
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vitro and in silico studies of the SARS-CoV-2 main protease inhibition and antioxidant activities of isolated compounds from Marrubium alysson L.†
Eman Ezza,
Ahmed R. Ali *b,
Mohamed Farid Lahlouba and
Amal F. Soliman*ac
*b,
Mohamed Farid Lahlouba and
Amal F. Soliman*ac
aDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: amalsoliman134@mans.edu.eg; amalsoliman134@yahoo.com
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Mansoura University, Mansoura 35516, Egypt. E-mail: ahmed_reda5588@mans.edu.eg; ahmed_reda551988@yahoo.com
cDepartment of Pharmacognosy, Faculty of Pharmacy, Mansoura National University, Gamasa 7731168, Egypt
First published on 22nd July 2025
Abstract
A phytochemical investigation of the aerial parts of Marrubium alysson L. yielded 10 compounds: β-sitosterol (1), glycerol monopalmitate (2), β-sitosterol glucoside (3), chrysoeriol (4), apigenin (5), (apigenin-7-O-(3′′,6′′-E-p-dicoumaroyl)-β-D-glucoside) (6), apigenin-7-O-(3′′-E-p-coumaroyl)-β-D-glucopyranoside (7), apigenin-7-O-(6′′-E-p-coumaroyl)-β-D-glucoside (8), apigenin-7-O-β-glucopyranoside (9) and verbascoside (10). Their structures were established using 1D and 2D NMR spectroscopic techniques. Compounds 1, 2, 6, 7 and 8 are reported from M. alysson L. for the first time in this study. The in vitro inhibitory activities against the SARS-CoV-2 main protease (Mpro) were evaluated using fluorogenic substrate assay. Compound 6 showed the highest inhibitory activity against the SARS-CoV-2 main protease (Mpro) with IC50 = 8.349 ± 0.35 μM, comparing favourably with the reference tipranavir (IC50 = 3.231 ± 0.14 μM). The antioxidant activities were determined using in vitro ABTS radical scavenging assay; it is noteworthy that compounds 6, 7 and 8 have potent antioxidant activity compared with L-ascorbic acid, while compound 10 has radical scavenging activity with IC50 = 25.58 ± 0.12 μM, more potent than the reference L-ascorbic acid (IC50 = 30.43 ± 0.14 μM). Molecular modelling studies of compound 6 showed that it is perfectly engaged in a wide range of hydrogen bonding with multiple residues, including Met49, Glu47, Thr24, Thr26, Gly143, and Gln 189 in the active site of CoV-2-3CL protease. This is reflected by its promising binding affinity, which explains the observed biochemical activity of compound 6 for inhibiting SARS-CoV-2 main protease (Mpro). Assessments of the pharmacokinetics, drug likeness and medicinal chemistry friendliness of the isolated compounds were also conducted.
1. Introduction
The genus Marrubium is a genus of flowering plants in the family Lamiaceae, which contains about 40 species, and is located mainly in the temperate regions of the Eurasian continent and along the Mediterranean Sea.1 Since ancient times, Marrubium species were well known for their medicinal properties in folk medicine, and they were used against issues such as diseases affecting the digestive, respiratory and kidney systems, fevers, skin injuries, and inflammatory processes.2,3 Previous studies indicated that several species of the genus Marrubium afforded various classes of compounds such as labdane diterpenes, flavonoids, phenylpropanoids and essential oils.4–11 Extracts of different Marrubium species showed various biological activities, such as analgesic, antioxidant, anti-inflammatory, cytotoxic, antiplasmodial, antimicrobial, antiulcer, and hypoglycaemic activity.2,12–15 Marrubium alysson L. is an annual herb with round, coarsely crenate, fleece-covered leaves with a cuneate base, pink or bluish corolla, and fruit-calyx with five spreading, star-shaped spines.16 Previous studies reported the presence of various classes of compounds in M. alysson L., mainly phenylpropanoid glycosides, flavonoids and diterpenes.11,17–19 Extracts of M. alysson L. were reported to have anti-inflammatory, antioxidant, analgesic, cytotoxic and hypoglycaemic activity.13,14,20Respiratory syndrome coronavirus 2 (SARS-CoV-2) is a novel strain of the coronavirus family. It was declared a pandemic on March 11, 2020, by the World Health Organisation (WHO) due to its great pathogenicity and rapid global spread. Even though several clinical trials are being conducted worldwide to examine several well-known antiviral treatments, there are presently no antiviral therapies that are widely acknowledged as effective against SARS-CoV-2. To find natural, semi-synthetic, or synthetic COVID-19 therapies, a lot of research has been conducted recently. The SARS-CoV-2 main protease (Mpro), sometimes referred to as the chymotrypsin-like protease 3CLpro, is crucial for the virus's capacity to replicate and complete its life cycle. As a result, Mpro inhibition would thus alleviate COVID-19 symptoms by more successfully hindering viral replication.
Free radicals are extremely reactive chemical species that can seriously harm a person's health or even oxidise food to the point that it is degraded. They play a role in many different clinical diseases, such as cancer, atherosclerosis, arthritis, cardiovascular disorders and Alzheimer's disease.21 Antioxidant defence mechanisms in the body provide resistance against free radicals. As a result, oxidative stress results from an imbalance between the body's antioxidant defence system and the production of reactive oxygen species (ROS).22 There is a growing interest in using naturally occurring antioxidants in place of synthetic antioxidants in food, cosmetics, and pharmaceutical goods as they are safe.23 Flavonoids and phenolic substances have been reported to have potent antioxidant effects.24 The richness of flavonoids and phenolic compounds in Marrubium alysson L. prompted us to investigate its antioxidant activity. A previous study showed the promising antioxidant activity of M. alysson L. aerial part extracts.14
Plant-derived natural compounds with therapeutic significance have recently attracted a lot of attention in an effort to investigate effective natural drugs for a variety of disorders, due to their availability and relative safety. Several Marrubium species, notably M. peregrinum and M. deserti, have been reported to exhibit antiviral activity against a variety of viruses, including coronaviruses. This prompted us to assess the anti-SARS-CoV-2 activity of compounds isolated from Marrubium alysson L. plant cultivated in Egypt. In this study, the phytoconstituents of the aerial parts were isolated by column chromatography using different stationary and mobile phases and identified via various 1D and 2D NMR and mass spectroscopy techniques. The antioxidant activities were measured using ABTS assays and anti-SARS-CoV-2 abilities were evaluated using both in vitro and in silico studies.
2. Materials and methods
2.1. General experimental procedure
Chromatographic separations were performed using silica gel (E-Merck, Germany). TLC was performed on G60F254 silica gel (E-Merck, Germany). Nuclear magnetic resonance spectra (1H NMR, 13C NMR, APT, HMBC and HSQC), using TMS as an internal standard, were obtained on a Bruker Ascend™ 400 spectrometer, operating at 400 MHz for 1H NMR and 100 MHz for APT, and on a Bruker DRX spectrometer 600, operating at 600 MHz for 1H NMR and 150 MHz for 13C NMR. The NMR solvents used were CD3OD, DMSO-d6 and CDCl3. An infrared spectrophotometer (PerkinElmer 1430 ratio recording, USA) was used. ESI-MS was determined using an Advion compact mass spectrometer (USA). The MS instrument was operated using an ESI source in negative ionization mode, with survey scans acquired over an m/z range of 10–1200; the acquisition speed was 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m/z units per sec; the sensitivity (ESI) was as follows: 10 pg of reserpine (FIA – 5 μL injected at 100 μL min−1), 100:1 S/N (RMS) with SIM of m/z 609.3; the stability was 0.1 m/z units at m/z 1200 over a 12-hour operating period at a temperature of 20 ± 1 °C; and the polarity switching speed was 50 ms. GC/MS was carried out on a Thermo Scientific Trace GC Ultra & ISQ Single Quadruple MS with a TG-5MS nonpolar 5% phenyl methylpolysiloxane (length: 30 m, ID: 0.25 mm, film thickness: 0.25 μm) capillary column. The injector temperature was set to 250 °C. Helium was used as the carrier gas with a flow rate of 1 mL min−1. Using a splitless injection technique, 1 μL of sample was injected into diethyl ether. The chromatogram was obtained by keeping the oven temperature at 150 °C for 3 minutes with a gradual rise by 5 °C per minute from 150 °C to 280 °C, before holding it there isothermally for 5 minutes. The electron ionisation (EI) technique at 70 eV was used to conduct mass spectral investigations.
000 m/z units per sec; the sensitivity (ESI) was as follows: 10 pg of reserpine (FIA – 5 μL injected at 100 μL min−1), 100:1 S/N (RMS) with SIM of m/z 609.3; the stability was 0.1 m/z units at m/z 1200 over a 12-hour operating period at a temperature of 20 ± 1 °C; and the polarity switching speed was 50 ms. GC/MS was carried out on a Thermo Scientific Trace GC Ultra & ISQ Single Quadruple MS with a TG-5MS nonpolar 5% phenyl methylpolysiloxane (length: 30 m, ID: 0.25 mm, film thickness: 0.25 μm) capillary column. The injector temperature was set to 250 °C. Helium was used as the carrier gas with a flow rate of 1 mL min−1. Using a splitless injection technique, 1 μL of sample was injected into diethyl ether. The chromatogram was obtained by keeping the oven temperature at 150 °C for 3 minutes with a gradual rise by 5 °C per minute from 150 °C to 280 °C, before holding it there isothermally for 5 minutes. The electron ionisation (EI) technique at 70 eV was used to conduct mass spectral investigations.
