cell cycle arrest, modulation of protein expression, the visualization and quantification of reactive oxygen species (ROS), and the impact on certain metabolic pathways.18 Both halogenated and non-halogenated spirooxindoles have shown cytotoxic and apoptotic effects on different cell lines, including melanoma, hepatocarcinoma, pancreatic cancer, cervical cancer, prostate, acute promyelocytic leukemia, and adenocarcinoma.12,37 They also demonstrated low cytotoxicity on normal fibroblasts and lymphocytes, while human fibroblasts showed particular sensitivity to their treatment.17Based on the in vitro findings, some of these spirooxindoles were tested on xenografts in research organizations using animals certified for preclinical studies.40 These studies aim to evaluate the animals' responses to new treatments in terms of observable outcomes and severity limits in order to calculate the dosage to be tested in humans.41 Following promising results in the initial patient cohort, preclinical studies explored the combination of halogenated spirothiazolidinediones with established therapeutic drugs.42 To end, mice belonging to the tumor histotype were treated with daily or weekly administrations of a specific dose of the drug. The treatment must be well tolerated by the animals.43 Establishing the safe doses of new drugs or combinations of existing ones in animal models informs decisions that can avoid overdosing with consequent toxic effects or a dose that seems to be too low, as with certain substances.44 However, these preclinical studies in animal cancer models are an important factor that demonstrates their effectiveness in living organisms and, therefore, can be tested in humans according to the protocol established during the research process.45
Several series of spirooxindoles have been utilized for the development of kinase inhibitors, especially for anticancer drug discovery.7 These potent hinge-binding inhibitors exploit chemotherapy combined with pharmacological bypassing of alternative signaling pathways that are frequently enhanced in cancer.18 The molecules identified in this review demonstrate excellent kinase activity, either in vitro or in cell potency, or at least in silico.46 Some of them have displayed in vivo efficacy and selectivity.40 Finally, some are currently under clinical trial studies.41 Based on all these aspects, time-to-event studies should be performed, and the clinical trial results must be available, which will prove the efficacy of spirooxindole kinase inhibitors in cancer therapeutics.47 Anticancer drugs are primarily discovered by focusing on the inhibitory activity of these molecules against specific cancer-associated kinases and other anticancer targets through docking studies of high-throughput screening hit molecules.48,49 However, these methods have limitations due to the inaccurate protein kinase model, the non-valuable consideration of protein flexibility, and the neglect of conserved water molecules.50 All these factors limit the success of the drug, which leads to current cancer treatment. A reliable and straightforward way to discover a potential drug is through tedious biological screenings.51 At the same time, these challenges may also require chemical strategies, such as the synthesis of new small molecules. Consequently, several spirooxindole pharmacophores have been employed as selective, potent, and druggable kinase inhibitors.38
Mechanistic aspects of the halogenated spiro-oxindoles as anticancer agents
Due to their role in influencing key molecular pathways that advance cancer, halogenated spiro-oxindoles are considered viable anticancer therapeutics. These unique chemical substances showcase their capacity to suppress cell proliferation by utilizing different mechanisms, which involve inducing apoptosis, stopping the cell cycle, and activating ferroptosis, as clarified in (Fig. 1).51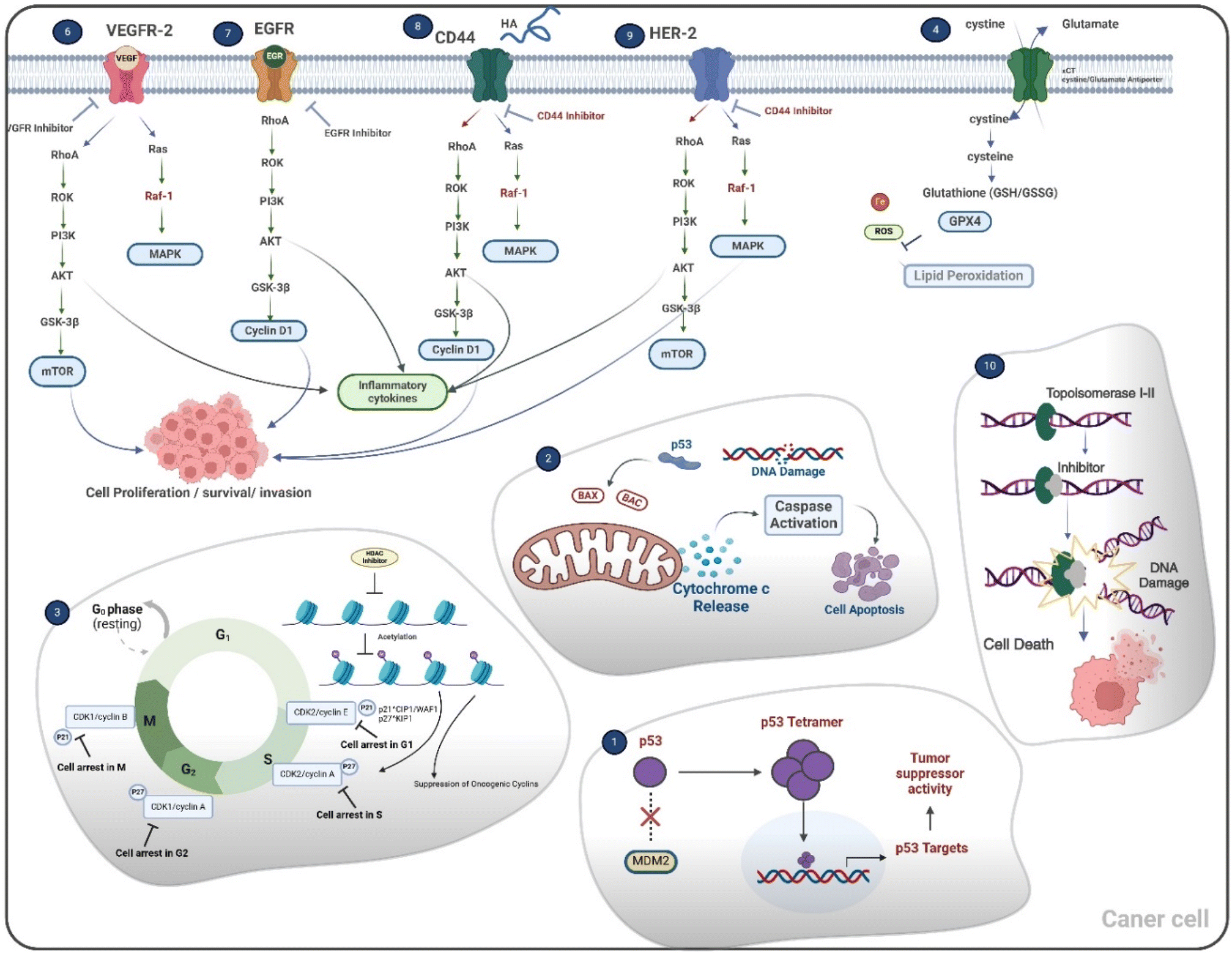 | ||
| Fig. 1 The mechanistic pathways for the therapeutic potential of halogenated spirooxindoles as anti-cancer agents.51 | ||
One of the major anticancer mechanisms of halogenated spiro-oxindoles involves disrupting the p53–MDM2 interaction; the tumor suppressor protein p53 plays a pivotal role in regulating cell cycle arrest and apoptosis.41 During normal physiological conditions, p53 performs negative modulation by MDM2, an E3 ubiquitin ligase that fosters the ubiquitination of p53, thereby resulting in its clearance through the proteasomal mechanism. Disruption of the p53–MDM2 interaction leads to stabilization and activation of p53, promoting apoptosis in cancer cells.52 Additionally, these specific agents have been shown to assist in apoptosis by inhibiting BCL-2 expression and promoting caspase activation, thereby causing the release of mitochondrial cytochrome c and the initiation of programmed cell death.53
Cell cycle regulation is another crucial target of halogenated spiro-oxindoles. These specific compounds have demonstrated their potential to inhibit cyclin-dependent kinase 2 (CDK2), thereby preventing the transition from the G1 phase to the S phase and effectively halting cellular proliferation.54 Furthermore, the inhibition of these compounds on histone deacetylase (HDAC) induces chromatin remodeling, suppressing oncogenic transcription and leading to cell cycle arrest.16
In addition to apoptosis, halogenated spiro-oxindoles have been implicated in inducing ferroptosis, an iron-dependent form of programmed cell death characterized by lipid peroxidation. Glutathione peroxidase 4 (GPX4) is a key enzyme that protects cells from lipid peroxidation, at which inhibiting GPX4 can induce ferroptosis, providing a novel approach to eliminating cancer cells. Although direct evidence of halogenated spiro-oxindoles targeting GPX4 is scarce, their potential to modulate oxidative stress pathways warrants further investigation.55
The ability of halogenated spiro-oxindoles to inhibit vascular endothelial growth factor receptor 2 (VEGFR2) and epidermal growth factor receptor (EGFR) is particularly significant in limiting tumor angiogenesis and metastasis. By blocking these receptors, the compounds reduce the availability of survival signals for cancer cells, thereby inhibiting the MAPK and PI3K/AKT signaling pathways that are frequently dysregulated in cancers, which promotes cell survival and proliferation. Targeting these pathways can enhance the efficacy of anticancer therapies.56 The RAS/RAF/MEK/ERK (MAPK) and PI3K/AKT/mTOR signaling pathways are frequently dysregulated in cancers, promoting cell survival and proliferation.57
Specific studies on halogenated spiro-oxindoles targeting Cluster of Differentiation Receptor (CD44) and Human Epidermal Growth Factor Receptor 2 (HER-2) are less available. However, they are also known to contribute to cancer cell survival and metastasis by inhibiting the RAS/RAF/MEK/ERK (MAPK) and PI3K/AKT/mTOR signaling pathways, so inhibiting these pathways could potentially disrupt cancer progression and enhance treatment outcomes.58,59
Halogenated spiro-oxindoles function as dual inhibitors of Topoisomerase I and II by interfering with the enzymes' ability to cleave DNA strands, thus converting them into physiological poisons that induce DNA damage and trigger cell death.60
Molecular perspective of spirooxindole scaffold as a promising kinase inhibitor
Targeting several members of the kinase family has raised the adequacy of the spirooxindole scaffold to fulfill the structural requirements and molecular landscape of the kinase catalytic domain.49 Comprising almost 300 amino acids, the overall folding and tertiary protein architecture of the kinase catalytic domain have been recognized as conserved across the kinase superfamily.61 This canonical domain of the kinase enzymes comprises two lobes (N- and C-terminal lobes) linked through a flexible hinge region. The large C-lobe is of predominant α-helix architecture (six helices), while the smaller catalytic domain (N-lobe) comprises singular α-helix (αC-helix) and five β-sheets (Fig. 2A). Kinase ATP-substrate binding site is typically a hydrophobic shallow surface groove being made by the residues of both structural lobes.62,63 This catalytic ATP-binding hydrophobic cleft is druggable for several small hydrophobic molecules and harbors the key structural motifs responsible for target activation.64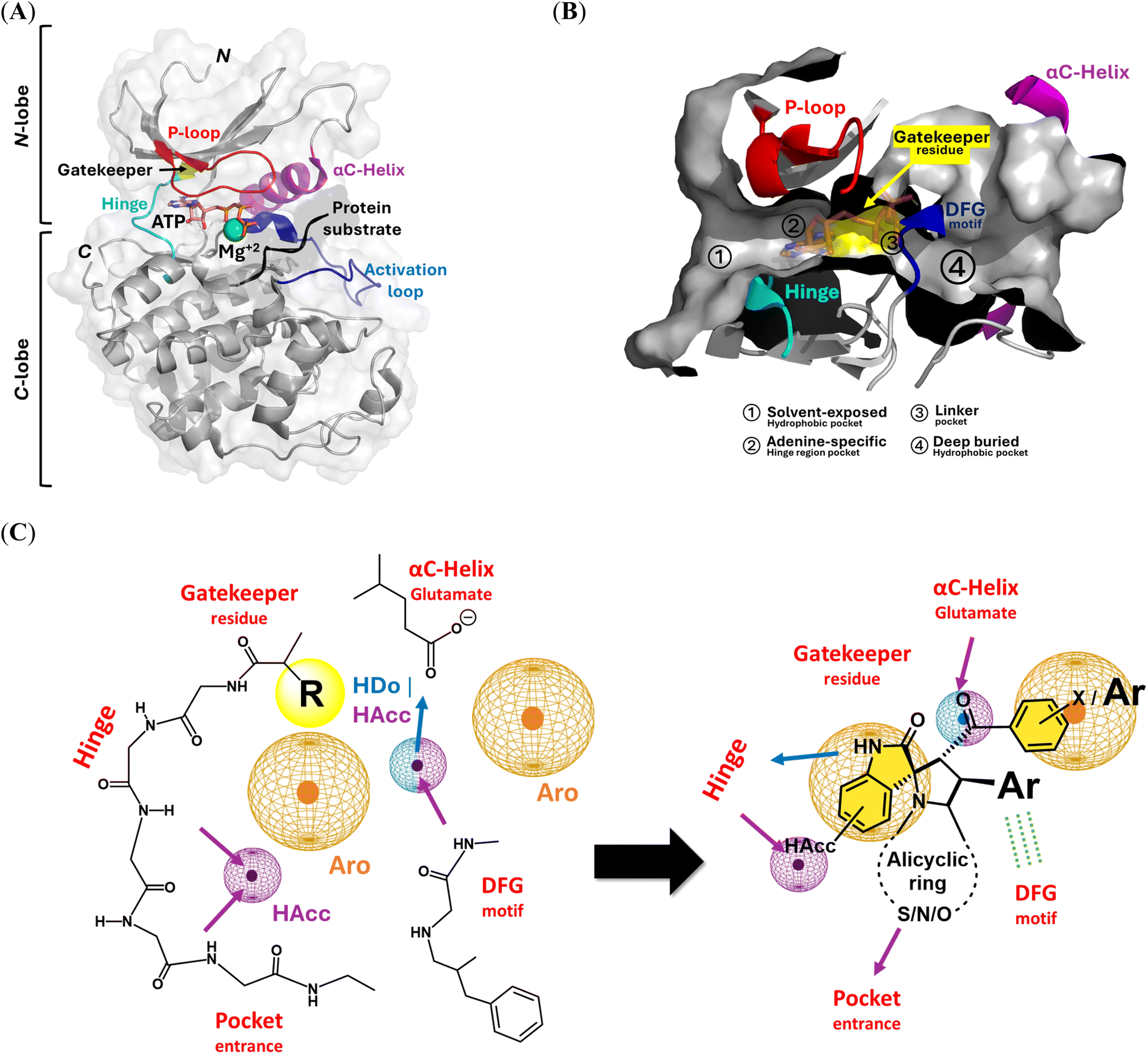 | ||
| Fig. 2 Architecture of kinase's catalytic domain and molecular aspects of spirooxindole scaffold binding at the ATP canonical binding site. (A) Cartoon/surface 3D-representation of the conserved folded tertiary architecture of an example kinase biotarget; CDK2 (PDB_ID: 1qmz) in complex with ATP molecule (orange sticks), Mg+2 ion (green sphere) and protein substrate (cartoon). Key kinase structural motifs and secondary structures are color-coded. The amino and carboxy-termini are denoted with letters N and C, respectively; (B) Detailed ATP-binding site showing the four main sub-pockets (①–④) for the recognition and binding to main scaffolds of the small molecule kinase inhibitors: (C) key pharmacophoric features of reported small molecule kinase inhibitors and the quite superimposition of the spirooxindole scaffolds over these features. Pharmacophoric features are shown in the 3D-spatial arrangement of color-coded meshed spheres (purple = hydrogen bond acceptor HAcc; blue = hydrogen bond donor HDo, and orange = aromatic rings Aro). Arrows denote the projection/directionality of the hydrogen bonds. | ||
Topological and structural analysis of the kinase catalytic site highlighted the following subregions for kinase inhibitor binding: a solvent-exposed hydrophobic region, adenine-specific pocket, bridge portion linker site, and deep buried hydrophobic region (Fig. 2B).65,66 The adenine-specific pocket is settled at the center of the catalytic site where inhibitors harboring purine isosteres are capable of mediating conserved hydrogen bonding towards the hinge region backbone residues. The solvent-exposed hydrophobic region is settled at the kinase pocket entrance, complementary to the non-polar aliphatic/aromatic structures of the kinase inhibitors that are decorated with hydrogen bonding accepting groups. The adenine-specific pocket is covered by a flexible loop endorsed between N-lobe sheets β1/β2 and harboring the glycine-rich structural motif (GXGXφG). The hydrophobic φ amino acid at this conserved Gly-rich loop provides non-covalent contacts with the ATP phosphates that coin its name as phosphate-binding loop (P-loop). Different kinase structural motifs, including the β3-sheet, conserved VIAK, and activation loop DFG motifs, support the placement of the bridge portion linker region near the kinase gatekeeper. The lysine residue at VIAK has been recognized as important for mediating kinase activation through the salt bridge with ATP phosphate and Cα-helix glutamate. Aspartate of the DFG motif mediates coordination between Mg+2 and ATP phosphates,67 while the phenylalanine residue either flapped in or out to limit or expand the deep buried hydrophobic region.68
Accommodation of these sub-regions, while depicting a conserved interaction towards the hinge region, both have been considered fundamental for developing potent kinase inhibitors.69 Binding interaction with the αC-helix glutamate sidechain and/or DFG structural motif vicinal to the linker site has been conserved across several FDA-approved kinase inhibitors.70 Additionally, a deep anchoring of compounds at the back side allosteric hydrophobic sub-pocket in case of DGF out-conformation would permit great compound's potent inhibition and target selectivity for several kinase inhibitors.71 Based on these molecular binding insights, a common pharmacophoric feature for kinase inhibitors can be proposed, where four main features are required to fulfill the binding requirement at the kinase catalytic pocket (Fig. 2C).72,73 Following a 3D spatial arrangement, a hydrogen bond acceptor feature is redeemed to satisfy the polar contacts at the solvent-exposed pocket and/or hinge region. This polar feature will be followed by an aromatic pharmacophoric ring resembling the ATP-purine isosteres for settling the kinase inhibitors at the adenine-specific site. A hydrogen bond donor/acceptor feature is to follow to maintain the key polar contacts towards the polar residues (VIAK, αC-helix glutamate, and DFG motifs) at the bridge portion linker. This polar feature is generally associated with urea, hydrazide, or carboxamide structural functionalities reported in several kinase inhibitors. The final pharmacophoric feature of kinase inhibitors is that another aromatic ring is complementary to the deep-buried hydrophobic region. Increasing the size of this terminal aromatic feature has been correlated with deeper anchoring at the allosteric hydrophobic pocket being accessible at DFG-out conformation.74–76
Literature reported that spirooxindole-based compounds showed comparable chemical architecture throughout several anti-cancer studies.77 The design of these reported spirooxindoles is generally based on four main structural aspects. First, an indole or even a thioindole ring is generally considered a relevant bioisostere of the ATP-purine core scaffold. This central indole core ring is suggested as beneficial for mediating hydrogen bonding towards hinge region residues, which could be further augmented by substituting hydrogen bond acceptors (i.e., fluorine). Such a core ring will be able to fulfill two of the main pharmacophoric features of the small kinase inhibitors at the hinge region and solvent-exposed pocket. The second structural aspect of spiro oxindole-based compounds is spiro cyclization using the saturated fused heterocyclic rings, which would satisfy the hydrogen bonding capability and non-polar contacts at the solvent-exposed hydrophobic site. The saturated property of the spirocyclized ring would introduce a relevant degree of structure flexibility and drug-likeness characteristics, which are valuable during target binding maneuvers and membrane permeation.78,79
Furthermore, several reports have highlighted that scraping the flatland by increasing the compounds' three-dimensional conformation would increase the success rates, binding frequencies, and selectivity indices.80 The spirocyclic ring represents a versatile point for structural diversity, as several reported compounds harbor both substituted aryl and aroyl groups. The aroyl groups fulfill the kinase aromatic and hydrogen bond donor/acceptor pharmacophores through deep anchoring at the buried hydrophobic pocket while maintaining key polar contacts with the linker site polar residues. The aroyl group could be substituted with benzene, fused aromatic rings, or bis-phenyl scaffolds, permitting enhanced anchoring at the kinase back pockets. Regarding the substituted aryl group, it is suggested to depict favored orientation towards the DFG motif permitting hydrophobic and/or π-driven contacts with close residues sidechains. Alignment of the reported spirooxindole scaffolds with the key pharmacophoric features of small molecule kinase inhibitors is shown in Fig. 2C, highlighting a quite relevant overlap.
