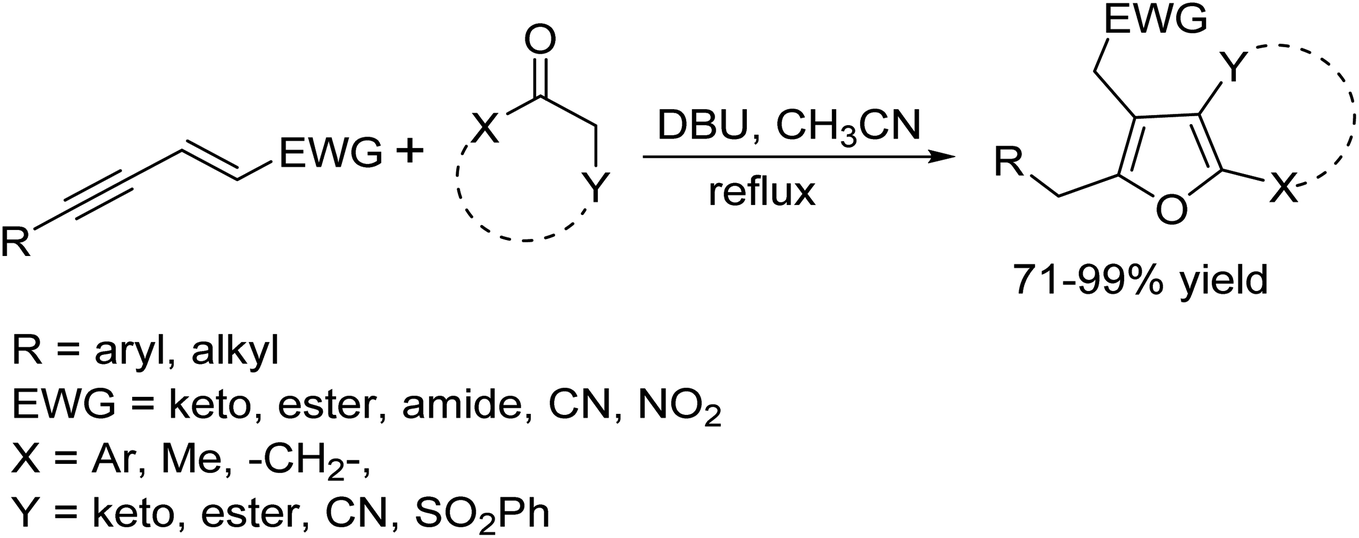
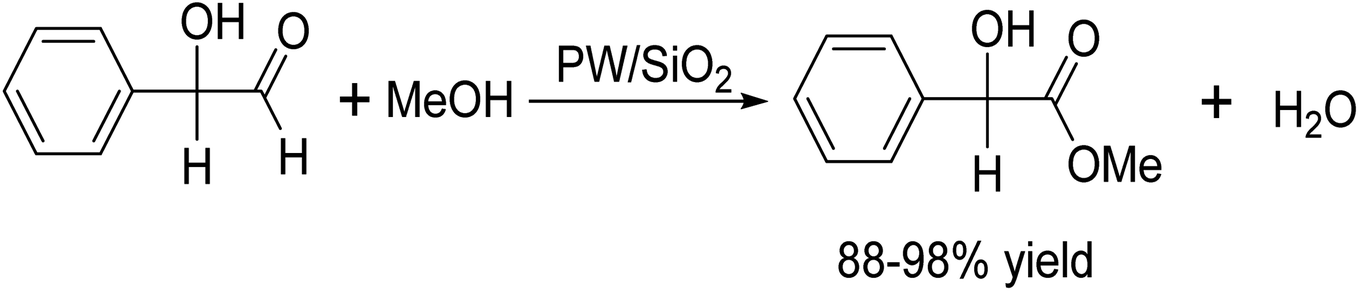
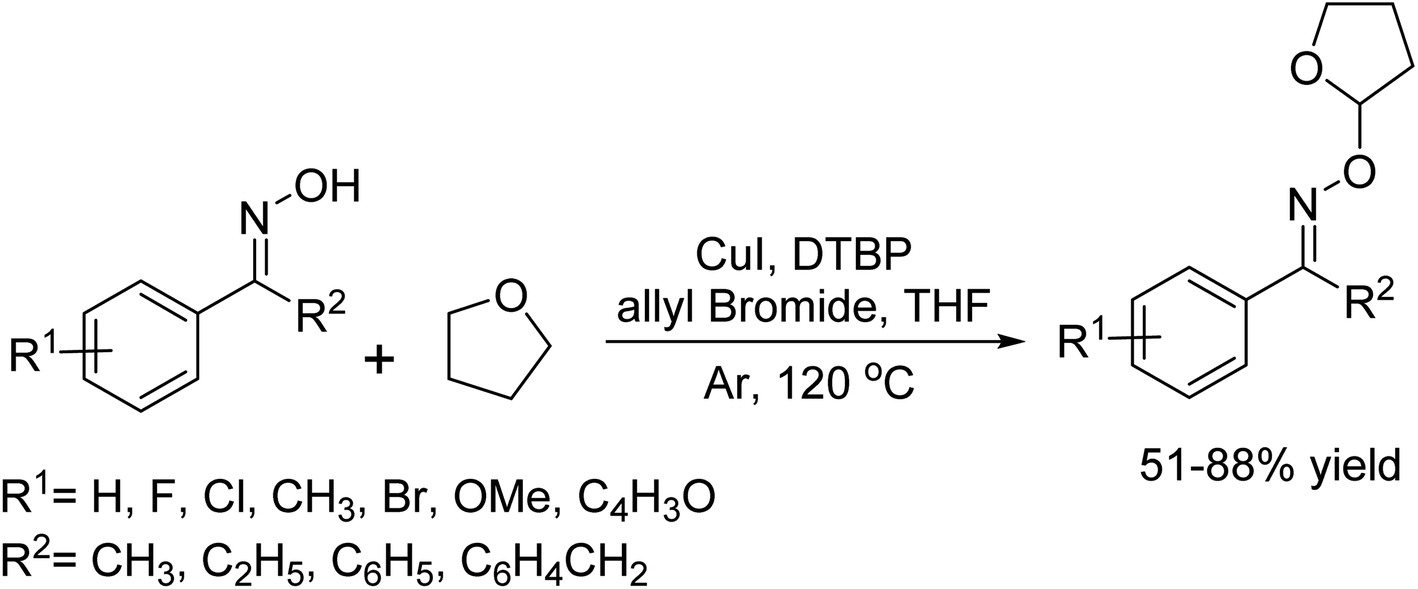
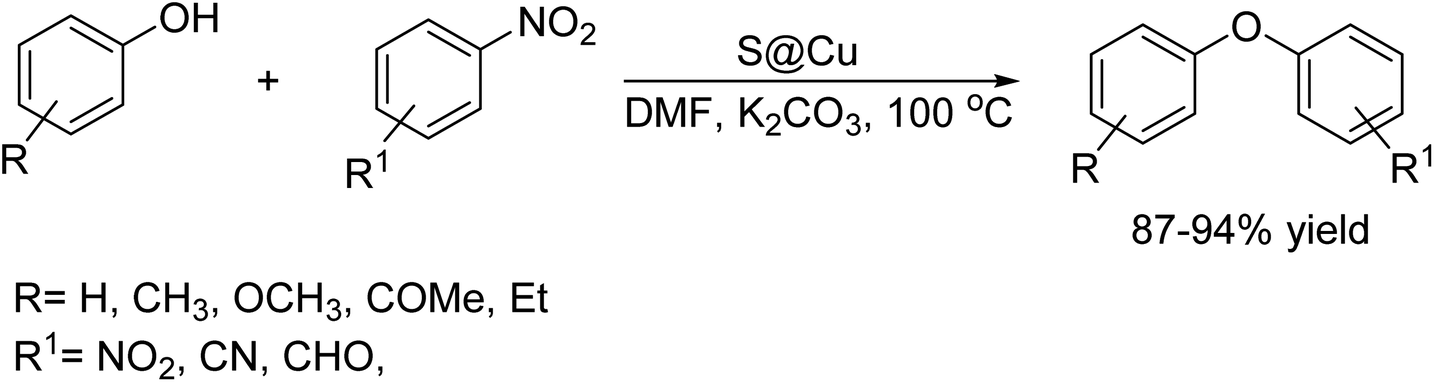
DOI:
10.1039/D5RA03124A
(Review Article)
RSC Adv., 2025,
15, 23704-23759
Frontiers in bond construction: innovative approaches to C–C and carbon–heteroatom (C–S, C–N, C–O, C–Se, C–P) transformations-a review
Received
3rd May 2025
, Accepted 21st June 2025
First published on 10th July 2025
Abstract
This comprehensive review explores the cutting-edge innovations driving modern bond construction, with a specific focus on C–C and carbon-heteroatom (C–S, C–N, C–O, C–Se, C–P) transformations. It critically examines how groundbreaking catalytic strategies including transition-metal catalysis, solid-face, photolytic protocols, and organocatalytic systems are revolutionizing synthetic methodologies by enabling unprecedented levels of selectivity, functional group tolerance, and operational simplicity under mild reaction conditions. The review dissects of these transformations, highlighting how advanced reaction protocol has paved the way for the rational design of next-generation catalytic methodology that overcomes classical synthetic limitations. Emphasis is placed on sustainable and environmentally benign approaches that integrate with green chemistry principles, thus broadening the applicability of these methods in the synthesis of complex molecules for pharmaceuticals, materials science, and other interdisciplinary applications. Future perspectives, challenges, and emerging trends are also discussed, offering valuable insights into the ongoing evolution of bond construction strategies and setting the stage for continued innovation in organic synthesis.
Introduction
The construction of molecular frameworks through precise bond formation stands at the very heart of organic synthesis.1 Among the myriad bonds that define organic molecules, the carbon–carbon (C–C) bond serves as the backbone of countless natural and synthetic compounds, while bonds formed between carbon and heteroatoms such as sulfur, nitrogen, oxygen, selenium, and phosphorus impart key functional attributes that dictate physical properties, biological activity, and reactivity profiles.2 In recent decades, the relentless pursuit of more efficient, selective, and sustainable synthetic routes has ignited a transformative wave of innovation in the methodologies used to forge these bonds.3 This review aims to survey and critically assess the latest advances in the construction of C–C as well as carbon-heteroatom bonds, providing an integrated perspective on catalytic innovations, and future directions in this dynamic field. Historically, the formation of C–C bonds was dominated by classical processes such as the Wurtz coupling, Grignard reactions, and aldol condensations methods that, while groundbreaking in their time, often suffered from limitations including harsh reaction conditions, limited selectivity, and functional group incompatibility.4 Similarly, the formation of carbon-heteroatom bonds has traditionally relied on nucleophilic substitution and electrophilic aromatic substitution reactions, which, although useful, sometimes necessitated forcing conditions that compromised yield and selectivity.5 Over time, these traditional methods have been augmented and in many cases supplanted by more modern, catalytic approaches that have revolutionized the field of synthetic chemistry. A major turning point came with the advent of transition metal catalysis. Pioneering cross-coupling reactions, such as those developed by Suzuki, Heck, and Negishi, transformed the synthetic landscape by offering chemists an unprecedented ability to construct intricate carbon networks under comparatively mild conditions.6 In parallel, the formation of bonds between carbon and heteroatoms has benefited from similar catalytic strategies that employ metals like palladium, nickel, copper, and even earth-abundant alternatives.7 These developments have unlocked a new dimension in the synthesis of complex molecules by affording chemoselectivity and stereo control that were once unattainable using conventional methods. In addition to transition metal catalysts, the last decade has witnessed a surge of interest in alternative activation strategies. Photolytic catalysis, for instance, leverages visible light to generate reactive radical intermediates in a controlled manner, enabling bond constructions that operate under mild conditions and in many cases display remarkable substrate tolerance.8 Similarly, organo catalysis has matured into a highly versatile tool, providing metal-free alternative protocols that are characterized by low toxicity and environmental impact.9 The successful integration of these modern methodologies has widened the toolbox available for the practical synthesis of molecules with densely functionalized architectures, including those featuring C–S, C–N, C–O, C–Se, and C–P bonds. The synthetic importance of carbon-heteroatom bonds cannot be overstated. Heteroatom incorporation into molecular frameworks plays a crucial role in modulating the electronic and steric environments of bioactive compounds, thereby directly impacting phenomena such as solubility, reactivity, and intermolecular interactions.10 For instance, the installation of oxygen or sulfur moieties can drastically alter the pharmacokinetic properties of a drug candidate, enhancing its potential as a therapeutic agent. In materials science, the introduction of heteroatoms into polymer backbones or conjugated systems can lead to significant changes in conductivity, luminescence, and other essential physical properties. Given these broad-ranging applications, the development of innovative and efficient routes to access such bonds is a research priority that bridges multiple disciplines including medicinal chemistry, materials science, and chemical biology. Despite the impressive advances achieved so far, several challenges persist in the realm of bond construction. One of the primary hurdles lies in the precise control of regio- and stereo selectivity. Traditional methods often require additional steps or protecting group strategies to achieve the desired selectivity, which in turn reduces overall efficiency. Modern catalytic systems have begun to address these issues by enabling direct functionalization and late-stage modifications of complex molecules.11 Moreover, the reaction conditions have been tuned to be more benign, thereby permitting the incorporation of sensitive functional groups that would otherwise be incompatible with harsher protocols. These achievements reflect not only the ingenuity in catalyst design but also the deep mechanistic understanding that has been achieved through experimental studies.12 Another significant dimension of current research is its commitment to sustainability.3 In today's environmentally conscious world, synthetic methodologies are increasingly evaluated not only for their efficiency and selectivity but also for their environmental impact.3 Innovations in bond construction now frequently incorporate principles of green chemistry, such as the use of renewable feed stocks, solvent-free conditions, or water as a benign reaction medium. By reducing waste, minimizing energy consumption, and employing recyclable catalysts, modern techniques are setting new standards for sustainable organic synthesis.3 In this context, the field is moving toward methods that balance creative synthetic solutions with an ever-growing emphasis on environmental stewardship. This review takes stock of the myriad approaches that have emerged at the frontiers of bond construction. It begins by tracing the evolution from traditional, often laborious methodologies to today's state of the art catalytic processes. Emphasis is placed on the mechanistic underpinnings that have allowed chemists to rationally design and optimize reactions for both C–C formation and carbon-heteroatom bond constructions.6,12 Particular attention is given to how innovative approaches ranging from transition metal catalysis to photolytic and organo catalysis have addressed longstanding challenges, such as limited substrate scope and functional group interference.8,9 At the heart of these advances is a trend toward the integration of diverse catalytic modalities. For example, dual catalytic systems that combine transition metal catalysis with photoredox activation have emerged as powerful strategies to achieve transformations that are both highly selective and operationally simple.7,8 Such synergistic approaches highlight the importance of combining mechanistic insights from different subfields to achieve overall process improvements.11,12 Furthermore, the development of efficient ligand frameworks and the application of computational modeling have provided further impetus to the rational design of catalytic systems, pushing the boundaries of what can be achieved in modern synthetic chemistry.11 Notably, the scope of these innovative strategies extends well beyond conventional small-molecule synthesis. In the pharmaceutical arena, for instance, new bond construction methodologies facilitate the rapid assembly of complex, multi-functional compounds, thereby accelerating drug discovery and development.10 Similarly, in the area of materials science, advances in the synthesis of heteroatom-rich frameworks directly translate into the creation of new polymers and functional materials with extraordinary properties.13 These interdisciplinary applications underscore how breakthroughs in bond construction are not isolated achievements but are integrally linked to technological and scientific progress across several domains. The review presented here aims to provide a detailed and critical overview of recent innovations in bond construction with a dual focus on C–C and carbon-heteroatom (C–S, C–N, C–O, C–Se, C–P) transformations. By delving into the technological and environmental aspects of these advances, this study not only highlights the current state of the art but also identifies the ongoing challenges and potential future trajectories in this vibrant field. The discussion is framed within the broader context of synthetic strategy development, emphasizing how the evolution of bond construction methodologies can unlock new possibilities for the design and synthesis of complex molecular architectures. Ultimately, by bridging classical approaches with modern innovations, this review seeks to illuminate the exciting frontiers in bond construction and to inspire further advancements that will continue to shape the future of organic synthesis.
Transition metal free reactions of C–C bond formation
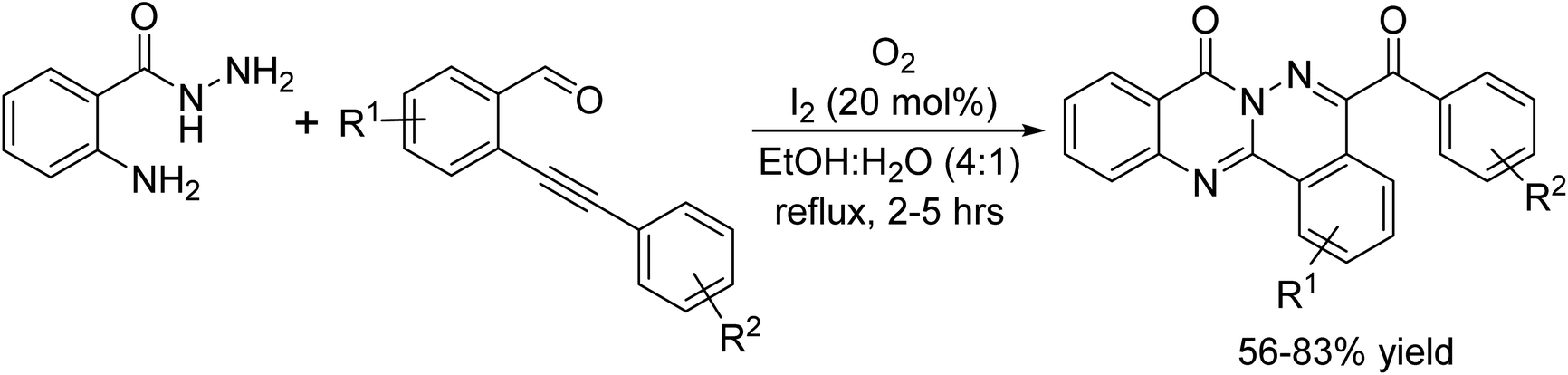
In 2016, Zhigang Lv et al. was reported an innovative protocol for the oxidative C–C bond forming reaction from C(sp3)–H and C(sp2)–H bonds has been used to contrive quinazoline framework from N,N′-disubstituted amidines catalyzed by I2/KI.14 The essential reactants are readily made ready from the corresponding RCOCl, RNH2, and C6H5CH2NH2 by successive amidation, chlorination, and amination reactions followed by oxidative cyclization, all these amidines were converted into the demand products in low to high yields. This is a greener approach that works well with crude amidine intermediates and the procedure is equally viable on a gram scale (Scheme 1).
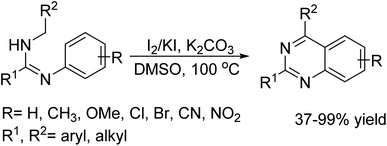 |
| | Scheme 1 I2/KI mediated oxidative C–C bond formation reaction. | |
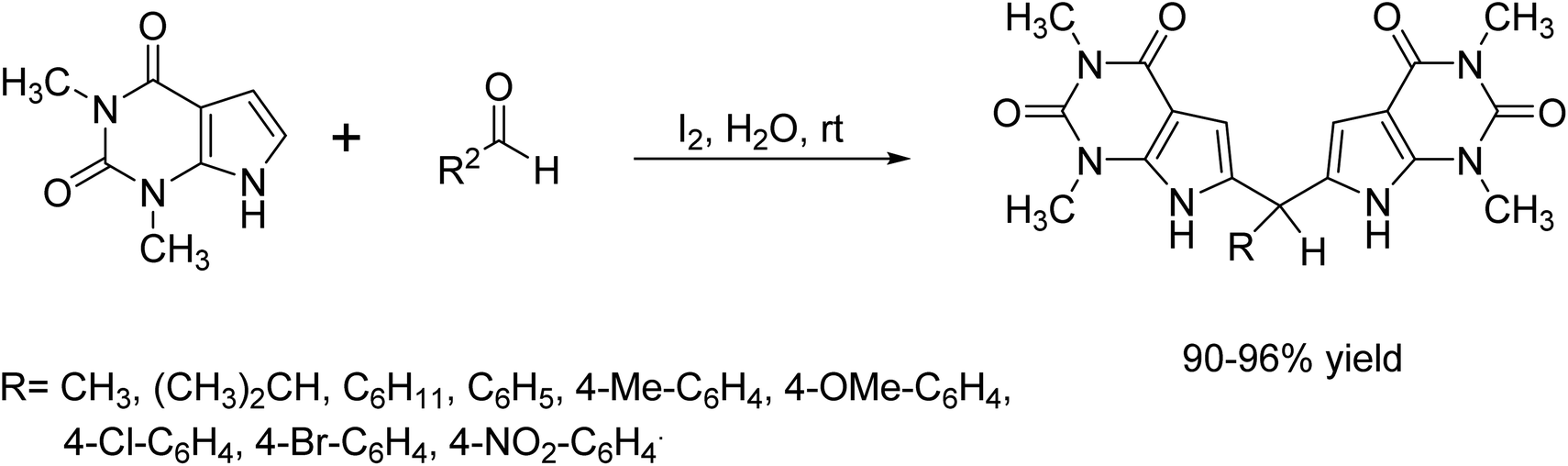
In 2014, M. Sharma et al.15 developed a green method for carbon–carbon bond formation in H2O medium. They synthesized a novel class of compounds called bis(pyrrolo[2,3-d]-pyrimidinyl)methanes from 1,3-dimethylpyrrolo[2,3-d]pyrimidine-2,4-dione and benzaldehyde in the presence of iodine as a catalyst (Scheme 2).
 |
| | Scheme 2 I2 catalyst carbon–carbon bond formation reaction in H2O medium. | |
In 2018, Constantin-Christian A. Voll and his team introduced a brilliant method for building extended aromatic structures using a clever dehydration-based C–C coupling process that starts with easily available diols.16 By treating these diols with a Brønsted acid, specifically p-TSA, the reaction sparks a nucleophilic addition of an arene or heteroarene, transforming the starting materials into fully aromatic compounds with impressive yields. This method works smoothly with a range of coupling partners such as thiophenes, furan, indole, and N,N-dimethylaniline. Even better, the reaction runs under gentle, open-flask conditions, maximizing atom efficiency and offering a refreshing alternative to traditional metal-catalyzed cross-coupling techniques (Scheme 3).
 |
| | Scheme 3 C–C bond formation extended π-conjugation catalyzed by p-TSOH·H2O. | |
In 2016, Alexander Lipp and colleagues unveiled an innovative, light-triggered method that forms new C–C bonds without using any transition metals.17 They demonstrated that 2-chlorobenzazoles can be effectively coupled with aliphatic carbamates, alcohols, and ethers using a simple, elegant setup. The reaction employs common and affordable reagents, sodium acetate, benzophenone, water and acetonitrile and uses a readily available, energy-saving 25 W UV-A lamp to drive the process at room temperature. This approach not only reduces reliance on expensive metal catalysts but also highlights a more sustainable path in synthetic chemistry (Scheme 4).
 |
| | Scheme 4 C–C bond formation from 2-chlorobenzazoles with alcohols. | |
In 2016, Zhengbao Xu et al.18 was reported a domino reaction initiated by free radicals, integrating radical addition, cyclization, and C(sp3)–C(sp3) bond formation. In this process, three new C–C bonds are formed sequentially. However, this method marks the first e.g., of cascade C–C bond formation accomplished through the selective functionalization of α-hydroxyl C(sp3)–H bonds in fluorinated alcohols (Scheme 5).
 |
| | Scheme 5 Ordered domino C–C bond formation reaction triggered by free-radical. | |
In 2014, Pramod B. Thakur et al.19 developed a novel protocol for a new C–C bond for the catalyst-free synthesized 3-hydroxy-2-oxindole scaffolds in water medium from isatin and 3-methyl-2-pyrazolin-5-one (Scheme 6).