2.2. Plant material
The aerial parts of M. alysson L. were collected from the North Coast of the Mediterranean coastal strip of Egypt in April 2019. The plant was authenticated by Prof. Ibrahim Mashaly, Professor of Ecology, Faculty of Science, Mansoura University, Egypt. Voucher specimens of the plant have been kept at Pharmacognosy Department, Faculty of Pharmacy, Mansoura University.2.3. Extraction and isolation of compounds
The air-dried aerial parts of M. alysson L (2.0 kg) were powdered and extracted with aqueous alcohol (70% methanol) and fractionated using different solvents with increasing polarities: petroleum ether followed by methylene chloride then ethyl acetate and, finally, n-butanol. The petroleum ether fraction of the aerial parts was subject to chromatography over a silica gel column. Gradient elution was started using petroleum ether:ethyl acetate (100:0 to 0:100). Collected fractions were monitored by TLC and similar fractions were collected. For fractions (49–65) eluted with petroleum ether-EtOAc (95:5), upon recrystallization from CH2Cl2/MeOH (1:1), white needles were obtained (compound 1; 45 mg). Fractions (180–199) eluted with petroleum ether-EtOAc (70:30) were resubjected to chromatography over a silica gel column using petroleum ether:ethyl acetate (80:20) for isocratic elution. Subfractions (19–25) were further purified over a Sephadex LH-20 column (40 × 0.6 cm) with CH2Cl2–MeOH, precipitating a white amorphous substance (compound 2; 5 mg). For fractions (295–307) eluted with petroleum ether-EtOAc (10:90), after repeated washing with n-hexane and then MeOH, a white amorphous powder was obtained (compound 3; 40 mg). The methylene chloride fraction of the aerial parts was chromatographed over VLC using silica gel. Gradient elution was started using petroleum ether:ethyl acetate (100:0 to 0:100). Collected fractions were monitored by TLC and similar fractions were collected for further investigation. Fractions eluted with petroleum ether:ethyl acetate (50:50 to 20:80) were then resubjected to chromatography over a silica gel column using petroleum ether-methylene chloride (1:1):ethyl acetate (100:0 to 0:100). Collected fractions were monitored by TLC and similar fractions were collected for further investigation. Subfractions (251–281) eluted with petroleum ether-methylene chloride (1:1):ethyl acetate (88:12) were resubjected to chromatography over a silica gel column using petroleum ether:ethyl acetate (25:75), when washed several times with methylene chloride, and a yellowish amorphous substance was obtained (compound 4; 3 mg). Fractions (291–298) eluted with petroleum ether-methylene chloride (1:1):EtOAc (86:14) were allowed to precipitate from CH2Cl2–MeOH (3:1), and a yellowish white amorphous substance was precipitated (compound 5; 21 mg).
Fractions (330–335) eluted with ethyl acetate (100%) were resubjected to chromatography over a silica gel column using CH2Cl2:MeOH for elution; the obtained subfractions (70–90) eluted with CH2Cl2–MeOH (96.5:3.5) precipitated a yellowish white amorphous substance (compound 6; 14 mg). The ethyl acetate fraction was chromatographed over a silica gel column. The column was packed with methylene chloride (100%) and eluted using gradient elution with CH2Cl2:MeOH (100:0 to 0:100). Collected fractions were monitored by TLC and similar fractions were collected. Fractions (85–99) eluted with CH2Cl2–MeOH (95:5) were resubjected to chromatography over a silica gel column using CH2Cl2:MeOH for elution, and the obtained subfractions (84–85) precipitated a yellowish white amorphous powder (compound 7; 4 mg). Fractions (111–114) eluted with CH2Cl2–MeOH (95:5) were washed several times with dichloromethane and then methanol, giving a white amorphous powder (compound 8; 18 mg). Fractions (161–193) eluted with CH2Cl2–MeOH (94:6) were resubjected to chromatography over a silica gel column using CH2Cl2:MeOH for elution, and the obtained subfractions (136–142) precipitated a yellowish white powder (compound 9; 6 mg). Fractions (221–226) eluted with CH2Cl2–MeOH (88:12) were purified using preparative silica gel TLC plates and an ethyl acetate:methanol:water (100:16.5:13.5) solvent system. Upon evaporation of the solvent, a yellowish amorphous powder (compound 10; 4 mg) was deposited.
2.4. Biological activities
:1, respectively), to produce ABTS˙+ stock solution; the mixture was kept and left to stand in the dark at R.T. overnight (or for roughly 12–16 hours in the dark). Then, to make the ABTS˙+ working solution, the ABTS˙+ stock solution was diluted in pure EtOH until it had an absorbance (Ablank) of 0.7 ± 0.02 (after three measurements) at a wavelength of 734 nm. The solution was then equilibrated in an incubator with the temperature set at 30 °C (Ablank was adjusted in this present assay to be exactly 0.7 before measuring the absorbance for all test compounds).The free radical scavenging activity was evaluated by combining 1.5 mL of the blue-green ABTS˙+ working solution with varied amounts ranging from 10 to 100 μg (in distilled H2O, pure EtOH, or a mixture of both of them according to the solubility of each compound) of all test compounds (1–10). After mixing, the change in absorbance at 734 nm was immediately observed after 0 s, 0.5 s, and 1 s, and then repeatedly at 5 s intervals until a steady-state value was attained. The absorbance value for each test chemical after it was added to the ABTS˙+ solution (Atest) was taken after 15 minutes of mixing, because the steady state was reached after that time in the current experiments. Values are the averages of three separate calculations (as all measurements were taken three times). The following equation was used to compute the percent decrease in absorbance, which represents the test compound's ability to scavenge ABTS˙+ radical cations:
| % inhibition = 100(Ablank − Atest)/Ablank |
After 15 minutes of reaction, the inhibitory concentration (IC50) of each test compound was calculated and compared to that of L-ascorbic acid (taken as the reference and standard antioxidant compound in this assay). IC50 is the concentration of any test compound required to reduce the absorption or amount of ABTS˙+ radical cations by 50% at a wavelength of 734 nm. Using GraphPad Prism 6 software (USA, 2015), the antioxidant or anti-ABTS˙+ IC50 value (for each test compound and the reference L-ascorbic acid) was computed. The lower the IC50 value, the more potent the test compound is as an antioxidant (i.e., the stronger the antioxidant activity of the test compound).
2.5. Computational studies
Docking analysis was conducted using the Online SwissDock server (http://www.swissdock.ch)28,29 with the EA-dock protocol and its default parameters. These settings effectively replicated the docking pose of the co-crystallized ligand, achieving an accuracy within 2 RMSD of the original coordinates. Protein targets were selected based on prior reviews related to the specific virus under study. The three-dimensional structures of these proteins were obtained in PDB format from the RCSB Protein Data Bank (source: https://www.rcsb.org/; accessed on September 24, 2023). Post-retrieval, PyMOL 2.3 was used to remove unnecessary water molecules and excess protein residues. Nonpolar hydrogen atoms were added to complete the structures, followed by energy minimization to their lowest states using the Swiss PDB Viewer tool. Following refinement and energy optimization, the macromolecules were stored in PDB format, paving the way for a detailed and thorough evaluation. The standard grid box dimensions were set at 20 Å × 20 Å × 20 Å with 0.375 Å spacing, centred on the co-crystallized ligand's location. UCSF Chimera v.1.10.2 was employed to analyze interaction patterns and visualize the docking poses. The pharmacokinetics, cellular permeability, and medicinal chemistry suitability of the isolated phytochemicals were evaluated using the Online Swiss ADME web tool (http://www.swissadme.ch).2.6. Statistical analysis
All values were expressed as mean ± S.D. The values were the average of three independent experiments. Statistical analysis was carried out using one-way analysis of variance (ANOVA) followed by Dunnett's post-hoc test in GraphPad Prism 6 software (USA, 2015).3. Results and discussion
3.1. Identification of the isolated compounds
In this study, the phytochemical investigation of the aerial parts of M. alysson L. yielded 10 compounds (Fig. 1). The isolated compounds can be classified as 2 sterols (1 and 3), 1 monoglyceryl fatty acid (2), 6 flavonoids (5–9) and 1 phenylpropanoid (10). The proton and carbon values of the isolated compounds are presented in the ESI file with the spectra (ESI, Fig. S1–S29†).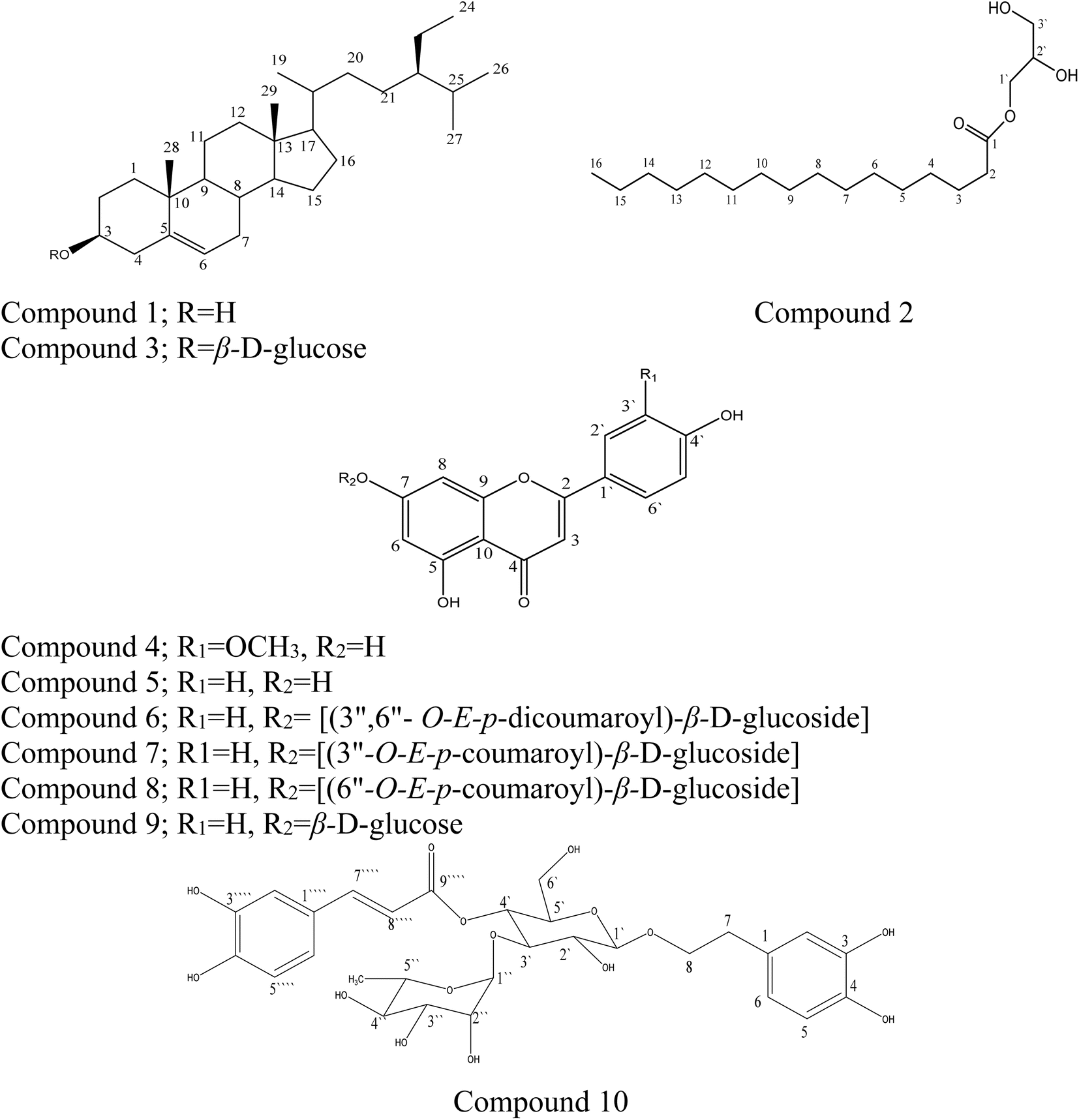 | ||
| Fig. 1 Chemical structure of the compounds isolated from Marrubium alysson. | ||
Compounds 1–3 were isolated from the petroleum ether fraction. Compounds 1 and 3 were confirmed based on comparing their IR spectra (KBr, νmax cm−1) with those reported in the literature,30,31 and also through comparing with authentic samples; they were identified as β-sitosterol (1) and its glucoside β-sitosterol 3-O-β-D-glucoside (3). Compound 1 was isolated for the first time from M. alysson L. Compound 2 was identified by analyzing its proton and carbon chemical shift values, and the HSQC, HMBC, and GC-MS data of the methyl ester of the fatty acid obtained after hydrolysis (ESI Fig. S2–S6†), which were consistent with literature reports.32,33 It was identified as glycerol monopalmitate 2, which has been isolated for the first time from the genus Marrubium. The spectral data of compounds 4 and 5 were compared with those reported in the literature.32,33 They were identified as chrysoeriol (4) and apigenin (5).