Chemical synthesis of halogenated spiro-oxindoles
Raposo et al. assessed the antiproliferative activity of selected spiropyrazoline derivatives against A2780 ovarian cancer cells.13 The synthesis of these compounds, as previously reported in the literature,81 involved the reaction of indolinone derivatives (1) with imine intermediates (2), which were generated in situ through the dehydrohalogenation of hydrazonyl chloride in the presence of triethylamine (TEA). This process proceeded via a 1,3-dipolar cycloaddition, affording the target spirooxindole derivatives (3a–e) with chemical yields ranging from 76% to 98%, as illustrated in Scheme 1.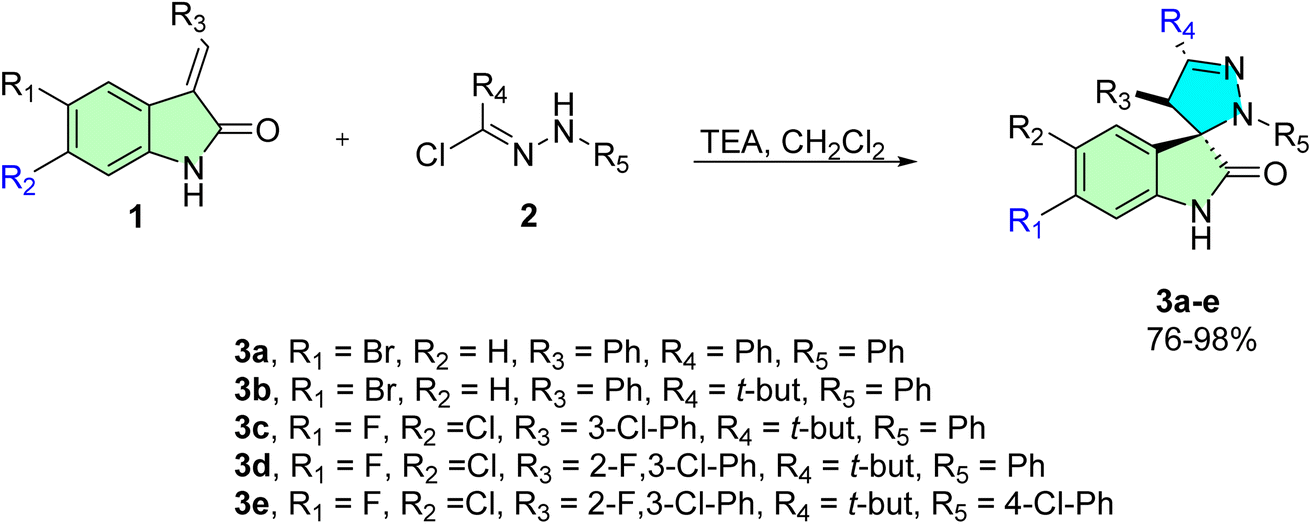 | ||
| Scheme 1 Synthesis of spiropyrazoline derivatives (3a–e). | ||
Compound 3a presented the highest inhibitory effect on the proliferation in the A2780 cell line with an IC50 of 8.5 ± 0.7 μM. In normal human primary fibroblasts, cell viability is almost unaffected, except for the highest concentrations, indicating that 3a is more selective for tumor A2780 cancer cells, as previously observed with this compound in breast and colon cells. Indeed, when exposing fibroblasts to 8.5 ± 0.7 μM of 3a (its IC50 concentration in A2780 cells), no change in viability is observed. Despite the higher IC50 of 3a in ovarian carcinoma compared to the standard chemotherapeutics drugs mentioned, the IC50 value is in the low micromolar range (<10 μM).13,81
Pan et al. synthesized novel spirocyclopropylthiooxindole hybrids featuring a stereochemically rich framework.82 Their approach involved an enantio- and diastereoselective strategy for the cyclopropanation of diazothiooxindoles 5, utilizing various Rh-catalyzed α-functionalized alkene systems 4. The incorporation of diverse substituents on the olefin scaffold facilitated the formation of the target optically active spirooxindoles 6, bearing a variety of pharmacophores within their skeleton, and achieved excellent chemical yields, as illustrated in Scheme 2.
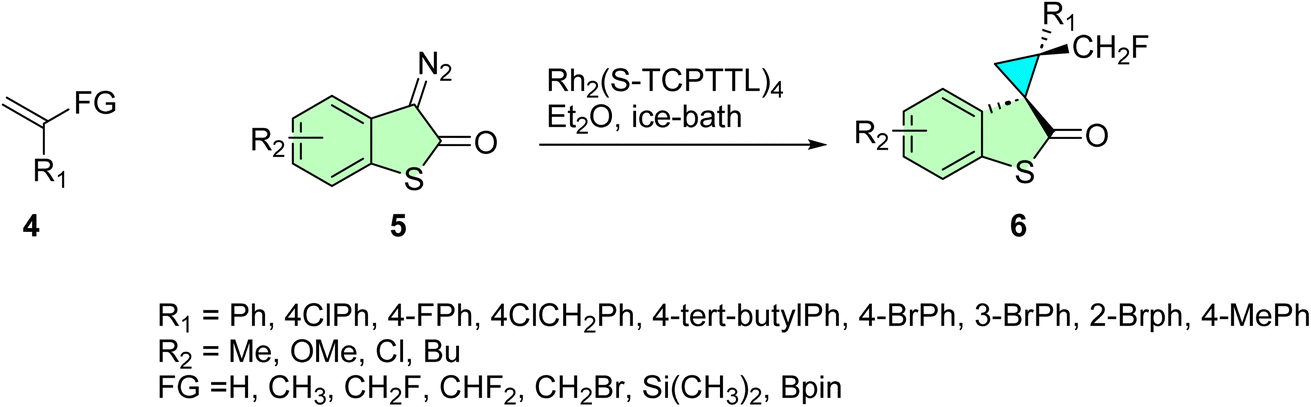 | ||
| Scheme 2 Synthesis of spirothiooxindoles 6. | ||
Compound 6j with CH2F as a substituent on the cyclopropane ring was the most effective inhibitor of SJSA-1 osteosarcoma cells (IC50 = 0.82 ± 0.08 μM), close to six times lower than cisplatin (IC50 = 4.85 ± 0.27 μM). Fluorine substitution of the scaffold at R3 also enhanced the activity in comparative cytotoxicity tests on MCF-7 breast cancer cells. Compound 3e, wherein the R1 and R2 groups remained the same, obtained an IC50 value of 1.09 μM, 38 times more potent. More importantly, these compounds exhibited comparatively less cytotoxic effects on normal cells at the same concentrations, offering a promising therapeutic index. These spirooxindole hybrids showed comparable low micromolar cytotoxicity against all cancer types, suggesting that their cytotoxicity values differed, supporting their potential as selective anticancer agents.82
Liu et al. synthesized a novel series of 3′-(nitroisoxazole)spiro[pyrrolidin-3,2′-oxindoles] bearing a CF3 moiety, which were evaluated for their potential as dual inhibitors of GPX4 and MDM2.83 The synthetic methodology of the most active compound, 6-chloro-3′-(3-methyl-4-nitroisoxazol-5-yl)-4′-phenyl-5′-(trifluoromethyl)spiro[indoline-3,2′-pyrrolidin]-2-one (9d), involved a reaction between 3-methyl-4-nitro-5-styrylisoxazole (7) and 6-chloro-isatin imine (8) in a basic medium of 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU). The reaction yield and time were highly solvent-dependent, with the optimal conditions achieved using dichloromethane (DCM) at room temperature, as shown in Scheme 3.83
 | ||
| Scheme 3 Synthesis of spiro derivative 9d. | ||
Compound 9 with strong GPX4 and MDM2 dual inhibition are important targets for breast adenocarcinoma. It inhibited the MDM2-mediated degradation of p53 and decreased GPX4 expression, leading to both ferroptosis and apoptosis in MCF-7 cells (IC50 = 0.12 μM). The process was related to ROS generation, lipid peroxidation, and caspase-9 processing. In vivo, 3D suppressed xenograft tumor growth in a mouse model without body weight loss. Immunohistochemistry confirmed enhanced p53 and TUNEL signals and reduced Ki-67 and GPX4 on dual suppression. These findings demonstrate that 3D is a selective, multitarget anticancer scaffold with submicromolar potency.83
Kanchrana et al. designed and synthesized a new analog of spirooxindolo-1,2,4-oxadiazole derivatives via a [3 + 2] cycloaddition reaction, employing a green methodology based on ultrasonication techniques.84 Various solvents and basic media were tested to optimize yield and minimize reaction time. The optimal conditions were achieved when N-hydroxycarbimidoyl chloride 11 and isatin Schiff bases 12 were reacted for 20 minutes at room temperature under ultrasonication, using CHCl3 as the solvent and TEA as the basic medium, as shown in Scheme 4. The resulting spiro derivatives were evaluated for their potential anticancer activity against six cancer cell lines: SK-OV-3, HeLa, HCT-116, DU-145, A549, and HEK-293.
 | ||
| Scheme 4 Synthesis of spirooxindolo-1,2,4-oxadiazoles 12a and 12b. | ||
5-Chloro-3′-(2-chloroquinolin-3-yl)-4′-phenyl-4′H-spiro[indoline-3,5′-[1,2,4]oxadiazol]-2-one (12a), 5-chloro-4′-(3-chlorophenyl)-3′-(2-chloroquinolin-3-yl)-4′H-spiro[indoline-3,5′-[1,2,4]oxadiazol]-2-one (12b): The compounds 12a and 12b exhibited significant activity against cervical cancer HeLa cell line with IC50 value 10.75 ± 0.39 μM and 12.43 ± 0.77 μM, respectively.84
Barakat et al. rationally designed and synthesized a novel series of spiro-indeno[1,2-b]quinoxaline derivatives incorporating a benzimidazole moiety.54 The synthetic approach involved a one-pot, multicomponent [3 + 2] cycloaddition (32CA) reaction of benzimidazolyl chalcones 13 with 11H-indeno[1,2-b]quinoxalin-11-one 14 and various amino acids, such as octahydroindole-2-carboxylic acid 15. The reaction was conducted in refluxed methanol for 8 hours, yielding the target spirooxindole derivatives in yields of 59–95% (16, 84%), as shown in Scheme 5.54
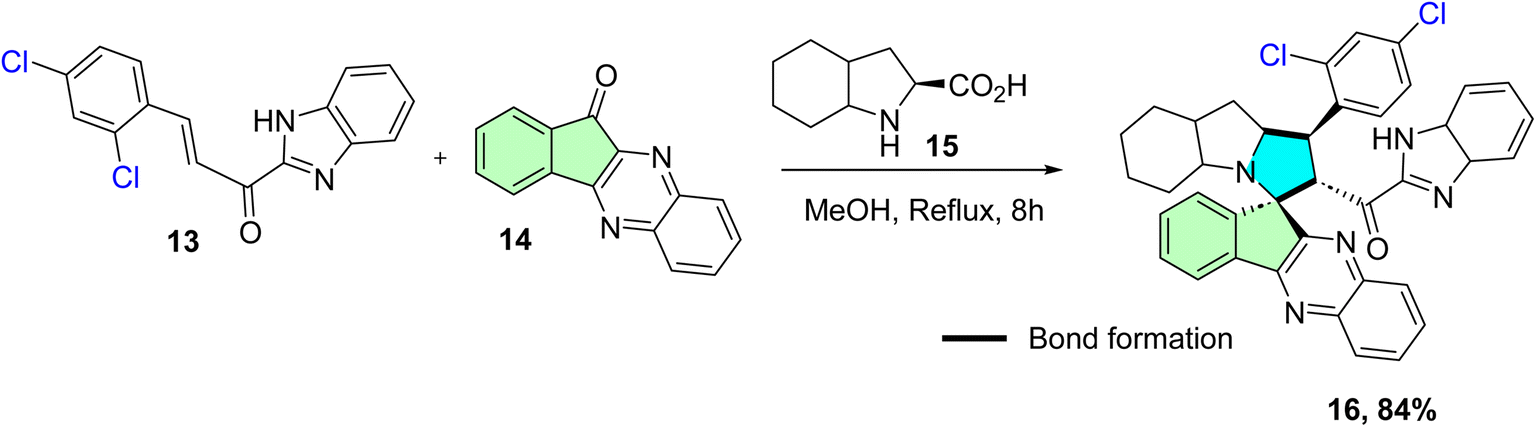 | ||
| Scheme 5 Synthesis of spirooxindole 16. | ||
The IC50 value of compound 16 on A549 human lung cancer cells was 0.054 ± 0.008 nM, which was much more potent than the reference drug fluorouracil (IC50 = 3.781 ± 0.52 μM). Normal lung fibroblasts (Wi-38) were 20–40 times less sensitive at this concentration, indicating significant selectivity towards cancer cells. This selectivity aligns with previous reports of spirooxindole compounds featuring benzimidazole and indenoquinoxaline moieties. Notably, normal fibroblasts did not exhibit significantly decreased viability at doses effective for A549 cells, indicating high selectivity of compound 16 against CDK2 and suggesting it is a potential anti-lung cancer drug candidate. The IC50 was shown to be in the nanomolar range, indicating a promising lead compound with high potency and low cytotoxicity toward normal cells.54
Korotaev et al. successfully carried out a regio- and stereoselective synthesis of a series of N-unsubstituted 3-aryl-4-(trifluoromethyl)-4H spiro[chromeno[3,4-c]-pyrrolidine-1,3′-oxindoles] (21a–l) through two synthetic approaches.85 The first approach involved a one-pot, three-component reaction employing isatin derivatives (17), 3-nitro-2-(trifluoromethyl)-2H-chromenes (18), and benzylamines (19). The reaction was optimized under reflux conditions in CH2Cl2 for 24 hours in the presence of a basic medium of 1,4-diazabicyclo[2.2.2]octane (DABCO), yielding the desired spiro derivatives (21a–l) with yields ranging from 24% to 79%, as shown in Scheme 6. The second synthetic approach involved a one-pot reaction of the chromene derivatives (18) with the azomethine ylides produced in situ by the reaction of the isatin derivatives (17) with L-phenyl glycine (20). The reaction was carried out in EtOH at 60 °C for 5 hours to produce the target spiro derivatives (21a–l) with yields between 32% and 71%, as shown in Scheme 6.
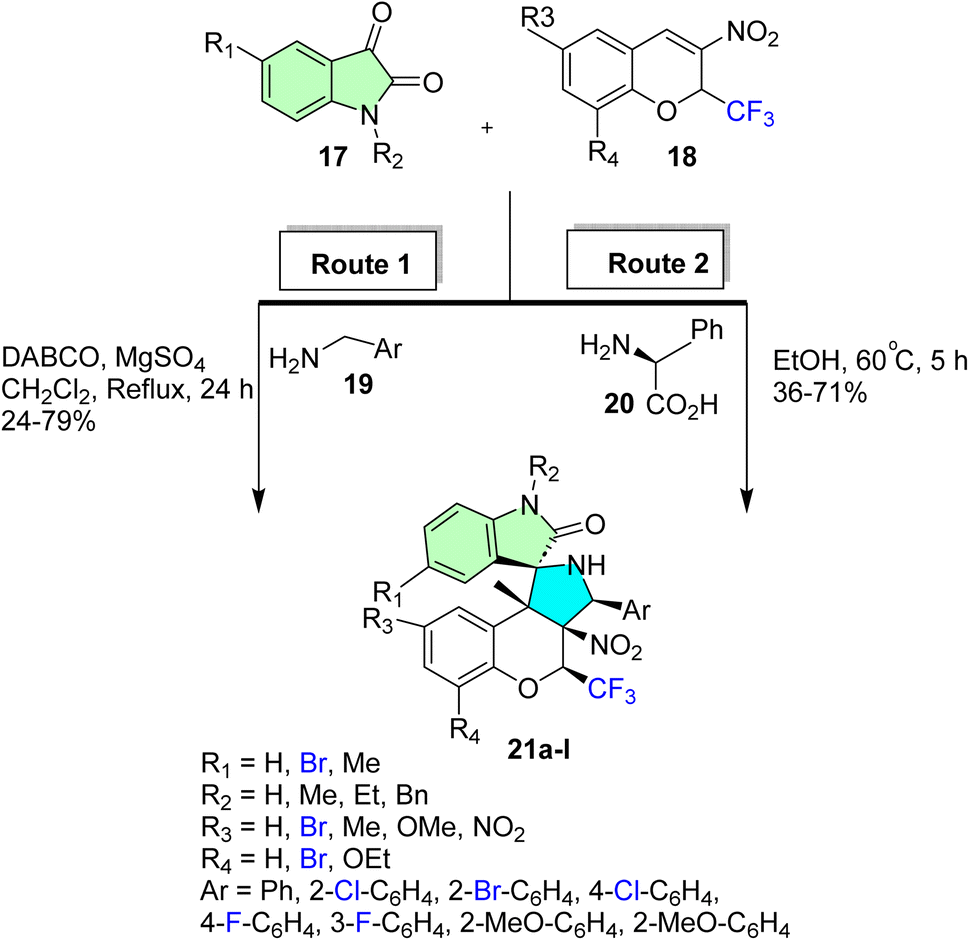 | ||
| Scheme 6 Synthetic routes of spirooxindole derivatives (21a–l). | ||
The regio- and stereoselective one-pot three-component reaction synthesized compounds 21a, 21e, and 21l, which displayed significant cytotoxicity towards HeLa cervical cancer cell lines. Compound 21l was the most potent compound, with an IC50 value of 0.71 ± 0.05 μM, and thus is greater than that of the reference camptothecin (IC50 = 1.66 ± 0.97 μM). Also, 21a and 21e were highly active, with IC50 = 1.74 ± 0.45 and 1.62 ± 0.52 μM, respectively. Notably, the three candidates demonstrated strong selectivity for cancer cells, and cytotoxicity tests revealed low toxicity towards normal human dermal fibroblasts (HDF) at the IC50 of each drug. This selectivity, combined with their low micromolar cytotoxic activities, qualifies these spirooxindole analogs as potential anticancer agents, especially for the selective attack of cervical cancer cells with low side effects on normal tissue.85
Al-Majid et al. developed a one-pot synthetic method to create a new series of di-spirooxindole derivatives featuring a cyclohexanone moiety via 32CA reaction.86 In this process, isatins (22a–f) and (2S)-octahydro-1H-indole-2-carboxylic acid 23 were reacted to generate azomethine ylides in situ, which were then reacted with cyclohexanone-based chalcones (24a–f) to yield the target di-spirooxindole derivatives (25a–n), as illustrated in Scheme 7.