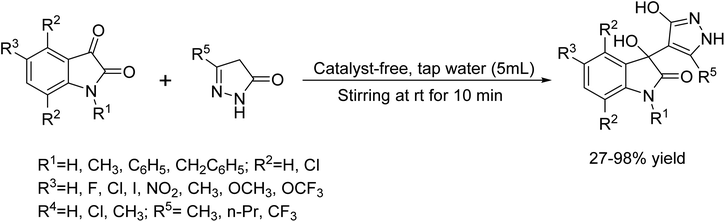 |
| | Scheme 6 Synthesis of 3-hydroxy-2-oxindole scaffolds in H2O medium condition. | |
In 2009, M. Bayat and colleagues introduced an unconventional method for the synthesis of 2,2′-aryl methylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one) derivatives.20 Their clever approach involves reacting dimedone with aromatic aldehydes in H2O medium at room temperature (Scheme 7). This protocol follows the normal work-up procedure and is refreshingly straightforward, bypassing the simple column chromatography. The synthesized compounds were characterized by FTIR, 1H NMR, 13C NMR spectroscopy, mass spectrometry, and elemental analysis.
 |
| | Scheme 7 Synthesis of 2, 2′-arylmethylene bis(3-hydroxy-5,5-dimethyl-2-cyclohexene-1-one in water. | |
In 2008, M. Lakshmi Kantam and colleagues presented a report demonstrating a recyclable and simple protocol21 conducted at ambient temperature, the direct alkylation of N-heterocycles, such as indoles and pyrroles, with epoxides through Friedel–Crafts reaction. This innovative protocol suggested to used the ionic liquid [bmim][OTf] as a functional catalyst and reaction medium (Scheme 8).
 |
| | Scheme 8 Synthesis of alkylated N-heterocycles and pyrrole through the ionic liquids catalyzed Friedel–Crafts reaction. | |
In 2012, Kumar et al. introduced an innovative protocol for the one-pot synthesis of 3-amino-alkylated indoles catalyzed by ingenious catalyst L-proline.22 This innovative method harnesses a three-component Mannich reaction, reacting 2°–amines with aldehydes, and indoles, under neat conditions at normal temperature. The process not only exemplifies efficiency but also emphasizes sustainable, green chemistry (Scheme 9).
 |
| | Scheme 9 Synthesis of 3-amino-alkylated indoles catalyzed by L-proline. | |
In 2007, an efficient and cost-effective protocol for the synthesis of bis(indol-3-yl)methanes from indoles and carbonyl compounds catalyzed by sulfamic acid at room temperature under neat conditions was demonstrated by An et al.23 (Scheme 10).
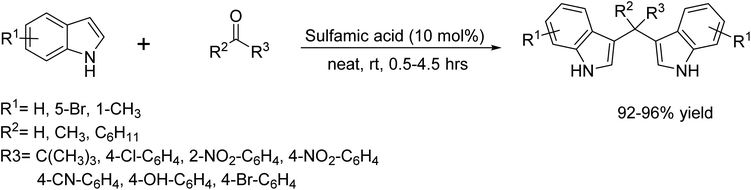 |
| | Scheme 10 Synthesis of bis(indol-3-yl)methanes catalyzed sulfamic acid. | |
In 2014, Freitas et al.24 developed a metal- and catalyst-free protocol for the efficient allylation of aldehydes to form homoallylic alcohols from aldehyde and potassium allyltrifluoroborate at room temperature under the influence of ultrasound irradiation (Scheme 11).
 |
| | Scheme 11 Ultrasound-assisted catalyst-free synthesis of homoallylic alcohols. | |
Narayanaperumal and colleagues have developed a solvent-free conjugate Michael addition method using 2,4-pentanedione and various nitroalkenes.25 In their approach, they utilized a catalytic amount of 1-methyl-3-(2-(piperidin-1-yl)ethyl)-1H-imidazole-3-ium chloride, a base-type, task-specific ionic liquid (TSIL) with ultrasonication. This clever setup efficiently produced the desired conjugate adducts in yields ranging from moderate to high (Scheme 12).
 |
| | Scheme 12 Ultrasound-assisted synthesis of Michael adducts catalyzed by Ionic liquid. | |
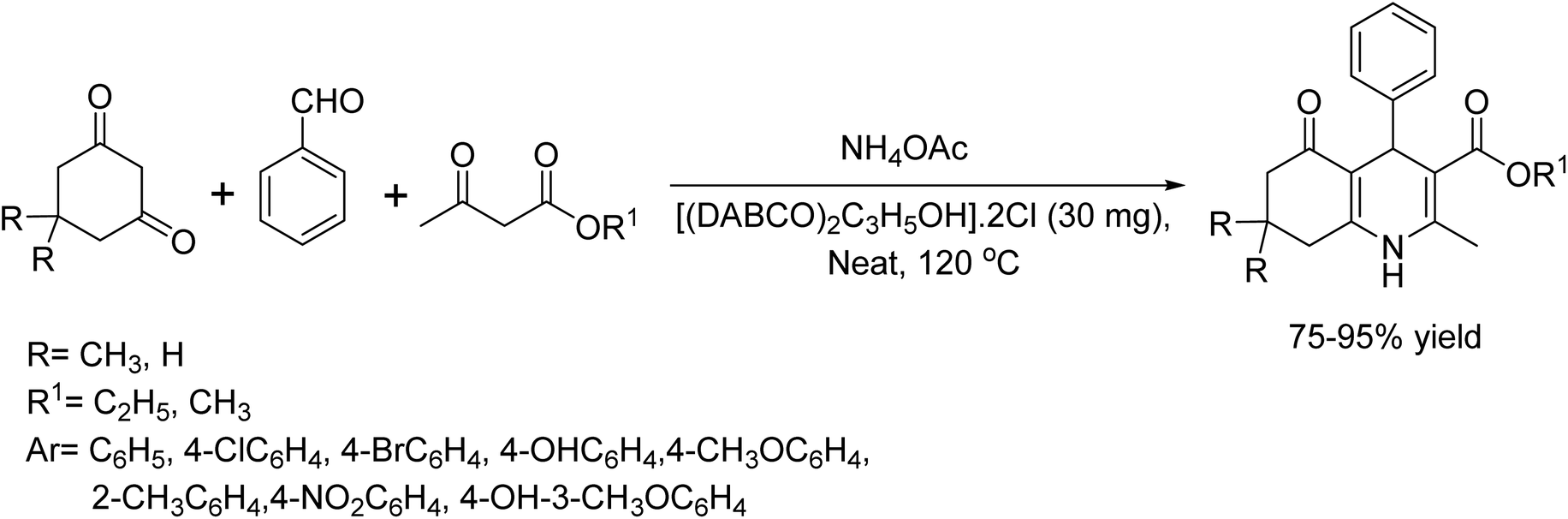
In 2020, Mehdi Zabihzadeh and colleagues synthesized polyhydroquinoline derivatives from aldehydes, 1,3-diketones, ethyl acetate, and NH4OAc using an affordable DABCO-based bis-dicationic ionic salt ([(DABCO)2C3H5OH]·2Cl) as the catalyst.26 The method offers several significant features, including ease of catalyst preparation and handling, high catalytic activity, short reaction times, absence of column chromatographic separation, and a straightforward work-up procedure. The advantageous feature of this method is to easily recover and reuse the catalyst for several cycles. It not only enhances the practicality of the process but also contributes to sustainability by minimizing waste. Researchers often seek such efficient and eco-friendly approaches in their synthetic method (Scheme 13).
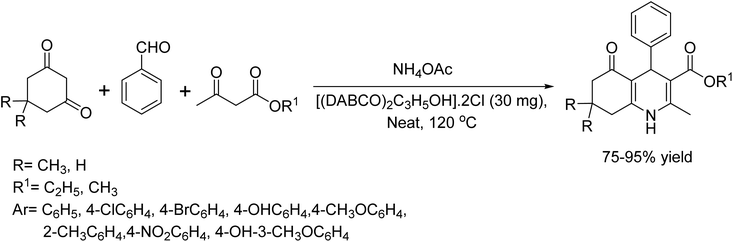 |
| | Scheme 13 [(DABCO)2C3H5OH]·2Cl catalyzed the synthesis of polyhydroquinoline derivatives. | |
In 2020, Dilip Aute and colleagues reported an environmentally viable and efficient protocol for synthesizing polyhydroquinolines.27 They utilized a sulfated polyborate catalyst in a Hantzsch four-component condensation involving aromatic aldehydes, dimedone, ammonium acetate, and either ethylacetoacetate or ethylcyanoacetate. The reaction occurred at 100 °C under solvent-free conditions. This innovative method truly shines with several standout features. It employs a catalyst with a gentle Brønsted acidic character, delivering impressive product yields of 85–94% in just 18–30 minutes. Operating under neat conditions, it boasts streamlined procedures and impressive versatility across a wide range of substrates (Scheme 14).
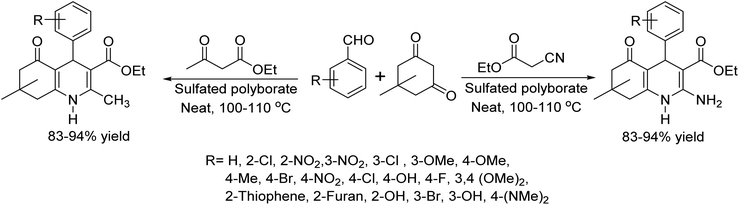 |
| | Scheme 14 Synthesis of polyhydroquinoline derivatives. | |
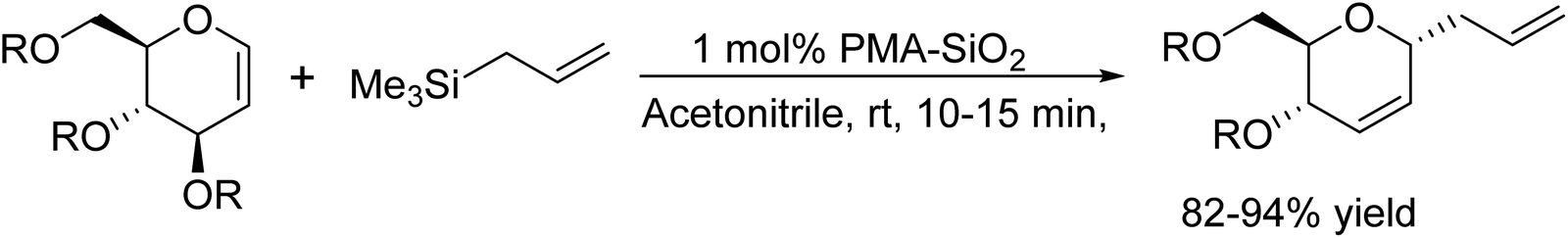
Brønsted acid catalysts have found widespread application in various organic transformations. To address limitations associated with toxicity, volatility, high cost, and hazardous properties of conventional methods, these catalysts are immobilized on silica gel. This approach offers several benefits, including ready availability, a straightforward work-up procedure, extended catalytic lifespan, environmental friendliness, and consistently high yields with recyclability. In this discussion, we explore examples of previously reported silica-supported organic syntheses. In 2006, Yadav and colleagues made a significant breakthrough in the synthesis of 2,3-unsaturated glycopyranosides using the Ferrier rearrangement.28 They employed phosphomolybdic acid (PMA) supported on silica gel as a mild, efficient, and reusable catalyst. Notably, PMA belongs to the class of heteropoly acids (HPAs), which exhibit significantly higher activity compared to traditional acids like H2SO4, TsOH, BF3·Et2O and ZnCl2. The use of HPAs often enables catalytic processes to be conducted at low concentrations and lower temperatures. In this specific case, PMA-SiO2 demonstrated remarkable selectivity, excellent product yields, minimal catalyst loading (only 1 mol%), and reusability. The synthetic versatility of phosphomolybdic acid supported on silica gel (PMA-SiO2) was further explored, leading to successful syntheses of 2,3-unsaturated allyl C-glycosides as well as O- and S-glycosides. The reactions proceeded smoothly at room temperature using allyltrimethylsilane. When alcohols and thiols replaced allyltrimethylsilane, the corresponding O- and S-glycosides were obtained in excellent yields within 10–15 minutes. Overall, utilizing PMA-SiO2 as a catalyst for this transformation offers distinct advantages, including high selectivity, efficient product formation, minimal catalyst requirements, and recyclability (Scheme 15).
 |
| | Scheme 15 Synthesis of 2,3-unsaturated glycopyranosides using PMA. | |
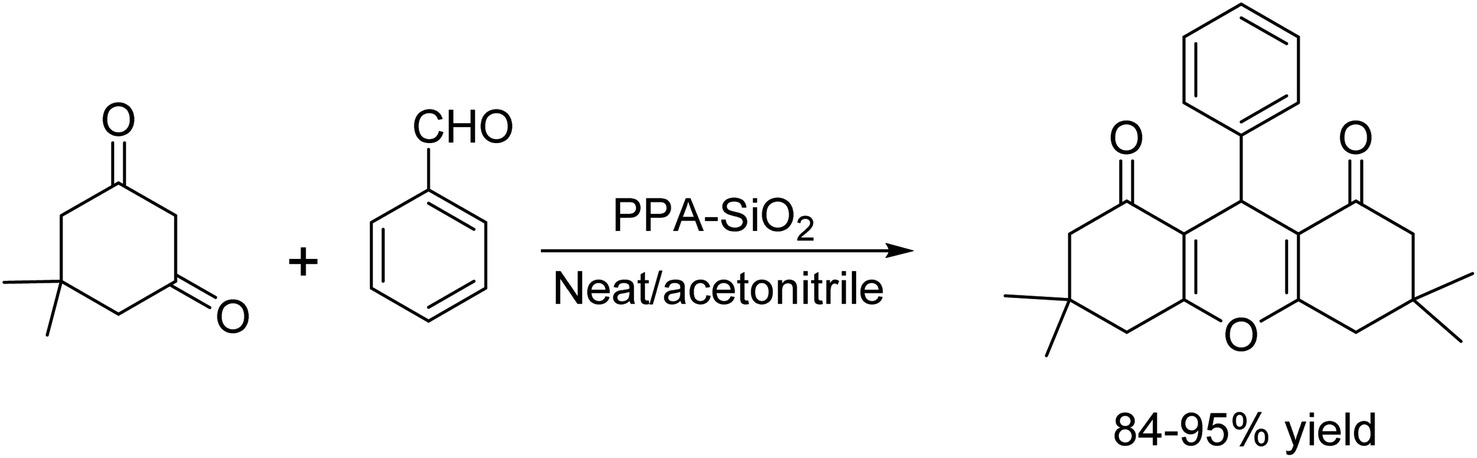
Kantevari et al. introduced an innovative method for the one-pot two-component synthesis of 1,8-dioxo-octahydroxanthene derivatives. This protocol effortlessly brings together three key reactions, Knoevenagel condensation, Michael addition, and cyclodehydration of dimedone with broad range of aldehydes as reactants, both in acetonitrile medium and under neat conditions.29 They utilized PPA-SiO2 (polyphosphoric acid supported on silica gel) as the catalyst, which resulted in the formation of 1,8-dioxo-octahydroxanthene with excellent yields. This method proved advantageous over previously reported synthetic methods that used Lewis acids, which were plagued by long reaction times, low yields, complex catalyst preparation, and poor selectivity. The benefits of using PPA-SiO2 included straightforward work-up procedures, cost-effectiveness of the catalyst, and its simple preparation, making it a superior alternative to conventional methods (Scheme 16).
 |
| | Scheme 16 Synthesis of 1,8-dioxo-octahydroxanthene using PPA/SiO2. | |
Khojastehnezhad and team synthesized polyhydroquinoline derivatives through a one-pot, four-component Hantzsch condensation reaction.30 This reaction involved ArCHO, dimedone, CH3COCH2COOEt, and NH4OAc, catalyzed by SiO2 supported polyphosphoric acid (SiO2- PPA), under neat conditions, resulting in high yields. The method offered superiority over other techniques, such as shorter reaction times, a clean reaction profile, and straightforward experimental and work-up procedures. Additionally, the catalyst could be recovered with excellent yield and reused three times without a significant loss of activity, highlighting its efficiency and sustainability (Scheme 17).
 |
| | Scheme 17 Synthesis of polyhydroquinoline derivatives using PPA-SiO2. | |
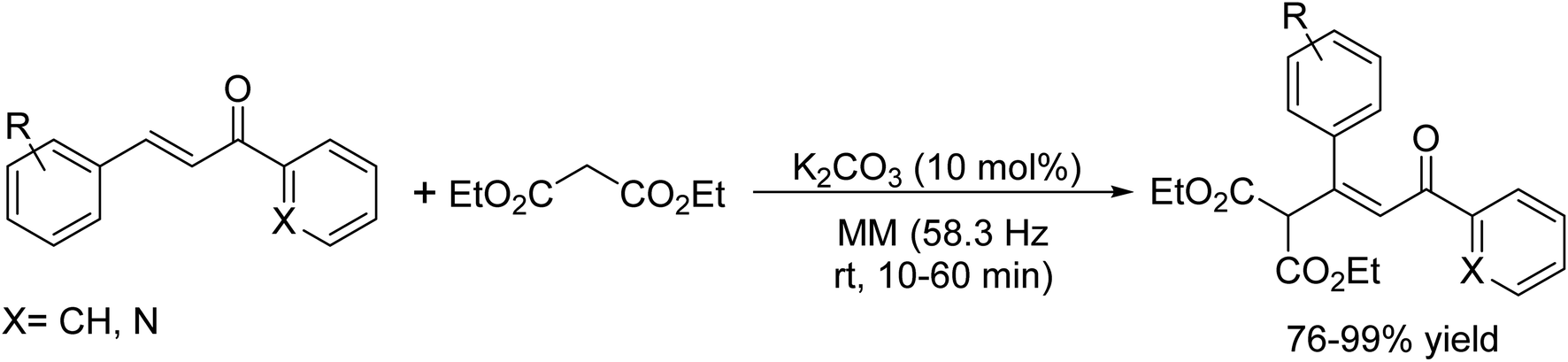
The Michael addition, traditionally performed in organic solvents with strong bases, was revolutionized by Wang and coworkers in 2004. They reported the first mechanochemical Michael reaction using a weak base, K2CO3, to catalyze the addition of 1,3-dicarbonyl compounds to chalcones and azachalcones.31 This solvent-free approach utilized diethyl malonate and an equimolar amount of chalcone or azachalcone, with 10 mol% K2CO3 in a high-speed vibration mill (HSVM). The reaction was conducted at a rate of 3500 rpm (58.3 Hz) for 10–60 minutes, resulting in Michael adducts with impressive yields ranging from 76% to 99% (Scheme 18).
 |
| | Scheme 18 Michael addition reaction of 1,3-dicarbonyl compounds to chalcones and azachalcones catalyzed by weak base K2CO3. | |
Building on the initial triumphs in organocatalytic enantioselective reactions, Bolm and colleagues delved deeper into the realm of green chemistry in 2006. They meticulously explored the proline-catalyzed aldol reaction between ketones and aromatic aldehydes using a ball mill, a technique that aligns with sustainable chemical practices.32 This solvent-free enantioselective aldol reaction, conducted at rotation speeds ranging from 250 to 400 rpm throughout 5 to 36 h, culminated in the predominant formation of anti-isomers. The process achieved impressive stereoselectivities, with diastereomeric ratios reaching up to 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 and enantiomeric excesses as high as 49:51% (Scheme 19).
:7 and enantiomeric excesses as high as 49:51% (Scheme 19).
 |
| | Scheme 19 Solvent-free enantioselective aldol reaction performed under mechanochemical condition. | |
Transition metal catalyzed reactions of C–C bond formation
In 2016, Bo Huang et al. was reported the para-quinone methides (p-QMs) can be activated by a Lewis acid, rendering them susceptible to nucleophilic attack by diazo compounds. This activation sets off a rearrangement in which N2 gas is extruded and a new C–C double bond is formed, effectively constituting a metathesis reaction process.33 Therefore, the diazoester is converted into tetrasubstituted alkenes, whereas the diazo-oxindoles dispatch the quinolinone products. In addition, 13C-labeling experiments were performed to shed light on the possible reaction mechanism (Scheme 20).
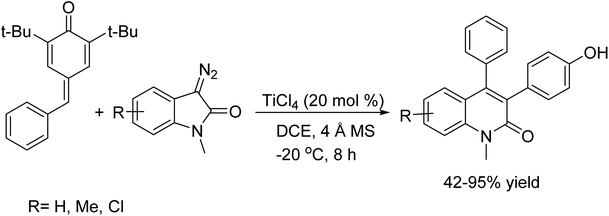 |
| | Scheme 20 Metathesis reaction for C–C bond formation reaction. | |
In 2022, Mingzhou Huang and colleagues demonstrated the successful synthesis of a extensive range of indolizines with allenes under ball-milling conditions.34 Through a [3 + 2] annulation process, they obtained numerous substituted pyrrolo[2,1,5-cd]indolizines in satisfactory yields. The key factor enhancing the reaction efficiency was the use of piezoelectric material as a charge transfer catalyst. The resulting pyrrolo[2,1,5-cd]indolizine compounds were also characterized for their photophysical properties. The mechanochemical reaction experiments were conducted using a model PMQW 04 unidirectional planetary ball mill. This mill operated at an optimum voltage of 220 V, an output power of 750 W, and speed of a rotor 550 rpm. Additionally, a blue LED from JIADENG (LS) was employed for irradiation, with a light power of 20 W and a light wavelength order of 450–460 nm (Scheme 21).
 |
| | Scheme 21 Ball milling induced dehydrogenation coupling and [3 + 2] cycloaddition of indolizines with allenes. | |
In 2015, Zhikun Zhang and his team unveiled a clever Cu(I) catalyzed cross-coupling reaction that not only builds a new C–C bond but also triggers a β-fluoride elimination.35 Their approach deftly marries terminal alkynes with trifluoromethyl ketone N-tosylhydrazones, offering an efficient pathway to synthesize 1,1-difluoro-1,3-enyne derivatives. At the heart of this transformation lies the generation of a copper carbene intermediate. This reactive species migrates into position and initiates the formation of the C–C bond. Once established, the reaction naturally progresses to a critical C–F bond cleavage, completing the synthesis in a beautifully orchestrated two-step mechanism (Scheme 22).
 |
| | Scheme 22 Cross-coupling of terminal alkynes catalyzed by Cu(I) catalyst. | |
In 2013, Guo-Bao Huang and his team presented an innovative Cu(OTf)2-catalyzed reaction that forges sp3–sp2 C–C bonds through the straight coupling of propargylic alcohols with terminal alkenes.36 This process operates under merciful conditions, even profitable in the appearance of air, which speaks volumes about its robustness and practical appeal. Its high atom economy aligns perfectly with the tenets of modern green chemistry, making it both efficient and environmentally friendly. Ultimately, this protocol unlocks a versatile route to a rich diversity of 1,4-enynes, delivered in high to excellent yields (Scheme 23).
 |
| | Scheme 23 Atom-economical C–C bond formation reaction catalyzed by Cu(OTf)2. | |
In 2014, Jing Peng and colleagues reported a breakthrough in the realm of copper-catalyzed organic transformations.37 They illustrated a Cu-catalyzed aryl carbocyclization reaction of alkynes using diaryliodonium salts, which interestingly accomplished C–C bond formation on a rather inert C(sp3)–H bond. This innovative method efficiently cyclizes alkyl alkynes to form carbocycles with excellent step economy, making the procedure concise. This truly stands out is the theoretical study accompanying this work. It revealed a fascinating, concerted copper-catalyzed pathway for the C–C bond formation, offering deep mechanistic insights that not only enhance our understanding but also open new avenues for designing related transformations (Scheme 24).
 |
| | Scheme 24 Cu-catalyst C–C bond formation on inert C(sp3)–H bond with diaryliodonium salts. | |
In 2004, Mona Hosseini Sarvari and Hashem Sharghi developed an efficient neat protocol38 for Friedel–Crafts acylation of aromatic compounds using nontoxic, inexpensive and reusable ZnO powder as a solid catalyst (Scheme 25).