A careful examination of the 1H NMR and APT spectra of compounds 6–9 (ESI, Fig. S13–S26†) indicated that these compounds were related to apigenin. Comparing the 1H NMR and APT spectra of compound 9 with those of compound 5 revealed that compound 5 is a unit structure in compound 9. A doublet signal at δH 5.05 ppm (1H, d, J = 7.3 Hz) corresponds to the anomeric proton of the glucose sugar (H-1′′), with the coupling constant (J = 7.3 Hz) confirming a β-linked configuration.34 The carbon signal at δC 100.3 ppm corresponds to the anomeric carbon of the sugar (C-1′′) which, together with five additional carbon signals at δC 73.5, 76.8, 70.0, 77.5, and 61.1 ppm, confirms the presence of a hexopyranosyl moiety. The chemical shifts of the protons and carbons of the sugar are comparable with those reported for O-linked β-D-glucose.35
The slight upfield shift of C-7 of compound 9 (δC 163.4 ppm) compared to apigenin aglycone (δC 164.2 ppm,35 and δC 164.3 ppm) of compound 4 indicated that compound 9 is glycosylated at C-7. Compound 9 was identified as apigenin-7-O-β-glucopyranoside.35,36 Careful analysis of the 1H NMR and APT spectra of compound 7 (ESI, Fig. S17 and S18†) revealed that compound 9 forms its core structure. Differences in the aromatic region suggested the presence of a p-coumaroyl moiety, which was confirmed by a conjugated carbonyl at δC 166.7 ppm and two trans-olefinic methine carbons at δC 145.1 and 115.1 ppm, corresponding to protons at δH 7.60 (H-7′′′) and 6.46 (H-8′′′) ppm with a large coupling constant (J = 16 Hz).
These observations suggested the presence of a p-coumaroyl moiety acylating the glucose. Both 1H and APT data indicated that the acylating p-coumaroyl moiety must be attached to the glucose at C-3′′. This could be proved by the relative downfield shift of H-3′′ at δH 5.11 ppm compared to that of apigenin-7-O-β-D-glucoside (the non-acylated sugar) which appeared at δH 3.14–3.39 ppm.36 Furthermore, the APT spectrum showed a relative downfield shift of C-3′′ (δC 77.7 ppm) compared to that of apigenin-7-O-β-D-glucoside (δC 76.5 ppm), while the signals of C-2′′ (δC 71.7 ppm) and C-4′′ (δC 67.9 ppm) representing the neighbouring carbons of C-3′′ showed a relative upfield shift compared to C-2′′and C-4′′ of apigenin-7-O-β-D-glucoside (δC 73.1 and 69.8 ppm, respectively).35,37 Compound 7 was identified as apigenin-7-O-(3′′-O-E-p-coumaroyl)-β-D-glucopyranoside.
A careful examination of the 1H NMR and APT spectra of compound 8 (ESI, Fig. S20 and S21†) indicated the presence of compound 9 as a unit structure in compound 8. Differences were observed in the aromatic region, proposing the presence of a p-coumaroyl moiety acylating the glucose. Both 1H and APT data indicated that the acylating p-coumaroyl moiety must be attached to the glucose at C-6′′. This could be proved by the relative downfield shift of H-6′′ at δH 4.17 and 4.47 ppm compared to that of apigenin-7-O-β-D-glucoside (the non-acylated sugar) which appeared at δH 3.55 and 3.73 ppm.36 Furthermore, the APT spectrum showed a downfield-shifted CH2 signal (C-6′′ of the sugar) at δC 63.9 ppm and an upfield shift (C-5′′ of the sugar) at δC 74.3 ppm (relative to δC 60.8 and 77.1 ppm, respectively, of apigenin-7-O-β-D-glucoside35), indicating the attachment of a p-coumaroyl moiety at the C-6′′ position,38 and this was also proved from the HMBC correlation between δH 4.17 (H-6′′) and δC 166.9 (C-9′′′) ppm.
Careful examination of the 1H NMR and APT spectra of compound 6 (ESI, Fig. S13 and S14†) indicated the presence of compound 9 as a unit structure in compound 6. Differences were observed in the aromatic region, and the presence of two p-coumaroyl moieties acylating the glucose was proposed; the positions of the trans coumaroyl group attachments to the sugar were determined from the downfield-shifted signals of 5.11, 4.23, and 4.47 ppm for H-3′′, H-6′′a and H-6′′b, respectively, compared to those of apigenin-7-O-β-D-glucoside (the non-acylated sugar) which appeared at δH 3.14–3.39 ppm (H-3′′) and δH 3.55 and 3.73 ppm (H-6′′).36
Furthermore, the APT spectrum showed relative downfield shifts in the C-3′′ (δC 77.3 ppm) and C-6′′ (δC 63.6 ppm) signals compared to those of apigenin-7-O-β-D-glucoside (δC 76.5 and 60.8 ppm for C- 3′′ and C-6′′, respectively), while the signals of C-2′′ (δC 71.5 ppm) and C-4′′ (δC 68.5 ppm), representing the neighbouring carbons of C-3′′, and the signal of C-5′′ (δC 74.6), representing the neighbouring carbons of C-6′′, showed a relative upfield shift compared to those of apigenin 7-O-β-D-glucoside (C-2′′, C-4′′, and C-5′′: δC 73.1, 60.8, and 77.1 ppm, respectively);35,37 this was also proved by the HMBC cross peaks between H-3′′ (5.11 ppm) and C-9′′′ (at 167.0 ppm) and between H-6′′ (4.23 ppm) and C-9′′ (at 166.9 ppm), further supporting the acyl group attachments to the glucose. The attachment of the sugar moiety to flavonoid C-7 was demonstrated by the association between H-1′′ (at 5.35 ppm) and C-7 (at 162.8 ppm). Additional analysis of the HMBC spectra revealed cross peaks between the hydrogens of the coumaroyl groups and their carbonyl carbons (H-7′′′/C-9′′′ and H-7′′′′/C-9′′′′), as well as between the hydrogens of the coumaroyl groups (H-8′′′ and H-8′′′′ and C-1′′′ and C-1′′′′, respectively).
Careful examination of the 1H NMR and 13C NMR spectra of compound 10 (ESI, Fig. S27 and S29†) revealed the following. The downfield shift of C-3′′ of the glucose unit (δC 81.7 ppm) indicated that the rhamnose moiety was acylated at the C-3′′ of the glucose. The acyl group (caffeoyl moiety) was positioned at the C-4′′ position of the glucose unit, based on the strong deshielding of the H-4′′ signal (δH 4.87 ppm, m) of the glucose unit. The downfield shifts of the aromatic signals (δC 146.3, 144.9, 148.8 and 149.9 ppm) allocated to C-3, 4, 3′′′′ and 4′′′′, respectively, proved their oxygenation. The shielded aromatic carbons resonated at δC 117.2, 116.3, 114.3 and 116.7 ppm, representing C-2, C-5, C-2′′′′ and C-5′′′′, respectively, which indicated their ortho positions to hydroxyl groups. Furthermore, the 1H NMR spectrum revealed the following: two sets of 1,3,4-trisubstituted aromatic ring signals at δH 6.96 (d, J = 1.6 Hz), δH 6.68 (d, J = 8.0 Hz), and δH 6.87 (dd, J = 1.6, 8.0 Hz) ppm for H-2′′′, H-5′′′, and H-6′′′ and δH 6.60 (d, J = 2.0 Hz), δH 6.58 (d, J = 8.0 Hz), and δH 6.47 (dd, J = 1.6, 8.0 Hz) ppm for H-2, H-5, and H-6; and two trans olefinic protons (CH) at δH 7.50 (H-7′′′) and 6.18 (H-8′′′) ppm with large J values of 16.0 Hz. The signals at δC 148.8 and 113.0 ppm in the 13C NMR spectrum were assigned as C-7′′′ and C- 8′′′ of the caffeic acid moiety; there was a benzylic methylene (CH2) signal at δH 2.69 ppm (2H, t, J = 7.2 Hz) and two non-equivalent proton signals at δH 3.50–3.70 and 3.94 ppm (each 1H, m). These results validate the presence of an (E)-caffeic acid unit and 3,4-dihydroxyphenethyl alcohol moiety. In addition, two anomeric proton signals at δH 4.28 (d, J = 8.0 Hz) and 5.08 (s) ppm were attributed to β-glucose and α-L-rhamnose units, respectively. The identity of the latter sugar was confirmed as rhamnose by the presence of a methyl group signal at δH 0.99 ppm (d, J = 6.0 Hz). This indicated a disaccharide structure for compound 10.
Compound 1 (β-sitosterol). IR (KBr, νmax cm−1): 3422 cm−1 (OH stretching), 2957, 2936, 2866 cm−1 (CH stretching), 1654 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching), 1463, 1243 cm−1 (CH2 bending), 1378 cm−1 (CH3 bending), 1051 cm−1 (C–O stretching), 961 cm−1 (=C–H bending).
C stretching), 1463, 1243 cm−1 (CH2 bending), 1378 cm−1 (CH3 bending), 1051 cm−1 (C–O stretching), 961 cm−1 (=C–H bending).
Compound 2 (glycerol monopalmitate). GC-MS m/z: 270.3 [M+]. 1H NMR (CDCl3, CD3OD (1:1), 600 MHz, J in Hz): δH 2.35 (2H, t, J = 7.8, H-2), 1.62 (2H, m, H-3), 1.28 (m, H-4-15), 0.89 (3H, t, J = 7.2, H-16), 4.08 (1H, dd, J1 = 11.4, J2 = 6.6, 1′a), 4.16 (1H, dd, J1 = 11.4, J2 = 4.8, 1′b), 3.83 (1H, m, 2′), 3.56 (1H, t, J = 4.8, 3′a), 3.67 (1H, m, 3′b) ppm. APT (CDCl3, CD3OD (1:1), 150 MHz): δC 175.7 (C-1), 35.1 (C-2), 26.1 (C-3), 30.2–30.8 (C-4-13), 33.1 (C-14), 23.8 (C-15), 14.6 (C-16), 66.6 (C-1′), 71.2 (C-2′), 64.1 (C-3′) ppm.