 | ||
| Scheme 7 Synthesis of di-spirooxindole derivatives (25a–n). | ||
Although all the tested compounds exhibited significant antiproliferative properties against the PC3 human prostate cancer cell line, the synthesized di-spirooxindole derivative 25b displayed the highest activity with an IC50 of 3.7 ± 1.0 μM in the low micromolar range. Compounds 25e and 25d exhibited profound cytotoxicity in cervical (HeLa) and triple-negative breast carcinoma (MDA-MB231) cancer cell lines, with IC50 values of 7.2 ± 0.5 and 7.63 ± 0.08 μM, respectively. These compounds were also highly selective between (at equivalent concentrations) normal and cancer cellular cytotoxicity. The structural rigidity of the di-spirooxindole skeleton may be responsible for the high affinity and selectivity for tumor-related targets. The potential dual antitumor effects in several cancers indicate the practical importance of this scaffold for probing into more anticancer drugs.86
Barakat et al. reported synthesizing a novel series of spirooxindole derivatives featuring thiochromene and pyrrolidine frameworks.87 The synthetic approach involved the formation of azomethine ylides through the condensation of L-proline with various isatin derivatives. These intermediates were subsequently reacted with various chalcones incorporating thiochromene scaffolds via a 1,3-dipolar cycloaddition reaction, forming the desired spirooxindole derivatives, as depicted in Scheme 8.
 | ||
| Scheme 8 Synthesis of spirooxindole derivatives (28a–m). | ||
1′-(4-Chloro-2H-thiochromen-3-yl)-5-fluoro-2′-(4-fluorobenzoyl)-1′,2′,5′,6′,7′,7a′-hexahyd rospiro[indoline-3,3′-pyrrolizin]-2-one (28d), 1′-(4-chloro-2 H -thiochromen-3-yl)-5-fluoro-2′-(4-nitrobenzoyl)-1′,2′,5′,6′,7′,7a′-hexahydr ospiro[indoline-3,3′-pyrrolizin]-2-one (28f), 5-chloro-1′-(4-chloro-2H-thiochromen-3-yl)-2′-(4-(trifluoromethyl)benzoyl)-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (28k): The anticancer assay showed promising results as good candidates for further studies. Compounds (28f, IC50 = 8.7 ± 0.7 μM) exhibited more potent activity against PC3, whereas hybrid (28k) was most active against cervical cancer HeLa (IC50 8.4 ± 0.5 μM) and for breast cancer MCF-7 cell lines (28d, IC50 = 7.36 ± 0.37 μM), whereas (28d, IC50 = 9.44 ± 0.32 μM) also appeared more active against MDA-MB231 breast cancer cell line.87
Aziz et al. employed an efficient synthetic strategy to design two novel spirooxindole derivatives targeting the p53–MDM2 interaction, which downregulates Bcl-2 signaling.53 The regio- and stereoselective approach involved the reaction of N-methyl-2-acetylpyrrole with substituted benzaldehydes via an aldol condensation reaction, yielding the corresponding N-methyl pyrrole chalcones (29a–b). These chalcones were subsequently subjected to a one-pot 32CA reaction with 5-chloroisatine and sarcosine/thioproline, resulting in the formation of the desired spirooxindole derivatives (30a and 30b), as depicted in Scheme 9.
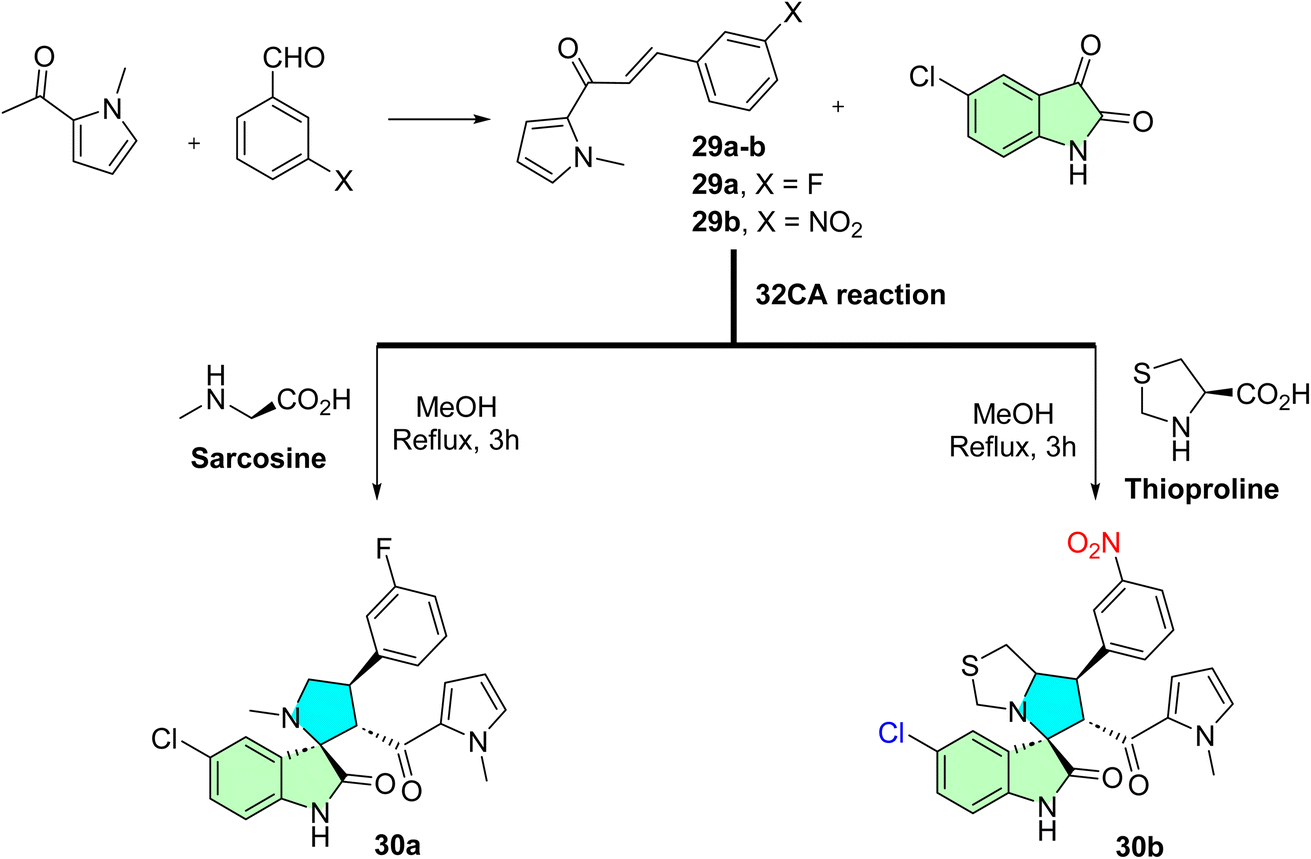 | ||
| Scheme 9 Synthesis of spirroxindole derivatives 30a–b. | ||
(3S,6′R,7′S)-5-Chloro-6′-(1-methyl-1H-pyrrole-2-carbonyl)-7′-(3-nitrophenyl)-1′,6′,7′,7a′-tetrahydro-3′H-spiro [indoline-3,5′-pyrrolo [1,2-c]thiazol]-2-one (30b): compound 30b showed significant anticancer activities via p53–MDM2 interaction and Bcl2 signaling, respectively. In MDA-MB-231 breast cancer cells, it induced p53 activation (47%), downregulation of the Bcl2 gene (1.25-fold), upregulation of p21 (2-fold), and 43.08% apoptosis, which was greater than 5-FU. For all cell lines, including MDA-MB-231, HepG2, and Caco-2, their IC50 values were in the low micromolar range, compared with 5-FU (IC50 = 7.4 μM). Molecular docking confirmed selective binding to the MDM2 pocket, stabilizing p53. This dual-target effect, simultaneously inhibiting antiapoptotic Bcl2 and reactivating tumor suppressor p53, positions compound 30b as a multitarget lead scaffold with potential for broad-spectrum anticancer therapy and minimal toxicity to normal cells.53
Mayakrishnan et al. reported the synthesis of new spirooxindole hybrids via a highly efficient and environmentally friendly in situ 1,3-dipolar cycloaddition reaction.88 This synthetic strategy entailed a one-pot reaction of quinoline-pyridine and quinoline-indole-based chalcones (31 and 33) with 5-bromoisatine derivatives in the presence of amino acids thioproline and sarcosine. The method demonstrated exceptional efficacy, producing the desired spirooxindole derivatives (32 and 34), respectively, with remarkable yields (88–90%), as illustrated in Scheme 10.
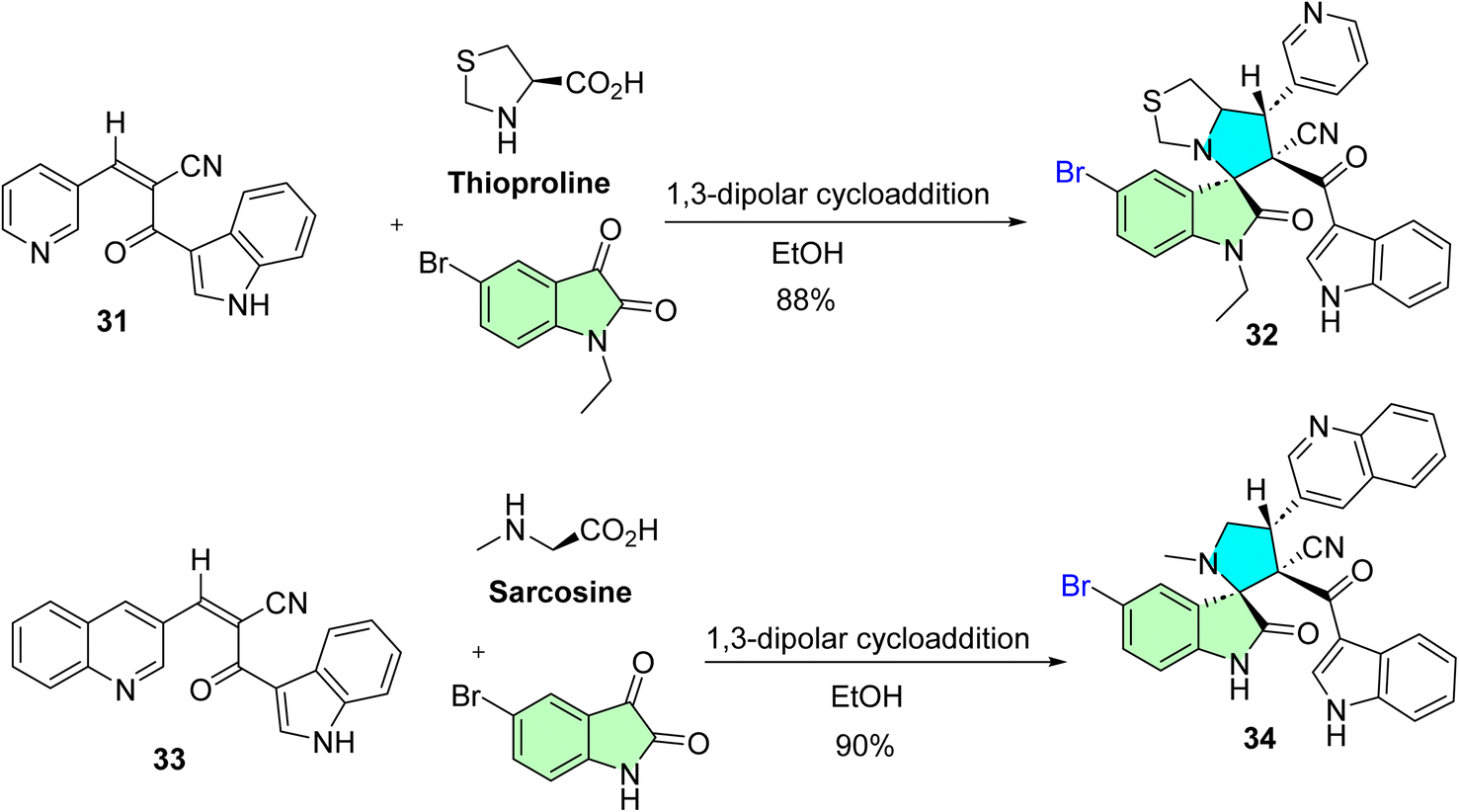 | ||
| Scheme 10 Synthesis of spirroxindole derivatives 32 and 34. | ||
(3S,6′R,7′R)-5-Bromo-1-ethyl-6′-(1H-indole-3-carbonyl)-2-oxo-7′-(pyridin-3-yl)-1′,6′,7′,7a′-tetrahydro-3′H-spiro[indoline-3,5′-pyrrolo[1,2-c]thiazole]-6′-carbonitrile (32), and (3S,3′R,4′R)-5-bromo-3′-(1H-indole-3-carbonyl)-1′-methyl-2-oxo-4′-(quinolin-3-yl)spiro[indoline-3,2′-pyrrolidine]-3′-carbonitrile (34): molecular docking results: 32 and 34 display excellent efficiency in inhibiting the Bcl-2 receptor. Among all the screened compounds, 32 and 34 displayed substantial cytotoxic activity against HepG-2 cells at less than 10 μg mL−1, with IC50 values of 9.0 and 8.0 μg mL−1, respectively. Molecular docking studies with the Bcl-2 receptor revealed that a higher binding energy was observed for 32 and 34, with a value of 6.56 and 8.41 kcal mol−1, respectively.88
Rajaraman et al. synthesized a new series of spirooxindole derivatives and evaluated their antioxidant activity against hydroxyl and superoxide radicals.89 The synthetic strategy involved the in situ generation of an azomethine ylide (AY) via the decarboxylative condensation of isatin 1 and 2-(piperazin-1-yl)ethanamine 2. These reactive intermediates were then undergoing 1,3-dipolar cycloaddition reaction with various chalcones, leading to the formation of the target spirooxindole derivatives (37a–g), as depicted in Scheme 11.89
 | ||
| Scheme 11 Synthesis of spirooxindole derivatives (37a–g). | ||
Both compounds 37d and 37f were identified as promising candidates for their anticancer efficacy against KB oral cancer cells. As indicated, for compound 37f, an excellent cytotoxic potency was obtained (IC50 = 9.5 μM), whereas compound 37d exerted only moderate inhibitory activity (IC50 = 32.5 μM). Despite the slightly elevated IC50 value for 37d, both compounds represent the overall potential of the spirooxindole scaffold to target epithelial-originated cancers. Notably, the structural diversity afforded by the 1,3-dipolar cycloaddition approach resulted in the selectivity of the tested compounds, which preserved low toxic effects on non-cancer cells even at these concentrations. The data indicate that 37f, which possesses sub-micromolar potency for the mechanism of action, has great potential for further structural and preclinical development as a highly selective anticancer drug.89
De Azevedo et al. successfully synthesized a novel series of 25 spirooxindole hybrids integrating phenylamino-pyrimidine-pyridine (PAPP) and isatin scaffolds, designed as potential anti-myeloproliferative agents against K562 cells.90 The synthetic pathway involved the condensation of PAPP with various isatin derivatives (38a–e) to form the corresponding Schiff base intermediates (38a–e), which were subsequently subjected to a 1,3-dipolar cycloaddition reaction with chloro-oxime (39). This approach yielded the target spirooxindole derivatives (40a–e) in good yields (64–86%), as illustrated in Scheme 12.
 | ||
| Scheme 12 Synthesis of spirooxindole derivatives (40a–c). | ||
5-Fluoro-4′-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)methyl)phenyl)-3′-(p-tolyl)-4′H-spiro[indoline-3,5′-[1,2,4]oxadiazol]-2-one (40a), 5-fluoro-3′-(4-fluorophenyl)-4′-(4-methyl-3-((4-(pyridin-3-yl)pyrimidin-2-yl)methyl)phenyl)-4′H-spiro[indoline-3,5′-[1,2,4]oxadiazol]-2-one (40b), and 5-bromo-4′-(4-fluorophenyl)-3′-(4-methyl-3-{[4-(pyridin-3-yl) pyrimidin-2-yl]amino}phenyl)-1,2-dihydro-3′H-spiro[indole-3,2′- [1,3,5]oxadiazol]-2-one (40e): spirooxindole derivatives with a phenylamino-pyrimidine-pyridine (PAPP) skeleton were tested for in vitro anti-leukemic activity in the chronic myelogenous leukemia line K562. Thirty-six compounds (40a–e) elicited significant antiproliferative activities against the EJ, LCFF, and SiHa cell lines, with the most potent CC50 value of 0.8 μM for compound 40c, closely followed by compound 40a (CC50 = 9.6 μM) and compound 40b (CC50 = 9.8 μM). These compounds also exhibited reasonable selectivity indices, indicating that they preferentially killed cancer cells compared to normal cells. Incorporating pyrimidine-pyridine moieties appears to have enhanced kinase-related inhibition, which aligns with the reported mechanisms of leukemia therapeutics. The sub-micromolar to low micromolar IC50 activity of 40c primarily supports the suitability of this scaffold for development toward targeted anti-myeloproliferative agents.90
Alshahrani et al. synthesized a novel series of spirooxindole derivatives incorporating a benzimidazole moiety.91 These compounds were regio- and stereoselectively obtained via a multicomponent one-pot [3 + 2] cycloaddition (32CA) reaction. The process involved the formation of an azomethine ylide intermediate through the reaction of 5-chloroisatin with (2S)-octahydro-1H-indole-2-carboxylic acid 42, followed by subsequent coupling with various chalcones 41a–o featuring the benzimidazole framework. This strategy yielded the targeted spirooxindoles 43a–o as cycloadducts with four asymmetric centers, as depicted in Scheme 13.