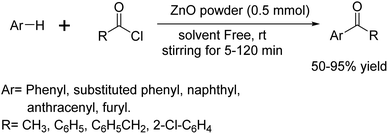 |
| | Scheme 25 Synthesis of substituted ketones via ZnO-catalyzed Friedel–Crafts acylation. | |
In 2018, Fahimeh Pakdaman et al.39 efficiently synthesized polyhydroquinolines using a novel Co(II) complex based on 5-nitro-N1-((pyridine-2-yl)methylene)benzene-1,2-diamine, (CoL2). This eco friendly catalyst provide a one-pot multi component Hantzsch condensation of dimedone, aldehydes, ethylacetoacetate, and ammonium acetate under both solvent medium and neat conditions. Notably, this method has many advantages such as easy product isolation, environmental friendliness, and excellent yields (Scheme 26).
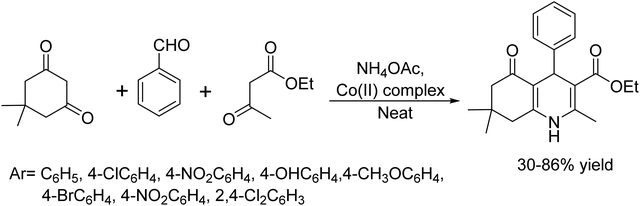 |
| | Scheme 26 Co(II) complex catalyzed synthesis of polyhydroquinolines. | |
In 2020, Guodong Shen and his team introduced a novel synthetic method to access fused chromenoquinolines using a unique Pd/Cu co-catalyzed strategy.40 This innovative approach takes advantage of a cascade method that initiated with the activated C–H bonds and breaking of C–C bond before reformation of new C–C bonds. By combining coumarin derivatives with anilines, the reaction efficiently produces a diverse array of fused chromenoquinoline derivatives through sequential C–C bond breaking, C–H functionalization, and subsequent C–C bond formation. Remarkably, the study also assessed the biocidal potential of these compounds. Using the MTT assay on human cervix tumor cells (HeLa), the evaluation revealed that specific derivatives, demonstrated a notable inhibitory effect on tumor cell growth. This finding highlights the potential of these metal-free analogs as candidates for further development in anticancer applications (Scheme 27).
 |
| | Scheme 27 Palladium/copper cocatalyzed C–C bond formation reaction. | |
In 2015, Junliang Wu and his colleagues unveiled a streamlined method for constructing quinolinone derivatives.41 Their approach utilizes a palladium-catalyzed cascade that marries C–H bond activation, C–C bond formation, and cyclization all starting from RNH2. This elegant strategy not only offers a practical route for synthesizing quinolinone-containing alkaloids and drug molecules but also shines in its efficiency and straightforward execution. The method's effectiveness was impressively showcased through a formal synthesis of tipifarnib, underscoring its potential impact in drug development (Scheme 28).
 |
| | Scheme 28 Pd-catalyzed C–H bond activation/C–C bond formation/cyclization cascade reaction. | |
In 2012, Gaikwad and Pore introduced a modified and eco-friendly method for the Matsuda Heck coupling reaction.42 Their approach involves the reaction of olefins with arene diazonium tetrafluoroborate salts under normal conditions. This methodology is the in situ generation of palladium nanoparticles, which serve as the catalyst. Notably, the reaction is conducted in water at room temperature and utilizes Triton X-100 as a surfactant, contributing significantly to both its environmental friendliness and operational simplicity (Scheme 29). This method not only demonstrates advancement in sustainable chemistry but also offers a practical route for executing the Heck coupling with high efficiency and minimal environmental impact.
 |
| | Scheme 29 Matsuda-Heck coupling of olefins. | |
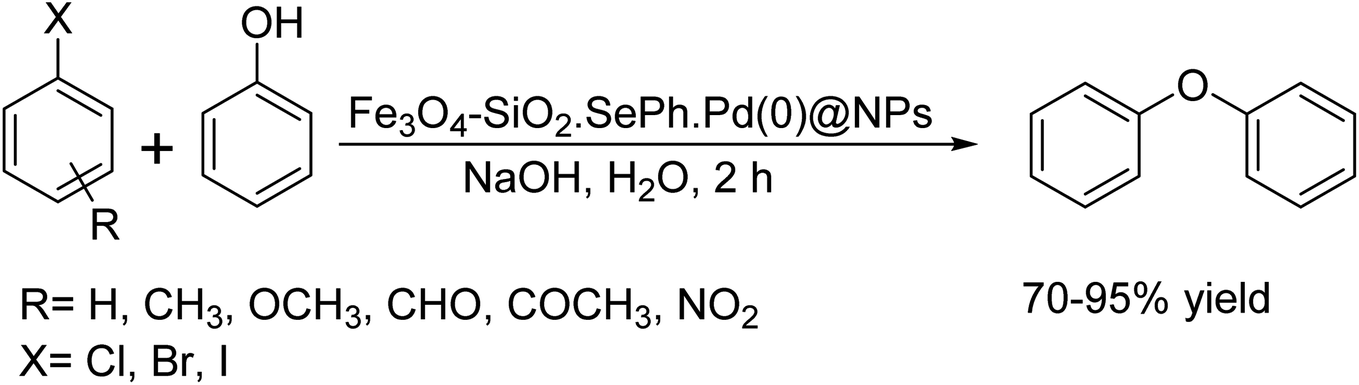
In 2023, M. A. Pawar et al.43 demonstrated a modified Sonogashira coupling by catalyzing the reaction of aryl halide and substituted phenylacetylene with K2CO3 using magnetically separable Pd-Fe3O4@GO in IPA medium (Scheme 30).
 |
| | Scheme 30 Modified Sonogashira cross-coupling reaction catalyst by Pd–Fe3O4@GO. | |
The first Pd-catalyzed coupling reaction under ball-milling conditions was indeed reported in 2000 by Peters and coworkers. This innovative approach addressed the challenge of reaction mixture stickiness, which is a common issue when milling soft or waxy substances. They tend to adhere to the milling chamber and balls, hindering the milling process.44 To overcome this, inert sodium chloride was added to the reaction mixture to reduce stickiness and facilitate a more powdery consistency. In the context of the Suzuki reaction, the choice of base was critical for optimizing yields. Among the bases tested, potassium carbonate (K2CO3) proved to be the most effective. The reaction protocol involved milling a mixture of ArX (1 equivalent), phenylboronic acid (2 equivalents), K2CO3 (3 equivalents), and palladium tetraphenylphosphine (Pd(PPh3)4) catalyst (5 mol%) in a Fritsch Planetary Micro Mill Pulverisette. The milling duration ranged from 30 to 60 minutes, resulting in the successful synthesis of the desired cross-coupling products with yields reaching up to 96% (Scheme 31).
 |
| | Scheme 31 Pd-catalyzed C–C coupling under ball-milling condition. | |
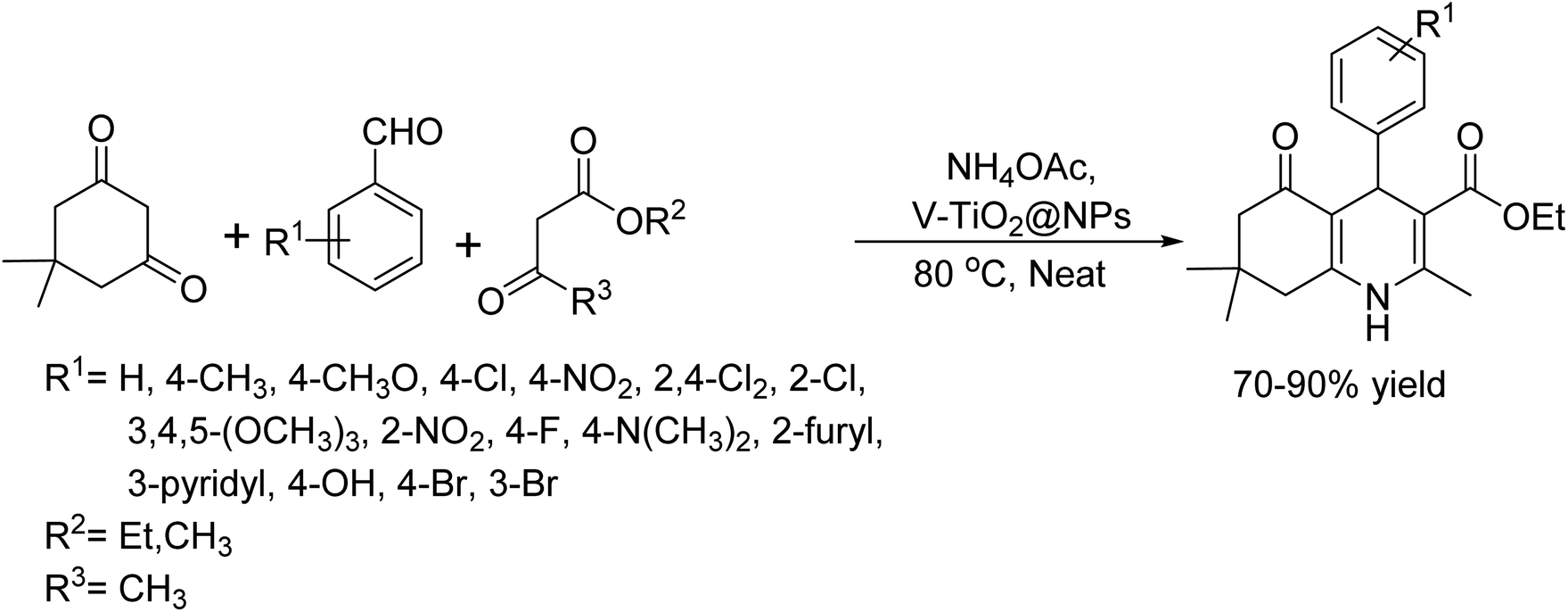
In 2017, G. B. Dharma Rao and colleagues synthesized polyhydroquinoline derivatives via the Hantzsch reaction using ArCHO, β-ketoester, dimedone, and NH4OAc at 80 °C under neat conditions.45 They employed mesoporous vanadium ion-doped titanium nanoparticles (V–TiO2) as a recyclable and robust heterogeneous catalyst for this one-pot multi component synthesis. The catalytic activity of V–TiO2 was compared with undoped commercial titanium nanocatalyst. Notably, this protocol successfully produced polyhydroquinoline derivatives from a wide range of structurally diverse arylaldehydes with β-ketoester, dimedone, and ammonium acetate. Key features include operational simplicity, short reaction time, and satisfactory yields. Additionally, the catalyst could be easily recycled and reused without any observable decrease in catalytic activity (Scheme 32).
 |
| | Scheme 32 V–TiO2@NPs promoted synthesis of polyhydroquinoline derivatives. | |
In 2014, Nasr-Esfahani and his co-workers reported the preparation of magnetic Fe3O4 nanoparticles as an efficient and recoverable nanocatalyst for synthesizing biologically active polyhydroquinoline derivatives.46 The structural analysis confirmed that the magnetic Fe3O4 nanoparticles have a spherical shape with an average size of 10–15 nm. Optimal conditions were successfully achieved through precise catalyst loading, and the nanoparticles exhibited remarkable recyclability, remaining efficient for at least 5 consecutive cycles (Scheme 33).
 |
| | Scheme 33 Fe3O4 MNPs catalyzed the synthesis of polyhydroquinolines. | |
In 2016, Ramazani and colleagues introduced an efficient protocol for synthesizing polyhydroquinolines using magnetic nickel ferrite nanoparticles (NiFe2O4@NPs) as a catalyst.47 They characterized these nanoparticles using spectroscopic and imaging techniques, including FT-IR, SEM, TEM, and XRD. They identified catalyst loading and microwave power as crucial parameters to achieve optimal reaction conditions. Under neat conditions and with microwave irradiation, the NiFe2O4@NPs catalyzed a multi-component reaction involving RCHO, dimedone, CH3COCH2COOEt, and NH4OAc. This process, neatly depicted in Scheme 34, enabled the rapid synthesis of polyhydroquinoline derivatives with moderate to high yields. Remarkably, the catalyst could be recycled several times without any loss of selectivity or catalytic activity, underscoring its robustness and potential for sustainable applications in organic synthesis.
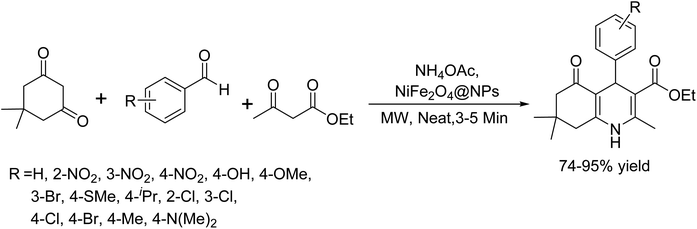 |
| | Scheme 34 Nickel ferrite@NPs catalyzed the synthesis of polyhydroquinolines under microwave irradiation in solvent-free conditions. | |
Khashi et al.48 also developed BiFeO3 magnetic nanoparticles, prepared via a chemical co-precipitation process, as an innovative, efficient, and recyclable catalyst for synthesizing polyhydroquinoline derivatives. The structure of the papered BiFeO3 magnetic nano-composite was characterized using FT-IR and XRD spectroscopy techniques. Notably, the expected product does not formed in the absence of the catalyst. Under neat conditions, catalyst amount and temperature played pivotal roles in determining the optimized conditions (as depicted in Scheme 35). Importantly, the catalyst could be recycled and reused up to three times.
 |
| | Scheme 35 BiFeO3@NPs catalyzed for synthesis of polyhydroquinolines. | |
Taghavi Fardood and his team have developed an eco-friendly approach to synthesizing Ni–Cu–Mg ferrite nanoparticles. They used tragacanth gum as a natural bio-template and metal nitrates as the metal source, employing the sol–gel method. They characterization of the catalyst has done the help of spectroscopic techniques such as FT-IR, XRD, VSM, and SEM. Excitingly, SEM and XRD studies revealed that the nanoparticles, averaging 19 nm in size, have a uniform spherical shape with a narrow size distribution. These Ni–Cu–Mg ferrite nanoparticles proved highly effective as catalysts in the multi-component Hantzsch synthesis of polyhydroquinoline derivatives,49 functioning under microwave irradiation and without the need for solvents (Scheme 36). A key advantage of this catalyst is its recyclability, it can be easily collected using an external magnet and reused for up to six cycles without any noticeable decline in catalytic performance.
 |
| | Scheme 36 Ni–Cu–Mg ferrite@NPs catalyzed the synthesis of polyhydroquinolines. | |
In 2019, Lumin Zhang and the team uncovered a fascinating twist in gold-catalyzed photocatalysis.50 Traditionally, [Au2(μ-dppm)2]Cl2 reactions kick off with ultraviolet A (UVA) light, but they discovered that blue light-emitting diodes (LEDs) can also serve as the excitation source, a setup that hadn't been reported before. This unexpected red shift in the absorption wavelength happens because of how [Au2(μ-dppm)2]Cl2 interacts with ligands like Ph3P or mercaptan. Building on this breakthrough, they developed a gold-catalyzed, reductive desulfurizing C–C bond forming reaction that works by coupling electrophilic radicals with styrenes under blue LED light is a feat not efficiently achievable with earlier methods. The reaction stands out for its mild conditions and impressive efficiency, using mercaptans both as electron-deficient alkyl radical precursors and as the hydrogen source. What's even more exciting is that the researchers successfully applied this strategy to modify amino acids and demonstrated its potential in polymer synthesis. They also showcased that the reaction can be scaled up to the gram level, all while providing keen insights into the underlying mechanism (Scheme 37).
 |
| | Scheme 37 C–C bond formation by desulfurizing gold-catalyzed photoreactions. | |
Transition metal free reactions of C–S bond formation
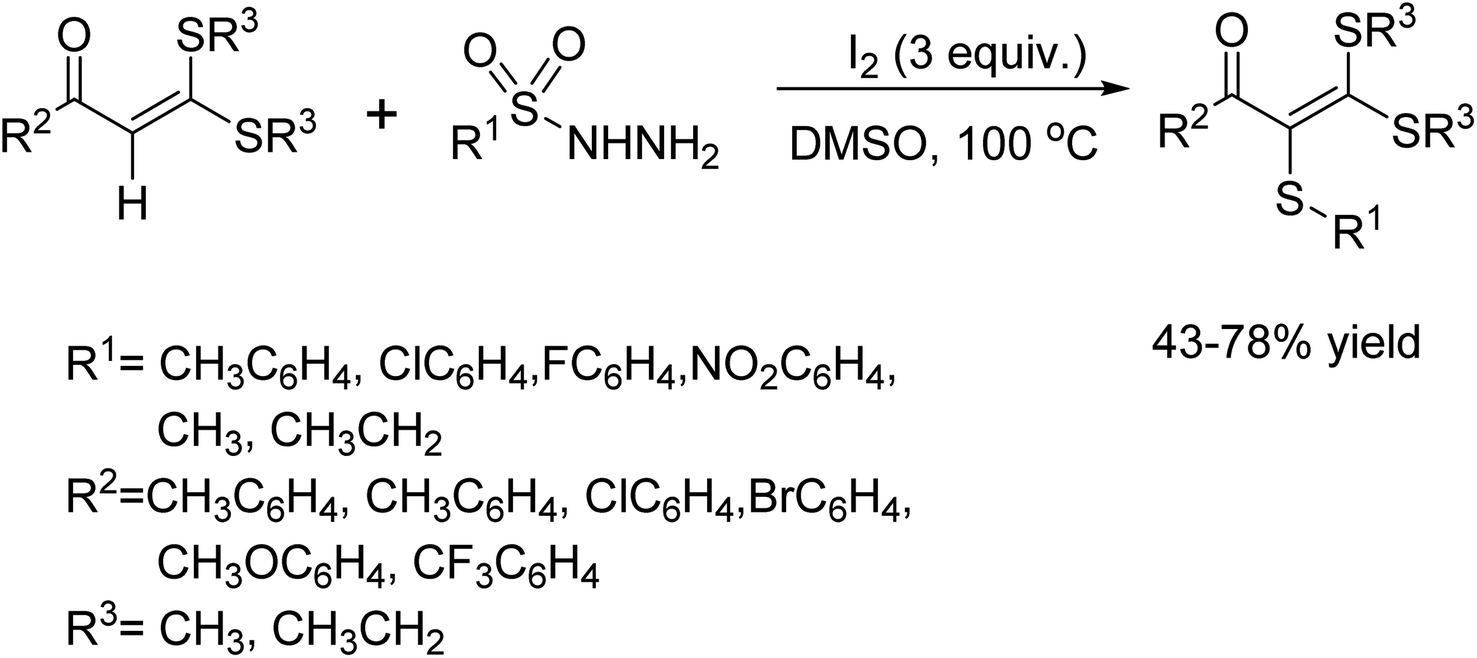
In 2018, the Lee group reported a new method to create sulfides through a dehydrative C–S bond formation.51 This method involves using 2-(hydromethyl)phenol and thiols in the presence of I2. The reaction is gentle yet efficient, taking only 4 h to complete at room temperature. It's worth noting that the protocol is compatible with both aryl thiol and aliphatic thiol. In fact, slightly higher selectivity was observed with aliphatic thiols in competition experiments. Additionally, products with good yields can be obtained on a gram scale by using long-chain aliphatic thiol (Scheme 38).
 |
| | Scheme 38 Dehydrative C–S coupling of 2-(hydromethyl)phenols and thiols. | |
In 2018, Leiling et al. demonstrated a method for sulfenylation of the vinyl C–H bond in ketene dithioacetals.52 This process ultimately led to the formation of polythiolated alkenes through the catalytic action I2. Additionally, this reaction was compatible with alkyl thiols, which underwent the C–H bond elaboration reaction to form alkylthiolated ketene dithioacetals in a DMSO solvent (Scheme 39).
 |
| | Scheme 39 Metal-free vinyl C–H sulfenylation/alkyl thiolation of ketene dithioacetals. | |
In 2019, Kazumasa Tanimoto and colleagues introduced an eco-friendly, two-component, metal-free catalytic oxidative sulfenylation of pyrazolones with thiols, utilizing biomimetic flavin and iodine (Scheme 40).53 This method was also successfully applied to the sulfenylation of pyrazoles and electron-rich benzenes, yielding a variety of thioethers in good yields.
 |
| | Scheme 40 Oxidative sulfenylation of pyrazolones catalyzed by metal-free flavin-iodine catalysis. | |
In 2020, Jingyang Qin and colleagues pioneered a mechanochemical electrophilic C–H sulfenylation of indoles using iodine catalysis.54 This groundbreaking method enables the rapid synthesis of indolyl sulfides with diverse structures under aerobic oxidation conditions, all while minimizing solvent usage and avoiding the need for metal catalysts. Mechanistic investigations further demonstrate the synergistic effect of solid and liquid grinding auxiliaries in facilitating this process (Scheme 41).
 |
| | Scheme 41 Electrophilic C–H sulfenylation of indoles with disulfides under ball milling. | |
In 2015, Wannian Zhao and team developed a method to generate ArS-substituted flavone derivatives using aryl thiols as sulfenylating agents. This innovative approach utilized ammonium iodide salt as an inducer, bypassing the need for conventional iodine/oxidant combinations.55 The technique facilitated the production of a range of ArS-substituted flavone derivatives with good to excellent yields, all under environmentally friendly conditions, thus making a significant contribution to the field of flavone chemistry (Scheme 42).
 |
| | Scheme 42 Synthesis of ArS-substituted flavone derivatives catalysis by NH4I. | |
In 2018, Liu and his research team introduced a novel approach for modifying the vinyl C–H bond of ketene dithioacetals.56 They successfully developed both aryl sulfenylation and alkyl thiolation using I2 as a catalyst for C–H bond coupling functionalization. Notably, a control experiment with TEMPO, a free-radical scavenger, yielded only a trace amount of the product strongly indicating that the reaction likely follows a free-radical pathway (Scheme 43).
 |
| | Scheme 43 Vinyl C–H sulfenylation of ketene dithioacetals catalyzed by I2. | |
In 2023, A. Islam and his team introduced a method of C(sp2)–H sulfenylation of β-enaminones under ball-milling conditions.57 Remarkably, only a catalytic amount of molecular iodine is used on a SiO2 surface, without any external heating. Compared to traditional solution-based methods, this approach significantly reduces reaction time. The frictional energy generated by ball-milling on mesoporous SiO2 materials has garnered interest for its role in heterogeneous catalysis. Their extensive surface area and well-defined porous structure enhance I2 catalytic efficiency in this method. Furthermore, the team investigated the antimicrobial properties of the synthesized compounds against both Gram-positive bacteria (Staphylococcus aureus and Bacillus cereus) and Gram-negative bacteria (Escherichia coli and Klebsiella pneumonia). To explore their potential as antimalarial agents, molecular docking studies were conducted, complemented by DFT analysis to assess their chemical reactivity and kinetic stability (Scheme 44).
 |
| | Scheme 44 Molecular iodine catalyzed sulfenylation of enaminone under mechanochemical condition. | |
In 2017, Pengfei Sun et al.58 demonstrated a groundbreaking method for the direct sulfenylation of sp2 C–H bonds using sulfinic acids as odorless sulfur reagents. This innovative approach was catalyzed by visible light and Eosin B, operating under transition metal-free conditions at room temperature. The process facilitated the formation of assorted heteroaryl sulfides in low to high yields. Notably, this is the first example of sulfenylation of Sp2 C–H bonds operated by arylsulfinic acids under visible light-induced conditions (Scheme 45).
 |
| | Scheme 45 Visible light-initiated C-3 sulfenylation of imidazopyridines with sulfinic acids. | |
In 2021, Goutam Brahmachari and colleagues demonstrated a method for the visible light-driven photochemical synthesis of a new series of biologically interesting 3-(alkyl/benzylthio)-4-hydroxy-2H-chromen-2-ones.59 This synthesis was carried out using a method known as cross-dehydrogenative C–H sulfenylation. In this process, 4-hydroxycoumarins were reacted with thiols at normal temperature, with Rose Bengal serving as a catalyst in acetonitrile under an oxygen-rich atmosphere (Scheme 46).