Compound 3 (β-sitosterol 3-O-β-D-glucoside). IR (KBr, νmax cm−1): 3421 cm−1 (OH stretching), 2958 and 2870 cm−1 (CH stretching), 1651 cm−1 (CC stretching), 1461 cm−1 (CH2 bending), 1375 cm−1 (CH3 bending), and 1163, 1025 cm−1 (C–O stretching).
Compound 4 (chrysoeriol). 1H NMR (DMSO-d6, 400 MHz, J in Hz): δH: 6.87 (1H, s, H-3), 6.17 (1H, s, H-6), 6.48 (1H, s, H-8), 7.55 (1H, s, H-2′), 6.94 (1H, d, J = 8.0 Hz, H-5′), 7.56 (1H, d, J = 8.0 Hz, H-6′), 3.90 (3H, s, O–Me), 12.97 (5-OH) ppm. APT (DMSO-d6, 100 MHz): δC 164.0 (C-2), 103.6 (C-3), 161.9 (C-5), 99.6 (C-6), 164.3 (C-7), 94.7 (C-8), 157.9 (C-9), 104.1(C-10), 122.0 (C-1′), 110.6 (C-2′), 148.6 (C-3′), 150.5 (C-4′), 116.3 (C-5′), 120.8 (C-6′), 56.4 (O–Me) ppm.
Compound 5 (apigenin). ESI-MS m/z: 268.8 [M–H]−. 1H NMR (DMSO-d6, 400 MHz, J in Hz): δH: 6.8 (1H, s, H-3), 6.2 (1H, d, J = 1.9 Hz, H-6), 6.49 (1H, d, J = 1.9 Hz, H-8), 7.94 (2H, d, J = 8.8 Hz, H-2′,6′), 6.94 (2H, d, J = 8.8 Hz, H-3′,5′), 12.98 (5-OH) ppm. APT (DMSO-d6, 100 MHz): δC 164.8 (C-2), 103.3 (C-3), 182.2 (C-4), 161.9 (C-5), 99.4 (C-6), 164.2 (C-7), 94.5 (C-8), 157.8 (C-9), 104.1 (C-10), 121.6 (C-1′), 128.9 (C-2′,6′), 116.4 (C-3′,5′), 161.7 (C-4′) ppm.
Compound 6 (apigenin-7-O-(3′′,6′′-E-p-dicoumaroyl)-β-D-glucoside). ESI-MS m/z: 722.9 [M–H]−. 1H NMR (DMSO-d6, 400 MHz, J in Hz)′′: δH 6.81 (1H, s, H-3), 6.51 (1H, b.s, H-6), 6.86 (1H, b.s, H-8), 7.94 (2H, d, J = 8.8 Hz, H-2′,6′), 6.92 (2H, d, J = 8.8 Hz, H-3′,5′), 5.35 (1H, d, J = 8.0 Hz, H-1′′), 3.35 (3H, 2′′,4′′,5′′), 5.11 (1H, t, H-3′′), 4.23 (1H, m, H-6′′a), 4.47 (1H, d, J = 11.4 Hz, H-6′′b), 7.57 (2H, d, J = 8 Hz, H-2′′′,6′′′), 6.82 (2H, d, J = 8 Hz, H-3′′′,5′′′), 7.60 (1H, d, J = 16 Hz, H-7′′′), 6.44 (1H, d, J = 16 Hz, H-8′′′), 7,36 (2H, d, J = 8.4 Hz, H-2′′′′,6′′′′), 6.68 (2H, d, J = 8.4 Hz, H-3′′′′,5′′′′), 7.49 (1H, d, J = 16 Hz, H-7′′′′), 6.33 (1H, d, J = 16 Hz, H-8′′′′) ppm. APT (DMSO-d6, 100 MHz): δC 164.9 (C-2), 103.4 (C-3), 182.4 (C-4), 162.0 (C-5), 100.3 (C-6), 162.8 (C-7), 95.2 (C-8), 157.4 (C-9), 104.1 (C-10), 121.6 (C-1′), 128.9 (C-2′,6′), 116.4 (C-3′,5′), 161.7 (C-4′), 99.5 (C-1′′), 71.5 (C-2′′), 77.3 (C-3′′), 68.5 (C-4′′), 74.0 (C-5′′), 63.6 (C-6′′), 125.6 (C-1′′′), 130.7 (C-2′′′,6′′′), 116.6 (C-3′′′,5′′′), 160.2 (C-4′′′), 145.5 (C-7′′′), 115.1 (C-8′′′), 167.0 (C-9′′′), 125.3 (C-1′′′′), 130.6 (C-2′′′′,6′′′′), 116.3 (C-3′′′′,5′′′′), 160.2 (C-4′′′′), 145.2 (C-7′′′′), 114.0 (C-8′′′′), 166.9 (C-9′′′′) ppm.
Compound 7 (apigenin-7-O-(3′′-O-E-p-coumaroyl)-β-D-glucoside). ESI-MS: m/z 577.0 [M–H]−. 1H NMR (DMSO-d6, 400 MHz, J in Hz)′′: δH 6.91 (1H, s, H-3), 6.49 (1H, d, J = 1.6 Hz, H-6), 6.89 (1H, b.s, H-8), 7.98 (2H, d, J = 8.8 Hz, H-2′, 6′), 6.96 (2H, d, J = 8.8 Hz, H-3′,5′), 5.30 (1H, d, J = 7.6 Hz, H-1′′), 3.30–3.77 (5H, 2′′,4′′,5′′, 6′′), 5.11 (1H, t, H-3′′), 7.59 (2H, d, J = 8 Hz, H-2′′′,6′′′), 6.82 (2H, d, J = 8 Hz, H-3′′′,5′′′), 7.60 (1H, d, J = 16 Hz, H-7′′′), 6.46 (1H, d, J = 16 Hz, H-8′′′) ppm. APT (DMSO-d6, 100 MHz): δC 164.8 (C-2), 103.7 (C-3), 182.5 (C-4), 161.8 (C-5), 100.2 (C-6), 163.1 (C-7), 95.2 (C-8), 157.4 (C-9), 105.9 (C-10), 121.5 (C-1′), 129.2 (C-2′,6′), 116.3 (C-3′,5′), 161.6 (C-4′), 99.9 (C-1′′), 71.7 (C-2′′), 77.3 (C-3′′), 67.9 (C-4′′), 74.6 (C-5′′), 60.9 (C-6′′), 125.7 (C-1′′′), 130.7 (C-2′′′,6′′′), 116.5 (C-3′′′,5′′′), 160.2 (C-4′′′), 145.1 (C-7′′′), 115.1 (C-8′′′), 166.7 (C-9′′′) ppm.
Compound 8 (apigenin-7-O-(6′′-O-E-p-coumaroyl)-β-D-glucoside). ESI-MS m/z: 576.9 [M–H]−. 1H NMR (DMSO-d6, 400 MHz, J in Hz)′′: δH 6.85 (1H, s, H-3), 6.49 (1H, d, J = 2 Hz, H-6), 6.83 (1H, d, J = 2 Hz, H-8), 7.96 (2H, d, J = 8.8 Hz, H-2′,6′), 6.95 (2H, d, J = 8.8 Hz, H-3′,5′), 5.18 (1H, d, J = 7.6 Hz, H-1′′), 3.20–3.40 (3H, 2′′,3′′,4′′), 3.85 (1H, t, J = 7.6 Hz, H-5′′), 4.17 (1H, dd, J = 11.9/7.1 Hz, H-6′′a), 4.47 (1H, d, J = 10.6 Hz, H-6′′b) 7.38 (2H, d, J = 8 Hz, H-2′′′,6′′′), 6.68 (2H, d, J = 8 Hz, H-3′′′,5′′′), 7.50 (1H, d, J = 16 Hz, H-7′′′), 6.34 (1H, d, J = 16 Hz, H-8′′′), 12.99 (5-OH), 10.46 (7-OH), 10.04 (4′-OH) ppm. APT (DMSO-d6, 100 MHz): δC 164.7 (C-2), 103.5 (C-3), 182.5 (C-4), 161.6 (C-5), 99.9 (C-6), 163.2 (C-7), 95.2 (C-8), 157.4 (C-9), 105.8 (C-10), 121.4 (C-1′), 129.0 (C-2′,6′), 116.5 (C-3′,5′), 161.9 (C-4′), 99.9 (C-1′′), 73.4 (C-2′′), 76.7 (C-3′′), 70.4 (C-4′′), 74.3 (C-5′′), 63.9 (C-6′′), 125.4 (C-1′′′), 130.6 (C-2′′′,6′′′), 116.1 (C-3′′′,5′′′), 160.3 (C-4′′′), 145.4 (C-7′′′), 114.2 (C-8′′′), 166.9 (C-9′′′) ppm.
Compound 9 (apigenin-7-O-β-D-glucopyranoside). ESI-MS m/z: 431.0 [M–H]−. 1H NMR (DMSO-d6, 400 MHz, J in Hz)′′: δH 6.86 (1H, s, H-3), 6.45 (1H, d, J = 2.0 Hz, H-6), 6.85 (1H, b.s, H-8), 7.96 (2H, d, J = 8.8 Hz, H-2′,6′), 6.95 (2H, d, J = 8.8 Hz, H-3′,5′), 5.05 (1H, d, J = 7.3 Hz, H-1′′), 3.17–3.92 (6H, 2′′,3′′,4′′,5′′,6′′) ppm. APT (DMSO-d6, 100 MHz): δC 164.9 (C-2), 103.5 (C-3), 182.5 (C-4), 100.0 (C-6), 163.4 (C-7), 95.3 (C-8), 157.4 (C-9), 105.7 (C-10), 121.5 (C-1′), 129.1 (C-2′,6′), 116.5 (C-3′,5′), 161.8 (C-4′), 100.3 (C-1′′), 73.5 (C-2′′), 77.5 (C-3′′), 70.0 (C-4′′), 76.8 (C-5′′), 61.1 (C-6′′) ppm.