 | ||
| Scheme 13 Synthesis of spirooxindole derivatives (43a–o). | ||
The anticancer reactivity for the tested compounds showed IC50 (μM) in the range between 3.80–6.88 μM, and compound (43d) with IC50 = 3.80 ± 0.21 μM was the most active candidate between the series.91
Islam et al. developed a novel series of spirooxindole derivatives featuring a spiro[3H-indole-3,2′-pyrrolidin]-2(1H)-one framework conjugated to a pyrazole moiety, aiming to explore their potential as MDM2 inhibitors.14 The synthesis strategy involved the in situ formation of an azomethine ylide through the reaction of isatin with L-thioproline, followed by a one-pot multicomponent [3 + 2] cycloaddition reaction with various chalcones (44a–o). This approach efficiently yielded the desired spirooxindole derivatives (45a–o), as illustrated in Scheme 14.14
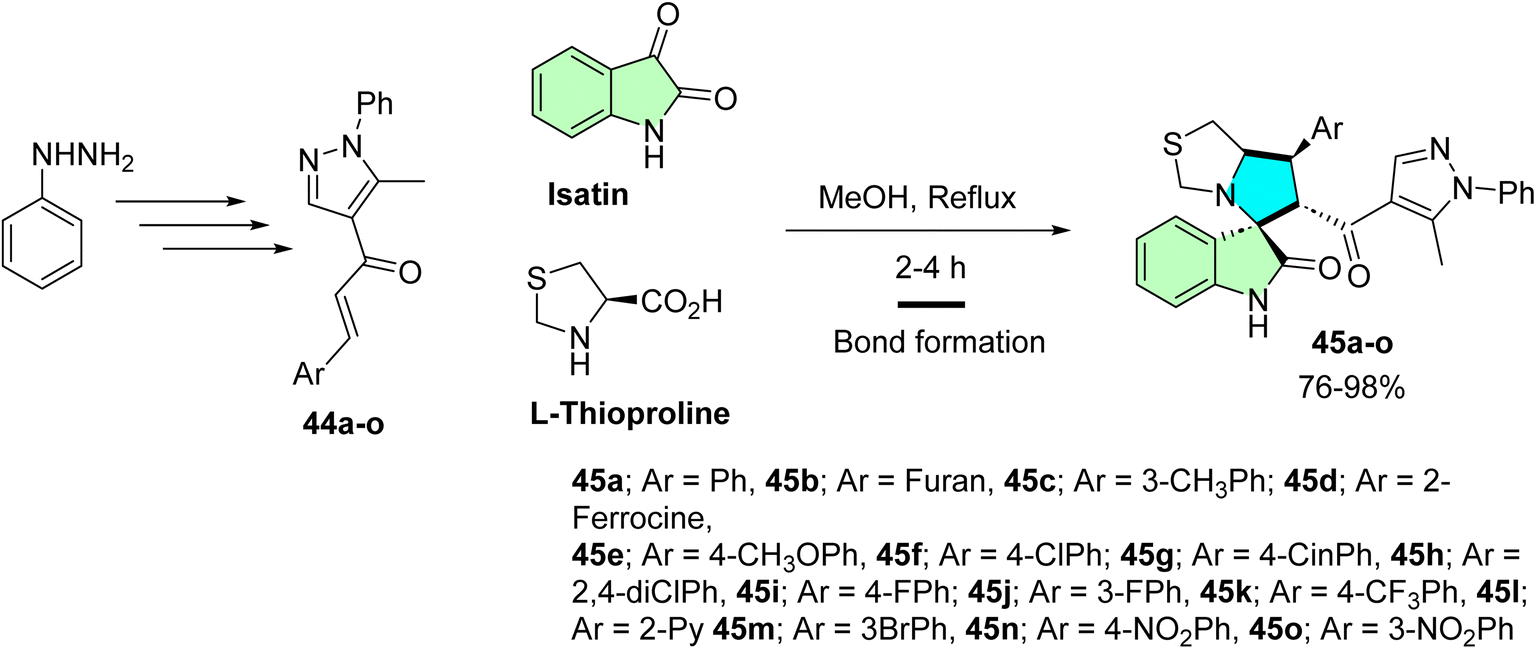 | ||
| Scheme 14 Synthesis of spirooxindole derivatives (45a–o). | ||
A new series of spirooxindoles (45a–o) was prepared to investigate their anti-MDM2 properties and in vitro antitumorigenic activity on various cell lines. Some of the compounds tested, such as 45h, were also found to have potent cytotoxic activity against A2780 ovarian cancer cells (IC50 = 10.30 ± 1.10 μM), and 45m was very active against A549 lung cancer cells (IC50 = 17.70 ± 2.70 μM). Compound 45k was also active against MDA-MB-453 breast cancer cells with an IC50 value of 21.40 ± 1.30 μM. These compounds showed improved p53-induced apoptotic activity due to their structural complementarity of the p53–MDM2 interaction pocket. The IC50 values were only moderate, in the micromolar range, and the selectivity against cancer cells, along with the ability to restore p53 tumor suppression, indicate this group's potential, especially in multi-target cancer treatment approaches.14
Korotaev et al. successfully employed a regio- and stereoselective approach to synthesize novel spiro[chromeno[3,4-c]pyrrolidine-1,3′-oxindoles.85 The optimized reaction conditions involved a three-component, one-pot reaction of a chromene derivative (9a–g), isatin derivatives (11a–f), and a benzylamine derivative (12) in the presence of MgSO4, using CH2Cl2 as the solvent at 40 °C for 24 hours. This method yielded the desired spirooxindole derivatives (14a–l) with a maximum chemical yield of 79%, as depicted in Scheme 15.
 | ||
| Scheme 15 Synthesis of spirooxindole derivatives 48a–l. | ||
The N-unsubstituted 3-aryl-4-(trifluoromethyl)-4H-spiro[chromeno[3,4-c]pyrrolidine-1, 3′-oxindoles] prepared showed extremely potent anticancer activities, especially against HeLa cervical cancer cells. Compounds 48d, 48j, and 48l were highly potent, and the IC50s were lower than that of the reference drug camptothecin, with 48l emerging as the most potent (IC50 = 0.71 ± 0.05 μM). The derivatives exerted their effect predominantly by inhibiting cancer cell proliferation and exhibited a high degree of selectivity, with little or no toxicity to normal human dermal fibroblasts (HDF). The primary molecular targets are atherosclerosis and inflammation, while the cell cycle and apoptotic pathways are affected through the use of the trifluoromethyl group. The spiro-oxindole core ensures the required regio- and stereoselectivity, which is important for bioactivity, demonstrating strong potential for the treatment of cervical and perhaps other cancers.85
Kumar et al. designed and synthesized a novel spirooxindole derivative incorporating a pyrrolidine motif using a one-pot [3 + 2] cycloaddition reaction.92 The synthesis protocol began with N-Alkylation of N-unsubstituted bisarylmethylidene-tetrahydropyridinone hybrids (49a–h) with 2-(chloromethyl)pyridine hydrochloride, affording the desired chalcones (50a–h) in high yields (85–90%). In the next steps, (5a–h) underwent a one-pot cycloaddition reaction with an azomethine ylide, generated in situ via the decarboxylative condensation of isatin with phenylglycine. This approach efficiently yielded the target novel spirooxindole derivatives (51a–h), as illustrated in Scheme 16.
 | ||
| Scheme 16 Synthesis of spirooxindole derivatives (8a–h). | ||
A new family of spirooxindole derivatives featuring a pyrrolidine nucleus has been prepared and tested for their time-dependent antiproliferative potential. Of these, compound 51f showed the most potent activity at 24 h, with an IC50 of approximately 43.46 μg mL−1. However, after 48 h on the trend line, 51c turned out to be the most effective of the active compounds, 51g, with an IC50 of around 12.79 μg mL−1, and time exposure showed a time-dependent increase in cytotoxic activity. While these IC50 values are higher than other low-micromolar hits, their potency increases over time, reflecting a possible cumulative or delayed mode of action. These results suggest that continued pharmacokinetic and structural optimization will be necessary to achieve sufficient potency and selectivity for effective anticancer use.92
Nafie et al. designed and synthesized a novel series of pyrrolidinyl-bis-spirooxindole-based rhodanine hybrids as potential anti-breast cancer agents.8 Following the reported method, the synthesis strategy involved utilizing previously prepared chalcones 52a–f, which incorporated a rhodanine moiety.93 These chalcones were then subjected to a one-pot [3 + 2] cycloaddition reaction with isatin derivatives 53a–e and thioproline, leading to the formation of the target bis-spirooxindole derivatives 54a–o, as illustrated in Scheme 17.
 | ||
| Scheme 17 Synthesis of bi-spirooxindole derivatives 54a–o. | ||
2-((3S,6′S,7′S,7a′S)-5-Bromo-7′-(4-chlorophenyl)-2,2′′,4′′-trioxo-7′,7a′- dihydro-1′H,3′H-dispiro [indoline-3,5′- pyrrolo [1,2-c]thiazole-6′,5′′-thiazolidin]-3′′-yl)acetic acid 54g, methyl (2-((3S,6′S,7′S,7a′S)-7′-(4-bromophenyl)-6-chloro-2,2′′,4′′-trioxo7′,7a′-dihydro-1′H,3′H-dispiro [indoline3,5′-pyrrolo [1,2-c]thiazole-6′,5′′- thiazolidin]-3′′-yl)acetyl)valinate 54l, methyl (2-((3S,6′S,7′S,7a′S)-6-chloro-7′-(4- chlorophenyl)-2,2′′,4′′-trioxo-7′,7a′- dihydro-1′H,3′H-dispiro [indoline-3,5′-pyrrolo [1,2-c]thiazole-6′,5′′-thiazolidin]- 3′′-yl)acetyl)alaninate 54n [8,93]. A new class of bis-spirooxindole derivatives (54a–o) with a pyrrolidinyl-thiazolidine skeleton was developed to provide a structure with improved anticancer activity. Compound 54g displayed the most potent cytotoxicity against MCF-7 and MDA-MB-231 breast cancer cells, with IC50 values of 2.80 μM and 23.50 μM, respectively. Compounds 54l and 54o also demonstrated markedly potent activities with IC50 values in the range of 3.40–4.50 μM toward MCF-7 and 4.30–8.40 μM toward MDA-MB-231. Compounds 54g, 54l, and 54n were especially selective with IC50 of >39 μM in WISH normal cells. Moreover, compounds 54l and 54n (with the validate and alaninate moieties, respectively) also showed good activity toward both MDA220-231 and HeLa cancer cell lines. These compounds also demonstrated potent dual EGFR and CDK-2 inhibition, and 54g significantly induced apoptosis in MCF-7 cells, confirming their promise as targeted anticancer agents.8
Shawish et al. synthesized triazole-spirooxindole derivatives and evaluated their anticancer potential against HepG2 and MDA-MB-231 cell lines.94 The initial synthetic approach aimed to construct chalcones featuring a triazole-s-triazine framework (57a–d)’; however, the triazine moiety was unexpectedly cleaved during the reaction, resulting in chalcones conjugated solely to the triazole motif as shown in Scheme 18. These chalcones 58a–d subsequently underwent a [3 + 2] reaction with an azomethine ylide (AY), generated in situ via the decarboxylative condensation of isatin derivatives 59a–c with Thioproline. This multi-component reaction efficiently produced nine spirooxindole derivatives (60a–i), as illustrated in Scheme 18.
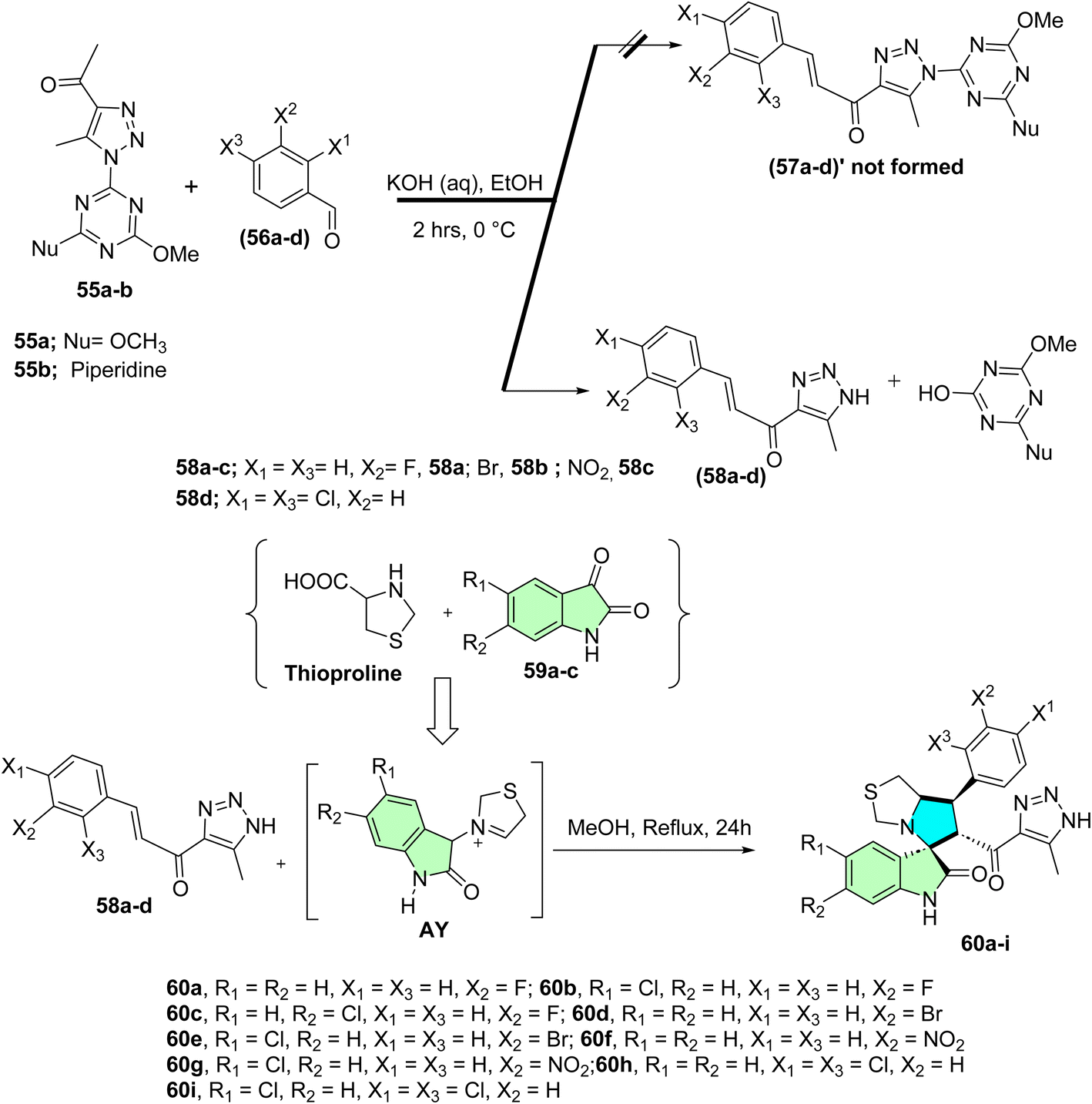 | ||
| Scheme 18 Synthesis of spirooxindole derivatives (60a–i). | ||
In this series of the prepared compound 60h, which contains a 2,4-dichlorophenyl substituent, is the most potent cytotoxic agent against MDA-MB-231 and HepG2 cell lines with IC50 values of 16.80 ± 0.37 and 17.00 ± 0.13 nM, respectively. Moreover, compound 60i showed significant activity with IC50 18.5 ± 0.74 μM against MDA-MB-231 and 13.50 ± 0.92 μM against HepG2. Molecular docking studies of 60h confirmed good binding interactions in the EGFR active site, indicating a potential for dual inhibition. Even though these analogs are weaker than sorafenib (IC50 = 2.60 μM for HepG2), they show significant selectivity and structural flexibility, from which their development as EGFR inhibitors in anticancer therapy can be pursued.94
A novel series of spirooxindole derivatives incorporating pyrrolothiazolidine and an additional structural framework16 was developed by Zhao et al. as dual MDM2/HDAC inhibitors for cancer therapy. The synthesis began with generating azomethine ylides via the condensation of secondary amino acids, such as thioproline, sarcosine, proline, and phenyl glycine, with substituted isatin derivatives. These intermediates were then subjected to a [3 + 2] cycloaddition reaction with various ethylene derivatives, yielding the target spirooxindole derivatives, as illustrated in Scheme 19. The nitro group on the benzene ring was subsequently reduced to an amine, which was then coupled with acid-linker derivatives representing the HDAC pharmacophore using peptide coupling reagents like HATU/DIEPA. The final step involved hydrolysis to produce the desired compounds, among which compound 65 emerged as the most active. Compound 65 exhibited significant inhibitory activity against MDM2 and HDAC, with 68% and 79% inhibition rates, respectively. Additionally, it demonstrated potent antiproliferative effects against MCF-7 cells, outperforming the reference drugs SAHA and Nutlin-3 in terms of efficacy.16
 | ||
| Scheme 19 Synthesis of spirooxindoles 65. | ||
The spirooxindole-based dual inhibitors were among the most potent compounds of the synthesized series; compound 65 was more potent (IC50 = 1.37 ± 0.45 μM) than hydroxamate (3.13 ± 0.78 μM) and Nutlin-3 (9.75 ± 2.97 μM) against MCF-7 breast cancer cells. It also exhibited strong enzyme inhibition activities, with 68% MDM2 and 79% HDAC inhibition. Compound 65 displayed selective inhibition against HDAC1 and HDAC2 (IC50 = 0.058 and 0.064 μM, respectively), and molecular docking studies confirmed strong binding to MDM2 and HDAC1. In addition, the expression of p53 and the acetylation of histone H4 were strongly induced by compound 65, leading to apoptosis in MCF-7 cells. These results suggest that compound 65 may be regarded as a promising anti-cancer agent for the simultaneous inhibition of HDAC and MDM2.16
El-Kalyoubi et al. developed a novel series of spirooxindole derivatives incorporating pyrimidines and in situ selenium nanoparticles (SeNPs) for certain derivatives Scheme 20,60 designed as dual Topo I/II inhibitors for cancer therapy. The synthesis commenced with the cyclo condensation of cyclic enaminones 66a,b with 6-chloroisatin, yielding polyheterocyclic spirocompounds 67a,b, as outlined in Scheme 20. The research team aimed to synthesize new SeNPs 68a NPs and 68b NPs, which exhibited suitable low reducing but high stabilizing properties during SeNPs formation. These organic heterocycle derivatives facilitated the reduction of Se+ cations to Se0 using ascorbic acid as a catalyst, which acted as an aldehyde equivalent to generate and stabilize the nanostructure of SeNPs. The designed compounds were also engineered to function as DNA intercalators.60
 | ||
| Scheme 20 Synthesis of spirooxindoles 68. | ||
Thirteen spirooxindole-based compounds, particularly the nanoformulated hybrids 68a NPs and 68b NPs, were evaluated in vitro against a panel of five human cancer cell lines (HepG2, HCT-116, Caco-2, A-549, and MCF-7). Both nanoparticles exhibited potent cytotoxic effects, with IC50 values ranging from 1.9 ± 0.1 μg mL−1 to 18.9 ± 2.4 μg mL−1. Mechanistically, compound 68a NPs and 68b NPs exhibited potent inhibition against topoisomerase I (IC50 = 0.119 μM and 0.042 μM) and topoisomerase II (IC50 = 4.469 μM and 1.172 μM) in both cases. Compound 68b NPs also significantly triggered S-phase cell cycle arrest and apoptosis in A549 lung cancer cells, suggesting them as dual-function anticancer agents that can intercalate with DNA and inhibit enzyme activity. These data highlight the potential therapeutic significance of SeNP and spirooxindole conjugates, as well as their cytotoxicity profile against different cancer cell lines due to the multi-mechanistic modes of action that target DNA topology and cell cycle progression.60
Ashraf et al. designed and synthesized a novel series of spirooxindole derivatives featuring a pyrazole motif (Scheme 21),95 targeting Ecto-5′-Nucleotidase (ecto-5′-NT) inhibitors for cancer therapy.95 The synthesis involved a one-pot, three-component reaction using equimolar amounts of isatin 69a–f, malononitrile, and substituted 3-aminopyrazoles 70a,b as shown in Scheme 21. The reaction was conducted at 100 °C on a sand bath in a reaction flask, yielding twelve spiroindoline-pyrazolo[3,4-b]pyridine derivatives 71a–l. These compounds were evaluated for their inhibitory activity against purified recombinant human and rat ecto-5′-NT isozymes. Among the synthesized derivatives, compound 71f (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) F3CO) emerged as a potent inhibitor of human ecto-5′-NT, exhibiting an IC50 value of 0.15 ± 0.02 μM. This compound demonstrated a remarkable 280-fold greater inhibitory potency compared to the reference standard, sulfamic acid, highlighting its potential as a promising candidate for further development in cancer therapy.95
F3CO) emerged as a potent inhibitor of human ecto-5′-NT, exhibiting an IC50 value of 0.15 ± 0.02 μM. This compound demonstrated a remarkable 280-fold greater inhibitory potency compared to the reference standard, sulfamic acid, highlighting its potential as a promising candidate for further development in cancer therapy.95
 | ||
| Scheme 21 Synthesis of spirooxindoles 71. | ||
An in vitro biological study of these compounds was the inhibition of the recombinant human and rat ecto-5′-NT isozymes, which have recently emerged as targets for cancer immunotherapy. Compound 71f, which has a trifluoromethoxy (F3CO) group, seemed to be the most potent inhibitor within the series with an IC50 value of 0.15 ± 0.02 μM towards human ecto-5′-NT. This in vitro potency represents a 280-fold improvement versus reference sulfamic acid, demonstrating a significant structural advance on the scaffold. The more potent inhibitory activity is ascribed to the F3CO group as an electron-withdrawing substituent, causing better binding at the enzyme's active site. This discovery positions the pyrazole-linked spirooxindoles as promising drug candidates for future anti-cancer research, particularly in approaches that target purinergic signaling in the modulation of tumor immunosuppression.95