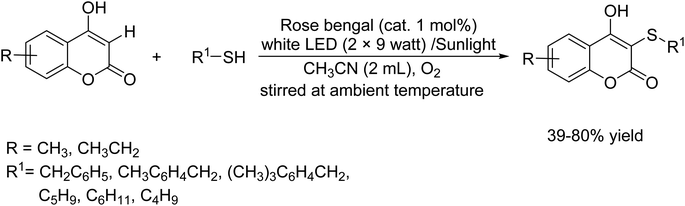 |
| | Scheme 46 Visible light-driven C–H sulfenylation of 4-hydroxycoumarins with thiols using rose bengal as a photosensitizer. | |
In 2018, Rajjakfur Rahaman and his team introduced a metal-free, visible-light-driven approach for regioselective C-3 sulfenylation of imidazo[1,2-a]pyridines and indoles using thiols.60 This method, based on C(sp2)–H functionalization, offers direct access to a broad range of structurally diversify 3-sulfenylimidazopyridines with potential biocidal substance. This technique particularly appealing is its simplicity in operation, reliance on eco-friendly energy sources, and excellent atom efficiency. Additionally, the use of green solvents under ambient conditions enhances its sustainability, making it a promising advancement in modern organic synthesis (Scheme 47).
 |
| | Scheme 47 Visible-light-promoted regioselective C-3 sulfenylation of imidazo[1,2-a]pyridines. | |
In 2019, Qing-Hu Teng and his team introduced a catalyst-free method for sulfenylating quinoxalinones using visible light and thiols through cross-dehydrogenative coupling.61 This innovative protocol allows direct access to the assorted 3-sulfenylquinoxalin-2(1H)-ones under transition metal free conditions, with atmospheric oxygen (O2) serving as the oxidant. A notable highlight of this approach is its versatility, readily available thiols and quinoxalin-2(1H)-ones were effectively utilized, making it a practical and efficient strategy for synthesizing 3-substituted quinoxalinones (Scheme 48).
 |
| | Scheme 48 Direct C–H sulfenylation of quinoxalinones with thiols under visible-light. | |
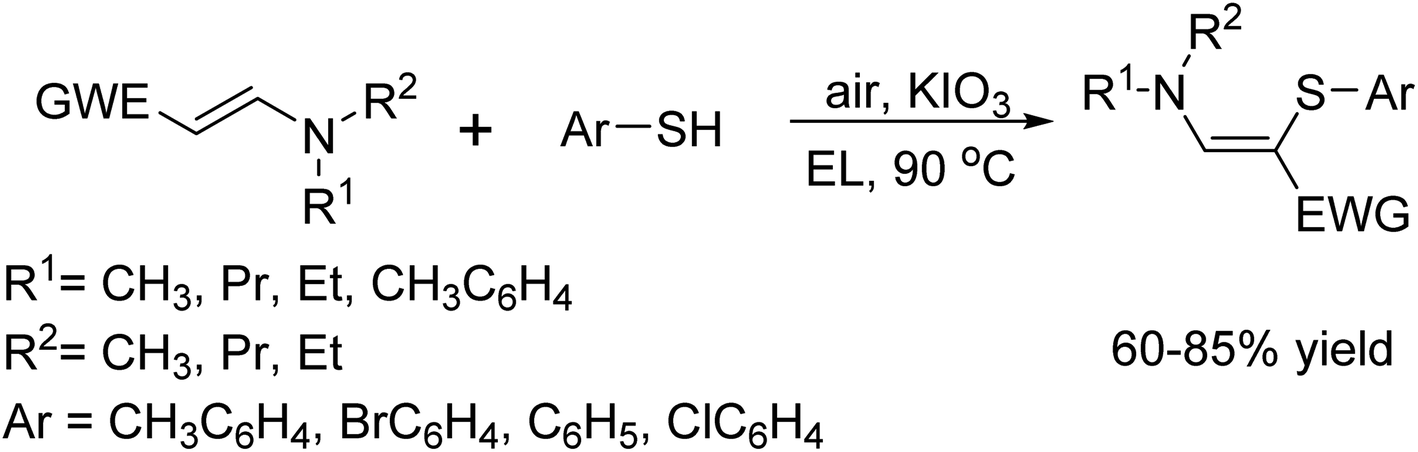
In 2016, Jie-Ping Wan and his team successfully synthesized polyfunctionalized aminothioalkenes through simple C–H sulfenylation of enaminones and related enamines.62 Impressively, these cross-coupling reactions were catalyzed using KIO3 under aerobic conditions, completely avoiding the need for transition metal catalysts or additional oxidants. Adding to the sustainability of this method, they utilized ethyl lactate (EL), a bio-based green solvent, as the reaction medium making this approach both efficient and environmentally friendly (Scheme 49).
 |
| | Scheme 49 C–H sulfenylation of enaminones catalyst by KIO3. | |
In 2023, Xiao-lin Cui and coworkers introduced a regioselective, metal-free sulfenylation of imidazoheterocycles with heterocyclic thiols or thiones.63 This innovative method, promoted by KBrO3, was carried out in a water medium, showcasing a significant advancement in cross-dehydrogenative coupling techniques (Scheme 50).
 |
| | Scheme 50 Synthesis of imidazo[1,2-a]pyridines derivatives promoted by KBrO3. | |
In 2023, Suvam Paul and colleagues introduced a visible-light-induced cross-dehydrogenative methodology for the regioselective sulfenylation of pyrazolo[1,5-a]pyrimidine derivatives.64 Essential components for this photocatalytic transformation include rose bengal, blue LEDs, KI, K2S2O8, and DMSO. The protocol facilitates the synthesis of a diverse library of 3-(aryl/hetero arylthio)pyrazolo[1,5-a]pyrimidine derivatives, showcasing broad functional capabilities (Scheme 51).
 |
| | Scheme 51 Visible-light-induced regioselective C–H sulfenylation of pyrazolo[1,5-a]pyrimidines. | |
Transition metal catalyzed reactions of C–S bond formation
In 2011, Li and colleagues documented the direct sulfanylation of anilines using diorganoyl disulfides with FeF3/I2 in acetonitrile at 80 °C. The process achieved moderate to high yields under optimal conditions, displaying notable regioselectivity at the para position in most instances.65 The reaction yielded low outputs without FeF3 and moderate outputs with I2 alone. The superior yields attained with both reagents led the authors to suggest a mechanism involving the in situ generation of RSI species through the oxidation of diorganoyl disulfides by I2, followed by electrophilic aromatic substitution with electron-rich anilines. Fe(III)'s function was linked to the replenishment of RSI species (Scheme 52).
 |
| | Scheme 52 Fe(III)-catalysed direct sulfanylation of anilines with diorganoyl disulfides. | |
In 2006, Ying-Chieh Wong and colleagues introduced an innovative approach to synthesizing thioethers through cobalt-catalyzed coupling.66 This method involves the reaction of aryl halides with thiophenols and alkanethiols, utilizing a catalytic system comprising 1–2 mol% of CoI2(dppe) and zinc. The process is notable for its ability to produce a diverse array of aryl sulfides with excellent yields under relatively mild conditions. This advancement offers a valuable alternative to existing methods for thioether synthesis, enhancing the efficiency and scope of these chemical transformations (Scheme 53).
 |
| | Scheme 53 Cobalt-catalyzed aryl-sulfur bond formation. | |
In 2019, Rina Sikari and her team introduced a streamlined and effective method for the C–S cross-coupling of an extensive array of (hetero)aryl thiols with (hetero)aryl halides. This process, predominantly conducted at room temperature, is facilitated by well-defined singlet diradical Ni(II) catalysts equipped with redox noninnocent ligands.67 By harnessing ligand-centered redox processes, the team successfully circumvented the need for the energetically demanding Ni(0)/Ni(II) or Ni(I)/Ni(III) redox transitions typically required in the catalytic cycle. The synergistic action of the Ni and its associated ligands functional part during the oxidative addition and reductive elimination phases enabled the execution of the catalytic reactions under gentle conditions (Scheme 54).
 |
| | Scheme 54 Nickel catalyzed C–S cross-coupling reaction. | |
In 2007, A. Saxena et al. reported an interesting cross-coupling reaction using Ni nanoparticles (15–18 nm).68 Additionally, the efficient synthesis of thioethers by the reaction of thiols with alcohols under normal conditions at room temperature has also been observed. This method successfully incorporated a diverse range of aryl and alkyl thiols, along with various alcohols, delivering impressive yields (Scheme 55).
 |
| | Scheme 55 Synthesis of thiol ethers catalyst by 15–18 mm Ni nanoparticles. | |
In 2009, Xin Ku and colleagues pioneered an innovative iron and copper co-catalyzed C–S cross-coupling reaction. This method employs N,N,N′,N′-tetramethylethylenediamine (TMEDA) as a catalyst under microwave (MW) irradiation. The technique has proven versatile, and applicable to a broad spectrum of substrates, including aliphatic thiols, heterocyclic thiols, a variety of substituted aryl thiols, as well as aryl and heterocyclic halides (Scheme 56).69
 |
| | Scheme 56 Efficient iron/copper cocatalyzed S-arylations of thiols with aryl halides. | |
In 2014, Yiming Li and coworkers demonstrated a highly efficient Cu-catalyzed S-transfer reaction from an amine to a sulfide (Scheme 57).70
 |
| | Scheme 57 Cu-catalyzed s-transfer reaction from amine to sulfide. | |
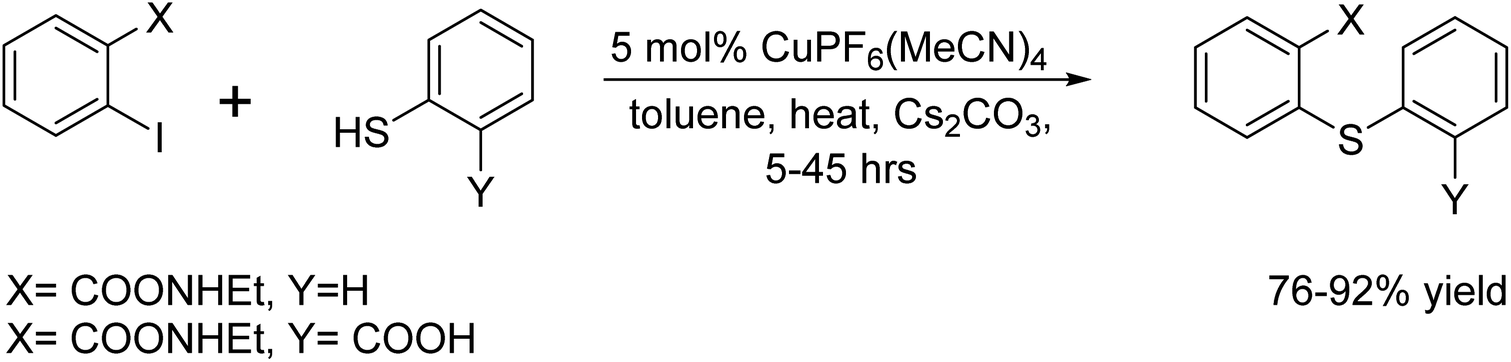
In 1999, A. V. Kalinin and his colleagues reported the use of a catalytic system incorporating the CuPF6(MeCN)4 complex.71 This system was used as a catalyst to synthesize diaryl thioethers from o-substituted aryl iodides and aryl thiols in the presence of Cs2CO3 under heating conditions (Scheme 58).
 |
| | Scheme 58 C–S coupling reaction catalyst by CuPF6(MeCN)4 catalyst. | |
In 2010, H. Firouzabadi and his team explored the use of thiourea as a thiol surrogate in thioether synthesis by the reaction of aryl halides with alkyl bromides catalyzed by CuI.72 This reaction was done in wet PEG-200 at 80–100 °C under an inert atmosphere, offering a excellent approach to thioetherification with easily available alkyl bromides. Beyond this, thiourea in wet PEG-200 also performed as a sulfur source in reactions involving various alkyl halides and electron-deficient alkenes. Additionally, intra- and intermolecular C–S cross-coupling reactions with substituted thiourea derivatives led to the synthesis of 2-(arylthio) arylcyanamides, alongside CuO-mediated transformations involving selenourea (Scheme 59).
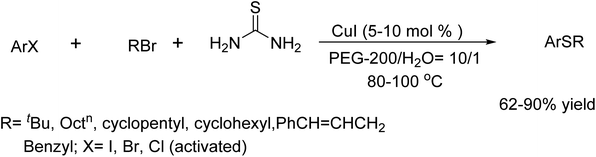 |
| | Scheme 59 Synthesis of thioethers catalyzed by CuI in PEG-200/H2O. | |
In 2019, Z. Taherinia et al. reported an innovative, modified multi-component reaction for synthesizing unsymmetrical diaryl sulfides.73 Their method leverages the CuI-catalyzed coupling of aromatic acids, aryl halides, and elemental sulfur, representing a noteworthy advance in efficient and selective C–S bond construction (Scheme 60).
 |
| | Scheme 60 Thio arylation of aromatic acid with elemental sulfur catalyst by CuI. | |
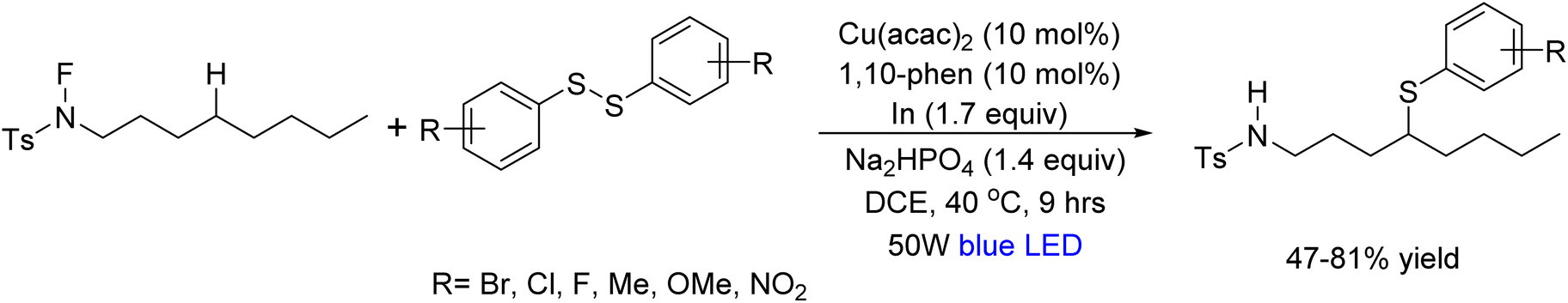
In 2020, Yuman Qin et al.74 demonstrated a chemical reaction that forms a bond between carbon and sulfur atoms through C(Sp2)-H sulfenylation of alkyl tosylamides at the δ-position. This reaction is referred to as C–S coupling. Under blue LED irradiation at 40 °C, the reaction proceeded by combining N-fluoro-tosylamide with diaryl disulfide in the presence of Cu(acac)2, 1,10-phenanthroline, Na2HPO4, and indium powder. Notably, the protocol exhibited broad substrate compatibility, effectively accommodating various diaryl disulfides along with dibenzyl disulfides and diphenyl diselenides (Scheme 61).
 |
| | Scheme 61 Sulfenylation of tosylamides with disulfides. | |
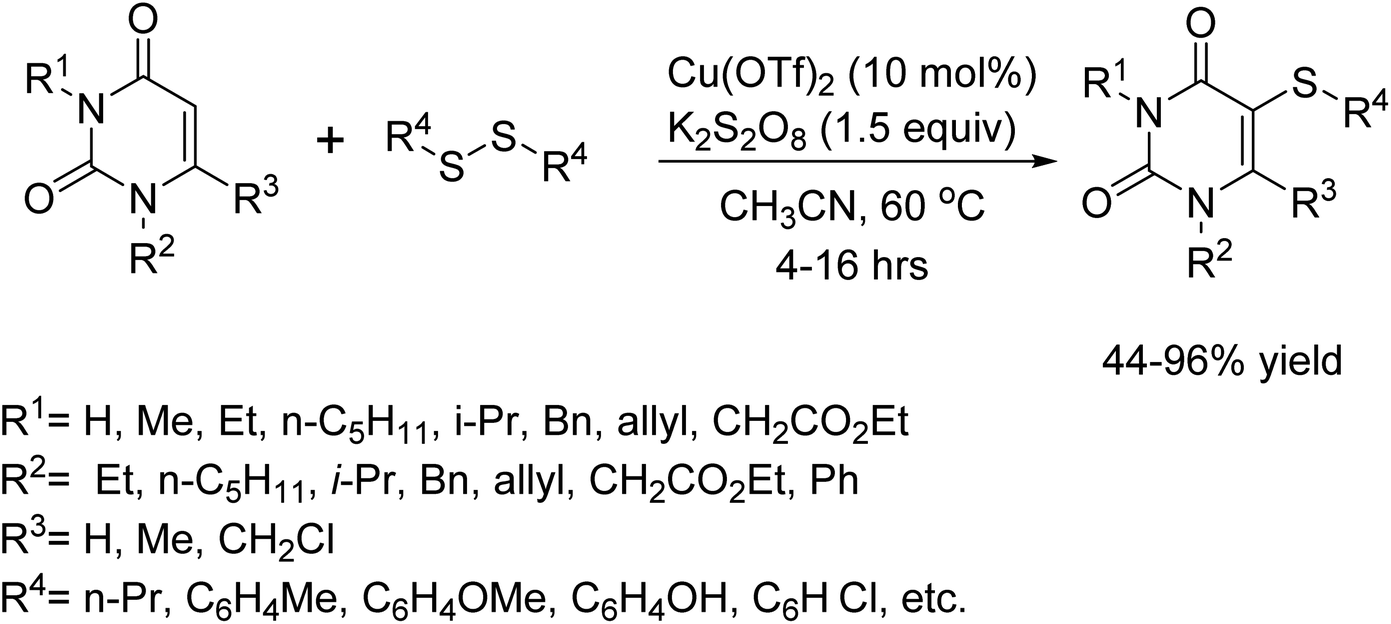
In 2019, the Yotphan group unveiled a groundbreaking method for selectively introducing sulfur-containing groups onto uracils.75 This groundbreaking achievement was made possible by utilizing copper as a catalyst and either disulfide or a specific compound referred to as NH4SCN. The reaction was successfully carried out in acetonitrile, with Cu(OTf)2 and K2S2O8 serving as co-catalysts, resulting in consistently excellent outcomes. Remarkably, the process exhibited remarkable tolerance towards a diverse range of functional groups. Furthermore, it was observed that the reaction proved to be notably more efficient with electron-rich diaryl disulfides than with their electron-deficient counterparts. Rigorous control experiments unequivocally established that the transformation followed a radical mechanism, solidifying the method's validity and potential impact (Scheme 62).
 |
| | Scheme 62 C(Sp2)-H thiolation of uracils with disulfides. | |
In 2002, Craig G. Bates and his team reported a gentle, palladium-free synthetic method for the cross-coupling of aryl iodides with thiols.76 This protocol employs 10 mol% CuI and 10 mol% neocuproine, using sodium tert-butoxide (NaOtBu) as the base, and is conducted in toluene at 110 °C. The method proved to be highly effective, yielding a diverse array of aryl sulfides from readily accessible iodides and thiols (Scheme 63).
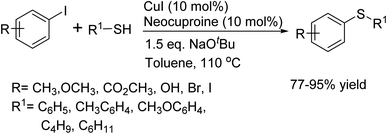 |
| | Scheme 63 General method of C–S coupling catalyzed by CuI. | |
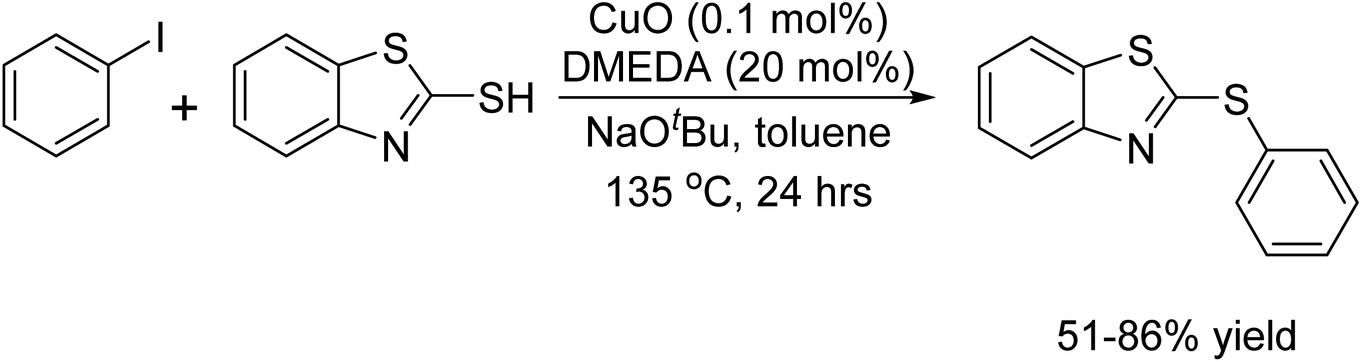
In 2009, Bolm and colleagues reported a notable instance of copper-catalyzed C–S bond formation, utilizing a mere 0.1 mol% of CuO as the copper source. Impressively, the process required 20 mol% of a ligand to produce the desired product in substantial yield.77 The researchers highlighted the critical role of the ligand in preventing metal aggregation, thereby averting the potential deactivation of the copper species (Scheme 64).
 |
| | Scheme 64 C–S bond formation catalyzed by Cu–DMEDA. | |
In 2002, George Y. Li demonstrated the C–S bond formation reaction from 1-chloro-1-cyclopentene generating thioethers coupling with thiophenol catalyzed by the air-stable palladium phosphinous acid complexes (Scheme 65).78
 |
| | Scheme 65 Palladium-catalyzed C–S bond formation of vinyl chlorides. | |
In 1980, Toshihiko Migita et al.79 demonstrated a successful reaction for forming a C–S bond from ArX and thiol derivatives catalyzed by the Pd(Ph3)4 complex (Scheme 66).