Compound 10 (verbascoside or acteoside). ESI-MS m/z: 623.0 [M–H]−. 1H NMR (CD3OD, 400 MHz, J in Hz)′′: (aglycone) δH 6.60 (1H, d, J = 2.0 Hz, H-2), 6.58 (1H, d, J = 8.0 Hz, H-5), 6.47 (1H, dd, J = 1.6/8 Hz, H-6), 2.69 (2H, t, J = 7.2 Hz, H-7), 3.94 (1H, m, H-8a), 3.5–3.7 (1H, H-8b); (β-D-glucose) 4.28 (1H, d, J = 8.0 Hz, H-1′), 3.30–3.80 (6H, 2′,3′,4′,5′,6′); (α-L-rhamnose) 5.09 (1H, s, H-1′′), 3.30–3.80 (3H, 2′′,4′′,5′′), 5.10 (1H, m, H-3′′), 0.99 (3H, d, J = 6.0 Hz, H-6′′); (acyl moiety) 6.96 (1H, d, J = 1.6 Hz, H-2′′′), 6.68 (1H, d, J = 8.0 Hz, H-5′′′), 6.87 (1H, dd, J = 1.6/8.0 Hz, H-6′′′), 7.50 (1H, d, J = 16 Hz, H-7′′′), 6.18 (1H, d, J = 16 Hz, H-8′′′) ppm. 13C-NMR (CD3OD, 100 MHz, J in Hz): (aglycone) δC 131.5 (C-1), 117.2 (C-2), 146.3 (C-3), 144.9 (C-4), 116.3 (C-5), 121.4 (C-6), 36.6 (C-7), 72.4 (C-8); (β-D-glucose) 104.3 (C-1′), 76.3 (C-2′), 81.7 (C-3′), 70.5 (C-4′), 76.2 (C-5′), 62.4 (C-6′); (α-L-rhamnose) 103.1 (C-1′′), 72.3 (C-2′′), 72.1 (C-3′′), 73.9 (C-4′′), 70.5 (C-5′′), 18.5 (C-6′′); (acyl moiety) 126.1 (C-1′′′), 114.3 (C-2′′′), 147.3 (C-3′′′), 149.9 (C-4′′′), 116.7 (C-5′′′), 123.8 (C-6′′′), 148.8 (C-7′′′), 113.0 (C-8′′′), 168.7 (C-9′′′) ppm.
3.2. Biological activities
| Compound (code) | Compound (name) | In vitro COV-Mpro IC50 (μM) |
|---|---|---|
| a Tipranavir was used as a standard, with IC50 3.231 ± 0.14 μM; DMSO was used as a solvent control; n = 3. | ||
| (Standard) | Tipranavir | 3.231 ± 0.14 |
| 1 | β-Sitosterol | 88.23 ± 3.75 |
| 2 | Glycerol monopalmitate | 29.2 ± 1.24 |
| 3 | β-Sitosterol-3-O-β-D-glucoside | 137.2 ± 5.83 |
| 4 | Chrysoeriol | 15.14 ± 0.64 |
| 5 | Apigenin | 38.76 ± 1.65 |
| 6 | Apigenin 7-O-(3′′,6′′-O-E-p-dicoumaroyl)-β-D-glucoside | 8.349 ± 0.35 |
| 7 | Apigenin 7-O-(3′′-O-E-p-coumaroyl)-β-D-glucoside | 83.35 ± 3.54 |
| 8 | Apigenin 7-O-(6′′-O-E-p-coumaroyl)-β-D-glucoside | 224.8 ± 9.55 |
| 9 | Apigenin 7-O-β-D-glucoside | 14.3 ± 0.61 |
| 10 | Verbascoside | 39.76 ± 1.69 |
The isolated compounds 1–10 demonstrated varying degrees of free radical scavenging activity toward the ABTS radical, with decreasing activity in the following order, according to a thorough analysis of the results in Table 2: 10 >6>7>8>5>9>4>3>1>2. Concerning the major compounds isolated from the ethyl acetate fraction, verbascoside 10 is the most effective, with an IC50 value higher than L-ascorbic acid (IC50 = 30.43 ± 0.14 μM) in relation to in vitro antiradical effects (IC50 = 25.58 ± 0.12 μM), which may be due to the presence of a pair of catechol groups which appeared to be crucial for high antioxidant activity, like the effect of the catechol group in ring B of flavonoids;39 other compounds showed activity that was either close to or lower than that of L-ascorbic acid (such as compounds 6, 7 and 8, with IC50 values of 36.61 ± 0.19, 36.67 ± 0.16, and 42.64 ± 0.20 μM, respectively). This may be due to the presence of a p-coumaroyl moiety, which appears to be crucial for high antioxidant activity.40
| Compound (code) | Compound (name) | ABTS IC50 (μM) |
|---|---|---|
| a Ascorbic acid (ABTS) is used as a standard antioxidant with IC50 = 30.43 ± 0.14 μM. | ||
| Standard | Ascorbic acid | 30.43 ± 0.14 |
| 1 | β-Sitosterol | 110.50 ± 0.67 |
| 2 | Glycerol monopalmitate | 131.60 ± 0.68 |
| 3 | β-Sitosterol 3-O-β-D-glucoside | 96.98 ± 0.53 |
| 4 | Chrysoeriol | 87.12 ± 0.42 |
| 5 | Apigenin | 52.92 ± 0.24 |
| 6 | Apigenin 7-O-(3′′,6′′-O-E-p-dicoumaroyl)-β-D-glucoside | 36.61 ± 0.19 |
| 7 | Apigenin 7-O-(3′′-O-E-p-coumaroyl)-β-D-glucoside | 36.67 ± 0.16 |
| 8 | Apigenin 7-O-(6′′-O-E-p-coumaroyl)-β-D-glucoside | 42.64 ± 0.20 |
| 9 | Apigenin 7-O-β-D-glucoside | 55.26 ± 0.28 |
| 10 | Verbascoside | 25.58 ± 0.12 |
Compounds 5 and 9 showed moderate antioxidant activity, with IC50 values of 52.92 ± 0.24 and 55.26 ± 0.28 μM, respectively, comparing with standard ascorbic acid, but the activity of compound 9 is less than 5 due to the blockage of 7-OH by glycosylation.41 Compound 4 has lower radical scavenging activity than apigenin, with an IC50 value of 87.12 ± 0.42 μM due to the presence of 3′-OCH3, which decreases the radical scavenging activity of flavonoids.39 The other compounds 1–3 demonstrated the lowest levels of free radical scavenging activity toward the ABTS radical, poor in comparison with L-ascorbic acid and the other compounds, with decreasing activity in the following order: 3<1<2, showed IC50 values of 96.98 ± 0.53, 110.50 ± 0.67, and 131.60 ± 0.68 μM, respectively. The weak antioxidant activities of compounds 1–3 may be due to the lack of a free phenolic hydroxy group.
3.3. Molecular modelling study
The docking type was selected to be rigid and accurate, and for each docking experiment the results were calculated using 250 different sequential runs. Those binding models possessing the most favourable energies were estimated via the fast analytical continuum treatment of solvation. The clustering of binding modes was based on a calculation of the average full fitness of the contributing elements. The interactions, number of hydrogen bonds and visualization of the obtained docking poses were analysed using UCSF Chimera v.1.10.2,44 and PyMOL.45 Validation of the docking protocol was done via the initial re-docking of the co-crystallized ligand (myricetin). It was proved that the EA-dock workflow implemented in the SwissDock server was able to reproduce a docking pose within 2-Å RMSD (1.17 Å) from the myricetin coordinates with the SARS-CoV-2 3CLpro crystal structure (PDB ID: 7DPP). A summary of the estimated binding energies with the residues of SARS-CoV-2 3CLpro of the isolated compounds is shown in Table 3.
| Compound | CoV-2-3CL protease IC50 (μM) | Estimated ΔG (kcal mol−1) |
|---|---|---|
| 1 | 88.23 | −7.56 |
| 2 | 29.2 | −8.11 |
| 3 | 137.2 | −7.93 |
| 4 | 15.14 | −7.47 |
| 5 | 38.76 | −7.11 |
| 6 | 8.349 | −9.45 |
| 7 | 83.35 | −7.44 |
| 8 | 224.8 | −7.85 |
| 9 | 14.3 | −8.04 |
| 10 | 39.76 | −8.26 |
| Myricetin | ND | −7.31 |
| Tipranavir | 3.231 | −8.25 |
We decided to study the docking scores for the most active compounds in more detail. We compared them with other isolated compounds that did not show high activity in the enzymatic assays. This would help to deduce the major differences in the interaction patterns and what is needed to achieve tight binding inside the active site. We planned to do so via examining the electrostatic and hydrophobic surfaces of the docked poses while they are aligned over myricetin. In addition, the 3D interaction pattern for each compound was checked to make sure it satisfies the essential binding requirements with key amino acids in CoV-2 3CLpro, using PyMOL. An analysis of the contact maps of the clustered poses was done for tipranavir and isolated compounds. This would help to track the binding affinity and shed light on the contributions of residues in the SARS-CoV-2 3CLpro active site and how they affected the observed activities in the biochemical assays.
Fig. 2 illustrates the docking poses of the top three active compounds (2, 6, and 10), tipranavir, and myricetin in the SARS-CoV-2 3CLpro active site (PDB ID: 7DPP). Compound 2 exhibits hydrogen-bonding interactions with Phe140, Gly143, Gly143, His163 and Glu166, showing high binding affinity (ΔG = −8.11 kcal mol−1). Compound 6 exhibits an extended network of hydrogen-bonding interactions with Thr24, Met49, Glu47, Asn142, Gly143, and Gln189, along with H-π stacking with Thr25, leading to its high binding affinity (ΔG = −9.45 kcal mol−1). Compound 10 can form H-π stacking interactions with Thr25 alongside hydrophobic interactions with Met165 for stabilization in binding to sub-pocket D. Compound 10 can also engage in H-bonding with Thr26, Cys145 and His164 and showed ΔG = −8.26 kcal mol−1. Tipranavir and myricetin act as reference ligands, where myricetin (ΔG = −7.31 kcal mol−1) forms multiple H-bonding interactions with His41, Leu141, Gly143, Ser144, Cys145, His163 and Gln189. Tipranavir (ΔG = −8.25 kcal mol−1) created hydrogen bonds with Arg188 and Gln189 and hydrophobic interactions with Thr45 and Ala191, supported by π–π stacking with Thr190.
 | ||
| Fig. 2 2D poses of identified phytochemical compounds with the highest docking scores alongside tipranavir and myricetin in the SARS-CoV-2 3CLpro active site (PDB ID: 7DPP). | ||
Interaction analysis derived from electrostatic/hydrophobic surface mapping (Fig. 3) revealed five potential sub-pockets (A–E) with different natures. Site A has a hydrophilic nature with Arg-188 and Gln-189 (positively charged region). Sub-pocket B is greasy, like sub-pocket C. Also, sub-pockets C and E are glutamate-rich which is reflected by their tendency to have negative charge. The central sub-pocket D is greasy to a great extent with some hydrophilic character due to the presence of several histidine residues (His164/His41). Tipranavir shows an IC50 value of 3.231 mM by occupying only 3 out of the 5 sub-pockets (A, B and D) in the CoV-2 3CLpro active site. This demonstrates the significance of those 3 sub-pockets in achieving tight and strong binding, which is translated into potent inhibition in the biochemical assay. Looking at the 3D interaction pattern, tipranavir could form hydrogen bonds with both Arg188 and Gln189 and π–π stacking interactions with the His41 side chain.