Zhou et al. successfully designed and synthesized a novel series of 5′-CF3-substituted 3,2′-pyrrolidinyl spirooxindoles with high yields (up to 89%) and high diastereoselectivities (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) and exceptional diastereoselective 1,3-dipolar [3 + 2] cycloaddition of N-2,2,2-trifluoroethylisatin-based ketimines with chalcones facilitated by DBU.96 These compounds were evaluated for their in vitro anticancer potential. The synthesis pathway was conducted through a Michael/Mannich-type cascade in which azomethine ylides were generated through a DBU-promoted method. The synthesis utilized a one-pot, two-component reaction involving N-2,2,2-trifluoroethylisatin ketimines 72 and various activated alkenes 73 through a [3 + 2] cycloaddition reaction. This cyclization process was carried out smoothly in ethyl acetate (EtOAc) at 28 °C using DBU as a catalyst, resulting in the production of twenty-four 5′-CF3-substituted 3,2′-pyrrolidinyl spirooxindole derivatives 74 as shown in Scheme 22.96
:1 dr) and exceptional diastereoselective 1,3-dipolar [3 + 2] cycloaddition of N-2,2,2-trifluoroethylisatin-based ketimines with chalcones facilitated by DBU.96 These compounds were evaluated for their in vitro anticancer potential. The synthesis pathway was conducted through a Michael/Mannich-type cascade in which azomethine ylides were generated through a DBU-promoted method. The synthesis utilized a one-pot, two-component reaction involving N-2,2,2-trifluoroethylisatin ketimines 72 and various activated alkenes 73 through a [3 + 2] cycloaddition reaction. This cyclization process was carried out smoothly in ethyl acetate (EtOAc) at 28 °C using DBU as a catalyst, resulting in the production of twenty-four 5′-CF3-substituted 3,2′-pyrrolidinyl spirooxindole derivatives 74 as shown in Scheme 22.96
 | ||
| Scheme 22 Synthesis of spirooxindoles 74. | ||
The antitumor activity of the prepared compounds was tested in vitro against human gastric cancer SCG7901 cells through MTT assay. Among selected compounds, 74(b, d, g, h, l, x) demonstrated cytotoxicity with IC50 values ranging from 29.97 to 34.07 μM, which suggested moderate activity. Although not sub-micromolar, the biological data underpin the pharmacological significance of the 5′-CF3 motif and spirooxindole scaffold. These findings confirm synthetic compounds as promising hits that can be used for lead optimization and further exploration of structure–activity relationships. Donor–acceptor optimizations are easily synthesized, and as such, their good scaffold flexibility, trifluoromethyl substitution, and synthetic versatility enhance their applications in drug discovery.96
Filatov et al. successfully designed and synthesized a novel series of bis-aryl spirooxindole-β-lactams through a Staudinger ketene-imine cycloaddition reaction.97 The synthesis utilized N-aryl-2-oxopyrrolidine-3-carboxylic acids 75 as a ketene source to react with isatin derivatives 76, achieving high yields and excellent diastereoselectivity Scheme 23. The resulting dispirooxindoles 77 Scheme 23 were evaluated for their biological activity, demonstrating moderate cytotoxicity in MTT assays against A549, MCF7, HEK293, and VA13 cell lines.97
 | ||
| Scheme 23 Synthesis of spirooxindoles 77. | ||
The cytotoxicity screening of compounds 77a–f revealed a structure-dependent inhibition of cancer (MCF7, A549) as well as normal cell (VA13, HEK293t) lines. Both compounds 77e and 77f showed the highest activity on all tested cancer cell lines with IC50 values below 8 μM and were, at the same time, much more active than former mono-spiro analogs. This also suggests that they have higher activity due to the right choice of aryl substitution and the increase in log P values, which affects membrane permeability and target affinity. Moreover, compound 77f exhibited selective antibacterial potency against the LPTD strain of E. coli (MIC = 1.3 μM), likely due to perturbation of outer membrane permeability. These results indicate that dispirooxindole-β-lactams do not lose and, in several situations, improve the therapeutic profile of the spirooxindole core, and their development as anticancer and antimicrobial leads is warranted.97
Ren et al. successfully designed and synthesized a new series of spirooxindole–indenoquinoxaline derivatives 78 Scheme 24, which act as inhibitors of tryptophanyl-tRNA synthetase (TrpRS).98 The synthesis involved a 1,3-dipolar cycloaddition reaction of azomethine ylides, generated in situ from isatin and amino acids such as proline, phenyl glycine, and sarcosine. These compounds were evaluated for their biochemical TrpRS inhibitory activity through in vitro experiments, testing against various Gram-positive and Gram-negative bacterial strains, as well as diffuse large B-cell lymphoma (DLBCL) cell lines.98
 | ||
| Scheme 24 Synthesis of spirooxindoles 78. | ||
Compound 78e was identified as the most active inhibitor of hmTrpRS and EoffTrpRS (IC50 = 225 nM and 74 nM, respectively). It also demonstrated excellent antibacterial activity against Staphylococcus aureus (MIC90 = 4 μg mL−1). Compound 78e exhibited significant anti-proliferative activities of 2.97–4.85 μM toward DLBCL cell lines in cancer models while remarkably inducing bacterial autolysis in MRSA strains, suggesting it may be a tool compound for the discovery of dual antibacterial and anticancer agents, and structure–activity relationship (SAR) should be improved for the development of potential therapeutic agents.98
Fawazy et al. successfully designed and synthesized a novel series of 1′′-(alkylsulfonyl)-dispiro[indoline-3,2′-pyrrolidine-3′,3′′-piperidine]-2,4′′-dione derivatives 79, which exhibit anti-SARS-CoV-2 properties.99 The synthesis was achieved through a 1,3-dipolar cycloaddition reaction involving azomethine ylides derived from 1-(alkylsulfonyl)-3,5-bis(ylidene)-piperidin-4-ones, generated in situ from isatin and amino acids such as proline, phenyl glycine, and sarcosine Scheme 25. These compounds were screened for their activity against several human cancer cell lines, including MCF7, HCT116, A431, and PaCa2, while demonstrating a favorable selectivity index toward normal RPE1 cells.99
 | ||
| Scheme 25 Synthesis of spirooxindoles 79. | ||
The dispirooxindole derivatives 79a–m were evaluated for anticancer, antiviral, and cholinesterase inhibitory activities. Compound 79m displayed the most potent anti-proliferative impact, equivalent to 5-fluorouracil and sunitinib for the MCF7, HCT116, A431, and PaCa2 cell lines. Moreover, apoptosis and necrosis in the case of compound 79l with IC50 values of 3.60 μM (MCF-7), 3.24 μM (HCT116), and 2.43 μM (A431) and 79m with IC50 = 8.83 μM against PaCa-2 cells were mediated by EGFR and VEGFR-2 inhibition. In anti-SARS-CoV-2 activity, 79k was also the most potent, with 3.3-fold and 4.8-fold superiority to chloroquine and hydroxychloroquine, respectively. In addition, a handful of the analogs exhibited significant dual activity against AChE and BChE. These results indicate that dispirooxindoles 79 can be regarded as highly promising multi-target therapeutics for cancer, viruses, and neurodegenerative diseases.99
Budovská et al. report an alternative synthetic approach for producing the anticancer molecule (S)-(−)-spirobrassinin and its 2′-amino analogs.100 The synthesis involves CrO3-mediated oxidative cyclization of chiral derivatives of brassinin, followed by removal of the chiral auxiliary, achieving high enantiomeric purity (93%, ee). Additionally, the study describes a one-step synthesis of 2′-phenylamino analogs of spirobrassinin and 1-methoxyspirobrassinin from thioureas using CrO3 as depicted in Scheme 26. These compounds 80 were evaluated for antiproliferative activity against six human cancer cell lines, with the CF3-containing analogs showing significant potency, particularly against leukemia cells, while exhibiting low toxicity to nonmalignant endothelial cells.
 | ||
| Scheme 26 Synthesis of spirooxindoles 80. | ||
The synthesized spirobrassinin and its (±)-2′-amino analogs were screened against six human cancer cell lines: Jurkat and CEM (T-cell acute lymphoblastic leukemia), MCF-7 and MDA-MB-231 (breast adenocarcinoma), HeLa (cervical carcinoma), and A-549 (non-small cell lung carcinoma). The parent molecule spirobrassinin (±)-3 showed weak activity (IC50 > 100 μM) across all lines except Jurkat (IC50 = 63.4 μM). Amino analogs (±)-41 to (±)-46 exhibited selective activity primarily toward leukemic cell lines, especially Jurkat and CEM (e.g., (±)-44 had IC50 = 27 μM on Jurkat, 34 μM on CEM). Importantly, 47 (±) and 48 (±) with CF3 and bis-CF3 groups displayed broad-spectrum potency against all cancer cell lines tested, IC50 26.1–35.5 μM. For instance, compound 80 derivatives [(±)-47] were active to IC50 = 26.1 μM (Jurkat), 33.6 μM (MCF-7 & HeLa), and 32.2 μM (A-549) while not toxic towards the normal cells HUVEC (IC50 > 100 μM). These data highlight the significant role of trifluoromethyl substitution in improving anti-cancer potency and selectivity.100
Odeh et al. report the synthesis of new spiro-indoline-pyrazolo[3,4-b]pyridines (81a-f) via a one-pot, three-component reaction of indoline-2,3-diones with 3-oxo-3-aryl propanonitriles and 1,3-diphenyl-1H-pyrazol-5-amine as shown in Scheme 27.101 The reactions were performed in aqueous acetic acid using reflux to yield the spiro compounds in good yields. The compounds exhibited spiro-fused heterocyclic scaffolds suitable for application in anticancer drug discovery. The simplicity and efficiency of this synthetic approach provide a general method for structurally diverse frameworks containing oxindole and pyrazolopyridine residues as suitable ligands for protein–protein interactions, such as p53–MDM2, and offer an excellent basis for bioactivity screening.101
 | ||
| Scheme 27 Synthesis of spirooxindoles 81f. | ||
Compound 81f was determined to be the most potent compound, showing 3.05 ± 0.12 μM of IC50 in the blockade of p53–MDM2 interaction, superior to Nutlin-1 (IC50 = 8.21 ± 0.25 μM). western blot analysis showed that 81b, 81e, and 81f up-regulated p53 and p21 protein expressions in leukemia (MOLT-4) and non-small cell lung cancer (HOP92) cell lines. In addition, they were able to cause a high degree of early and late apoptosis of MCF-7 breast cancer cells. Molecular docking analyses revealed that the active compounds could bind favorably in the MDM2 cleft, and their mechanism of action as p53–MDM2 inhibitors could be therapeutically relevant. Screening of the anti-cancer profile of the synthesized spiro-indoline-pyrazolo[3,4-b]pyridines 6a–f and 7a–f showed interesting profiles in the NCI-60 human tumor cell lines panel.101
Eichhorst et al. designed and synthesized a spirooxindole-1,3-oxazines, a class of compounds structurally related to the spirooxindole alkaloid family.102 The synthetic route started with dioxyreserpine 82, a naturally occurring spirooxindole alkaloid, as the starting material. Through a sequence of chemical transformations, they generated a diverse set of novel derivatives. These compounds were then evaluated for their anticancer activity against six selected cancer cell lines. Among them, the derivative 10-(4-trifluoromethylphenyl)trioxyreserpine 85 showed the most promising activity. The synthesis pathway of this potent derivative involved an initial bromination of dioxyreserpine 82 at the C-10 position, yielding compound 83. This was followed by an aryl substitution reaction to produce compound 84. Subsequently, selective oxidation of the C–H bond at the C-14 position of (16) under optimized photooxygenation conditions was carried out to get the target spirooxindole 85, as detailed in Scheme 28.102
 | ||
| Scheme 28 Synthesis of spirroxindole 85. | ||
The spiro-oxindoline compound 85, a 10-(4-trifluoromethylphenyl)trioxyreserpine derivative, had strong antitumor activity in 6 human cancer cell lines with selectivity. It demonstrated IC50 in the range of 0.61 ± 0.38 μM (NALM-6), 0.32 ± 0.06 μM (RS4; 11), 1.14 ± 0.20 μM (SU-DHL-4), 1.62 ± 0.13 μM (SUP-T1), 1.90 ± 0.88 μM (MIA-PaCa-2), and 4.21 ± 0.95 μM (Capan-1), being more potent than its parent compound dioxyreserpine. Compound 85 significantly reduced metabolic activity and induced apoptosis (up to 82.6% late apoptosis), as confirmed by Annexin-V/PI staining. It also caused G1 phase arrest and cell fragmentation. While its precise molecular targets remain unspecified, its functional modifications at C-10 and C-14 enhance hydrophobic interactions and oxidative stress pathways, contributing to apoptosis and cell cycle disruption. There is no hemolytic activity, illustrating the selectivity and safety. Its effect comprises decreased cell proliferation, increased cell fragmentation, and detritus formation, indicating potent anti-tumor activity for both hematological and solid tumors.102
A novel series of spirooxindole derivatives was synthesized by Westphal et al. using a green multicomponent domino reaction.103 These derivatives were subsequently investigated in vitro against different cancer cell lines. The microwave-assisted synthesis of the most active derivative in this series 86 involved the reaction of 6-bromo isatin, malononitrile, and barbituric acid in ethanol as the solvent, with 1-methylimidazolium chloride serving as the catalyst, as shown in Scheme 29.103
 | ||
| Scheme 29 Synthesis of spirroxindole 86. | ||
In vitro antiproliferative activities of the spirocycles against four types of human cancer cell lines, including HCT116 (human colon carcinoma), PC3 (prostate carcinoma), HL60 (promyelocytic leukemia), and SNB19 (astrocytoma), were screened by MTT-based assay. It is noteworthy that spiro compound 1c inhibited the four cell lines tested with the lowest IC50 values: 52.81 μM for HCT116, 74.40 μM for PC3, 101.00 μM for SNB19, and 49.72 μM for HL60.103
More recently, Islam et al., designed a novel spirooxindoles to target NSCLC and bacteria. By combining structural features from marine antitumor and antibacterial agents, a new series of pyrazole-linked spirooxindoles was synthesized Scheme 30.104
 | ||
| Scheme 30 Synthesis of spirooxindoles 90a–v to target both NSCLC and bacteria. | ||
The marine natural product-inspired compound 90p showed potent anticancer (towards non-small cell lung carcinoma/NSCLC) and antibacterial (against Staphylococcus aureus) dual effects. The TIC was highly cytotoxic in A549 lung cancer cells with IC50 0.042 μM and a selectivity index (SI) of 58.28, which means that they are highly tumor-specific. It also exhibited antibacterial activity towards S. aureus (MIC = 25.00 μg mL−1). Calf thymus DNA and S. aureus DNA put a low amount of biosensor to the probe by DNA damage test in terbium(III) chloride, while the concentration of the biosensor and the concentration of the results will show the damaging effect. Docking studies indicated intercalative binding within DNA strands. Compound 90p could also inhibit tumor cell proliferation by arresting A549 cells at the G1/S phase of the cell cycle and promoting cell apoptosis by 33.65%, indicating that it might be a dual-purpose drug.104
Table 1 summarizes the biological findings of some recently synthesized halogenated spirooxindoles derivatives as anticancer agents, highlighting the effective molecular target. Halogenated spirooxindoles demonstrate significant anticancer activity through diverse mechanisms, including apoptosis induction, cell cycle arrest, and modulation of key oncogenic signaling pathways. Their structural rigidity and halogen substituents enhance binding affinity to molecular targets, improving therapeutic potential.