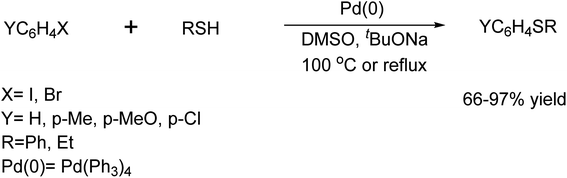 |
| | Scheme 66 Pd(Ph3)4 catalysed C–S bond formation reaction. | |
In 2008, J. Lee and colleagues conducted a chemical reaction that involved the formation of a C–S bond using indiumtri(organothiolate) as the nucleophilic coupling partner.80 This procedure allowed for the creation of a wide range of sulfides including aryl-aryl, aryl-alkyl, aryl-vinyl, and alkyl-vinyl sulfides in good to excellent yields. When alkenyl halides were used in the reactions, the double-bond geometry was retained, as shown in Scheme 67.
 |
| | Scheme 67 Pd-catalyzed C–S bond formation reaction using indiumtri(organothiolate). | |
In 2006, Michael C. Willis and colleagues made a significant breakthrough in organic synthesis by developing a method for synthesizing benzothiophenes derivatives.81 The procedure involves an intramolecular thio-enolate S-arylation reaction using the DPE-phos ligand. Enolates are produced from o-haloaryl-substituted thio-ketones, and the reaction is catalyzed by Pd2dba3, resulting in the formation of products in moderate to good yields (Scheme 68).
 |
| | Scheme 68 Synthesis of benzothiophenes catalyzed by Pd2dba3 in the DPE-phos ligand. | |
In 2004, M. Murata and colleagues introduced an efficient method for the C–S coupling reaction of aryl and alkyl thiols with aryl bromides or iodides, catalyzed by palladium.82 The key feature of this protocol is the use of DiPPF (1,1-bis(diisopropylphosphino)ferrocene), L as an active ligand (Scheme 69).
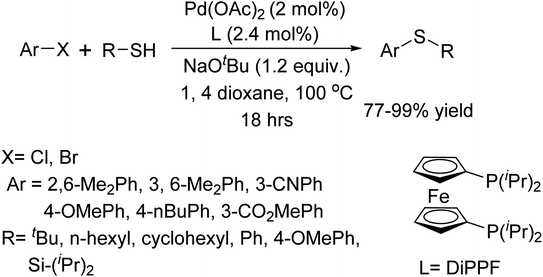 |
| | Scheme 69 C–S coupling of thiols with aryl halides catalyzed by Pd–DiPPF. | |
In 2001, U. Schopfer and colleagues reported the use of DPEphos (bis-[2-(diphenylphosphino)phenyl]ether, L), a ligand with an electronic effect akin to xantphos but with a more flexible backbone.83 This ligand was utilized in the palladium-catalyzed C–S coupling reaction of aryl iodides and thiols (Scheme 70).
 |
| | Scheme 70 C–S coupling reaction catalyzed by Pd–DPEphos. | |
In 2012, Chen and colleagues presented a direct sulfanylation method for electron-rich arenes and diorganoyl disulfides, utilizing air as an eco-friendly oxidant.84 This process was efficiently catalyzed by FeBr3 in a DMF solvent system (Scheme 71).
 |
| | Scheme 71 Fe(III)-catalysed direct sulfanylation of electron-rich arenes with diorganoyl disulfides. | |
In 2015, Chou and colleagues demonstrated the sulfanylation of quinones by the reaction of diaryl disulfides catalyzed by Ag(I), resulting in quinonyl aryl thioethers (Scheme 72).85 Optimization of the reaction conditions, the AgOAc and dppp (1,3-bis(diphenylphosphino)propane), with (NH4)2S2O8 acting as the oxidant. The addition of Bu4NBF4 was necessary to enhance reagent solubility. Remarkably, the reaction yielded superior outcomes when diaryl disulfides bearing electron-donating groups at the p-position or electron-withdrawing groups at the m-position were employed. In contrast, diaryl disulfides with electron-withdrawing groups at the p-position provided only satisfactory yields, and dialkyl disulfides proved ineffective in producing the desired product.
 |
| | Scheme 72 Ag(I)-catalysed direct sulfanylation of quinones with diaryl disulfides. | |
In 2018, Yotphan and team detailed sulfanylation of pyrazolones at the C-4 position catalyzed by Ag(I).86 This technique was utilized for the incorporation of organosulfur structures into antipyrine and various pyrazolones under ambient conditions, with a brief reaction duration and in the presence of air. Nonetheless, diaryl disulfides with straightforward electron-donating or electron-withdrawing groups necessitated elevated temperatures and prolonged reaction periods to achieve satisfactory product yields. The authors also discovered that adding 2 mol% DABCO (1, 4-diazabicyclo[2.2.2]octane) markedly decreased the reaction time and enhanced the yields (Scheme 73).
 |
| | Scheme 73 Ag(I)-mediated direct sulfanylation of pyrazolones. | |
In 2010, C. J. Weiss et al.87 reported the application of organozirconium(IV)-mediated intermolecular hydrothiolation of terminal alkynes with aliphatic, aromatic, and benzylic thiols using various precatalysts. The reaction displays remarkable selectivity, attaining up to 99% selectivity with yields that consistently exceed 90%. Moreover, when scaled up, the method has achieved an impressive 72% isolated yield while maintaining a 99% Markovnikov selectivity. They also reported indicating that the activation parameters for CGCZrMe2-mediated 1-pentanethiol hydrothiolation of 1-hexyne, measured between 50-80 °C, are in line with a turnover-limiting alkyne insertion into the Zr-SR bond, followed by a thiol-induced Zr–C protonolysis (Scheme 74).
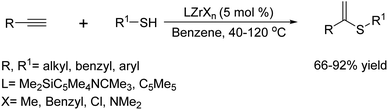 |
| | Scheme 74 Lanthanide-mediated selective hydrothiolation of terminal alkynes with thiols. | |
Kuniyasu and colleagues explored the mechanistic intricacies of alkynes' carbochalcogenation, assessing its utility in synthetic applications. Their findings revealed that unsymmetrical alkynes undergo decarbonylative aryl thiolation mediated by thioethers under catalyzed platinum.88 The suggested mechanism highlights the critical steps of alkyne insertion into the metal–sulfur (M–S) bond, followed by reductive elimination. Additionally, they exhibit Pt-catalyzed thiolation modification, distinctively furyl, thienyl, and pyridyl thiolation of alkynes. A particularly innovative development derived from their work is the Pd-catalyzed regioselective iminothiolation of alkynes (Scheme 75).
 |
| | Scheme 75 Pt-catalyzed decarbonylative aryl thiolation of unsymmetrical alkynes. | |
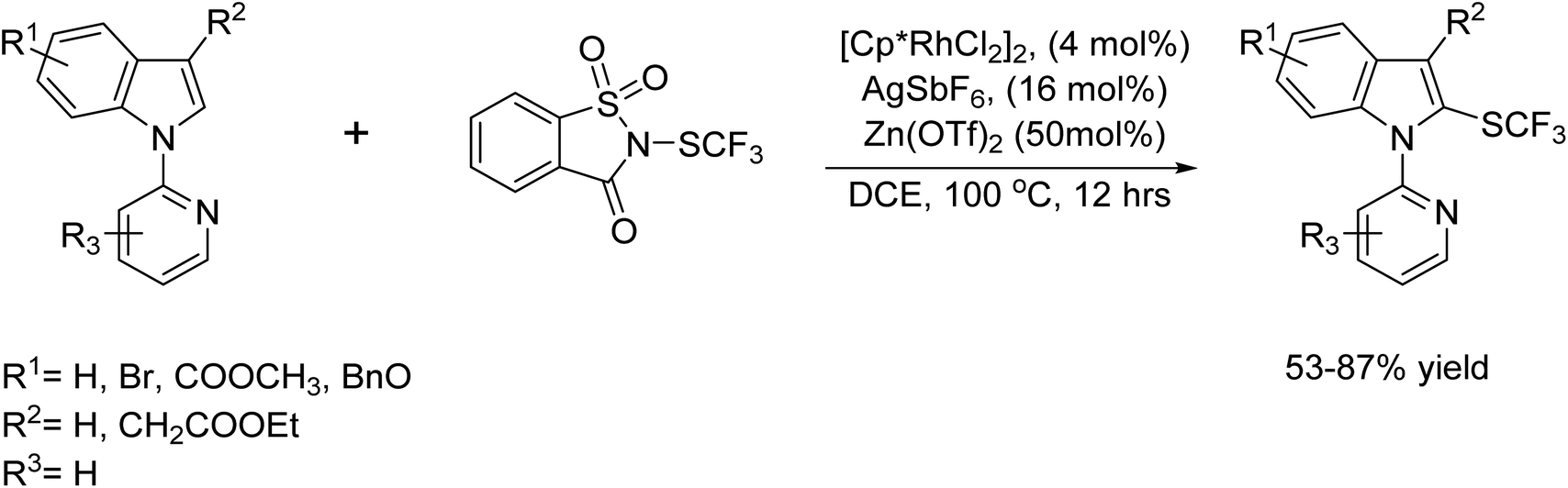
In 2015, Li and colleagues unveiled a Rh(III)-catalyzed C-2 direct trifluoromethyl thiolation of indoles using N-(trifluoromethylthio)saccharin as the sulfur donor. The 2-pyridyl group guided the reaction at the C-2 position of the indole nucleus, yielding trifluoromethylthiolated products with high efficiency.89 Typically, small quantities of Ag(I) were necessary to activate the [Cp*RhCl2]2 catalyst. Additionally, Zn(II) Lewis acid played a crucial role in effectively activating N-(trifluoromethylthio)saccharin (Scheme 76).
 |
| | Scheme 76 Rh(III)-catalysed C-2 direct trifluoromethylsulfanylation of indoles. | |
In 2019, Ji and colleagues revealed a Rh(III)-catalyzed direct sulfanylation of acrylic acids at the β-position using diorganoyl disulfides.90 The subtle coordination by a carboxylic acid group was sufficient for this chelation-assisted sulfanylation, as evidenced by the exclusive Z-configuration of the products, confirming its role as a directing group. The reaction exhibited no significant electronic effects concerning the acrylic acids; however, steric hindrance was notable. For instance, cinnamic acid yielded only minimal amounts of the anticipated products. Additionally, diaryl disulfides with strong electron-withdrawing groups were unsuccessful in producing the targeted sulfanylation products (Scheme 77).
 |
| | Scheme 77 Rh(III)-catalysed direct sulfanylation of acrylic acids at the β-position. | |
In 2009, Vutukuri Prakash Reddy and colleagues introduced an efficient method for the ligand-free C–S cross-coupling of aryl halides with aromatic/alkyl thiols. This innovative approach utilizes a catalytic amount of nanocrystalline indium oxide as a recyclable catalyst; with potassium hydroxide (KOH) serving as the base.91 The reaction takes place in dimethyl sulfoxide (DMSO) at a temperature of 135 °C. Notably, this protocol allows the synthesis of various aryl sulfides in excellent yields (Scheme 78).
 |
| | Scheme 78 C–S cross-coupling reaction catalyst by nano indium oxide. | |
In 2016, Min Jiang and colleagues unveiled a straightforward and proficient method for the visible-light photo-redox arylation of thiols with aryl halides, conducted at room temperature.92 This protocol notably includes various aryl chlorides as effective arylating agents (Scheme 79).
 |
| | Scheme 79 Room-temperature photoredox arylation of thiols. | |
In 2012, P. Malik and colleagues showcased a method for C–S bond formation that leverages a combination of Bi2O3 and N, N-dimethylethane-1, 2-diamine as an effective catalytic system.93 This innovative approach facilitates the coupling of aryl thiols with aryl bromides and iodides. Notably, the reactions were conducted in water, highlighting the protocol's environmentally friendly aspect (Scheme 80).
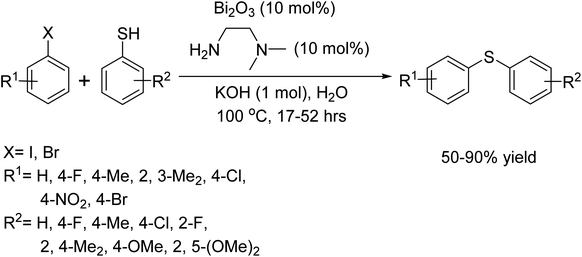 |
| | Scheme 80 Synthesis of diaryl thioethers from the reaction of aryl thiols with aryl iodides and bromides catalyzed by Bi. | |
Transition metal free reactions of C–N bond formation
In 2014, Z. Begum and colleagues made a significant contribution to the field of glycochemistry by documenting the amidoglycosidation of tri-O-acetyl-D-glucal with various N-nucleophiles, such as t-butyl carbamate and N-benzyl carbamate.94 Utilizing an equimolar quantity of molecular iodine under neutral and mild conditions, they successfully synthesized the corresponding N-glycosyl amides with good yields. Notably, the reactions exhibited preferential α-anomeric selectivity (Scheme 81).
 |
| | Scheme 81 Synthesis of amidoglycosidation of tri-O-acetyl-D-glucal with different N-nucleophiles catalyzed by I2. | |
In 2014, F. M. Moghaddam and colleagues demonstrated an efficient method for synthesizing fused pyrimido[4,5-d]pyrimidine-2,4-dione derivatives.95 The process involved the reaction of 6-aminouracils with N,N′–bis(arylmethylidene)arylmethane, catalyzed by I2. This readily available catalyst facilitated the formation of the desired fused pyrimidine derivatives (Scheme 82).
 |
| | Scheme 82 Synthesis of fused pyrimido[4,5-d]pyrimidine-2,4-dione catalyzed by I2. | |
In 2015, M. Krishnamurthy and colleagues developed an efficient method for coupling N-α-protected hydroxamic acids with an amino component. This reaction was catalyzed by molecular iodine under mild conditions, with a reaction time ranging from 5 to 30 minutes. The process proceeded through an unstable yet highly reactive acyl nitroso intermediate.96 Notably, this method boasts several key advantages, straightforward operational procedure, normal reaction conditions, and a remarkably short period of time. Furthermore, it demonstrates excellent tolerance toward a variety of protecting groups such as tert-butyl oxycarbonyl (Boc), benzyl chlorocarbonate (Cbz), and fluorenylmethoxycarbonyl (Fmoc) rendering it particularly attractive for intricate synthetic applications (Scheme 83).
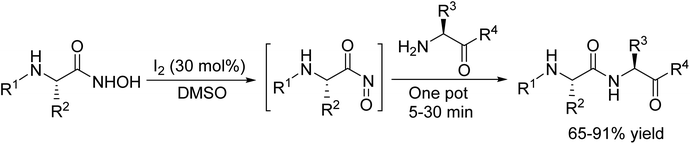 |
| | Scheme 83 Coupling of N-α-protected hydroxamic acids with an amino component catalyzed by I2. | |
In 2015, P. Niu and their team made a significant contribution to the field of heterocyclic chemistry by documenting the synthesis of 2-amino-substituted 1, 3, 4-oxadiazoles. They innovatively used molecular iodine to mediate oxidative C–O or C–S bond formation, starting from aldehydes and semicarbazide or thiosemicarbazide.97 The reactions were effectively carried out in the presence of potassium carbonate, using 1,4-dioxane as the solvent at a temperature of 80 °C. This method proved to be versatile, being applicable to a variety of aliphatic, aromatic, and cinnamic aldehydes, leading to the successful synthesis of the desired 2-amino substituted oxadiazoles and thiadiazoles (Scheme 84).
 |
| | Scheme 84 Synthesis of 2-amino-substituted 1, 3, 4-oxadiazoles catalyzed by molecular iodine. | |
In 2018, S. Angapelly and team presented a novel one-pot domino synthesis of (1,3,4-oxadiazol-2-yl)anilines, by the reaction of isatins and hydrazides catalyzed by I2.98 This method is characterized by a domino process that includes condensation, hydrolytic ring cleavage, and intramolecular decarboxylation, ultimately leading to the formation of a new C–O bond. This approach is noteworthy for its transition metal-free and practical application in synthesizing diversified aniline derivatives (Scheme 85).
 |
| | Scheme 85 Synthesis of (1, 3, 4-oxadiazol-2-yl)anilines catalyzed by I2. | |
In 2018, J.-Q. Liu and colleagues achieved the synthesis of 5-benzoyl-8H-phthalazino[1,2-b]quinazolin-8-ones through a one-pot reaction.99 Under the catalytic influence of I2, 2-aminobenzohydrazides were seamlessly combined with 2-alkynyl benzaldehydes to afford the desired products in excellent yields. The transformation proceeds through an elegantly orchestrated sequence, encompassing an initial condensation, followed by sequential addition and hydroamination steps, and finally culminating in an oxidation process that constructs aromatic rings. Notably, this entire transformation occurred under metal-free reaction conditions, with oxygen serving as the oxidant (Scheme 86).
 |
| | Scheme 86 Synthesis of 5-benzoyl-8H-phthalazino[1,2-b]quinazolin-8-ones catalyzed by I2. | |
In 2020, Q.-W. Gui and colleagues developed an efficient method for synthesizing α-cyanopyrrolines through a one-pot, three-component reaction. This innovative approach utilized alkenes, TMSCN, and N,N-substituted formamides, catalyzed by 10 mol% of I2 under neat conditions.100 The transformations were conducted in the presence of ambient air as the oxidant, eliminating the need for transition metal catalysis. This protocol resulted in excellent conversions and high chemoselectivity, yielding the final products in excellent yields (Scheme 87).
 |
| | Scheme 87 Synthesis of α-cyanopyrrolines from alkenes catalyst by I2. | |
In 2009, Shaterian and colleagues synthesized 2H-indazolo[2,1-b]phthalazine-trione derivatives through a three-component condensation reaction. They used phthalhydrazide, dimedone, and aromatic aldehydes with a neat reaction setup. This resulted in excellent yields and short reaction times. The catalyst used was reusable silica-supported polyphosphoric acid (PPA-SiO2) at a loading of 0.05 mmol. This method offered several advantages, including low cost, easy preparation, catalyst recyclability, and environmentally friendly conditions. The PPA-SiO2 catalyst demonstrated high activity, ease of handling, and recoverability, making it a practical choice for this synthesis (Scheme 88).101
 |
| | Scheme 88 Synthesis of 2H-indazolo[2,1-b]phthalazine-trione using PPA–SiO2. | |
Li and Pan have successfully utilized chiral N-phosphonylimines in the stereoselective synthesis of various substituted chiral propargyl amines, reacting with lithium aryl/alkyl acetylides. These acetylides were synthesized in situ by treating terminal alkynes with LDA. Subsequently, the resulting alkynyl lithium was combined with the chiral N-phosphonylimines, yielding propargyl derivatives with a high diastereomeric ratio. The phosphonyl group was then removed under acidic conditions, producing propargylamines/amides (Scheme 89).102
 |
| | Scheme 89 Synthesis of propargylamines/amides by addition of lithium acetylides to chiral N-phosphonylimines. | |
In 2023, A. Islam et al. introduced an innovative, one-pot, two-component greener protocol for synthesized biologically significant 1,4-benzothiazine derivatives.103 This protocol, catalyzed by PEG-200, facilitates by the reaction of 2-aminobenzothiols and 1,3-dicarbonyl compounds, providing a sustainable and metal-free alternative for organic synthesis. Furthermore, polyethylene glycol (PEG-200) has emerged as an innovative class of green and biocompatible reaction media, reinforcing its potential in environmentally friendly chemical processes (Scheme 90).