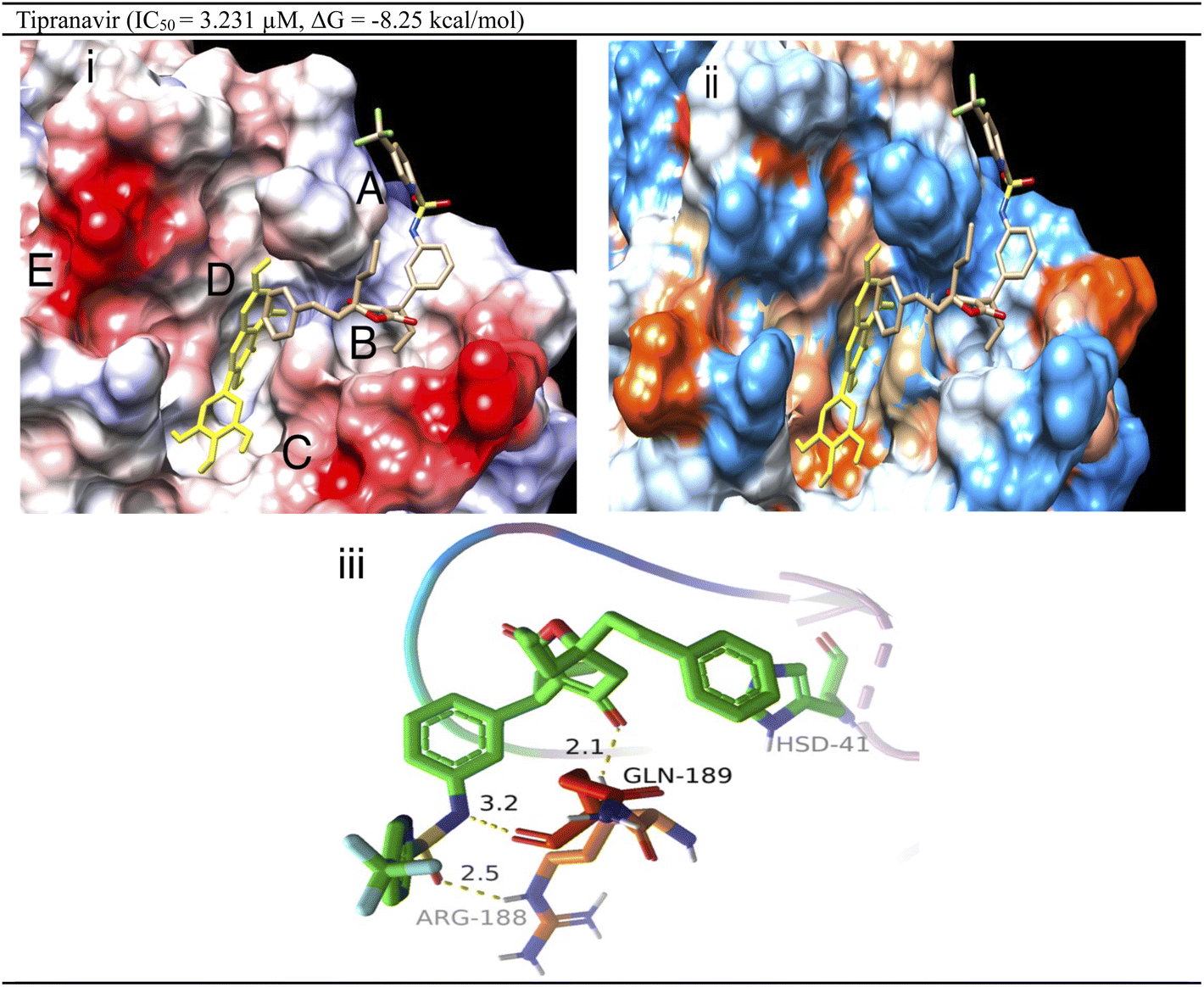 | ||
| Fig. 3 (i) The electrostatic surface via coulombic forces for tipranavir aligned over myricetin (yellow-coloured) in SARS-CoV-2 3CLpro grey-coloured regions are lipophilic, electrostatically positive regions (donor) are coloured blue, and negative regions (acceptor) are shown in red; (ii) the hydrophobic surface of tipranavir aligned over myricetin (yellow-coloured) (green-coloured regions are lipophilic, while blue and reddish-coloured (donor) areas are hydrophilic); and the (iii) 3D interaction pattern of tipranavir in the SARS-CoV-2 3CLpro active site with non-covalent bond distances shown. | ||
Regarding the docking poses for compounds 3 and 6, we could see that neither occupied sub-pocket A (Fig. 4). In addition, we aligned the docked poses over myricetin (yellow-coloured) and we could find that myricetin is going deep in the central sub-pocket D. Compound 3 showed a CoV-2-3CL protease IC50 value of 137.2 μM with an estimated free energy of binding ΔG of −7.93 kcal mol−1. Compound 3 occupies only the central sub-pocket D without deep insertion into it, similar to myricetin and tipranavir. This is evidenced by the absence of any π–π stacking interactions with His41/His164 side chains. Compound 3 has its aliphatic side chain extending into the greasy sub-pocket C and its sugar part into the hydrophilic negatively charged region E. As a result, it undergoes hydrogen bonding with Glu166 and His163.
 | ||
| Fig. 4 (i) The electrostatic surfaces via coulombic forces for compounds 3 and 6 aligned over myricetin (yellow-coloured) in SARS-CoV-2 3CLpro grey-coloured regions are lipophilic, electrostatically positive regions (donor) are coloured blue, and negative regions (acceptor) are shown in red; (ii) the hydrophobic surfaces of compounds 3 and 6 aligned over myricetin (yellow-coloured) (green-coloured regions are lipophilic, while blue and reddish-coloured (donor) areas are hydrophilic); and (iii) the 3D interaction patterns of compounds 3 and 6 in the SARS-CoV-2 3CLpro active site with non-covalent bond distances shown. | ||
Although compound 3 still occupies 3 out of the 5 sub-pockets, like tipranavir, this resulted in weak inhibition in the biochemical assay. This signifies the necessity for a CoV-2-3CL protease inhibitor to occupy sub-pockets A, B and D. This concept is further proved through comparison with the improved potency of compound 6 (IC50 = 8.349 μM, ΔG = −9.45 kcal mol−1). Compound 6 has fragments well-oriented to occupy sub-pockets B, C and D. On one hand, compound 6 does not extend to region A or even have π–π stacking interactions with His41. On the other hand, compound 6, as shown in the 3D interaction pattern, is perfectly engaged in an extensive network of hydrogen bonding with the surrounding residues: Thr24, Thr26, Gly143, and Gln 189 in sub-pocket B; and Met49 and Glu47 in sub-pocket C. Several hydrogen bonds between amino acid residues in CoV-2-3CL protease and compound 6 might explain the observed biochemical activity. The IC50 value is less than 3 times that of the tipranavir activity without engagement with the essential His41 or region A.
Regarding the docking poses for compounds 4 and 5, we found that both compounds occupy the central sub-pocket D with minimal extension to other regions in the active site (Fig. 5). We were interested in those analogs due to their relative potency towards CoV-2-3CL protease. Compound 4 has an IC50 value equal to 15.14 μM and ΔG = −7.47 kcal mol−1, while compound 5 has IC50 = 38.76 μM and ΔG = −7.11 kcal mol−1. They have minimal interactions in the active site, which was reflected by the low binding energies. On the other hand, we could see that both compounds 4 and 5 possess a flavonoid scaffold, which acts as a structural determinant promoting their binding with the protease.
 | ||
| Fig. 5 (i) The electrostatic surfaces via coulombic forces for compounds 4 and 5 aligned over myricetin (yellow-coloured) in SARS-CoV-2 3CLpro grey-coloured regions are lipophilic, electrostatically positive regions (donor) are coloured blue, and negative regions (acceptor) are shown in red; (ii) the hydrophobic surfaces of compounds 4 and 5 aligned over myricetin (yellow-coloured) (green-coloured regions are lipophilic, while blue and reddish-coloured (donor) areas are hydrophilic); and (iii) the 3D interaction patterns of compounds 4 and 5 in the SARS-CoV-2 3CLpro active site with non-covalent bond distances shown. | ||
The orientations of compounds 4 and 5 at the centre of the binding site result in distinct ligand–protein interaction patterns. Notably, the chromone group of compounds 4 and 5 forms π–π stacking interactions with His41/His164. This would serve to stabilize the complex formed between compound 4/5 and CoV-2-3CL protease. The above observations confirm our initial docking model for achieving tight binding and potent inhibitory activity. The observed moderate enzymatic activities of compounds 4 and 5 by occupying one sub-pocket demonstrate the pivotal role of π–π stacking with His41/His164 during binding for chromone-based inhibitors.
Regarding docking poses for compounds 8 and 9, when compared to myricetin, we could deduce that both compounds lie in the central sub-pocket D with the occupation of an additional region in the active site (Fig. 6). Compound 8 has an IC50 value equal to 224.8 μM and ΔG = −7.85 kcal mol−1, while compound 9 has IC50 = 14.3 μM and ΔG = −8.04 kcal mol−1. We were interested in setting up a comparison between these closely related analogues. This comparison was done for the sake of finding out the reasons behind the significant differences in the observed enzymatic activities. We approach this with the aid of molecular interactions in the CoV-2-3CL active site.
 | ||
| Fig. 6 (i) The electrostatic surfaces via coulombic forces for compounds 8 and 9 aligned over myricetin (yellow-coloured) in SARS-CoV-2 3CLpro grey-coloured regions are lipophilic, electrostatically positive regions (donor) are coloured blue, and negative regions (acceptor) are shown in red; (ii) the hydrophobic surfaces of compounds 8 and 9 aligned over myricetin (yellow-coloured) (green-coloured regions are lipophilic, and blue and reddish-coloured (donor) areas are hydrophilic); and (iii) the 3D interaction patterns of compounds 8 and 9 in the SARS-CoV-2 3CLpro active site with non-covalent bond distances shown. | ||
Compound 8 has a chromone nucleus existing in the central D sub-pocket with the sugar part and its attached cinnamate moiety pointing towards region C. Looking at the central sub-pocket in the case of compound 8, the chromone ring of the flavonoid compound is not sticking in deep enough to achieve the vital π–π stacking with His41/His164. Rather than achieving the essential interactions, compound 8 forms several hydrogen bonds with Thr24, Ser46, Gln189 and Gln192. This explains the observed binding energy and weak protease inhibitory activity. Compound 9 differs from 8 by the absence of the cinnamate ester, which resulted in a 15-fold improvement in protease inhibitory activity. This could be explained by it having a different binding mode compared to compound 8. Compound 9 has its chromone nucleus pointed deep within the central sub-pocket D to form π–π stacking with His41/His164. In compound 9, we could see the sugar moiety pointed in sub-pocket B rather than C as is the case for compound 8. The efficient occupation of sub-pocket D and maintaining π–π stacking interactions helps to differentiate the binding affinity of compound 8 vs. that of compound 9.