| Cpd no. | Structure | Anti-cancer activity | Molecular target | Ref. | |||
|---|---|---|---|---|---|---|---|
| 3a |  |
IC50 [μM] | — | 13 | |||
| Cell line | Treatment | Cisplatin | Doxorubicin | ||||
| A2780 | 8.50 ± 0.70 | 1.90 ± 0.20 | 0.60 ± 0.03 | ||||
| 6J |  |
IC50 [μM] | — | 82 | |||
| Cell line | Treatment | Cisplatin | |||||
| SJSA-1 | 0.82 ± 0.08 | 4.85 ± 0.27 | |||||
| 9 |  |
IC50 [μM] | MDM2 and GPX4 | 83 | |||
| Cell line | Treatment | Nutlin-3 | ML210 | ||||
| MCF-7 | 0.12 | 14.63 | 0.076 | ||||
| 12a |  |
IC50 [μM] | Histone deacetylases enzyme | 84 | |||
| Cell line | Treatment | Doxorubicin | |||||
| HeLa | 10.75 ± 0.39 | 1.73 ± 0.08 | |||||
| 12b |  |
IC50 [μM] | Histone deacetylases enzyme | 84 | |||
| Cell line | Treatment | Doxorubicin | |||||
| HeLa | 12.43 ± 0.77 | 1.73 ± 0.08 | |||||
| 16 |  |
IC50 [nM μM−1] | CDK2 | 54 | |||
| Cell line | Treatment [nM] | Fluorouracil [μM] | |||||
| A549 | 0.05 ± 0.01 | 3.78 ± 0.52 | |||||
| 21a |  |
IC50 [μM] | — | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 1.74 ± 0.45 | 1.66 ± 0.97 | |||||
| 21e |  |
IC50 [μM] | — | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 1.62 ± 0.52 | 1.66 ± 0.97 | |||||
| 21l |  |
IC50 [μM] | — | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 0.71 ± 0.05 | 1.66 ± 0.97 | |||||
| 25b |  |
IC50 [μM] | MDM2 | 86 | |||
| Cell line | Treatment | Doxorubicin | |||||
| PC3 | 3.70 ± 1.00 | 1.90 ± 0.40 | |||||
| 25e |  |
IC50 [μM] | MDM2 | 86 | |||
| Cell line | Treatment | Doxorubicin | |||||
| HeLa | 7.20 ± 0.50 | 0.90 ± 0.14 | |||||
| 25d | 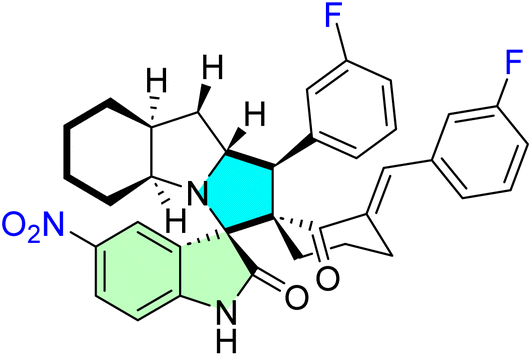 |
IC50 [μM] | MDM2 | 86 | |||
| Cell line | Treatment | Doxorubicin | |||||
| MDA-MB231 | 7.63 ± 0.08 | 0.32 ± 0.00 | |||||
| 28f |  |
IC50 [μM] | — | 87 | |||
| Cell line | Treatment | Doxorubicin | |||||
| PC3 | 8.70 ± 0.70 | 1.90 ± 0.40 | |||||
| 28k | 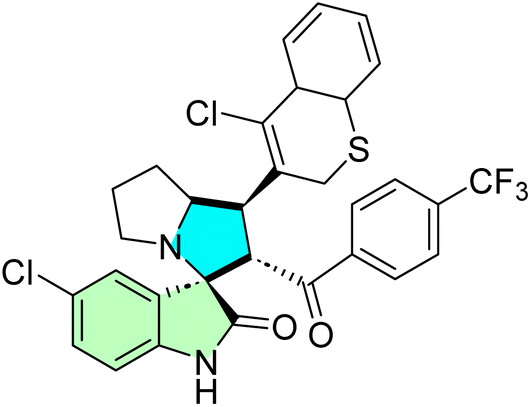 |
IC50 [μM] | — | 87 | |||
| Cell line | Treatment | Doxorubicin | |||||
| HeLa | 8.40 ± 0.50 | 0.90 ± 0.14 | |||||
| 28d |  |
IC50 [μM] | — | 87 | |||
| Cell line | Treatment | Doxorubicin | |||||
| MCF-7 | 7.36 ± 0.37 | 0.79 ± 0.05 | |||||
| MDA-MB231 | 9.44 ± 0.32 | 0.32 ± 0.00 | |||||
| 30b |  |
IC50 [μM] | p53–MDM2 and Bcl2 | 53 | |||
| Cell line | Treatment | 5-Fluorouracil (5-FU) | |||||
| MDA-MB 231 | 0.002 ± 0.00 | 7.05 ± 0.20 | |||||
| HepG-2 | 0.057 ± 0.00 | 4.83 ± 0.30 | |||||
| Caco-2 | 0.003 ± 0.00 | 1.05 ± 0.16 | |||||
| 32 |  |
IC50 [μg mL−1] | Bcl-2 | 88 | |||
| Cell line | Treatment | Cisplatin | |||||
| HepG-2 | 9.00 ± 0.00 | — | |||||
| 34 | 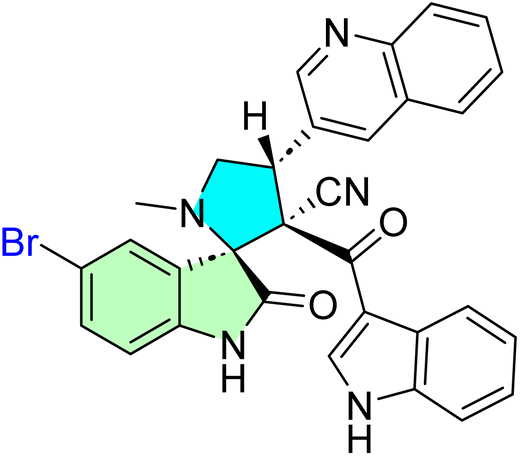 |
IC50 [μg mL−1] | Bcl-2 | 88 | |||
| Cell line | Treatment | Cisplatin | |||||
| HepG-2 | 8.00 ± 0.00 | — | |||||
| 37d |  |
IC50 [μM] | — | 89 | |||
| Cell line | Treatment | — | |||||
| KB | 32.50 ± 0.00 | — | |||||
| 37f |  |
IC50 [μM] | — | 89 | |||
| Cell line | Treatment | — | |||||
| KB | 9.50 ± 0.00 | — | |||||
| 40a |  |
IC50 [μM] | Kinase-related inhibition | 90 | |||
| Cell line | Treatment | Imatinib | |||||
| K562 | 9.60 ± 0.00 | 0.08 ± 0.00 | |||||
| 40b |  |
IC50 [μM] | Kinase-related inhibition | 90 | |||
| Cell line | Treatment | Imatinib | |||||
| K562 | 9.80 ± 0.00 | 0.08 ± 0.00 | |||||
| 40c | 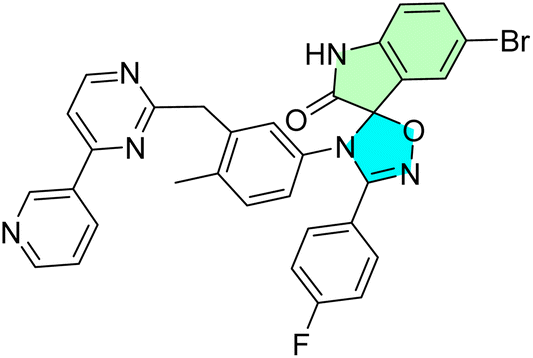 |
IC50 [μM] | Kinase-related inhibition | 90 | |||
| Cell line | Treatment | Imatinib | |||||
| K562 | 0.80 ± 0.00 | 0.08 ± 0.00 | |||||
| 43d |  |
IC50 [μM] | — | 91 | |||
| Cell line | Treatment | — | |||||
| MDA-MB 231 | 3.80 ± 0.21 | — | |||||
| 45h | 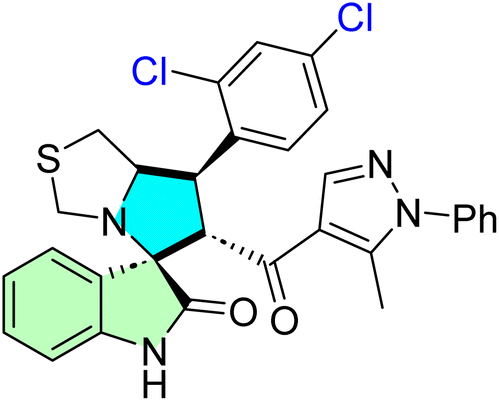 |
IC50 [μM] | p53–MDM2 | 14 | |||
| Cell line | Treatment | Doxorubicin | |||||
| A2780 | 10.30 ± 1.10 | 7.70 ± 3.90 nM | |||||
| HepG2 | 18.60 ± 2.40 | 127.00 ± 18.00 nM | |||||
| 45m |  |
IC50 [μM] | p53–MDM2 | 14 | |||
| Cell line | Treatment | Doxorubicin | |||||
| A549 | 17.70 ± 2.70 | 61.30 ± 15.20 nM | |||||
| 45k |  |
IC50 [μM] | p53–MDM2 | 14 | |||
| Cell line | Treatment | Doxorubicin | |||||
| MDA-MB-453 | 21.40 ± 1.30 | 178.00 ± 26.00 nM | |||||
| 48d |  |
IC50 [μM] | Atherosclerosis and inflammation | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 0.32 ± 0.05 | 1.66 ± 0.97 | |||||
| 48j |  |
IC50 [μM] | Atherosclerosis and inflammation | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 0.47 ± 0.04 | 1.66 ± 0.97 | |||||
| 48l |  |
IC50 [μM] | Atherosclerosis and inflammation | 85 | |||
| Cell line | Treatment | Camptothecin | |||||
| HeLa | 0.71 ± 0.05 | 1.66 ± 0.97 | |||||
| 51f |  |
IC50 [μg mL−1] | — | 92 | |||
| Cell line | Treatment | Melphalan | |||||
| HepG2 | 43.46 ± 2.10 | — | |||||
| 51g |  |
IC50 [μg mL−1] | — | 92 | |||
| Cell line | Treatment | Melphalan | |||||
| HepG2 | 12.79 ± 2.60 | — | |||||
| 54g |  |
IC50 [μM] | EGFR CDK2 | 8 | |||
| Cell line | Treatment | Erlotinib | |||||
| MCF-7 | 2.80 ± 0.40 | 2.14 ± 0.30 | |||||
| MDA-MB-231 | 23.50 ± 0.90 | 3.25 ± 0.50 | |||||
| 54l |  |
IC50 [μM] | EGFR CDK2 | 8 | |||
| Cell line | Treatment | Erlotinib | |||||
| MCF-7 | 3.40 ± 0.50 | 2.14 ± 0.30 | |||||
| MDA-MB-231 | 8.43 ± 0.60 | 3.25 ± 0.50 | |||||
| 54n |  |
IC50 [μM] | EGFR CDK2 | 8 | |||
| Cell line | Treatment | Erlotinib | |||||
| MCF-7 | 3.90 ± 0.30 | 2.14 ± 0.30 | |||||
| MDA-MB-231 | 5.30 ± 0.40 | 3.25 ± 0.50 | |||||
| 60h | 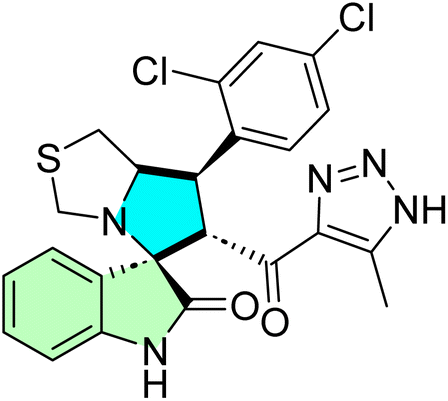 |
IC50 [μM] | EGFR | 94 | |||
| Cell line | Treatment | Sorafenib | |||||
| MDA-MB-231 | 16.80 ± 0.37 | — | |||||
| HepG2 | 17.00 ± 0.13 | 2.60 ± 0.00 | |||||
| 60i | 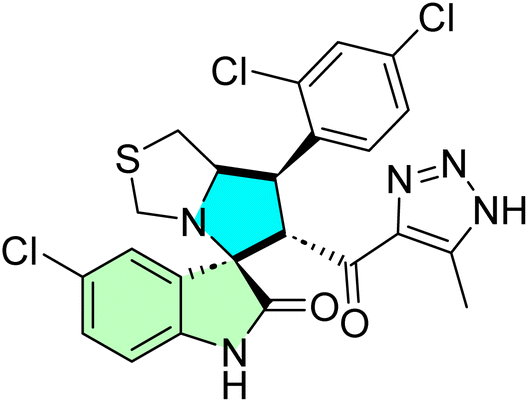 |
IC50 [μM] | EGFR | 94 | |||
| Cell line | Treatment | Sorafenib | |||||
| MDA-MB-231 | 18.50 ± 0.74 | — | |||||
| HepG2 | 13.50 ± 0.92 | 2.60 ± 0.00 | |||||
| 65 |  |
IC50 [μM] | p53 expression and histone H4 acetylation; MDM2, and HDAC | 16 | |||
| Cell line | Treatment | Nutlin-3 | Hydroxamate | ||||
| MCF-7 | 1.37 ± 0.45 | 9.75 ± 2.97 | 3.13 ± 0.78 | ||||
| 68b | 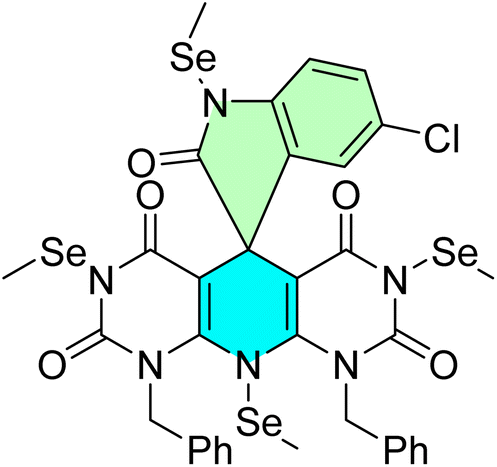 |
IC50 [μg mL−1 and μM] | Topo I Topo II | 60 | |||
| Cell line | Treatment [μg mL−1] | Topo I [μM] | |||||
| HepG2 | 3.76 ± 0.40 | 0.119 ± 0.00 | |||||
| HCT-116 | 5.90 ± 3.40 | 0.042 ± 0.00 | |||||
| Caco-2 | 18.90 ± 2.40 | Topo II [μM] | |||||
| A-549 | 1.90 ± 0.10 | 4.469 ± 0.00 | |||||
| MCF7 | NA | 1.172 ± 0.00 | |||||
| 71f |  |
IC50 [μM] | h-ecto-5′-NT | 95 | |||
| Cell line | Treatment | Sulfamic acid | |||||
| In vitro | 0.15 ± 0.02 | 42.10 ± 7.80 | |||||
| 74h |  |
IC50 [μM] | — | 96 | |||
| Cell line | Treatment | — | |||||
| SCG7901 | 29.97 ± 0.00 | — | |||||
| 77e |  |
IC50 [μM] | — | 97 | |||
| Cell line | Treatment | — | |||||
| Mcf7 | 5.60 ± 0.60 | — | |||||
| HEK293 t | 6.10 ± 0.40 | — | |||||
| 77f | 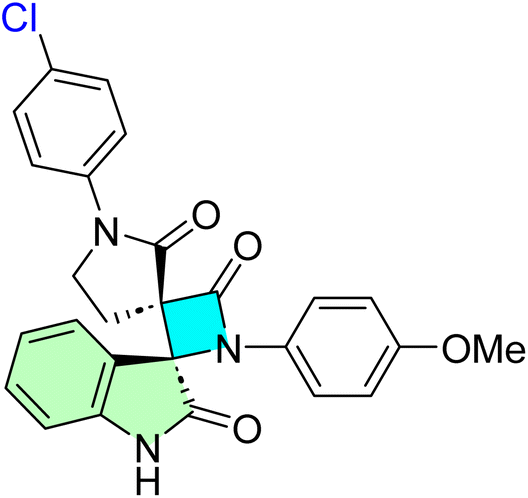 |
IC50 [μM] | — | 97 | |||
| Cell line | Treatment | E. coli MIC [μM] | |||||
| A549 | 6.40 ± 0.40 | 1.30 ± 0.00 | |||||
| 78e |  |
IC50 [μg mL−1 and μM] | hmTrpRS and EoffTrpRS | 98 | |||
| Cell line | Treatment [μM] | S. aureus MIC90 [μg mL−1] | |||||
| OCI-Ly1 | 4.85 ± 0.51 | 4.00 ± 0.00 | |||||
| SU-DHL4 | 2.97 ± 0.25 | — | |||||
| U2932 | 2.44 ± 0.18 | — | |||||
| 79l |  |
IC50 [μM] | — | 99 | |||
| Cell line | Treatment | 5-Fluorouracil | Sunitinib | ||||
| MCF-7 | 3.60 ± 0.19 | 3.15 ± 0.44 | 3.97 ± 0.32 | ||||
| HCT116 | 3.24 ± 0.27 | 20.43 ± 1.99 | 9.67 ± 0.22 | ||||
| A431 | 2.43 ± 0.18 | 23.44 ± 2.09 | — | ||||
| 79m | 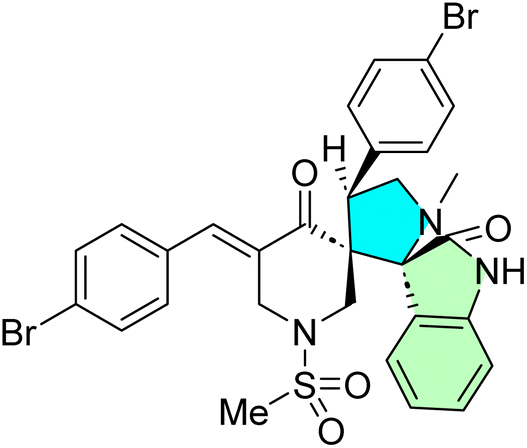 |
IC50 [μM] | EGFR and VEGFR-2 | 99 | |||
| Cell line | Treatment | 5-Fluorouracil | Sunitinib | ||||
| PaCa-2 | 8.83 ± 0.51 | — | 16.91 ± 0.95 | ||||
| 79k |  |
IC50 [μM] | EGFR and VEGFR-2 | 99 | |||
| Cell line | Treatment | 5-Fluorouracil | Sunitinib | ||||
| PaCa-2 | 8.83 ± 0.70 | — | 16.91 ± 0.95 | ||||
| 80 |  |
IC50 [μM] | — | 100 | |||
| Cell line | Treatment | Cisplatin | |||||
| Jurkat | 26.10 ± 0.00 | 12.00 ± 0.00 | |||||
| MCF-7 | 33.60 ± 0.00 | 11.40 ± 0.00 | |||||
| MDA-MB-231 | 35.50 ± 0.00 | 14.70 ± 0.00 | |||||
| HeLa | 33.60 ± 0.00 | 7.70 ± 0.00 | |||||
| CEM | 25.80 ± 0.00 | 4.40 ± 0.00 | |||||
| A-549 | 33.20 ± 0.00 | 12.20 ± 0.00 | |||||
| 81f |  |
IC50 [μM] | p53–MDM2 protein–protein interaction | 101 | |||
| Cell line | Treatment | Nutlin-1 | |||||
| MCF7 | 3.05 ± 0.12 | 8.21 ± 0.25 | |||||
| 85 |  |
IC50 [μM] | G1 phase arrest and cell fragmentation | 102 | |||
| Cell line | Treatment | — | |||||
| SU-DHL-4 | 1.14 ± 0.20 | — | |||||
| SUP−T1 | 1.62 ± 0.13 | — | |||||
| SUP−T1 | 0.61 ± 0.38 | — | |||||
| RS4; 11 | 0.32 ± 0.06 | — | |||||
| MIA-PaCa-2 | 1.90 ± 0.88 | — | |||||
| Capan-1 | 4.21 ± 0.95 | — | |||||
| 86 |  |
IC50 [μM] | — | 103 | |||
| Cell line | Treatment | — | |||||
| HCT116 | 52.81 ± 0.00 | — | |||||
| PC3 | 74.40 ± 0.00 | — | |||||
| SNB19 | 101.00 ± 0.00 | — | |||||
| HL60 | 49.72 ± 0.00 | — | |||||
| 90p |  |
IC50 [μg mL−1 and μM] | G1/S phase cell cycle arrest | 104 | |||
| Cell line | Treatment [μM] | S. aureus MIC [μg mL−1] | |||||
| A549 | 0.042 ± 0.00 | 25.00 ± 0.00 | |||||
Conclusion
Halogenated spirooxindoles exhibit potent anticancer activity by precisely modulating molecular targets critical for cancer cell survival and proliferation. They have demonstrated promising in vitro and in vivo potency and selectivity across various cancer cell lines, with highlighted mechanisms of action. One of the promising molecular targets is protein kinases, which are involved in mechanisms of self-sufficiency in growth signals, evasion of growth suppressors, enabling replicative immortality, angiogenesis, and inducing invasion and metastasis. The synergistic effects of incorporating heterocyclic scaffolds and halogen bonding with the spiro scaffold's rigidity enhance their anticancer activity as effective and selective chemotherapeutics. Moreover, innovative synthetic strategies should be further explored to enhance anticancer efficacy while minimizing toxicity in cancer cells. Hopefully, novel, unique compounds prepared in this direction will be used in subsequent clinical studies to develop collaboration between basic and clinical cancer researchers.Expert opinion
Halogenated spirooxindoles represent a significant advancement in targeted cancer therapy, particularly for p53-dependent cancers and those resistant to treatment. These compounds combine structural ingenuity with mechanistic precision, offering new opportunities for therapeutic development. The findings highlight the potential for medicinal chemists to optimize further halogen placement, scaffold rigidity, and hybrid designs, which could lead to the creation of novel, target-oriented chemotherapeutics. Halogen substitution may increase the ADME pharmacokinetics, binding affinities towards molecular targets, and enhance metabolic stability. Based on these promising results, halogenated spirooxindoles scaffolds are up-and-coming candidates for further development as anticancer agents. Therefore, continued research is essential to investigate the effects of various substituents on the drug-like properties of these scaffolds, aiming to overcome existing challenges and optimize their potential as targeted chemotherapeutics.Data availability
All data associated with this manuscript will be available upon reasonable request from the corresponding authors.Conflicts of interest
The authors declare that they have no financial or personal interests.Acknowledgements
Dr Mohamed S. Nafie appreciates to the Seed Research Project No. (24021440154) funded by the Research and Graduate Studies at the University of Sharjah, United Arab Emirates. Additionally, he acknowledges the electronic library sources at the University of Sharjah, which provide full access to published papers and enables searching on Chemistry databases. Dr Sherif Ashraf Fahmy acknowledges the financial support and sponsorship from the Alexander von Humboldt Foundation, Germany. Dr Ihab Shawish acknowledges Prince Sultan University, for their support. Additionally, the authors acknowledge MUST University for the institutional access to web-based resources.References
- L. Li, T. Shan, D. Zhang and F. Ma, Nowcasting and forecasting global aging and cancer burden: analysis of data from the GLOBOCAN and Global Burden of Disease Study, J. Natl. Cancer Cent., 2024, 4, 223–232, DOI:10.1016/j.jncc.2024.05.002.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries, Ca-Cancer J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- G. R. Bhat, I. Sethi, H. Q. Sadida, B. Rah, R. Mir, N. Algehainy, I. A. Albalawi, T. Masoodi, G. K. Subbaraj, F. Jamal, M. Singh, R. Kumar, M. A. Macha, S. Uddin, A. S. A.-S. Akil, M. Haris and A. A. Bhat, Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance, Cancer Metastasis Rev., 2024, 43, 197–228, DOI:10.1007/s10555-024-10172-z.