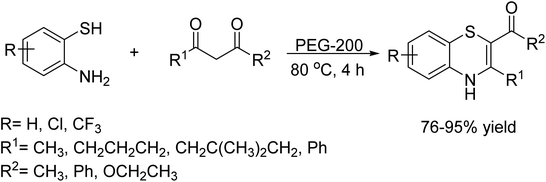 |
| | Scheme 90 Metal-free synthesis of functionalized 1,4-benzothiazines. | |
In 2009, Lamaty and colleagues advanced peptide synthesis by using a ball milling method to create peptides under solvent-free conditions. They successfully synthesized dipeptides and a tripeptide by opening urethane-protected α-amino acid N-carboxyanhydride (UNCA) derivatives with α-amino acid derivatives.104 This approach addresses the challenge of minimizing solvent usage in peptide synthesis, contributing to greener chemistry practices (Scheme 91).
 |
| | Scheme 91 Peptides synthesis under mechanochemical condition. | |
In 2021, R. Singha et al. introduced a neat, efficient, and straightforward one-pot, multi-component approach for synthesizing chromeno[4,3-b]quinolin-6-one derivatives.105 This method, catalyzed by graphene oxide (GO), enables the reaction between 4-hydroxycoumarin, aldehydes, and aromatic amines, providing a streamlined and environmentally friendly synthetic route. Notably, GO has emerged as an innovative heterogeneous carbocatalyst, demonstrating remarkable reusability retaining its catalytic efficiency even after five consecutive runs. The protocol also features broad substrate applicability (Scheme 92).
 |
| | Scheme 92 Solvent-free graphene oxide catalysed synthesis of chromeno-[4,3-b]quinolin-6-one derivatives from 4-hydroxycoumarin with aldehydes and aromatic amines. | |
Transition metal catalyzed reactions of C–N bond formation
Zou, Zhang, and their team introduced a straightforward and efficient method for the synthesizing indolo[1,2-c]quinazolines catalyzed by Cu, utilizing air as the oxidant. Their approach employs readily available 2-(2-halophenyl)-1H-indoles and (aryl)methanamines, offering a practical and sustainable route to these valuable heterocyclic compounds (Scheme 93).106
 |
| | Scheme 93 Synthesis of indolo[1,2-c]-quinazolines using Cu-catalyzed and air as oxidant. | |
In 2012, Bao and his colleagues synthesized a series of 6H-isoindolo[2,1-a]indol-6-ones through a one-pot two-component reaction. The first step in this reaction is a C–N coupling cyclization catalyzed by Cu, followed by Pd-catalyzed C–H activation.107 They initiated the transformation by reacting o-gem-dibromo vinyl anilines and substituted C6H5COCl promoted by CuBr (10%), N,N-dimethyl ethylenediamine (DMEDA) (20%) as a ligand, and K2CO3 (1.0 equiv.) in toluene at 120 °C. This reaction resulted in the formation of (2-bromo-1H-indol-1-yl) (phenyl)methanone in moderate to good yields (Scheme 94).
 |
| | Scheme 94 Synthesis of 6H-isoindolo[2,1-a]indol-6-ones. | |
In 2012, R. Berrino and colleagues developed a method to synthesize 2-quinolones through an intra-molecular process that involved C–H functionalization and C–N bond formation.108 The process involved treating 3,3-diaryl acryl amides with CuI (10 mol%), PPh3 (20 mol%), and tBuOK in o-xylene at 100 °C for 5 to 24 h under experimental conditions. The 2-quinolones were obtained in good yields using this method (Scheme 95).
 |
| | Scheme 95 Synthesis of 2-quinolones based on an intra-molecular C–H functionalization/C–N bond forming process. | |
Yao, Ma, and their co-workers reported in 2012 that 2-(2,6-dimethylphenylamino)-2-oxoacetic acid (DMPAO)L3 can be used as an inexpensive and aryl amination of aryl halides and amines catalyzed Cu-complex of powerful ligand, producing high yields of N-arylated products (Scheme 96).109
 |
| | Scheme 96 Synthesis of N-arylated product catalyzed by CuI, (DMPAO) L3. | |
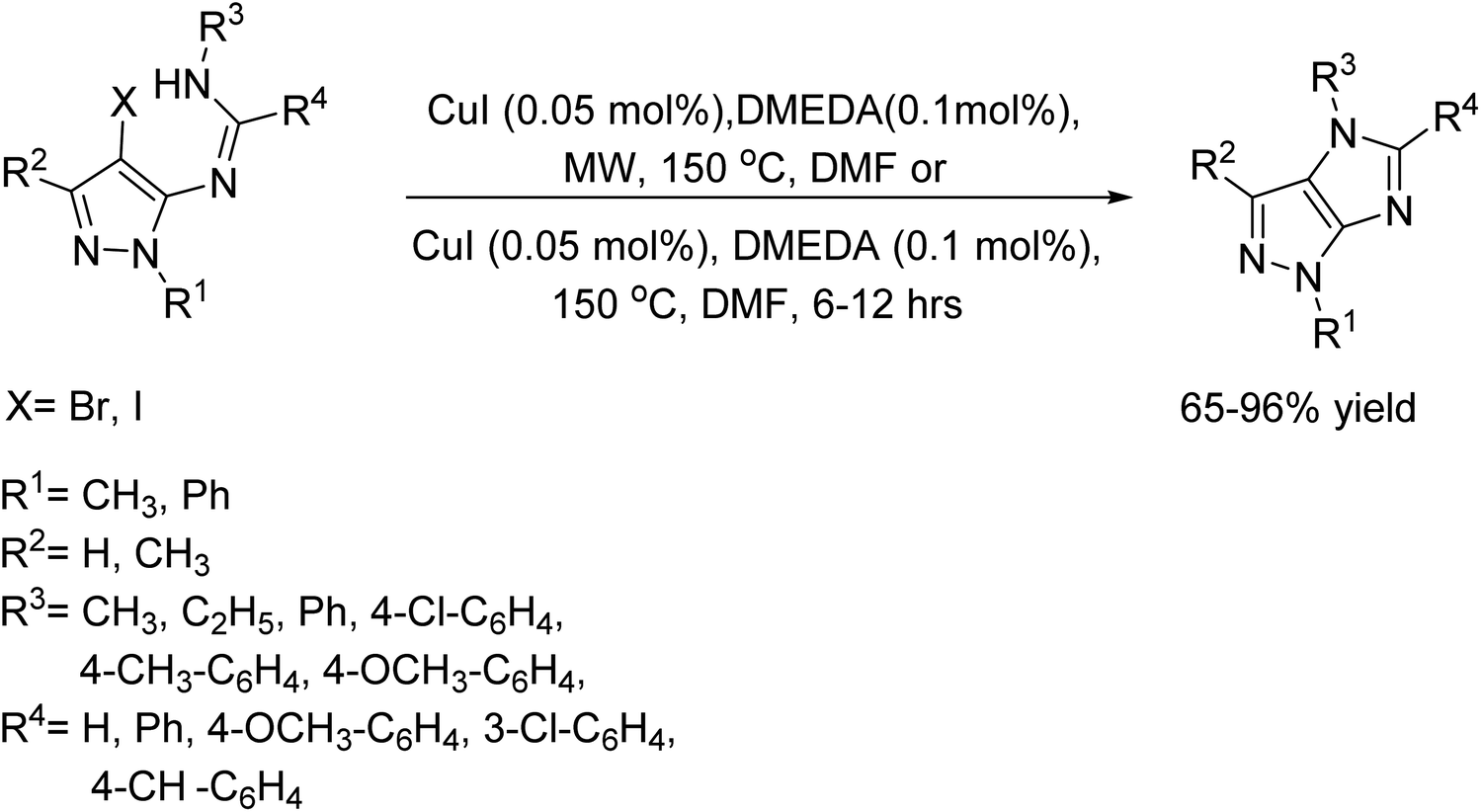
In 2012, Liubchak, Tolmachev, and their team introduced an innovative method for creating 4-substituted imidazo-[4,5-c]pyrazoles.110 They proposed that by using copper-catalyzed cross-coupling reactions, N-(4-halopyrazol-5-yl)amidines could undergo cyclization to form the expected compounds. The reaction was carried out using 0.05 mol% of CuI, 0.1 mol% of DMEDA and an excess of K2CO3. The reaction was performed using both microwave and conventional heating methods, with DMF as a solvent (Scheme 97).
 |
| | Scheme 97 Synthesis of 4-substituted imidazo[4,5-c]pyrazoles under MW and conventional heating condition. | |
In 2021, Xianjun Xu and colleagues developed a highly efficient synthetic route to produce unsymmetric 1, 4-diamino-2-butynes. This approach uses microwave energy along with a copper iodide catalyst to drive both a A3-coupling and a decarboxylative coupling reaction. Essentially, two distinct amines combine with formaldehyde and propiolic acid in a single, seamlessly orchestrated domino cascade. Ultimately, this one-pot multicomponent reaction produces a variety of target compounds with yields ranging from low to high, all while maintaining impressive chemoselectivity (Scheme 98).111
 |
| | Scheme 98 Microwave-assisted CuI-catalyzed synthesis of unsymmetrical 1,4-diamino-2-butynes. | |
In 2012, Arndtsen and colleagues developed a copper-catalyzed Petasis-type reaction utilizing imines, acid chlorides and organoboranes to access alpha-substituted amides (Scheme 99).112
 |
| | Scheme 99 Synthesis of α-substituted amides catalyzed by Cu. | |
In 2012, Evano and colleagues reported a successful synthesis of ynimines through a Cu-mediated oxidative cross-coupling of imines with terminal alkynes (Scheme 100).113
 |
| | Scheme 100 Synthesis of ynimines via Cu-mediated oxidative cross-coupling of imines with terminal alkynes. | |
In 2012, A.B. Sheremetev et al. developed a robust method for coupling various furazanyl iodides with 1, 2, 4, 5-tetrazines.114 This study stands out it's the only report showing that electron-deficient, nitrogen-rich heterocyclic iodides can successfully couple with similarly electron-deficient, nitrogen-rich heterocyclic amines. To pull this off, aminotetrazine was reacted with substituted iodofurazans, all in the presence of copper(II) acetate and 2-acetyl cyclohexanone, which served as the key supporting ligand. The reaction was carried out at 50 °C for 4 h, resulting in the formation of coupled secondary amines in good yields (Scheme 101).
 |
| | Scheme 101 Coupling reaction of furazanyl iodides with s-tetrazinylamines. | |
Stolle and colleagues made a significant contribution to the field of mechanochemistry with their groundbreaking report on a ligand- and solvent-free 1,3-dipolar cycloaddition reaction. In this innovative process, copper(II) acetate [Cu(OAc)2] served as the catalyst, and a planetary ball mill operated at 800 rpm (13.3 Hz) for 10 minutes. The versatility of this method was evident in its ability to facilitate reactions between various alkynes and azides, including the successful coupling of propargyl-functionalized polystyrene with decylazide.115 Moreover, the team achieved click polymerization of 1,12-diazidododecane with bis-ethynyl compounds within the ball mill. Notably, the efficacy of the mechanochemical protocol extended beyond [Cu(OAc)2]; other copper salts, such as cuprous iodide and cupric sulfate, also yielded comparable results. This approach exemplifies the promise of green chemistry practices by eliminating the need for solvents and ligands, thereby reducing the environmental impact of chemical synthesis (Scheme 102).
 |
| | Scheme 102 Click reaction under mechanochemical condition. | |
In 2024, A. Islam and colleagues reported a coupling reactions catalyzed by the Cu-complex. These C–N bond-forming reactions have drawn considerable attention because they are essential for synthesizing biocidal molecules(Scheme 103).116 Among the various catalytic systems available, organo-Cu complexes stand out due to their cost-effectiveness and the use of stable, readily accessible ligands. Building on this momentum and their experimental studies in the field, successfully synthesized and thoroughly characterized a novel organo–Cu complex using advanced spectroscopic techniques (NMR, FT-IR) and single crystal X-ray diffraction (XRD).
 |
| | Scheme 103 Synthesis of polyhydroquinoline derivatives catalyst by Cu(II)-complex in H2O medium. | |
In 2010, G. Shore and colleagues innovatively crafted a thin layer of copper, which, upon SEM analysis, revealed a porous aggregation of copper nanoparticles (Cu@NPs). This discovery led to the development of a novel technique for synthesizing propargylamine. The method, known as MACOS, utilizes a capillary coated with Cu(0), demonstrating a significant advancement in the field of organic synthesis (Scheme 104).117
 |
| | Scheme 104 Synthesis of propargylamine by MACOS technique. | |
In 2007, Kidwai and colleagues were the first to report an A3 reaction catalyzed by copper(0) nanoparticles. The initial model reaction was skillfully conducted using benzaldehyde, morpholine, and phenylacetylene, marking a significant milestone in the field of catalysis (Scheme 105).118
 |
| | Scheme 105 Synthesis of propargylamine catalyzed by Cu@NPs. | |
In 2019, Digambar B. Bankar and colleagues masterfully synthesized propargylamine. This synthesis was achieved through the reaction of aromatic aldehydes, amines, and phenylacetylene, catalyzed by ZnCl2 loaded on TiO2 nanomaterial. Remarkably, the process was conducted under solvent-free conditions at a temperature of 100 °C, demonstrating both the efficacy and environmental consideration of this approach (Scheme 106).119
 |
| | Scheme 106 Synthesis of propargylamines catalyzed by ZnCl2 loaded TiO2 nanomaterial. | |
In 2006, Yan and colleagues presented an eco-friendly synthesis of silver nanoparticles (Ag0NPs) using polyethylene glycol (PEG), highlighting ethylene glycol's environmentally benign nature compared to other reducing agents like sodium borohydride, hydrazine, and DMF. The process involves introducing silver nitrate (AgNO3) into PEG and introducing hydrogen gas (H2), which results in the formation of AgNPs. These nanoparticles have been effectively employed as catalysts in reactions involving aldehydes, amines, and alkynes. While aromatic and aliphatic aldehydes, along with alkynes, successfully yielded the anticipated products, crotonaldehyde notably did not lead to the formation of propargylamine using the same protocol (Scheme 107).120
 |
| | Scheme 107 A3-coupling reaction catalyst by Ag@NPs in PEG. | |
In 2007, Kidwai and colleagues made a pioneering discovery in the field of Nanocatalysts, reporting the synthesis of propargylamine catalyzed by gold nanoparticles (Au@NPs) derived from benzaldehyde, piperidine, and phenylacetylene. The Au@NPs were synthesized within the aqueous core of reverse miceller droplets, exhibiting a size distribution between 15 to 70 nm. A notable observation was the correlation between particle size and reaction rate; specifically, particles larger than 20 nm exhibited a gradual decrease in reaction rate. Impressively, Au@NPs sized at 18 ± 2 nm achieved a 97% conversion rate within 5 hours, while the larger 70 nm particles reached only a 78% conversion rate after 15 h. This study highlights the critical impact of nanoparticle size on the efficiency of catalytic processes (Scheme 108).121
 |
| | Scheme 108 Au@NPs catalyzed synthesis of propargylamine. | |
Various strategies exist for immobilizing Fe3II/IIIO4 nanoparticles (NPs) on different supports. However, the direct utilization of Fe3O4@NPs as catalysts in organic chemistry remains relatively uncommon. In 2010, Li and colleagues purchased and employed Fe3O4@NPs and Fe2IIIO3@NPs with sizes less than 50 nm for a three-component A3 coupling reaction (Scheme 109).122
 |
| | Scheme 109 A3 coupling catalyzed by Fe3O4@NPs. | |
In a notable study from 2012, Poisson and colleagues detailed the synthesis of chiral propargylamines with impressive yield and diastereoselectivity by adding lithiated ynol ethers to chiral N-sulfinyl imines. The lithium alkoxy alkynes, crucial for this reaction, were synthesized in situ from dichloroenolethers, facilitated by the addition of two equivalents of n-BuLi. In a fascinating twist, pre-complexing the N-sulfinyl imines with BF3–Et2O led to a complete inversion of selectivity, resulting in amines as the predominant product. This outcome, which saw the chirality at the C-1 stereocenter reversed, aligns with the findings of Galvez's research (Scheme 110).123
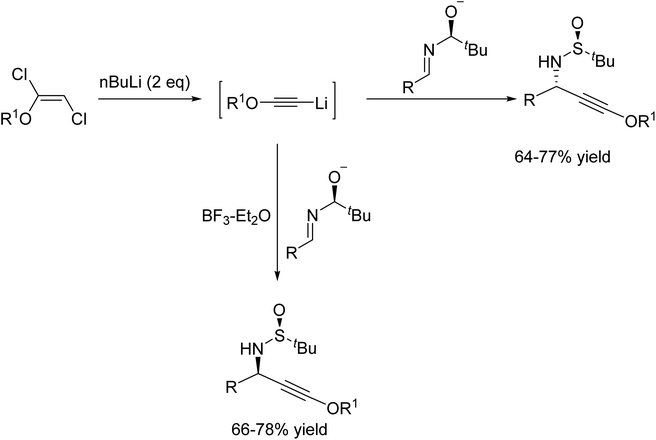 |
| | Scheme 110 Synthesis of chiral propargylamines. | |
In 2020, Pavneet Kaur and her team introduced a method for the decarboxylative A3-coupling of o-hydroxybenzaldehydes, 2°-amines, and alkynoic acids, without any solvents.124 This sophisticated, zero-waste method efficiently synthesizes hydroxylated propargylamines, serving as crucial precursors for constructing a broad array of biocidal heterocyclic scaffolds (Scheme 111).
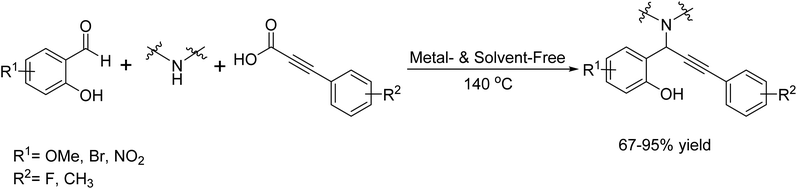 |
| | Scheme 111 Metal free multicomponent decarboxylative A3-coupling for the synthesis of propargylamines. | |
In 2006, the Frejd group expanded on the mechanochemical protocol to synthesize a variety of amino- and hydroxy-substituted dehydrophenylalanine derivatives under conditions free from solvents and phosphine. They explored how electron-withdrawing groups and the position of the heteroatom substituent about the halide affected the reactions. Their findings revealed that ortho-amino dehydrophenylalanine derivatives with appropriate substitutions could undergo a cyclization–elimination reaction, leading to the formation of 2-substituted indoles, especially when acidic conditions were applied (Scheme 112).125
 |
| | Scheme 112 Synthesis of amino-and hydroxy-substituted dehydrophenylalanine derivatives under ball-milling condition. | |
Transition metal free reactions of C–O bond formation
In 2018, G. Q. Rong and colleagues reported an efficient synthesis of indolylchromeno[2,3-b]indoles, facilitated by molecular iodine.126 This synthesis involved a Friedel–Crafts alkylation and oxidative coupling reaction between indoles and salicylaldehydes. The process was carried out in ethanol under reflux conditions. This method stands out for its use of molecular iodine, which acts as a catalyst, streamlining the reaction and enhancing its efficiency (Scheme 113).
 |
| | Scheme 113 Synthesis of indolylchromeno[2,3-b]indoles by the molecular iodine. | |
In 2019, L. Xing and colleagues introduced a straightforward method to synthesize a series of 4-chloroisocoumarins starting from o-alkynylbenzoates.127 The key to their approach was a PhICl2-mediated intramolecular cyclization carried out under metal-free conditions. In this reaction, PhICl2 serves two essential roles; it acts as the oxidant to trigger the oxidative C–O bond formation and also supplies the chlorine atom. The reaction conditions, the method delivers high-yielding products, features easy purification, and is even scalable to gram-level synthesis (Scheme 114).
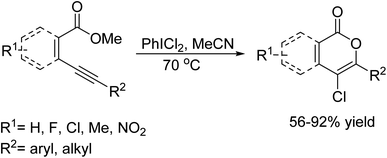 |
| | Scheme 114 PhICl2-mediated C–O bond formation reaction. | |
In 2016, Yunhe Lv et al. introduced an innovative protocol for the intermolecular C–O cross coupling reaction promoted by Selectfluor; enabling the synthesis of N-hydroxyimide esters.128 This protocol successfully couples readily accessible aryl and alkyl aldehydes with N-hydroxyphthalimide (NHPI) and N-hydroxysuccinimide (NHSI), making the protocol highly advantageous. Notably, the resulting active esters can be directly converted into amides in a single, streamlined step (Scheme 115).
 |
| | Scheme 115 Metal-free intermolecular C–O cross-coupling reactions. | |
In 2014, C. R. Reddy and colleagues introduced an innovative and efficient method for synthesizing a wide variety of functionalized furans.129 Their technique employs a DBU-mediated tandem process that combines Michael addition with 5-exo-dig cyclo isomerization of enynes and keto methylenes. Notably, this [3 + 2]-annulation reaction is operationally simple, proceeds under metal-free conditions, achieves 100% atom economy, and accommodates a broad range of substrates (Scheme 116).
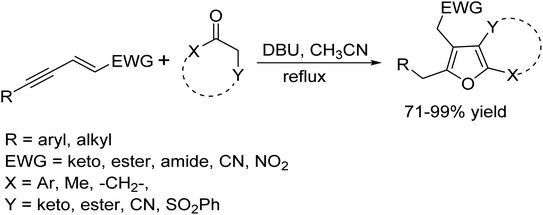 |
| | Scheme 116 Greener approach of C–O bond formation. | |
In 2006, Lana and colleagues introduced the method for the synthesis of 1,8-cineole and 1, 4-cineole through the isomerization of α-terpineol, catalyzed by the heteropoly acid H3PW12O40 (PW). This process, which is beneficial for flavoring and pharmaceutical applications, demonstrated that PW has superior catalytic activity and selectivity compared to traditional acid catalysts like H2SO4 and Amberlyst-15. The research highlighted the efficacy of PW, the most potent heteropoly acid in the Keggin series, in both homogeneous and solid acid catalyst forms for the isomerization reaction.130 Notably, the heterogeneous system proved more efficient, yielding 35% of 1,8-cineole and 25% of 1, 4-cineole at a conversion rate of 70–100% in a cyclohexane solution. Furthermore, the use of silica-supported PW as a solid acid catalyst offered the advantage of recyclability (Scheme 117).
 |
| | Scheme 117 Synthesis of 1,8-cineole & 1,4-cineole using PW/SiO2 as catalyst. | |
In 2008, Rafiee and colleagues probe the catalytic efficiency of various supported heteropoly acids for synthesizing mandelates from mandelic acid and alcohols.131 Their study aimed to classify the most effective catalyst, comparing dodecatungestophosphoric acid (PW) supported on SiO2 with molybdophosphoric acid (PMo) on different solid materials. Traditional approaches to mandelate synthesis often face challenges, including harsh acidic conditions, complex purification steps, low yields, long reaction times, and side reactions such as carbonization, oxidation, and etherification. Notably, PW supported on SiO2 outperformed other systems, delivering high yields in a short time while offering a cleaner, milder, and environmentally friendly alternative with outstanding selectivity and efficiency. Additionally, this catalyst proved stable, highly efficient, reusable, and cost-effective. The results highlight that SiO2 is the optimal support material for heteropoly acids compared to other options. PW/SiO2 represents an environmentally friendly, inexpensive, non-toxic, and easily workable catalyst (Scheme 118).
 |
| | Scheme 118 Synthesis of mandelates using dodecatungestophosphoric acid. | |
In 2010, Li and colleagues developed an innovative method for deprotecting oximes to their corresponding carbonyl compounds. The system utilized solid-supported acid (SSA), surfactant, and paraformaldehyde.132 Notably, this approach allowed the reaction to proceed with excellent yields at a moderate temperature of 50 °C in water, facilitated by ultrasound irradiation. Compared to previous techniques for cleaving oximes (such as acid-catalyzed hydrolysis and various forms of deoximation), this method addressed several limitations. These included the need for high temperatures, complex work-up procedures, harsh conditions, prolonged reaction times, suboptimal yields, and the use of toxic reagents. By employing SSA as a catalyst, Li and colleagues achieved several advantages: prevention of acid-sensitive functional group destruction, elimination of toxic solvents and expensive reagents, stability, reusability, cost-effectiveness, and ease of handling. Additionally, the method provided higher yields, was readily available, and enabled shorter reaction times and milder conditions (Scheme 119).
 |
| | Scheme 119 Deprotection of oximes to carbonyl compounds using H2SO4–SiO2. | |
Transition metal catalyzed reactions of C–O bond formation
In 2018, Bo Yang and colleagues introduced an environment friendly and efficient approach for synthesizing 2-arylbenzoxazoles using a Fe-catalyzed C–N/C–O cross-coupling reaction.133 This innovative method effectively tackles common challenges associated with Pd and Cu-catalyzed synthesis, such as limited substrate scope and prolonged reaction times, offering a practical alternative with improved efficiency and sustainability, (Scheme 120).
 |
| | Scheme 120 Iron-catalyzed domino C–N/C–O cross-coupling reaction. | |
In 2016, Yang Lin et al. introduced an innovative protocol for the heterogeneous C–O coupling reaction between ArI and aliphatic alcohols, carried out in either neat alcohol or toluene at 110 °C.134 The process utilized 10 mol% of an MCM-41-immobilized 1,10-phenanthroline copper(I) complex, [MCM-41-1,10-phen–CuI]) alongside Cs2CO3 as a base, effectively yielding a range of aryl alkyl ethers in good to excellent yields. Notably, this new heterogeneous copper catalyst is easily synthesized using readily available and cost-effective reagents. It can be recovered simply by filtering the reaction mixture and reused at least eight cycles without any significant drop in activity (Scheme 121).
 |
| | Scheme 121 MCM-41-immobilized 1,10-phenanthroline-copper(I) catalyst C–O coupling reaction. | |
In 2016, Z.-H. Ren and colleagues introduced an efficient method of oxidative tetrahydrofuranylation of oximes catalyzed by CuI.135 This reaction demonstrates excellent functional group tolerance, allowing various substituted ketoximes and aldoximes to react smoothly with tetrahydrofuran (THF), yielding the corresponding tetrahydrofuran-2-yl oxime ethers in excellent yields (Scheme 122).