Structure–activity relationship (SAR) studies demonstrated that diacylation leads to much stronger binding affinity and inhibition potency toward SARS-CoV-2 3CLpro. Compound 6 (apigenin-7-O-(3′′,6′′-O-E-p-dicoumaroyl)-β-D-glucoside; ΔG = −9.45 kcal mol; IC50 = 8.349 ± 0.35 μM) bears two coumaroyl residues on β-D-glucoside that work together to form a long and strong hydrogen bonding network with vital residues such as Thr24, Thr26, Met49, Glu47, Gly143, and Gln189, therefore anchoring the ligand across sub-pockets A and D of the active site. The monoacylated counterparts—compound 7 (apigenin-7-O-(6′′-O-E-p-coumaroyl)-β-D-glucoside; ΔG = −7.92 kcal mol; and IC50 = 65.43 ± 2.78 μM) and compound 8 (apigenin-7-O-(3′′-O-E-p-coumaroyl)-β-D-glucoside; ΔG = −7.85 kcal mol; and IC50 = 224.8 ± 9.55 μM)—have fewer hydrogen bonds (mostly with Gly143 and Gln189), and they are less stable than compound 6 because of the presence of only one coumaroyl unit. This additional acyl group in compound 6 reinforces the hydrophobic interactions with Met49 and possibly π–π stacking, which perfects the orientation and helps improve the potency.
The binding energy (ΔG) values of the isolated compounds (Table 3) generally correlate with the in vitro SARS-CoV-2 3CLpro inhibitory activities reported (Table 1), like in the case of compound 6 (ΔG = −9.45 kcal mol; IC50 = 8.349 ± 0.35 μM). However, dissimilarities were noted for some compounds, especially compound 10 (verbascoside), which shows an energetic advantage for binding (ΔG = −8.26 kcal mol−1), comparable to tipranavir (ΔG = −8.25 kcal mol−1, IC50 = 3.231 ± 0.14 μM), but shows low inhibitory potency (IC50 = 39.76 ± 1.69 μM). There could be several reasons for this discrepancy; firstly, the disaccharide part of verbascoside (β-glucose and α-L-rhamnose) and the caffeoyl group seem to be involved in a network of hydrogen bonding, favorably affecting ΔG, but they do not interact with critical residues like His41/His164 for the π–π stacking that is quite important for strong inhibition, as noted for compounds 4 and 9. Secondly, compound 10 is expected to show a large topological polar surface area (TPSA > 200 Å2), which, together with three Lipinski rule violations, indicates that it has poor cellular permeability and bioavailability which may hinder its effective concentration obtained from the in vitro assay. Similar discrepancies are noted for compound 8 (ΔG = −7.85 kcal mol; IC50 = 224.8 ± 9.55 μM), where a lack of π–π stacking with His41/His164 and poor occupancy of the sub-pocket reduced its activity, whereas π–π stacking was clearly favorable for compound 4 (ΔG = −7.47 kcal mol; IC50 = 15.14 ± 0.64 μM), enhancing its potency. Some other factors could also have contributed to these discrepancies, for example, the solubility of the compound, its stability under assay conditions, or restrictions relating to the rigid docking model.
The data derived from molecular docking studies was used with a rigid protein model of SARS-CoV-2 3CLpro (PDB ID: 7DPP) in the EA-dock workflow of the SwissDock server. Although it has been used to test binding mode predictions for the ungated compounds 1–10, through myricetin re-docking (RMSD = 1.17 Å), it may, however, restrict accuracy in some cases without protein flexibility. Rigid docking does not accommodate any adaptation by active site residues that could be alterably attached during ligand binding, which could be a cause of inconsistencies between docking energies and in vitro activities, as represented by compound 10 (ΔG = −8.26 kcal mol; IC50 = 39.76 ± 1.69 μM). A flexible docking method or MD simulations would have assured better dynamism around protein-ligand interactions, but this is out of the scope of this study because of computational resource constraints; besides, the main goal is bringing together in vitro and in silico data. Such stringent limitations can be solved partially by making use of a high-resolution crystal structure (7DPP), validating the docking protocol, and correlating it with in vitro Mpro inhibitory activities (Table 1). It would be possible to use flexible docking or MD simulations to further refine binding predictions and improve the accuracy of the computational model in future studies.
| Compound no | Molar refractivity | Fraction C sp3a | TPSAb (Å2) | iLOGPc | LogSd |
GI absorption | BBB permeant | No. of Lipinskie violations | No. of PAINSf alerts | Bioavailability score | Synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Fraction of sp3 carbons.b Total polar surface area.c Lipophilicity measure.d Aqueous solubility.e Number of violations of Lipinski's rules.f Number of alerts from the PAINS filter. | |||||||||||
| 1 | 133.23 | 0.93 | 20.23 | 5.16 | −7.90 | Low | No | 1 | 0 | 0.55 | 6.30 |
| 2 | 97.06 | 0.95 | 66.76 | 3.93 | −4.69 | High | Yes | 0 | 0 | 0.55 | 3.88 |
| 3 | 169.33 | 0.94 | 90.15 | 4.99 | −8.06 | Low | No | 2 | 0 | 0.17 | 7.83 |
| 4 | 80.48 | 0.06 | 100.13 | 2.44 | −4.06 | High | No | 0 | 0 | 0.55 | 3.06 |
| 5 | 73.99 | 0.00 | 90.90 | 1.89 | −3.94 | High | No | 0 | 0 | 0.55 | 2.96 |
| 6 | 192.58 | 0.17 | 213.42 | 3.79 | −7.40 | Low | No | 3 | 0 | 0.17 | 6.45 |
| 7 | 151.21 | 0.23 | 187.12 | 3.12 | −5.64 | Low | No | 3 | 0 | 0.17 | 5.76 |
| 8 | 151.21 | 0.23 | 187.12 | 3.63 | −5.30 | Low | No | 3 | 0 | 0.17 | 5.77 |
| 9 | 109.83 | 0.32 | 160.82 | 2.17 | −3.77 | Low | No | 1 | 0 | 0.55 | 5.03 |
| 10 | 148.42 | 0.48 | 245.29 | 2.15 | −2.87 | Low | No | 3 | 1 | 0.17 | 6.37 |
Lipophilicity is described via the partition coefficient between n-octanol and water. iLOGP is an in-house physics-based method which was developed by the Swiss Institute of Bioinformatics. It uses a generalized-Born and solvent-accessible surface area (GB/SA) model that depends on the free energies of solvation in n-octanol and water. iLOGP was benchmarked using two drug or drug-like external sets and performed equally as well as or better than six well-established predictors.48 The consensus logPo/w is the arithmetic mean of the values predicted by the five proposed methods. The saturation index is indicated by the fraction of carbons in an sp3 hybridization state; it is preferable for this to be not less than 0.25. Only compounds 4–8 have low fractions of sp3 carbons.
Water solubility is one of the characteristics that allows the ease of formulation of future drug candidates. Aqueous solubility is a major factor that influences absorption, especially for drug discovery projects targeting oral administration. The ESOL model for logS calculations is based on a fragmental method corrected by molecular weight. LogS is preferred not to be higher than 6, and our isolated compounds were soluble to poorly soluble.49 Predictions for passive gastrointestinal absorption (HIA) and permeation through the blood–brain barrier (BBB) depend on the readout from the BOILED-Egg model.47 The isolated compounds have low GIT permeability, except for 2, 4 and 5, while all the synthesized analogues exhibited low BBB permeability, except for compound 2.
Drug-likeness could be seen as an estimate in qualitative terms of the possibility for a synthesized molecule to become an oral drug candidate with respect to bioavailability. All the isolated compounds showed 0–1 violations of Lipinski's Rule of Five,50 except for compounds 3, 6, 7, 8 and 10. The Abbot bioavailability score focuses on an estimation of a compound's chances to have at least 10% oral bioavailability in a Caco-2 permeability or rat model.51 This score uses the TPSA, total charge and violations of Lipinski's rules to give compounds different probabilities of 11%, 17%, 56% or 85%. All our isolated compounds showed a score of either 0.17 or 0.55.
Medicinal chemistry friendliness is used as a guide for medicinal chemists in their endeavours to discover new drug entities. The recognition of potentially problematic pattern/fragments is a primary step in drug discovery projects to prioritize the compounds to be synthesized and screened. PAINS (pan assay interference compounds, promiscuous compounds, frequent hitters) are molecules with built-in substructures showing potent responses in multiple assays, irrespective of the target protein. Such fragments would result false positives in six orthogonal assays and be considered as a potential cause of promiscuous compounds.52 SwissADME flags warnings if such fragments are detected in the molecule of interest. All isolated compounds showed zero alerts for the PAINS filter, with the exception of compound 10. Another factor is the synthetic accessibility (SA), which helps medicinal chemists to select the most promising hypothetical molecules to be synthesized and tested in biological assays. It ranges from 1 (very easy) to 10 (very difficult) based on contributions from 1024 fragmental blocks modulated by size and complexity.53 The isolated compounds have various SA scores ranging from 2.96 to 7.83 (Table 4).
4. Limitations of the study
The present work does bear limitations, which should be considered while interpreting the results. In vitro assays of Mpro inhibition and antioxidant activity do not necessarily translate in vivo due to other factors, including bioavailability and metabolic or off-target effects. Consider, for example, the low oral bioavailability of glycosylated compounds (e.g., 6 and 10), shown by the high TPSA values and violations of Lipinski's rules (Table 4). Their clinical translation could be hampered. The small number of isolated compounds (n = 10) prohibits any wider-range structure–activity relationship investigations, including QSAR modelling. On a brighter note, notwithstanding the limitations, the validated docking protocol in the study (RMSD = 1.17 Å for myricetin), high-resolution crystal structure used, and in vitro results provide a solid foundation for the conclusions presented in this work.5. Conclusions
We investigated the phytochemical contents of Marrubium alysson L., which revealed 10 substances, including acylated apigenin glucosides and β-sitosterol, which were identified as new isolates. Compound 6 (apigenin-7-O-(3′′,6′′-E-p-dicoumaroyl)-β-D-glucoside) demonstrated superior inhibition over the control drug tipranavir when its potency against SARS-CoV-2 protease (Mpro) was tested using a tetranucleotide probe. This indicates the in vitro inhibition of Mpro with an IC50 value of 8.349 ± 0.35 μM, whereas tipranavir's IC50 value was 3.231 ± 0.14 μM. It was shown to bind by coordination through hydrogen-bonding interactions with the target during docking. Based on the ABTS antioxidant activity, verbascoside exhibited a strong IC50 value of 25.58 ± 0.12 μM compared to L-ascorbic acid (IC50 = 30.43 ± 0.14 μM); compounds 6–8 also showed favourable performance due to their p-coumaroyl moieties. Biomedical studies pointed out that the oral administration of some of these molecules is not plausible due to poor bioavailability, while chrysoeriol and apigenin were quite promising. This investigation shows the importance of M. alysson as a future optimizable source of anti-SARS-CoV-2 and antioxidant candidates.Future works should concentrate on: (1) molecular dynamics (MD) simulations for studying the dynamic stability of compounds like 6 inside the 3CLpro active site, to address the limitations of rigid docking;54,55 (2) quantitative structure–activity relationship (QSAR) analyses to further elucidate the structural determinants of the Mpro inhibition and antioxidant activities of the isolated compounds, potentially guiding the optimization of lead candidates;56 (3) the optimization of glycosylated compounds (e.g., 6 and 10) by deglycosylation or selective modification to increase bioavailability while maintaining activity; and (4) the execution of in vivo studies in animal models to confirm the therapeutic efficacy and safety of these compounds. These routes would prove to be advantageous in the design of more selective antiviral agents, with better pharmacokinetic profiles for clinical development, and validation of their efficacy toward COVID-19 and diseases related to oxidative stress, thus moving one step forward in developing the therapeutic prospects of Marrubium alysson L. compounds.