- R. Stojchevski, E. A. Sutanto, R. Sutanto, N. Hadzi-Petrushev, M. Mladenov, S. R. Singh, J. K. Sinha, S. Ghosh, B. Yarlagadda, K. K. Singh, P. Verma, S. Sengupta, R. Bhaskar and D. Avtanski, Translational Advances in Oncogene and Tumor-Suppressor Gene Research, Cancers, 2025, 17, 1008, DOI:10.3390/cancers17061008.
- Y.-Q. Liu, Y. Wu, B. Li, X. Tang and C. Chen, Recent advances in the organocatalytic synthesis of chiral C3-spiro-cyclopentaneoxindoles, Org. Biomol. Chem., 2025, 23, 757–773, 10.1039/D4OB01773K.
- P. K. Warghude, A. Bhowmick and R. G. Bhat, Stereoselective synthesis of spirooxindole scaffold, in Spirooxindole, Elsevier, 2024, pp. 283–308, DOI:10.1016/B978-0-443-22324-2.00011-4.
- M. S. Islam, R. M. Al-Jassas, A. M. Al-Majid, M. Haukka, M. S. Nafie, M. M. Abu-Serie, M. Teleb, A. El-Yazbi, A. M. A. Alayyaf, A. Barakat and M. M. Shaaban, Exploiting spirooxindoles for dual DNA targeting/CDK2 inhibition and simultaneous mitigation of oxidative stress towards selective NSCLC therapy; synthesis, evaluation, and molecular modelling studies, RSC Med. Chem., 2024, 15, 2937–2958, 10.1039/D4MD00337C.
- M. S. Nafie, A. M. Al-Majid, M. Ali, A. A. Alayyaf, M. Haukka, S. Ashraf, Z. Ul-Haq, A. El-Faham and A. Barakat, Exploring pyrrolidinyl-spirooxindole natural products as promising platforms for the synthesis of novel spirooxindoles as EGFR/CDK2 inhibitors for halting breast cancer cells, Front. Chem., 2024, 12, 1364378, DOI:10.3389/fchem.2024.1364378.
- M. S. Islam and V. P. Verma, Medicinal applications of spirooxindole and its derivatives, in Spirooxindole, Elsevier, 2024, pp. 537–585, DOI:10.1016/B978-0-443-22324-2.00023-0.
- S. Nasri, M. Bayat and F. Mirzaei, Recent Strategies in the Synthesis of Spiroindole and Spirooxindole Scaffolds, Top. Curr. Chem., 2021, 379, 25, DOI:10.1007/s41061-021-00337-7.
- D. M. Patel, P. J. Patel and H. M. Patel, Catalytic Stereoselective Multicomponent Reactions for the Synthesis of Spiro Derivatives: Recent Progress, Eur. J. Org Chem., 2022, 2022, e202201119, DOI:10.1002/ejoc.202201119.
- N. K. Chouhan, M. N. Talati, M. Sharma and S. Pabbaraja, Anticancer effect of spirooxindole derivatives, in Spirooxindole, Elsevier, 2024, pp. 587–604, DOI:10.1016/B978-0-443-22324-2.00024-2.
- L. R. Raposo, A. Silva, D. Silva, C. Roma-Rodrigues, M. Espadinha, P. V. Baptista, M. M. M. Santos and A. R. Fernandes, Exploiting the antiproliferative potential of spiropyrazoline oxindoles in a human ovarian cancer cell line, Bioorg. Med. Chem., 2021, 30, 115880, DOI:10.1016/j.bmc.2020.115880.
- M. S. Islam, A. M. Al-Majid, E. N. Sholkamy, A. Barakat, M. Viale, P. Menichini, A. Speciale, F. Loiacono, M. Azam, V. P. Verma, S. Yousuf and M. Teleb, Optimized spirooxindole-pyrazole hybrids targeting the p53–MDM2 interplay induce apoptosis and synergize with doxorubicin in A549 cells, Sci. Rep., 2023, 13, 7441, DOI:10.1038/s41598-023-31209-3.
- S. Chahal, J. Sindhu and P. Kumar, Medicinal application of spirooxindole and its derivatives: an introduction, in Spirooxindole, Elsevier, 2024, pp. 369–385, DOI:10.1016/B978-0-443-22324-2.00014-X.
- Q. Zhao, S.-S. Xiong, C. Chen, H.-P. Zhu, X. Xie, C. Peng, G. He and B. Han, Discovery of spirooxindole-derived small-molecule compounds as novel HDAC/MDM2 dual inhibitors and investigation of their anticancer activity, Front. Oncol., 2022, 12, 972372, DOI:10.3389/fonc.2022.972372.
- M. S. Altowyan, S. M. Soliman, M. Haukka, N. H. Al-Shaalan, A. A. Alkharboush and A. Barakat, Synthesis, Characterization, and Cytotoxicity of New Spirooxindoles Engrafted Furan Structural Motif as a Potential Anticancer Agent, ACS Omega, 2022, 7, 35743–35754, DOI:10.1021/acsomega.2c03790.
- I. Shawish, A. M. Al-Majid and A. Barakat, Spirooxindole derivatives as an anticancer agent, in Spirooxindole, Elsevier, 2024, pp. 411–438, DOI:10.1016/B978-0-443-22324-2.00016-3.
- S. Nishal, V. Jhawat, S. Gupta and P. Phaugat, Utilization of kinase inhibitors as novel therapeutic drug targets: areview, Oncol. Res., 2022, 30, 221–230, DOI:10.32604/or.2022.027549.
- A. K. Rout, B. Dehury, S. N. Parida, S. S. Rout, R. Jena, N. Kaushik, N. K. Kaushik, S. K. Pradhan, C. R. Sahoo, A. K. Singh, M. Arya and B. K. Behera, A review on structure-function mechanism and signaling pathway of serine/threonine protein PIM kinases as a therapeutic target, Int. J. Biol. Macromol., 2024, 270, 132030, DOI:10.1016/j.ijbiomac.2024.132030.
- E. V. Koroleva, A. L. Ermolinskaya, Z. V. Ignatovich, Y. V. Kornoushenko, A. V. Panibrat, V. I. Potkin and A. M. Andrianov, Design, in silico Evaluation, and Determination of Antitumor Activity of Potential Inhibitors Against Protein Kinases: Application to BCR-ABL Tyrosine Kinase, Biochemistry, 2024, 89, 1094–1108, DOI:10.1134/S0006297924060099.
- R. Moreddu, Nanotechnology and Cancer Bioelectricity: Bridging the Gap Between Biology and Translational Medicine, Adv. Sci., 2024, 11, 2304110, DOI:10.1002/advs.202304110.
- J. Calder, A. Nagelberg, J. Luu, D. Lu and W. W. Lockwood, Resistance to BET inhibitors in lung adenocarcinoma is mediated by casein kinase phosphorylation of BRD4, Oncogenesis, 2021, 10, 27, DOI:10.1038/s41389-021-00316-z.
- A. Javed, M. Yarmohammadi, K. S. Korkmaz and T. Rubio-Tomás, The Regulation of Cyclins and Cyclin-Dependent Kinases in the Development of Gastric Cancer, Int. J. Mol. Sci., 2023, 24, 2848, DOI:10.3390/ijms24032848.
- W. Wei, M. Valerio, N. Ma, H. Kang, L. X. T. Nguyen, G. Marcucci and N. Vaidehi, Disordered C-Terminus Plays a Critical Role in the Activity of the Small GTPase Ran, Biochemistry, 2025, 64, 1393–1404, DOI:10.1021/acs.biochem.4c00484.
- P. Ghosh, T. Das, A. Chattopadhyay and P. Sahoo, Differential detection of aspartic acid in MCF-7 breast cancer cells, Org. Biomol. Chem., 2023, 21, 7018–7023, 10.1039/D3OB01072D.
- X. Cao, W. Wu, D. Wang, W. Sun and S. Lai, Glycogen synthase kinase GSK3α promotes tumorigenesis by activating HIF1/VEGFA signaling pathway in NSCLC tumor, Cell Commun. Signaling, 2022, 20, 32, DOI:10.1186/s12964-022-00825-3.
- S. Ali, M. Alam and Md. I. Hassan, Kinase inhibitors: an overview, in Protein Kinase Inhib, Elsevier, 2022, pp. 1–22, DOI:10.1016/B978-0-323-91287-7.00026-0.
- J. M. Valverde, G. Dubra, M. Phillips, A. Haider, C. Elena-Real, A. Fournet, E. Alghoul, D. Chahar, N. Andrés-Sanchez, M. Paloni, P. Bernadó, G. Van Mierlo, M. Vermeulen, H. Van Den Toorn, A. J. R. Heck, A. Constantinou, A. Barducci, K. Ghosh, N. Sibille, P. Knipscheer, L. Krasinska, D. Fisher and M. Altelaar, A cyclin-dependent kinase-mediated phosphorylation switch of disordered protein condensation, Nat. Commun., 2023, 14, 6316, DOI:10.1038/s41467-023-42049-0.
- Y. Wang, J. Zhang, C. Zheng, Z. Huang, W. Zhang, Y. Long, G. Gao, Y. Sun, W. W. Xu, B. Li and Q. He, C20orf24 promotes colorectal cancer progression by recruiting Rin1 to activate Rab5-mediated mitogen-activated protein kinase/extracellular signal-regulated kinase signalling, Clin. Transl. Med., 2022, 12, e796, DOI:10.1002/ctm2.796.
- L. Miao, D. Pan, J. Shi, J. Du, P. Chen, J. Gao, Y. Yu, D.-Z. Shi and M. Guo, Role and Mechanism of PKC-δ for Cardiovascular Disease: Current Status and Perspective, Front. Cardiovasc. Med., 2022, 9, 816369, DOI:10.3389/fcvm.2022.816369.
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Trends in kinase drug discovery: targets, indications and inhibitor design, Nat. Rev. Drug Discovery, 2021, 20, 839–861, DOI:10.1038/s41573-021-00252-y.
- X. Li, X. Li, F. Liu, S. Li and D. Shi, Rational Multitargeted Drug Design Strategy from the Perspective of a Medicinal Chemist, J. Med. Chem., 2021, 64, 10581–10605, DOI:10.1021/acs.jmedchem.1c00683.
- D. Bora, A. Kaushal and N. Shankaraiah, Anticancer potential of spirocompounds in medicinal chemistry: a pentennial expedition, Eur. J. Med. Chem., 2021, 215, 113263, DOI:10.1016/j.ejmech.2021.113263.
- P. Saraon, S. Pathmanathan, J. Snider, A. Lyakisheva, V. Wong and I. Stagljar, Receptor tyrosine kinases and cancer: oncogenic mechanisms and therapeutic approaches, Oncogene, 2021, 40, 4079–4093, DOI:10.1038/s41388-021-01841-2.
- S. Waliany, J. J. Lin and J. F. Gainor, Evolution of first versus next-line targeted therapies for metastatic non-small cell lung cancer, Trends Cancer, 2025, 11, 245–257, DOI:10.1016/j.trecan.2025.01.005.
- R. Sharma, L. Yadav, A. A. Nasim, R. K. Yadav, R. H. Chen, N. Kumari, F. Ruiqi, A. Sharon, N. K. Sahu, S. K. Ippagunta, P. Coghi, V. K. W. Wong and S. Chaudhary, Chemo-/Regio-Selective Synthesis of Novel Functionalized Spiro[pyrrolidine-2,3′-oxindoles] under Microwave Irradiation and Their Anticancer Activity, Molecules, 2023, 28, 6503, DOI:10.3390/molecules28186503.
- D. H. Dawood, Therapeutic Insights of Indole Scaffold-Based Compounds as Protein Kinase Inhibitors, ChemistrySelect, 2025, 10, e202404767, DOI:10.1002/slct.202404767.
- N. N. Chahat, Y. Murti, S. Yadav, P. Rawat, S. Dhiman and B. Kumar, Advancements in targeting tumor suppressor genes (p53 and BRCA 1/2) in breast cancer therapy, Mol. Divers., 2024, 1–26, DOI:10.1007/s11030-024-10964-z.
- V. F. Batista, D. C. G. A. Pinto and A. M. S. Silva, Recent in vivo advances of spirocyclic scaffolds for drug discovery, Expert Opin. Drug Discov., 2022, 17, 603–618, DOI:10.1080/17460441.2022.2055544.
- A. S. Girgis, Y. Zhao, A. Nkosi, N. S. M. Ismail, M. S. Bekheit, D. R. Aboshouk, M. N. Aziz, M. A. Youssef and S. S. Panda, The Therapeutic Potential of Spirooxindoles in Cancer: A Focus on p53–MDM2 Modulation, Pharmaceuticals, 2025, 18, 274, DOI:10.3390/ph18020274.
- G. Ranjan, S. Ranjan, P. Sunita and S. P. Pattanayak, Thiazolidinedione derivatives in cancer therapy: exploring novel mechanisms, therapeutic potentials, and future horizons in oncology, Naunyn. Schmiedebergs Arch. Pharmacol., 2024, 398, 4705–4725, DOI:10.1007/s00210-024-03661-z.
- M. K. Kumawat, N. Yadav and K. Kumar, Anticancer properties of spirooxindole derivatives, in Spirooxindole, Elsevier, 2024, pp. 333–367, DOI:10.1016/B978-0-443-22324-2.00013-8.
- M. Panchal, S. Kaur, B. Pawar, T. Gupta, N. Vasdev, M. Tekade and R. K. Tekade, Importance of dose selection in toxicity studies, in Public Health Toxicol. Issues Drug Res., Elsevier, 2024, vol. 2, pp. 87–119, DOI:10.1016/B978-0-443-15842-1.00018-1.
- H. Guo, X. Xu, J. Zhang, Y. Du, X. Yang, Z. He, L. Zhao, T. Liang and L. Guo, The Pivotal Role of Preclinical Animal Models in Anti-Cancer Drug Discovery and Personalized Cancer Therapy Strategies, Pharmaceuticals, 2024, 17, 1048, DOI:10.3390/ph17081048.
- R. M. Al-Jassas, M. S. Islam, A. M. Al-Majid, M. S. Nafie, M. Haukka, A. F. M. M. Rahman, A. M. A. Alayyaf and A. Barakat, Synthesis and SARs study of novel spiro-oxindoles as potent antiproliferative agents with CDK-2 inhibitory activities, Arch. Pharm., 2023, 356, 2300185, DOI:10.1002/ardp.202300185.
- S. Kaur, J. Kaur, B. A. Zarger, N. Islam and N. Mir, Quantitative structure-activity relationship and ADME prediction studies on series of spirooxindoles derivatives for anti-cancer activity against colon cancer cell line HCT-116, Heliyon, 2024, 10, e35897, DOI:10.1016/j.heliyon.2024.e35897.
- S. Sengupta, L. Maji, P. K. Das, G. Teli, M. Nag, N. Khan, M. Haque and G. S. P. Matada, Explanatory review on DDR inhibitors: their biological activity, synthetic route, and structure–activity relationship, Mol. Divers., 2025, 1–31, DOI:10.1007/s11030-024-11091-5.
- R. M. Al-Jassas, M. S. Islam, A. M. Al-Majid, M. Haukka, M. S. Nafie, M. M. Abu-Serie, M. Teleb, M. M. Shaaban, A. M. A. Alayyaf, L. R. Domingo, S. Ashraf, Z. Ul-Haq, Y. M. Abdel Aziz and A. Barakat, Marine-Inspired Spirooxindole PIM-1 Kinase Inhibitors Endowed with Concomitant TRKA/CDK2 Inhibition for Multifaceted Non-Small Cell Lung Cancer Apoptotic Induction, ChemMedChem, 2025, 2500028, DOI:10.1002/cmdc.202500028.
- P. Chunarkar-Patil, M. Kaleem, R. Mishra, S. Ray, A. Ahmad, D. Verma, S. Bhayye, R. Dubey, H. Singh and S. Kumar, Anticancer Drug Discovery Based on Natural Products: From Computational Approaches to Clinical Studies, Biomedicines, 2024, 12, 201, DOI:10.3390/biomedicines12010201.
- BioRender, BioRender.com, Toronto, Canada: BioRender, 2025, Available from: https://biorender.com Search PubMed.
- S. Shangary, K. Ding, S. Qiu, Z. Nikolovska-Coleska, J. A. Bauer, M. Liu, G. Wang, Y. Lu, D. McEachern, D. Bernard, C. R. Bradford, T. E. Carey and S. Wang, Reactivation of p53 by a specific MDM2 antagonist (MI-43) leads to p21-mediated cell cycle arrest and selective cell death in colon cancer, Mol. Cancer Ther., 2008, 7, 1533–1542, DOI:10.1158/1535-7163.MCT-08-0140.