 |
| | Scheme 122 CuI-catalyzed oxidative cross coupling of oximes for direct access to tetrahydrofuran-2-yl oxime ethers. | |
In 2021, L. Ai and colleagues introduced an efficient method for synthesizing benzothieno[3,2-b]benzofurans via intramolecular dehydrogenative C–O coupling.136 This approach delivers moderate to high yields (64–91%), regardless of whether the substituents are electron-donating or electron-withdrawing. It enables the construction of fused thienofuran systems containing three to six rings. Mechanistic studies revealed that 1,1-diphenylethylene completely discourage the reaction, confirming a radical pathway initiated by single-electron transfer between the hydroxyl of the substrate and the Cu catalyst (Scheme 123).
 |
| | Scheme 123 Copper-mediated intramolecular dehydrogenative C–O coupling reaction. | |
In 2016, Tanmoy Maity et al.137 demonstrated Heterogeneous O-arylation of nitroarenes with substituted phenols over a copper-immobilized mesoporous silica catalyst (S@Cu) in DMF medium (Scheme 124).
 |
| | Scheme 124 S@Cu catalysed O-arylation reaction of nitroarene with aryl alcohols. | |
In 2019, J. Wang and colleagues introduced a groundbreaking strategy for synthesizing 2,5-disubstituted oxazoles.138 Their method involved reacting aryl acetylenes with α-amino acids in the presence of I2 and Cu(NO3)2. This protocol commenced with the transformation of aryl acetylene into α-iodo acetophenone. This was followed by a sequence of reactions including Kornblum oxidation, condensation to form imine, decarboxylation, annulation, and finally oxidation, which culminated in the creation of 2, 5-disubstituted oxazoles (Scheme 125).
 |
| | Scheme 125 Synthesis of 2, 5- disubstituted oxazoles from aryl acetylenes and α-amino acids catalyst by I2. | |
In 2020, A. K. Sharma and colleagues introduced a method for synthesizing Pd(0)-layered nanoparticles, starting with nano-sized magnetic Fe3O4. The particles were first coated with SiO2, then sequentially treated with PhSeCl in N2 atmosphere and PdCl2 in H2O medium. The resulting Fe3O4–SiO2.SePh.Pd(0)@NPs are magnetically retrievable and represent the first instance where the outermost Pd(0) layer is primarily stabilized by selenium. ICP-AES analysis confirmed the Pd content at 1.96%. Characterization by TEM, SEM-EDX, XPS, and powder XRD demonstrated their efficiency as catalysts for C–O and C–C (Suzuki–Miyaura) coupling reactions involving ArX (X = Br, Cl) in water.139 XPS studies indicated a palladium oxidation state of zero in NPs ranging from 12 to 18 nm in size. Impressively, these catalysts are recyclable over seven cycles without significant loss of activity. The method stands out for its normal reaction conditions, simplicity, and efficiency, as the catalyst can be easily separated from the reaction mixture using an external magnet and reused in subsequent cycles. Optimal Pd loading ranged from 0.1–1.0 mol% for O-arylation and 0.01–1.0 mol% for Suzuki–Miyaura coupling, with higher amounts required for ArCl compared to ArBr. The catalytic system operates primarily through a heterogeneous mechanism (Scheme 126).
 |
| | Scheme 126 Fe3O4@SiO2@SePh@Pd(0)-catalyzed O-arylation of phenol. | |
In 2014, Deqiang Liang and colleagues introduced a protocol of oxidative C–O cross-coupling reaction of active alkenes with carboxylic acids.140 Using Pd(OAc)2 as a catalyst and PhI(OAc)2 as an oxidant, ketene dithioacetals underwent smooth acyloxylation in a carboxylic acid water solution, yielding a variety of vinyl esters with high selectivity, efficiency, and excellent functional group tolerance. A proposed mechanism suggests that a vinyl iodonium species acts as a key intermediate in the process (Scheme 127).
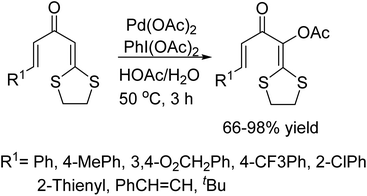 |
| | Scheme 127 Palladium-catalyzed oxidative C–O cross-coupling of ketene dithioacetals and carboxylic acids. | |
In 2014, Mona Hosseini-Sarvari and colleagues developed an efficient ligand-free Pd-supported ZnO nanoparticle system for O-arylation of phenols using ArX (X = Cl, Br, I).141 The Pd–ZnO@NPs catalyst was readily synthesized and characterized, with ICP analysis confirming a Pd content of 9.84 wt% (0.005 g of catalyst containing 462 × 10−8 mol% of Pd). These nanoparticles, averaging 20–25 nm in size with a specific surface area of 40.61 m2 g−1, function as a reusable heterogeneous catalyst for C–O bond formation in organic synthesis. Notably, the protocol yields arylated products efficiently without requiring an inert nitrogen or argon atmosphere. Moreover, the catalyst can be recovered and recycled multiple times without significant loss of catalytic activity (Scheme 128).
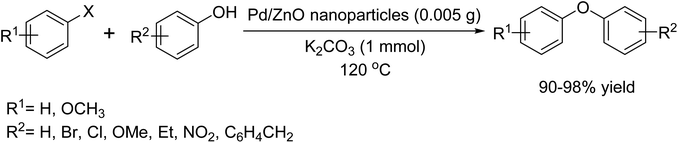 |
| | Scheme 128 Coupling reaction catalyst by highly active recyclable heterogeneous Pd/ZnO nanoparticle. | |
In 2015, Firouz Matloubi Moghaddam et al.142 synthesized magnetic nanoparticles of NiFe2O4 and CoFe2O4 and demonstrated that these catalysts facilitate the formation of a C–O bond when phenol reacts with aryl halides. Both spinel systems effectively catalyzed the coupling of diverse ArX and phenol derivatives. Additionally, the catalysts can be effortlessly separated from the reaction mixture and reused in multiple reaction cycles. Under optimized conditions, all the reactions yielded moderate to high results, although those catalyzed by NiFe2O4 reached completion in noticeably short periods of time. Furthermore, analyses indicated that the reusability of the two catalysts is more or less equivalent (Scheme 129).
 |
| | Scheme 129 C–O bond formation reaction catalyst by NiFe2O4 and CoFe2O4 nanoparticles. | |
In 2018, the Zhang and Huang group designed a novel donor–acceptor (D–A) fluorophore, 4-DPAPN, featuring precisely tuned triplet energy levels. Leveraging its photocatalytic properties, they introduced a photoinduced Ni-catalyzed C–O coupling reaction between ArBr and carboxylic acids under ambident conditions.143 The catalytic system employed Ni(NO3)2·6H2O in combination with dtbbpy (Scheme 130).
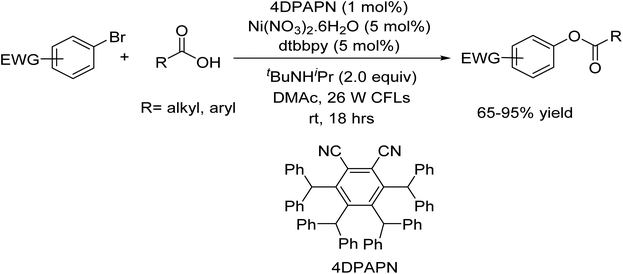 |
| | Scheme 130 C–O coupling reactions of aryl halide with carboxylic acids using Ni(NO3)2·6H2O/dtbbpy as the catalyst. | |
Transition metal free reactions of C–Se bond formation
In 2018, C. S. Pires reported a highly selective one-pot synthesis of 5-amino-4-(arylselanyl)-1H-pyrazoles. The method involved a three-component condensation, combining diversely substituted benzoylacetonitriles, arylhydrazines, and diaryldiselenides. Remarkably, the reactions were conducted in refluxing acetonitrile under the atmosphere of air as the oxidant. The protocol exhibited excellent functional group tolerance, accommodating both electron-donating and electron-accepting substituents, as well as bulky groups. The resulting yields ranged from good to excellent, highlighting the versatility and efficiency of this synthetic approach (Scheme 131).144
 |
| | Scheme 131 Selective synthesis of 5-amino-4-(arylselanyl)-1H-pyrazoles catalyst by I2. | |
In a study conducted in 2015, Gondru and colleagues devised a novel and expeditious methodology for synthesizing 2,4-disubstituted 1,3-selenazoles. This was accomplished by reacting α-bromoketone with selenourea at a temperature of 25 °C within an aqueous medium, facilitated by ultrasonic irradiation. The process yielded analytically pure products in a remarkably brief duration of 10 to 60 seconds, with the outcomes being of exceptional yield (Scheme 132).145
 |
| | Scheme 132 Synthesis of 2,4- disubstituted 1,3-selenazole in H2O medium under ultrasonic irradiation. | |
In 2023, Ji-Hong Xia and colleagues introduced a metal-free strategy for the regioselective selenation of chromones using Selectfluor in ambident reaction conditions.146 This protocol accommodates a broad range of substrates, delivering 3-selenylated chromones with high selectivity and satisfactory yields. Additionally, the approach successfully integrates KSCN with enaminones for the synthesis of thiocyano chromones within this transformation (Scheme 133).
 |
| | Scheme 133 Selectfluor-mediated tandem cyclization of enaminones with diselenides to form 3-selenylated chromones. | |
In 2019, Jyun-Cyuan Liou et al. introduced an innovative protocol for the C–H selenation of aldehydes with diselenides promoted by DTBP, conducted under entirely metal-free and neat conditions.147 This approach demonstrates broad functional group compatibility, efficiently accommodating substituents such as bromo, trifluoromethyl, chloro, and amine groups, as well as heterocycles like thiophene and furan. Both the diaryl and dialkyl diselenides reacted flawlessly with aldehydes, produced selenoesters habitually excellent yields (Scheme 134).
 |
| | Scheme 134 Synthesis of selenoesters from diaryl diselenides and aldehydes catalyst by DTBP. | |
In 2021, Wei-Long Xing et al. demonstrated an innovative protocol for the construction of C–Se bonds from aryl diselenides, highly adaptable substrates under normal reaction conditions.148 Furthermore, this approach was adeptly applied in the late-stage refinement of pharmaceutical carboxylates, display extraordinary chemoselectivity and remarkable tolerance toward a wide array of functional groups (Scheme 135).
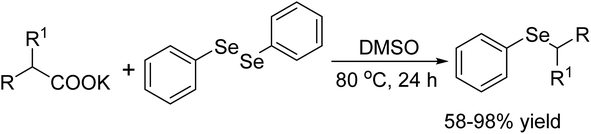 |
| | Scheme 135 Aliphatic carboxylates for C–Se coupling. | |
Transition metal catalyzed reactions of C–Se bond formation
In 2014, N. V. Orlov and his team introduced an innovative nickel-catalyzed reaction that streamlined the production of entirely new monoseleno-substituted 1,3-dienes all in one step.149 They started with simple, readily available terminal alkynes and benzeneselenol, then used a common catalyst precursor, Ni(acac)2, paired with a carefully chosen phosphine ligand, PPh2Cy. This smart combination paved the way for the selective formation of C–Se bonds, exclusively building the unique s-gauche diene framework. What's really exciting is that these diene products turned out to be exceptionally stable under normal conditions, retaining their s-gauche structure both as solids and in solution, as confirmed by X-ray crystallography and NMR spectroscopy. Detailed mechanistic investigations using ESI-MS further revealed the key nickel species driving this transformation (Scheme 136).
 |
| | Scheme 136 Ni-catalyzed alkyne hydroselenation. | |
In 2020, Oleksandr Vyhivskyi and his team unveiled a captivating method to form carbon-selenium bonds using an Ullmann-type cross-coupling reaction catalyzed by CuI.150 Their approach lets a wide variety of ArI fluently react with inconsistent disubstituted 2-selenohydantoins under normal reaction conditions, yielding Se-arylated imidazolines in moderate to excellent quantities. Detailed computational studies indicate that the reaction likely follows a pathway where oxidative addition is followed by an intramolecular reductive elimination, making it the most energy-efficient route. In addition, the researchers assessed the anticancer potential of these compounds by testing their cytotoxic activity in vitro against MCF7 and A549 cancer cell lines, using VA13 and MCF10a cells as controls (Scheme 137).
 |
| | Scheme 137 Ullmann-type C–Se cross-coupling. | |
In 2010, Li et al. introduced a highly efficient and ligand-free CuS-catalyzed coupling reaction between aryl halides and diaryldiselenides. This reaction was accelerated by the addition of Fe powder and could be completed in only 3–12 hours at 110 °C under an argon atmosphere.151 The purpose of this reaction was to synthesize unsymmetrical diaryl selenides with good to excellent yields (Scheme 138).
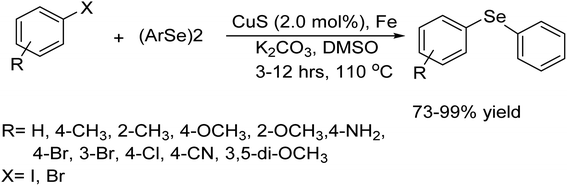 |
| | Scheme 138 Synthesis of unsymmetrical diaryl selenides using Fe catalysis. | |
In 2014, S. Roy et al. developed an eco-friendly method for phenylselenylation of aryl boronic acid. This method was proceed on a nitrogen-rich porous covalent imine network (CuII–CIN-1), catalyzed by a highly recyclable heterogeneous Cu(II) catalyst in PEG-600 solvent.152 The catalyst was successfully used to catalyze the cross-coupling reaction between a diverse set of aryl boronic acids and diphenyl diselenide. This resulted in the synthesis of good to excellent yields of the corresponding unsymmetrical organoselenides in PEG-600 solvent in the presence of K2CO3 at 80 °C for 8 h (Scheme 139).
 |
| | Scheme 139 CuII–CIN-1 catalysed phenyl selenylation of aryl boronic acid. | |
In 2016, Matsumura and colleagues reported a copper-catalyzed synthesis of symmetrical diaryl selenides from triarylbismuthanes and selenium under aerobic conditions.153 The reaction involved treating Ph3Bi, 1.5 equivalents of Se powder using 10 mol% Cu(OAc)2 and 1,10-phenanthroline as the catalytic system in DMSO medium at 100 °C. Remarkably, this transformation proceeded without the need for any base or inorganic reagent under aerobic conditions (Scheme 140).
 |
| | Scheme 140 Cu-catalysed procedure for the synthesis of symmetrical diaryl selenides. | |
In 2012, Nageswar's team unveiled a groundbreaking approach to C–Se cross-coupling reaction of organoboranes with PhSeBr to form the unsymmetrical diorganyl selenides catalyzed by bimetallic nano catalyst.154 They achieved this by employing CuFe2O4 as the catalyst in a PEG-400 medium, paving the way for a process that's both efficient and innovative. What makes this reaction truly stand out is its versatility. It smoothly handles both aryl and heteroaryl boronic acids (Scheme 141), increasing its applicability for diverse synthetic needs. Moreover, the heterogeneous nature of the catalyst means it can be easily recovered and reused several times with only a slight drop in performance, highlighting its promise for sustainable chemical synthesis.
 |
| | Scheme 141 Nano-CuFe2O4 catalyzed C–Se cross coupling organo boranes with PhSeBr. | |
In 2022, Yuki Murata and colleagues introduced an easy and versatile method to create 5-arylselanyluracils through a selective C–H selenation of uracils.155 In this approach, uracils are reacted with arylboronic acid and selenium powder in the presence of 10 mol% AgNO3 at 120 °C under air, resulting in various 5-arylselanyluracils. Significantly, the selanyl group is produced directly from the commonly available arylboronic acid and selenium powder during the reaction, making the method both straightforward and efficient. Notably, this is the first reported instance of synthesizing 5-arylselanyluracils using a multi-component system (Scheme 142).
 |
| | Scheme 142 Reaction of uracils with arylboronic acids, and selenium catalyzed by silver to form 5-arylselanyluracils. | |
Transition metal free reactions of C–P bond formation
In 2018, L. Liu and colleagues unveiled a clever, transition-metal-free method to phosphorylate cinnamic acids using P(O)H compounds introduced a radical-promoted decarboxylation process carried out under really mild conditions.156 This innovative approach makes it straightforward and efficient to produce valuable (E)-alkenylphosphine oxides across a diverse range of substrates while delivering satisfactory yields. The genius of this technique lies in its simplicity and versatility. Without relying on metal catalysts, it opens up a more sustainable and potentially cost-effective pathway for synthesizing these key organic compounds (Scheme 143).
 |
| | Scheme 143 TBPB catalyzed C–P bond formation by decarboxylative phosphorylation of cinnamic acids. | |
In 2020, Runmin Zhao and colleagues introduced a groundbreaking acid-catalyzed direct coupling strategy that merges a diverse range of unprotected 2,3-allenols with aryl phosphine oxides.157 This one-step approach efficiently generates structurally varied γ-keto phosphine oxides by promoting simultaneous formation of C–P and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, all while maintaining remarkable functional group tolerance and achieving complete atom economy without the need for metals or additives (Scheme 144).
O bonds, all while maintaining remarkable functional group tolerance and achieving complete atom economy without the need for metals or additives (Scheme 144).
 |
| | Scheme 144 TfOH-catalyzed phosphinylation of 2,3-allenols into γ-ketophosphine oxides. | |
In 2020, Jia Yuan and colleagues introduced an innovative protocol for the C–P bond formation reaction promoted by visible-light that notably eliminates the need for transition metals and photoredox catalysts.158 By leveraging readily available heteroaryl chlorides and bromides as substrates, their approach efficiently provided a spectrum of heteroaryl phosphine oxides in moderate to good yields. This method stands out as a straightforward and practical route to heteroaryl phosphine oxides, showcasing both simplicity and efficiency (Scheme 145).
 |
| | Scheme 145 Visible-light-promoted metal-free phosphinylation of heteroaryl halides in the presence of potassium tBuOK. | |
In 2017, I. Kim et al. introduced a noble protocol for the phosphonation of quinolinones promoted by the light irradiation that eliminates the external photocatalyst.159 This protocol reveals that both the reactant materials and the reaction products themselves can act as photosensitizers. When excited by compact fluorescent light, these molecules trigger the dissociation of the N–O bond in the pyridinium salt through a single electron transfer process. This elegant approach not only simplifies the reaction setup but also accommodates a wide range of quinolinone substrates under very mild conditions (Scheme 146).
 |
| | Scheme 146 Light-promoted phosphonation of quinolinones without any external photocatalyst. | |
In 2017, D. Liu and colleagues introduced a greener method for the direct difunctionalization of alkynoates, promoted by visible-light under remarkably mild conditions.160 Utilizing catalytic amounts of commercially available Eosin Y as the photocatalyst, along with tert-butyl hydroperoxide (TBHP) serving as the oxidant, they developed a radical tandem phosphorylation/cyclization reaction. This innovative approach successfully synthesizes a diverse array of 3-phosphorylated coumarins, delivering products with high functional group tolerance, moderate to good yields, and excellent regioselectivities (Scheme 147).
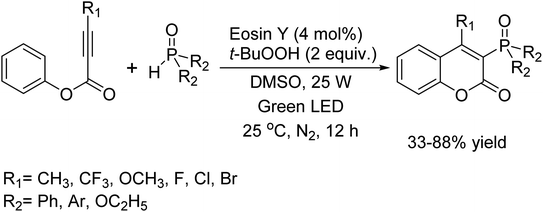 |
| | Scheme 147 Metal-free, visible-light promoted direct phosphorylation/cyclization of alkynoates catalyst by Eosin Y (EY) and (TBHP). | |
Transition metal catalyzed reactions of C–P bond formation
In 2017, Pan Zhou and colleagues introduced a novel protocol for the oxidative phosphonylation of N,N-dimethylenaminones with H-phosphonates using Mn(OAc)3.161 This approach features a one-step process that achieves both chemo- and regioselective cleavage of a Csp2–Csp2 bond and formation of a Csp3–P bond, successfully yielding functionalized β-ketophosphonates under ambident conditions. Additionally, the research team explored oxidative Csp3–H/P–H cross-coupling reactions via Csp3–C(CO) bond cleavage. Their preliminary mechanistic studies led to the proposal of a plausible pathway that accounts for the observed transformations. The protocol is especially notable for its operational simplicity, broad substrate scope, scalability, and the consistently moderate to high yields it produces (Scheme 148).
 |
| | Scheme 148 Mn(OAc)3-caytalyst oxidative Csp3–P bond formation through Csp2–Csp2 and P–H bond cleavage. | |
In 2016, Pan Xu and colleagues introduced a cutting-edge protocol for the oxidative C–H/P–H cross-coupling of aldehyde hydrazones with diphenylphosphine oxide, catalyzed by K2S2O8 and Cu(OAc)2.162 This method directly furnishes functionalized α-iminophosphine oxides with excellent functional group tolerance and operates seamlessly under normal conditions. The reaction's success is rooted in a net oxidative aminyl radical-polar crossover process, offering a strategic and efficient approach to C–P bond formation (Scheme 149).
 |
| | Scheme 149 Oxidative C(sp2)−H phosphonation of aldehyde hydrazones. | |
In 2021, Xingyu Chen and colleagues introduced an innovative protocol for the C–P bond formation reaction that employs acylphosphines as differential phosphination reagents for ArBr and ArOTf substrates catalyzed Pd2(dba)3.163 Despite being resistant to air and moisture, these acylphosphines display excellent reactivity with ArBr and ArOTf, making them highly effective in this context. The transformation directly yields trivalent phosphines in satisfactory yields and boasts a wide range of substrate scope along with impressive functional group tolerance. This work not only demonstrates the efficiency of the approach but also uncovers the potential of acylphosphines as novel phosphorus sources in the direct synthesis of trivalent phosphines (Scheme 150).
 |
| | Scheme 150 Palladium-catalyzed C–P(III) bond formation by coupling ArBr/ArOTf with acylphosphines. | |
In 2018, Ryota Isshiki et al. made an exciting method for the synthesis of C–P bond by the reaction of aromatic esters as distinctive aryl sources catalyzed by Ni(OAc)2. The key success of this protocol was an innovative use of a thiophene-based diphosphine ligand known as dcypt.164 This approach allowed a variety of aromatic esters, even those featuring heteroaromatic structures to be efficiently coupled with phosphine oxides and phosphates, yielding valuable aryl phosphorus compounds. To demonstrate the versatility of their method, applied the protocol to sequential coupling reactions, underscoring its broad synthetic potential (Scheme 151).
 |
| | Scheme 151 Ni catalyzed C–P bond formation via decarbonylative using aromatic esters and organophosphorus compounds. | |
In 2014, Tao Wang and his team introduced an outstanding Pd-catalyzed cross-coupling method that brings together triarylbismuths and various P(O)–H compounds.165 What makes this reaction especially appealing is that it runs effortlessly under normal conditions, no need to keep moisture or air at bay. This can reliably create a broad range of valuable aryl phosphonates, aryl phosphinates, and aryl phosphine oxides with excellent atom economy and straightforward operations, all while achieving excellent yields. Furthermore, this reaction is particularly groundbreaking as it represents the very first example of transition-metal-catalyzed C–P bond construction using triarylbismuth compounds as substrates (Scheme 152).