Data availability
Data supporting the submitted investigation are available in the manuscript and the ESI.†Author contributions
E. E., M. F. L. and A. F. S. designed the study. E. E. and A. F. S. carried out the laboratory work. A. R. A. carried out molecular modelling and pharmacokinetics studies. All the authors contributed to the analysis and interpretation of the results. E. E. and A. R. A wrote the manuscript in consultation with A. F. S. and M. F. L. All authors revised and approved the final manuscript and agreed to be responsible for all aspects of the work.Conflicts of interest
The authors declare no competing interests.References
- T. Mabry, K. R. Markham and M. B. Thomas, The Systematic Identification of Flavonoids, Springer Science & Business Media, 2012 Search PubMed.
- M. De Souza, R. De Jesus, V. Cechinel-Filho and V. Schlemper, Phytomedicine, 1998, 5, 103–107 CrossRef CAS PubMed.
- V. Schlemper, A. Ribas, M. Nicolau and V. Cechinel Filho, Phytomedicine, 1996, 3, 211–216 CrossRef CAS PubMed.
- H. Golmakani, H. Rabbani Nasab, M. Sharifan, H. Kamali and A. Yadollahi, J. Essent. Oil-Bear. Plants., 2016, 19, 963–971 CrossRef CAS.
- A. Karioti, J. Heilmann and H. Skaltsa, Phytochemicals, 2005, 66, 1060–1066 CrossRef CAS PubMed.
- A. Karioti, H. Skaltsa, J. Heilmann and O. Sticher, Phytochemicals, 2003, 64, 655–660 CrossRef CAS PubMed.
- D. M. Lazari, H. D. Skaltsa and T. Constantinidis, Flavour Fragrance J., 1999, 14, 290–292 CrossRef CAS.
- D. Rigano, N. A. Arnold, M. Bruno, C. Formisano, A. Grassia, S. Piacente, F. Piozzi and F. Senatore, Biochem. Syst. Ecol., 2006, 34, 256–258 CrossRef CAS.
- D. Rigano, G. Aviello, M. Bruno, C. Formisano, S. Rosselli, R. Capasso, F. Senatore, A. A. Izzo and F. Borrelli, J. Nat. Prod., 2009, 72, 1477–1481 CrossRef CAS PubMed.
- D. Rigano, A. Grassia, M. Bruno, S. Rosselli, F. Piozzi, C. Formisano, N. A. Arnold and F. Senatore, J. Nat. Prod., 2006, 69, 836–838 CrossRef CAS PubMed.
- G. Savona, F. Piozzi, L. M. Aranguez and B. Rodriguez, Phytochemicals, 1979, 18, 859–860 CrossRef CAS.
- U. Abdussalam, M. Aliyu and I. Maje, Trop. J. Nat. Prod. Res., 2018, 2, 132–135 CrossRef CAS.
- R. Alaa, M. Abd-Alhaseeb, E. Habib, A. Ibrahim and S. Ahmed, J. Chem. Pharm. Res., 2016, 8, 283–289 CAS.
- S. S. Essawy, D. M. Abo-Elmatty, N. M. Ghazy, J. M. Badr and O. Sterner, Saudi Pharm. J., 2014, 22, 472–482 CrossRef PubMed.
- M. Kozyra, A. Biernasiuk, R. Antonik, A. Malm and G. Zgórka, Acta Pol. Pharm.-Drug Res., 2018, 75, 937–950 CrossRef CAS.
- V. Tackholm and L. Boulos, Students' Flora of Egypt, Cairo university, 2nd edn, 1974 Search PubMed.
- İ. Çaliş, M. Hosny, T. Khalifa and P. Rüedi, Phytochemicals, 1992, 31, 3624–3626 CrossRef PubMed.
- N. H. Mohamed, A. S. Awaad, J. N. Govil, V. K. Singh, Recent Progress in Medicinal Plants, Studium Press LLC, 2010, vol. 27, pp. 185–193 Search PubMed.
- M. Saleh, T. Sarg, A. Metwally and A. Rakha, Planta Med., 1981, 41, 202–203 CrossRef CAS PubMed.
- H. A. Samir, M. S. Abbas, A. S. Soliman and R. A. Lotfy, Res. J. Pharm., Biol. Chem. Sci., 2018, 9(6), 82–94 CAS.
- C.-I. Choi, H. J. Eom and K. H. Kim, Russ. J. Bioorg. Chem., 2016, 42, 310–315 CrossRef CAS.
- B. R. Nallamilli, K. Nataraj and R. K. Jat, Int. J. Pharm. Pharm. Sci., 2018, 11, 1 Search PubMed.
- R. Kahl and H. Kappus, Z. Lebensm. Unters. Forsch., 1993, 196, 329–338 CrossRef CAS PubMed.
- M. Ambreen and M. Ahmad, Sci. Int., 2015, 27, 6087–6090 CAS.
- J. S. Morse, T. Lalonde, S. Xu and W. R. Liu, Chembiochem, 2020, 21, 730–738 CrossRef CAS PubMed.
- G. A. Wahab, W. S. Aboelmaaty, M. F. Lahloub and A. Sallam, RSC Adv., 2022, 12, 18412–18424 RSC.
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radic. Biol. Med., 1999, 26, 1231–1237 CrossRef CAS PubMed.
- A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS PubMed.
- M. Bugnon, U. F. Rohrig, M. Goullieux, M. A. S. Perez, A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2024, 52, W324–W332 CrossRef PubMed.
- J. Bulama, S. Dangoggo and S. Mathias, Int. J. Sci. Res., 2015, 5, 1–3 Search PubMed.
- H. López-Salazar, B. H. Camacho-Díaz, S. V. Ávila-Reyes, M. D. Pérez-García, M. González-Cortazar, M. L. Arenas Ocampo and A. R. Jiménez-Aparicio, Molecules, 2019, 24, 3926 CrossRef PubMed.
- P. K. Agrawal, R. S. Thakur and M. C. Bansal, Carbon-13 NMR of Flavonoids, Elsevier Science Publishing Company INC, 1989 Search PubMed.
- H. Itokawa, K. Suto and K. Takeya, Chem. Pharm. Bull., 1981, 29, 254–256 CrossRef CAS.
- E. Breitmaier and W. Voelter, Carbon 13-NMR Spectroscopy: High Resolution Methods and Application in Organic Chemistry and Biochemistry, 3rd edn, 1987, p. 381 Search PubMed.
- J. B. Harborne and T. J. Mabry, The Flavonoids: Advances in Research, Champan and Hall Ltd, 1982, p. 61 Search PubMed.
- Z. Güvenalp, H. Özbek, T. Ünsalar, C. Kazaz and L. Demirezer, Turk. J. Chem., 2006, 30, 391–400 Search PubMed.
- K. Yamasaki, R. Kasai, Y. Masaki, M. Okihara, O. Tanaka, H. Oshio, S. Takagi, M. Yamaki, K. Masuda and G. Nonaka, Tetrahedron Lett., 1977, 18, 1231–1234 CrossRef.
- P. Venturella, A. Bellino and M. L. Marino, Phytochemicals, 1995, 38, 527–530 CrossRef CAS PubMed.
- K. E. Heim, A. R. Tagliaferro and D. J. Bobilya, J. Nutr. Biochem., 2002, 13, 572–584 CrossRef CAS PubMed.
- P. Cos, M. Calomme, L. Pieters, A. Vlietinck and D. V. Berghe, in Studies in Natural Products Chemistry, Elsevier, 2000, vol. 22, pp. 307–341 Search PubMed.
- Z. E. Pápay and I. Antal, Eur. Sci. J., 2014, 3, 1859 Search PubMed.
- A. Grosdidier, V. Zoete and O. Michielin, Nucleic Acids Res., 2011, 39, W270–W277 CrossRef CAS PubMed.
- H. Su, S. Yao, W. Zhao, Y. Zhang, J. Liu, Q. Shao, Q. Wang, M. Li, H. Xie and W. Shang, Nat. Commun., 2021, 12, 3623 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- A. K. Agarwal, C. Xing, G. N. DeMartino, D. Mizrachi, M. D. Hernandez, A. B. Sousa, L. Martinez de Villarreal, H. G. dos Santos and A. Garg, Am. J. Hum. Genet., 2010, 87, 866–872 CrossRef CAS PubMed.
- P. Ertl, B. Rohde and P. Selzer, J. Med. Chem., 2000, 43, 3714–3717 CrossRef CAS PubMed.
- A. Daina and V. Zoete, ChemMedChem, 2016, 11, 1117–1121 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, J. Chem. Inf. Model., 2014, 54, 3284–3301 CrossRef CAS PubMed.
- J. S. Delaney, J. Chem. Inf. Comput. Sci., 2004, 44, 1000–1005 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Deliv. Rev., 2012, 64, 4–17 CrossRef.
- Y. C. Martin, J. Med. Chem., 2005, 48, 3164–3170 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- Y. Fukunishi, T. Kurosawa, Y. Mikami and H. Nakamura, J. Chem. Inf. Model., 2014, 54, 3259–3267 CrossRef CAS PubMed.
- A. Agrwal, S. Juneja, S. Dwivedi and V. Kasana, Mater. Today: Proc., 2022, 57, 2250–2254 CAS.
- S. Bhandari, A. Agrwal, V. Kasana, S. Tandon, Y. Boulaamane and A. Maurady, ChemistrySelect, 2022, 7(48), e202201572 CrossRef CAS.
- A. Agrwal, A. Verma, N. Chantola, S. Verma and V. Kasana, J. Environ. Sci. Health, Part B, 2022, 57, 379–420 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra03213j |
| This journal is © The Royal Society of Chemistry 2025 |