- Y. M. A. Aziz, G. Lotfy, M. M. Said, E. S. H. El Ashry, E. S. H. El Tamany, S. M. Soliman, M. M. Abu-Serie, M. Teleb, S. Yousuf, A. Dömling, L. R. Domingo and A. Barakat, Design, Synthesis, Chemical and Biochemical Insights Into Novel Hybrid Spirooxindole-Based p53–MDM2 Inhibitors With Potential Bcl2 Signaling Attenuation, Front. Chem., 2021, 9, 735236, DOI:10.3389/fchem.2021.735236.
- A. Barakat, S. Alshahrani, A. M. Al-Majid, A. S. Alamary, M. Haukka, M. M. Abu-Serie, L. R. Domingo, S. Ashraf, Z. Ul-Haq, M. S. Nafie and M. Teleb, New spiro-indeno[1,2-b]quinoxalines clubbed with benzimidazole scaffold as CDK2 inhibitors for halting non-small cell lung cancer; stereoselective synthesis, molecular dynamics and structural insights, J. Enzyme Inhib. Med. Chem., 2023, 38, 2281260, DOI:10.1080/14756366.2023.2281260.
- J. Lee and J.-L. Roh, Targeting GPX4 in human cancer: Implications of ferroptosis induction for tackling cancer resilience, Cancer Lett., 2023, 559, 216119, DOI:10.1016/j.canlet.2023.216119.
- Q. Li, Z. Li, T. Luo and H. Shi, Targeting the PI3K/AKT/mTOR and RAF/MEK/ERK pathways for cancer therapy, Mol. Biomed., 2022, 3, 47, DOI:10.1186/s43556-022-00110-2.
- D. Fu, Z. Hu, X. Xu, X. Dai and Z. Liu, Key signal transduction pathways and crosstalk in cancer: Biological and therapeutic opportunities, Transl. Oncol., 2022, 26, 101510, DOI:10.1016/j.tranon.2022.101510.
- S. M. Sagar, D. Yance and R. K. Wong, Natural Health Products That Inhibit Angiogenesis: A Potential Source for Investigational New Agents to Treat Cancer—Part 1, Curr. Oncol., 2006, 13, 14–26, DOI:10.3747/co.v13i1.77.
- J. M. Espejo-Román, B. Rubio-Ruiz, V. Cano-Cortés, O. Cruz-López, S. Gonzalez-Resines, C. Domene, A. Conejo-García and R. M. Sánchez-Martín, Selective Anticancer Therapy Based on a HA-CD44 Interaction Inhibitor Loaded on Polymeric Nanoparticles, Pharmaceutics, 2022, 14, 788, DOI:10.3390/pharmaceutics14040788.
- S. El-Kalyoubi, M. M. Khalifa, M. T. Abo-Elfadl, A. A. El-Sayed, A. Elkamhawy, K. Lee and A. A. Al-Karmalawy, Design and synthesis of new spirooxindole candidates and their selenium nanoparticles as potential dual Topo I/II inhibitors, DNA intercalators, and apoptotic inducers, J. Enzyme Inhib. Med. Chem., 2023, 38, 2242714, DOI:10.1080/14756366.2023.2242714.
- S. S. Taylor, D. R. Knighton, J. Zheng, L. F. Ten Eyck and J. M. Sowadski, Structural framework for the protein kinase family, Annu. Rev. Cell Biol., 1992, 8, 429–462, DOI:10.1146/annurev.cb.08.110192.002241.
- D. R. Knighton, J. H. Zheng, L. F. Ten Eyck, V. A. Ashford, N. H. Xuong, S. S. Taylor and J. M. Sowadski, Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase, Science, 1991, 253, 407–414, DOI:10.1126/science.1862342.
- J. Zheng, E. A. Trafny, D. R. Knighton, N. Xuong, S. S. Taylor, L. F. Ten Eyck and J. M. Sowadski, 2.2 Å refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor, Acta Crystallogr., Sect. D:Biol. Crystallogr., 1993, 49, 362–365, DOI:10.1107/S0907444993000423.
- N. M. Saleh, M. G. El-Gazzar, H. M. Aly and R. A. Othman, Novel Anticancer Fused Pyrazole Derivatives as EGFR and VEGFR-2 Dual TK Inhibitors, Front. Chem., 2020, 7, 917, DOI:10.3389/fchem.2019.00917.
- O. P. J. van Linden, A. J. Kooistra, R. Leurs, I. J. P. de Esch and C. de Graaf, KLIFS: A Knowledge-Based Structural Database To Navigate Kinase–Ligand Interaction Space, J. Med. Chem., 2014, 57, 249–277, DOI:10.1021/jm400378w.
- S. Röhm, A. Krämer and S. Knapp, Function, Structure and Topology of Protein Kinases, in Proteinkinase Inhibitors, ed. S. Laufer, Springer International Publishing, Cham, 2021, pp. 1–24, doi: DOI:10.1007/7355_2020_97.
- D. Fabbro, S. W. Cowan-Jacob and H. Moebitz, Ten things you should know about protein kinases: IUPHAR Review 14, Br. J. Pharmacol., 2015, 172, 2675–2700, DOI:10.1111/bph.13096.
- S. Müller, A. Chaikuad, N. S. Gray and S. Knapp, The ins and outs of selective kinase inhibitor development, Nat. Chem. Biol., 2015, 11, 818–821, DOI:10.1038/nchembio.1938.
- P. Cohen, D. Cross and P. A. Jänne, Kinase drug discovery 20 years after imatinib: progress and future directions, Nat. Rev. Drug Discovery, 2021, 20, 551–569, DOI:10.1038/s41573-021-00195-4.
- X.-J. Liu, H.-C. Zhao, S.-J. Hou, H.-J. Zhang, L. Cheng, S. Yuan, L.-R. Zhang, J. Song, S.-Y. Zhang and S.-W. Chen, Recent development of multi-target VEGFR-2 inhibitors for the cancer therapy, Bioorg. Chem., 2023, 133, 106425, DOI:10.1016/j.bioorg.2023.106425.
- P. J. Chaudhari, A. R. Nemade and A. A. Shirkhedkar, Recent updates on potential of VEGFR-2 small-molecule inhibitors as anticancer agents, RSC Adv., 2024, 14, 33384–33417, 10.1039/D4RA05244G.
- M. J. McGregor, A Pharmacophore Map of Small Molecule Protein Kinase Inhibitors, J. Chem. Inf. Model., 2007, 47, 2374–2382, DOI:10.1021/ci700244t.
- Q.-Q. Xie, H.-Z. Xie, J.-X. Ren, L.-L. Li and S.-Y. Yang, Pharmacophore modeling studies of type I and type II kinase inhibitors of Tie2, J. Mol. Graph. Model., 2009, 27, 751–758, DOI:10.1016/j.jmgm.2008.11.008.
- M. W. Karaman, S. Herrgard, D. K. Treiber, P. Gallant, C. E. Atteridge, B. T. Campbell, K. W. Chan, P. Ciceri, M. I. Davis, P. T. Edeen, R. Faraoni, M. Floyd, J. P. Hunt, D. J. Lockhart, Z. V. Milanov, M. J. Morrison, G. Pallares, H. K. Patel, S. Pritchard, L. M. Wodicka and P. P. Zarrinkar, A quantitative analysis of kinase inhibitor selectivity, Nat. Biotechnol., 2008, 26, 127–132, DOI:10.1038/nbt1358.
- L. T. Alexander, H. Möbitz, P. Drueckes, P. Savitsky, O. Fedorov, J. M. Elkins, C. M. Deane, S. W. Cowan-Jacob and S. Knapp, Type II Inhibitors Targeting CDK2, ACS Chem. Biol., 2015, 10, 2116–2125, DOI:10.1021/acschembio.5b00398.
- Z. Zhao, H. Wu, L. Wang, Y. Liu, S. Knapp, Q. Liu and N. S. Gray, Exploration of Type II Binding Mode: A Privileged Approach for Kinase Inhibitor Focused Drug Discovery?, ACS Chem. Biol., 2014, 9, 1230–1241, DOI:10.1021/cb500129t.
- T. T. Shivani and S. Chaudhary, Spirooxindole derivatives as an anticancer agents: Synthetic developments, structure–activity relationship, and biological applications, in Spirooxindole, Elsevier, 2024, pp. 387–409, https://linkinghub.elsevier.com/retrieve/pii/B9780443223242000151, accessed April 2, 2025 Search PubMed.
- H. A. Carlson, Protein flexibility and drug design: how to hit a moving target, Curr. Opin. Chem. Biol., 2002, 6, 447–452, DOI:10.1016/S1367-5931(02)00341-1.
- G. Caron, V. Digiesi, S. Solaro and G. Ermondi, Flexibility in early drug discovery: focus on the beyond-Rule-of-5 chemical space, Drug Discov. Today, 2020, 25, 621–627, DOI:10.1016/j.drudis.2020.01.012.
- F. Lovering, J. Bikker and C. Humblet, Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success, J. Med. Chem., 2009, 52, 6752–6756, DOI:10.1021/jm901241e.
- J. D. Amaral, D. Silva, C. M. P. Rodrigues, S. Solá and M. M. M. Santos, A Novel Small Molecule p53 Stabilizer for Brain Cell Differentiation, Front. Chem., 2019, 7, 15, DOI:10.3389/fchem.2019.00015.
- B.-W. Pan, Y. Shi, S.-Z. Dong, J.-X. He, B.-S. Mu, W.-B. Wu, Y. Zhou, F. Zhou and J. Zhou, Highly stereoselective synthesis of spirocyclopropylthiooxindoles and biological evaluation, Org. Chem. Front., 2022, 9, 2640–2646, 10.1039/D2QO00300G.
- S.-J. Liu, Q. Zhao, C. Peng, Q. Mao, F. Wu, F.-H. Zhang, Q.-S. Feng, G. He and B. Han, Design, synthesis, and biological evaluation of nitroisoxazole-containing spiro[pyrrolidin-oxindole] derivatives as novel glutathione peroxidase 4/mouse double minute 2 dual inhibitors that inhibit breast adenocarcinoma cell proliferation, Eur. J. Med. Chem., 2021, 217, 113359, DOI:10.1016/j.ejmech.2021.113359.
- M. Kanchrana, B. S. Allaka, G. R. Krishna and S. Basavoju, An ultrasound assisted synthesis of spirooxindolo-1,2,4-oxadiazoles via [3 + 2] cycloaddition reaction and their anti-cancer activity, Arkivoc 2023, 2023, DOI:10.24820/ark.5550190.p011.940.
- V. Yu. Korotaev, S. V. Barkovskii, I. B. Kutyashev, M. V. Ulitko, A. Yu. Barkov, N. S. Zimnitskiy, I. A. Kochnev and V. Ya. Sosnovskikh, Two approaches toward the regio- and stereoselective synthesis of N-unsubstituted 3-aryl-4-(trifluoromethyl)-4H-spiro-[chromeno[3,4-c]pyrrolidine-1,3′-oxindoles], Chem. Heterocycl. Compd., 2021, 57, 679–690, DOI:10.1007/s10593-021-02967-7.
- A. M. Al-Majid, M. Ali, M. S. Islam, S. Alshahrani, A. S. Alamary, S. Yousuf, M. I. Choudhary and A. Barakat, Stereoselective Synthesis of the Di-Spirooxindole Analogs Based Oxindole and Cyclohexanone Moieties as Potential Anticancer Agents, Molecules, 2021, 26, 6305, DOI:10.3390/molecules26206305.
- A. Barakat, M. S. Islam, M. Ali, A. M. Al-Majid, S. Alshahrani, A. S. Alamary, S. Yousuf and M. I. Choudhary, Regio- and Stereoselective Synthesis of a New Series of Spirooxindole Pyrrolidine Grafted Thiochromene Scaffolds as Potential Anticancer Agents, Symmetry, 2021, 13, 1426, DOI:10.3390/sym13081426.
- S. Mayakrishnan, D. Kathirvelan, Y. Arun, K. Saranraj, C. Balachandran, S. Aoki, P. Yuvaraj and N. U. Maheswarai, Design and synthesis of spirooxindole–pyrrolidines embedded with indole and pyridine heterocycles by multicomponent reaction: anticancer and in silico studies, New J. Chem., 2022, 46, 10089–10106, 10.1039/D1NJ05839H.
- D. Rajaraman, L. A. Anthony, G. Sundararajan, M. Shanmugam and A. Arunkumar, Synthesis, NMR, anti-oxidant, anti-cancer activity, Molecular docking, DFT Calculations and in silico ADME analysis of 3′-benzoyl-4′-phenyl-5′-(piperazin-1-ylmethyl)spiro[indoline-3,2′-pyrrolidin]-2-one derivatives, J. Mol. Struct., 2022, 1267, 133551, DOI:10.1016/j.molstruc.2022.133551.
- L. D. De Azevedo, D. I. Leite, A. P. De Oliveira, F. P. S. Junior, R. F. Dantas, M. M. Bastos, N. Boechat and L. C. F. Pimentel, Spirooxadiazoline-oxindoles derived from imatinib show antimyeloproliferative potential in K562 cells, Arch. Pharm., 2024, 357, 2400029, DOI:10.1002/ardp.202400029.
- S. Alshahrani, A. M. Al-Majid, M. Ali, A. S. Alamary, M. M. Abu-Serie, A. Dömling, M. Shafiq, Z. Ul-Haq and A. Barakat, Rational Design, Synthesis, Separation, and Characterization of New Spiroxindoles Combined with Benzimidazole Scaffold as an MDM2 Inhibitor, Separations, 2023, 10, 225, DOI:10.3390/separations10040225.
- R. S. Kumar, D. M. Al-thamili, A. I. Almansour, N. Arumugam and F. Mohammad, A One-Pot Three-Component Synthesis and Investigation of the In Vitro Mechanistic Anticancer Activity of Highly Functionalized Spirooxindole-Pyrrolidine Heterocyclic Hybrids, Molecules, 2020, 25, 5581, DOI:10.3390/molecules25235581.
- A. Barakat, M. Haukka, S. M. Soliman, M. Ali, A. M. Al-Majid, A. El-Faham and L. R. Domingo, Straightforward Regio- and Diastereoselective Synthesis, Molecular Structure, Intermolecular Interactions and Mechanistic Study of Spirooxindole-Engrafted Rhodanine Analogs, Molecules, 2021, 26, 7276, DOI:10.3390/molecules26237276.
- I. Shawish, S. Al Ayoubi, A. El-Faham, A. Aldalbahi, F. F. El-Senduny, F. A. Badria, M. Ríos-Gutiérrez, H. H. Hammud, S. Ashraf, Z. Ul-Haq and A. Barakat, Novel spirooxindole-triazole derivatives: unveiling [3+2] cycloaddition reactivity through molecular electron density theory and investigating their potential cytotoxicity against HepG2 and MDA-MB-231 cell lines, Front. Chem., 2024, 12, 1460384, DOI:10.3389/fchem.2024.1460384.
- A. Ashraf, Z. Shafiq, M. S. Khan Jadoon, M. N. Tahir, J. Pelletier, J. Sevigny, M. Yaqub and J. Iqbal, Synthesis, Characterization, and In Silico Studies of Novel Spirooxindole Derivatives as Ecto-5′-Nucleotidase Inhibitors, ACS Med. Chem. Lett., 2020, 11, 2397–2405, DOI:10.1021/acsmedchemlett.0c00343.
- F.-J. Zhou, B.-L. Zhu, Z.-H. Huang, N. Lin and Z.-W. Zhang, DBU-catalyzed diastereoselective 1,3-dipolar [3+2] cycloaddition of trifluoroethyl amine-derived isatin ketimines with chalcones: synthesis of 5′-CF3 -substituted 3,2′-pyrrolidinyl spirooxindoles, RSC Adv., 2024, 14, 548–551, 10.1039/D3RA08127C.
- V. E. Filatov, D. A. Iuzabchuk, V. A. Tafeenko, Y. K. Grishin, V. A. Roznyatovsky, D. A. Lukianov, Y. A. Fedotova, M. A. Sukonnikov, D. A. Skvortsov, N. V. Zyk and E. K. Beloglazkina, Dispirooxindole-β-Lactams: Synthesis via Staudinger Ketene-Imine Cycloaddition and Biological Evaluation, Int. J. Mol. Sci., 2022, 23, 6666, DOI:10.3390/ijms23126666.
- W. Ren, Q. Zhao, M. Yu, L. Guo, H. Chang, X. Jiang, Y. Luo, W. Huang and G. He, Design and synthesis of novel spirooxindole–indenoquinoxaline derivatives as novel tryptophanyl-tRNA synthetase inhibitors, Mol. Divers., 2020, 24, 1043–1063, DOI:10.1007/s11030-019-10011-2.
- N. G. Fawazy, S. S. Panda, A. Mostafa, B. M. Kariuki, M. S. Bekheit, Y. Moatasim, O. Kutkat, W. Fayad, M. A. El-Manawaty, A. A. F. Soliman, R. A. El-Shiekh, A. M. Srour, R. F. Barghash and A. S. Girgis, Development of spiro-3-indolin-2-one containing compounds of antiproliferative and anti-SARS-CoV-2 properties, Sci. Rep., 2022, 12, 13880, DOI:10.1038/s41598-022-17883-9.
- M. Budovská, V. Tischlerová, J. Mojžiš, O. Kozlov and T. Gondová, An alternative approach to the synthesis of anticancer molecule spirobrassinin and its 2′-amino analogues, Monatsh. Chem., 2020, 151, 63–77, DOI:10.1007/s00706-019-02528-x.
- D. M. Odeh, H. A. Allam, W. R. Mahmoud, M. M. Odeh, H. A. Abdel Aziz and E. R. Mohammed, One-pot three-component synthesis, in vitro anticancer activity and p53-MDM2 protein-protein interaction inhibition of novel spiro-oxindoline-based pyrazolo[3,4-b]pyridines, Egypt, J. Chem., 2024, 67(3), 495–513, DOI:10.21608/ejchem.2023.228742.8415.
- A. Eichhorst, M. Gallhof, A. Voss, A. Sekora, L. Eggers, L. T. Huyen, C. Junghanss, H. Murua Escobar and M. Brasholz, Spirooxindol-1,3-oxazine Alkaloids: Highly Potent and Selective Antitumor Agents Evolved from Iterative Structure Optimization, ChemMedChem, 2022, 17, e202200162, DOI:10.1002/cmdc.202200162.
- R. Westphal, E. Venturini Filho, L. B. Loureiro, C. F. Tormena, C. Pessoa, C. D. J. Guimarães, M. P. Manso, R. G. Fiorot, V. R. Campos, J. A. L. C. Resende, F. Medici and S. J. Greco, Green Synthesis of Spiro Compounds with Potential Anticancer Activity through Knoevenagel/Michael/Cyclization Multicomponent Domino Reactions Organocatalyzed by Ionic Liquid and Microwave-Assisted, Molecules, 2022, 27, 8051, DOI:10.3390/molecules27228051.
- M. S. Islam, A. Barakat, A. M. A. Alayyaf, M. Haukka, V. P. Verma, M. M. Abu-Serie, A. F. El-Yazbi, M. G. Shehat, M. Alseqely and M. Teleb, Synthesis of Marine-Inspired Multifaceted DNA Damaging Spirooxindoles Combating NSCLC and Associated Bacterial Infection, ACS Med. Chem. Lett., 2025, 5c00014, DOI:10.1021/acsmedchemlett.5c00014.
| This journal is © The Royal Society of Chemistry 2025 |