 |
| | Scheme 152 Pd-catalyzed C–P bond formation by the reaction of triarylbismuth with dialkyl H-phosphite. | |
In 2009, Rita G. de Noronha and her team unveiled a novel approach by using MoO2Cl2 as an innovative catalyst for C–P bond forming reaction, demonstrated through the hydrophosphonylation of aldehydes. They successfully prepared a series of R-hydroxyphosphonates in satisfactory yields using a catalytic system based on HP(O)(OEt)2 in combination with MoO2Cl2 (5 mol%). The reaction was carried out either under solvent-free conditions.166 Mechanistic insights provided by DFT calculations revealed that the PO group coordinates to molybdenum while simultaneously transferring the hydrogen from the P–H bond to one of the Mo–O oxygen atoms. This step generates the activated species reacts with the aldehyde to form the R-hydroxyphosphonates. Notably, the calculated activation barriers are around 20 kcal mol−1, showcasing the reaction's efficiency (Scheme 153).
 |
| | Scheme 153 MoO2Cl2 catalyzed C–P bond formation via hydrophosphonylation of aldehydes. | |
In 2014, Ying He and colleagues introduced an innovative approach for the P-arylation of aryl diazonium salts with H-phosphonates using a dual catalytic system that combines Au catalysis with photoredox activation.167 The transformation proceeds smoothly at ambident temperature without the need for any bases or additional additives, efficiently producing aryl phosphonates. Mechanistic investigations suggest that the key step involves a photoredox-promoted generation of an electrophilic aryl Au(III) intermediate, couples with the nucleophilic H-phosphonate to yield the final product. This method not only streamlines the synthesis by operating under mild conditions but also highlights the powerful synergy between Au and photoredox catalysis in enabling new reactivities (Scheme 154).
 |
| | Scheme 154 Synthesis of P-arylation of aryl diazonium salts with H-phosphonates catalyst by dual gold and photoredox. | |
In 2017, Linbin Niu and colleagues unveiled an external oxidant-free protocol for synthesizing α-aminophosphonates by ingeniously merging photocatalysis with proton-reduction catalysis.168 This innovative approach not only bypasses the need for traditional oxidants but also has demonstrated its experimental utility through a successful gram–scale reaction. Importantly, the strategy holds significant promise for the selective functionalization of oxidant-sensitive C(sp3)–H bonds, potentially opening new avenues in mild and efficient synthetic transformations (Scheme 155).
 |
| | Scheme 155 Oxidant-free synthesis of α-aminophosphonates by synergistically combining photocatalysis and proton-reduction catalysis. | |
In 2018, Yong Jian and colleagues reported a innovative protocol of visible-light induced denitrogenative phosphorylation of benzotriazoles, enabling the synthesis of a broad range of substituted aryl phosphonates in moderate to highly satisfactory yields.169 This efficient approach exhibits impressive tolerance toward a variety of functional groups. Additionally, the utility of this experimental method was displayed through a victorious gram-scale reaction (Scheme 156).
 |
| | Scheme 156 Visible-light-induced C(sp2)−P bond formation by denitrogenative coupling of benzotriazoles with phosphites. | |
Conclusion
This review has highlighted the transformative impact that innovative approaches to bond construction are having on modern synthetic chemistry. Advanced catalytic methods for forming C–C bonds, along with carbon-heteroatom bonds (C–S, C–N, C–O, C–Se, C–P), have ushered in new efficiencies and selectivities that overcome the limitations of traditional methods. Transition-metal catalysis now enables robust cross-coupling reactions under mild conditions, while photolytic and organocatalytic strategies offer complementary routes that work under ambient conditions with broad substrate tolerance. These combined innovations not only enhance the precision of synthetic methods but also promote sustainability through greener reaction conditions, such as solvent-free processes and recyclable catalysts. Despite significant progress, challenges remain, particularly in achieving precise control over reaction selectivity in densely functionalized systems. The continued integration of new catalytic strategies and sustainable practices promises to resolve these issues, paving the way for advancements in pharmaceuticals, materials science, and other applied fields. In essence, the evolution of bond construction techniques from traditional cross-coupling to innovative dual-catalytic and green methodologies marks a new era in chemical synthesis, one defined by heightened efficiency, selectivity, and environmental consciousness.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- K. C. Nicolaou and S. A. Snyder, Angew. Chem., Int. Ed., 2005, 44, 1012–1044 CrossRef CAS PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- P. T. Anastas, and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- F. A. Carey, and R. J. Sundberg, Advanced Organic Chemistry: Part A, Springer, New York, 2007 Search PubMed.
- M. B. Smith, and J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, John Wiley, New York, 2007 Search PubMed.
- A. Suzuki, J. Organomet. Chem., 2005, 690, 2454–2464 Search PubMed.
- C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- J. F. Hartwig, ACS Cent. Sci., 2017, 3, 692–700 CrossRef PubMed.
- S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101–110 CrossRef CAS PubMed.
- Z. Bao and Y. Zhang, Chem. Rev., 2016, 116, 12842–12892 Search PubMed.
- Z. Lv, B. Wang, Z. Hu, Y. Zhou, W. Yu and J. Chang, J. Org. Chem., 2016, 81, 9924–9930 CrossRef CAS PubMed.
- M. Sharma and P. J. Bhuyan, RSC Adv., 2014, 4, 15709–15712 RSC.
- C.-C. A. Voll and T. M. Swager, J. Am. Chem. Soc., 2018, 140, 17962–17967 CrossRef CAS PubMed.
- A. Lipp, G. Lahm and T. Opatz, J. Org. Chem., 2016, 81, 4890–4897 CrossRef CAS PubMed.
- Z. Xu, Z. Hang and Z.-Q. Liu, Org. Lett., 2016, 18, 4470–4473 CrossRef CAS PubMed.
- P. B. Thakur and H. M. Meshram, RSC Adv., 2014, 4, 6019 RSC.
- M. Bayat, H. Imanieh and S. H. Hossieni, Chin. Chem. Lett., 2009, 20, 656 CrossRef CAS.
- M. L. Kantam, R. Chakravarti, B. Sreedhar and S. Bhargava, Synlett, 2008, 1449 CrossRef CAS.
- A. Kumar, M. K. Gupta and M. Kumar, Green Chem., 2012, 24, 290 RSC.
- L.-T. An, F.-Q. Ding, J.-P. Zou, X.-H. Lu and L.-L. Zhang, Chin. J. Chem., 2007, 25, 822 CrossRef CAS.
- J. J. R. Freitas, T. R. Couto, I. H. Cavalcanti, J. C. R. Freitas, R. A. Oliveira and P. H. Menezes, Ultrason. Sonochem., 2014, 21, 1609 CrossRef CAS PubMed.
- S. Narayanaperumal, R. C. da Silva, K. S. Feu, A. F. de la Torre, A. G. Corrêa and M. W. Paixão, Ultrason. Sonochem., 2013, 20, 793 CrossRef CAS PubMed.
- M. Zabihzadeh, A. Omidi, F. Shirini, H. Tajik and M. S. N. Langarudi, J. Mol. Struct., 2020, 1206, 127 CrossRef.
- D. Aute, A. Kshirsagar, B. Uphade and A. Gadhave, J. Chem. Sci., 2020, 132, 147 CrossRef CAS.
- G. W. Wang, Y. W. Dong, P. Wu, T. T. Yuan and Y. B. Shen, J. Org. Chem., 2008, 73, 7089 Search PubMed.
- S. Kantevari, R. Bantu and L. Nagarapu, J. Mol. Catal. A:Chem., 2007, 269, 53 CrossRef CAS.
- A. Khojastehnezhad, F. Moeinpour and A. Davoodnia, Chin. Chem. Lett., 2011, 22, 807 CrossRef CAS.
- Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Synlett, 2004, 1, 61–64 CrossRef.
- B. Rodríguez, T. Rantanen and C. Bolm, Angew. Chem., Int. Ed., 2006, 45, 6924 CrossRef PubMed.
- B. Huang, Y. Shen, Z. Mao, Y. Liu and S. Cui, Org. Lett., 2016, 18, 4888–4891 CrossRef CAS PubMed.
- M. Huang, L. Deng, T. Lao, Z. Zhang, Z. Su, Y. Yu and H. Cao, J. Org. Chem., 2022, 87, 3265–3275 CrossRef CAS PubMed.
- Z. Zhang, Q. Zhou, W. Yu, T. Li, G. Wu, Y. Zhang and J. Wang, Org. Lett., 2015, 17, 2474–2477 CrossRef CAS PubMed.
- G.-B. Huang, X. Wang, Y.-M. Pan, H.-S. Wang, G.-Y. Yao and Y. Zhang, J. Org. Chem., 2013, 78, 2742–2745 CrossRef CAS PubMed.
- J. Peng, C. Chen, J. Chen, X. Su, C. Xi and H. Chen, Org. Lett., 2014, 16, 3776–3779 CrossRef CAS PubMed.
- M. H. Sarvari and H. Sharghi, J. Org. Chem., 2004, 69, 6953 CrossRef CAS PubMed.
- F. Pakdaman, S. Allameh and M. Shaker, J. Res. Med. Dent. Sci., 2018, 6, 192–195 Search PubMed.
- G. Shen, Z. Wang, X. Huang, M. Hong, S. Fan and X. Lv, Org. Lett., 2020, 22, 8860–8865 CrossRef CAS PubMed.
- J. Wu, S. Xiang, J. Zeng, M. Leow and X.-W. Liu, Org. Lett., 2015, 17, 222–225 CrossRef CAS PubMed.
- D. S. Gaikwad and D. M. Pore, Synlett, 2012, 23, 2631 CrossRef CAS.
- M. A. Pawar, A. V. Nakhate, S. S. Kadu and P. V. Tekade, Appl. Organomet. Chem., 2024, 38, e7405 CrossRef CAS.
- S. F. Nielsen, D. Peters and O. Axelsson, Synth. Commun., 2000, 30, 3501 CrossRef CAS.
- G. B. D. Rao, S. Nagakalyan and G. K. Prasad, RSC Adv., 2017, 7, 3611 RSC.
- S. Nasr-Esfahani, S. J. Hoseini, M. Montazerozohori, R. Mehrabi and H. Nasrabadi, J. Mol. Catal. A: Chem., 2014, 382, 99 CrossRef.
- H. Ahankar, A. Ramazani and S. W. Joo, Res. Chem. Intermed., 2016, 42, 2487 CrossRef CAS.
- M. Khashi, S. Allameh, S. A. Beyramabadi, A. Morsali, E. Dastmalchian and A. Gharib, Iran. J. Chem. Chem. Eng., 2017, 36, 45 CAS.
- S. T. Fardood, A. Ramazani and S. J. Moradi, Sol-Gel Sci. Technol., 2017, 82, 432 CrossRef CAS.
- L. Zhang, X. Si, Y. Yang, S. Witzel, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, ACS Catal., 2019, 9, 6118–6123 CrossRef CAS.
- R. S. Basha, C.-W. Chen, D. M. Reddy and C.-F. Lee, Chem.–Asian J., 2018, 13, 2475–2483 CrossRef CAS PubMed.
- L. Deng and Y. Liu, ACS Omega, 2018, 3, 11890–11895 CrossRef CAS PubMed.
- K. Tanimoto, R. Ohkado and H. Iida, J. Org. Chem., 2019, 84, 14980–14986 CrossRef CAS PubMed.
- J. Qin, H. Zuo, Y. Ni, Q. Yu and F. Zhong, ACS Sustainable Chem. Eng., 2020, 8, 12342–12347 CrossRef CAS.
- W. Zhao, P. Xie, Z. Bian, A. Zhou, H. Ge, B. Niua and Y. Ding, RSC Adv., 2015, 5, 59861 RSC.
- L. L. Deng and Y. Y. Liu, ACS Omega, 2018, 3, 11890–11895 CrossRef CAS PubMed.
- A. Islam, P. Choudhury, K. Sarkar, R. K. Das, M. Bhattacharya and P. Ghosh, Mol. Divers., 2023, 28, 1597–1607 CrossRef PubMed.
- P. Sun, D. Yang, W. Wei, M. Jiang, Z. Wang, L. Zhang, H. Zhang, Z. Zhang, Y. Wanga and H. Wang, Green Chem., 2017, 19, 4785–4791 RSC.
- G. Brahmachari, A. Bhowmick and I. Karmakar, J. Org. Chem., 2021, 86, 9658–9669 CrossRef CAS PubMed.
- R. Rahaman, S. Das and P. Barman, Green Chem., 2018, 20, 141 RSC.
- Q.-H. Teng, Y. Yao, W.-X. Wei, H.-T. Tang, J.-R. Li and Y.-M. Pan, Green Chem., 2019, 21, 6241–6245 RSC.
- J.-P. Wan, S. Zhong, L. Xie, X. Cao, Y. Liu and L. Wei, Org. Lett., 2016, 18, 584–587 CrossRef CAS PubMed.
- X.-L. Cui, H.-Y. Tian, S.-J. Wang, X. Chen and D.-L. Kong, J. Org. Chem., 2023, 88, 8576–8582 CrossRef CAS PubMed.
- S. Paul, S. Das, T. Choudhuri, P. Sikdar and A. K. Bagdi, J. Org. Chem., 2023, 88, 4187–4198 CrossRef CAS PubMed.
- X.-L. Fang, R.-Y. Tang, X.-G. Zhang and J.-H. Li, Synthesis, 2011, 1099 CAS.
- Y.-C. Wong, T. T. Jayanth and C.-H. Cheng, Org. Lett., 2006, 8, 5613–5616 CrossRef CAS PubMed.
- R. Sikari, S. Sinha, S. Das, A. Saha, G. Chakraborty, R. Mondal and N. D. Paul, J. Org. Chem., 2019, 84, 4072–4085 CrossRef CAS PubMed.
- A. Saxena, A. Kumar and S. Mozumdar, Appl. Catal., A, 2007, 317, 210 CrossRef CAS.
- X. Ku, H. Huang, H. Jiang and H. Liu, J. Comb. Chem., 2009, 11, 338–340 CrossRef PubMed.
- Y. Li, J. Pu and X. Jiang, Org. Lett., 2014, 16, 2692–2695 CrossRef CAS PubMed.
- A. V. Kalinin, J. F. Bower, P. Riebel and V. Snieckus, J. Org. Chem., 1999, 64, 2986 CrossRef CAS PubMed.
- H. Firouzabadi, N. Iranpoor and M. Gholinejad, Adv. Synth. Catal., 2010, 352, 119 CrossRef CAS.
- Z. Taherinia and A. Ghorbani-Choghamarani, Can. J. Chem., 2019, 97, 46–52 CrossRef CAS.
- Y. Qin, Y. Han, Y. Tang, J. Wei and M. Yang, Chem. Sci., 2020, 11, 1276–1282 RSC.
- M. Noikham and S. Yotphan, Eur. J. Org Chem., 2019, 2019, 2759–2766 CrossRef CAS.
- C. G. Bates, R. K. Gujadhur and D. V. Venkataraman, Org. Lett., 2002, 4, 2803–2806 CrossRef CAS PubMed.
- P.-F. Larsson, A. Correa, M. Carril, P.-O. Norrby and C. Bolm, Angew. Chem., Int. Ed., 2009, 48, 5691–5693 CrossRef CAS PubMed.
- G. Y. Li, J. Org. Chem., 2002, 67, 3643–3650 CrossRef CAS PubMed.
- T. Migita, T. Shimizu, Y. Asami, J.-i. Shiobara, Y. Kato and M. Kosugi, Bull. Chem. Soc. Jpn., 1980, 53, 1385 CrossRef CAS.
- J. Lee and P. H. Lee, J. Org. Chem., 2008, 73, 7413 CrossRef CAS PubMed.
- M. C. Willis, D. Taylor and A. T. Gillmore, Tetrahedron, 2006, 62, 11513 CrossRef CAS.
- M. Murata and S. L. Buchwald, Tetrahedron, 2004, 60, 7397–7403 CrossRef CAS.
- U. Schopfer and A. Schlapbach, Tetrahedron, 2001, 57, 3069–3073 CrossRef CAS.
- M. Zhang, S. Zhang, C. Pan and F. Chen, Synth. Commun., 2012, 42, 2844 CrossRef CAS.
- C. Zhang, J. McClure and C. J. Chou, J. Org. Chem., 2015, 80, 4919 CrossRef CAS PubMed.
- A. Thupyai, C. Pimpasri and S. Yotphan, Org. Biomol. Chem., 2018, 16, 424 RSC.
- C. J. Weiss and T. J. Marks, J. Am. Chem. Soc., 2010, 132, 10533 CrossRef CAS PubMed.
- F. Yamashita, H. Kuniyasu, J. Terao and N. Kambe, Org. Lett., 2008, 10, 101 CrossRef CAS PubMed.
- Q. Wang, F. Xie and X. Li, J. Org. Chem., 2015, 80, 8361 CrossRef CAS PubMed.
- C. Liu, Y. Fang, S.-Y. Wang and S.-J. Ji, ACS Catal., 2019, 9, 8910 CrossRef CAS.
- V. P. Reddy, A. V. Kumar, K. Swapna and K. R. Rao, Org. Lett., 2009, 11, 1697–1700 CrossRef CAS PubMed.
- M. Jiang, H. Li, H. Yang and H. Fu, Angew. Chem., Int. Ed., 2016, 55, 1–7 CrossRef.
- P. Malik and D. Chakraborty, Appl. Organometal. Chem., 2012, 26, 557–561 CrossRef CAS.
- Z. Begum, C. Kishore, V. V. Reddy and B. V. S. Reddy, Tetrahedron Lett., 2014, 55, 6048 CrossRef CAS.
- F. M. Moghaddam, M. R. Khodabakhshi and M. Aminaee, Tetrahedron Lett., 2014, 55, 4720 CrossRef CAS.
- M. Krishnamurthy, T. M. Vishwanatha, N. R. Panguluri, V. Panduranga and V. V. Sureshbabu, Synlett, 2015, 2565 CAS.
- P. Niu, J. Kang, X. Tian, L. Song, H. Liu, J. Wu, W. Yu and J. Chang, J. Org. Chem., 2015, 80, 1018 CrossRef CAS PubMed.
- S. Angapelly, P. V. S. Ramya, R. Sodhi, A. Angeli, K. Rangan, N. Nagesh, C. T. Supuran and M. Arifuddin, J. Enzyme Inhib. Med. Chem., 2018, 33, 615 CrossRef CAS PubMed.
- J.-Q. Liu, B.-B. Feng and X.-S. Wang, Tetrahedron, 2018, 74, 4746 CrossRef CAS.
- Q.-W. Gui, F. Teng, Z.-C. Li, X.-F. Jin, M. Zhang, J.-N. Dai, Y.-W. Linc, Z. Cao and W.-M. He, Org. Chem. Front., 2020, 7, 4026 RSC.
- H. R. Shaterian, A. Hosseinian and M. Ghashang, Arkivoc, 2009, 59 Search PubMed.
- P. Kaur, G. Shakya, H. Sun, Y. Pan and G. Li, Org. Biomol. Chem., 2010, 8, 1091–1096 RSC.
- A. Islam, R. Singha and P. Ghosh, ChemistrySelect, 2023, 8, e202203780 CrossRef CAS.
- V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318 CrossRef CAS PubMed.
- R. Singha, A. Islam and P. Ghosh, Sci. Rep., 2021, 11, 19891 CrossRef CAS PubMed.
- P. Sang, Y. Xie, J. Zou and Y. Zhang, Org. Lett., 2012, 14, 3894–3897 CrossRef CAS PubMed.
- H. He, S. Dong, Y. Chen, Y. Yang, Y. Le and W. Bao, Tetrahedron, 2012, 68, 3112–3116 CrossRef CAS.
- R. Berrino, S. Cacchi, G. Fabrizi and A. Goggiamani, J. Org. Chem., 2012, 77, 2537–2542 CrossRef CAS PubMed.
- Y. Zhang, X. Yang, Q. Yao and D. Ma, Org. Lett., 2012, 14, 3056–3059 CrossRef CAS PubMed.
- K. Liubchak, A. Tolmachev and K. Nazarenko, J. Org. Chem., 2012, 77, 3365–3372 CrossRef CAS PubMed.
- X. Xu, H. Feng and E. V. Van der Eycken, J. Org. Chem., 2021, 86, 14036–14043 CrossRef CAS PubMed.
- M. S. T. Morin, Y. Lu, D. A. Black and B. A. Arndtsen, J. Org. Chem., 2012, 77, 2013–2017 CrossRef CAS PubMed.
- A. Laouiti, M. M. Rammah, M. B. Rammah, J. Marrot, F. Couty and G. Evano, Org. Lett., 2012, 14, 6–9 CrossRef CAS PubMed.
- A. B. Sheremetev, N. V. Palysaeva, M. I. Struchkova, K. Y. Suponitsky and M. Y. Antipin, Eur. J. Org Chem., 2012, 2266–2272 CrossRef CAS.
- R. Thorwirth, A. Stolle, B. Ondruschka, A. Wild and U. S. Schubert, Chem. Commun., 2011, 47, 4370 RSC.
- A. Islam, K. Malakar, M. Velusamy and P. Ghosh, Tetrahedron, 2024, 167, 134278 CrossRef CAS.
- G. Shore, W.-J. Yoo, C.-J. Li and M. G. Organ, Chem. Eur. J., 2010, 16, 126–133 CrossRef CAS PubMed.
- M. Kidwai, V. Bansal, N. K. Mishra, A. Kumar and S. Mozumdar, Synlett, 2007, 1581 CrossRef CAS.
- D. B. Bankar, R. R. Hawaldar, S. S. Arbuj, M. H. Moulavi, S. T. Shinde, S. P. Takle, M. D. Shinde, D. P. Amalnerkar and K. G. Kanade, RSC Adv., 2019, 9, 32735–32743 RSC.
- W. Yan, R. Wang, Z. Xu, J. Xu, L. Lin, Z. Shen and Y. Zhou, J. Mol. Catal. A: Chem., 2006, 255, 81–85 CrossRef CAS.
- M. Kidwai, V. Bansal, A. Kumar and S. Mozumdar, Green Chem., 2007, 9, 742–745 RSC.
- T. Zeng, W.-W. Chen, C. M. Cirtiu, A. Moores, G. Song and C.-J. Li, Green Chem., 2010, 12, 570–573 RSC.
- C. Verrier, S. Carret and J.-P. Poisson, Org. Lett., 2012, 14, 5122–5125 CrossRef CAS PubMed.
- P. Kaur, B. Kumar, K. K. Gurjar, R. Kumar, V. Kumar and R. Kumar, J. Org. Chem., 2020, 85, 2231–2241 CrossRef CAS PubMed.
- E. Tullberg, F. Schacher, D. Peters and T. Frejd, Synthesis, 2006, 1183 CAS.
- G.-Q. Rong, J.-Q. Zhao, X.-M. Zhang, X.-Y. Xu, W.-C. Yuan and M.-Q. Zhou, Tetrahedron, 2018, 74, 2383 CrossRef CAS.
- L. Xing, Y. Zhang, B. Li and Y. Du, Org. Lett., 2019, 21, 1989–1993 CrossRef CAS PubMed.
- Y. Lv, K. Sun, W. Pu, S. Mao, G. Li, J. Niu, Q. Chen and T. Wang, RSC Adv., 2016, 6, 93486 RSC.
- C. R. Reddy and M. D. Reddy, J. Org. Chem., 2014, 79, 106–116 CrossRef CAS PubMed.
- E. J. L. Lana, K. A. da Silva Rocha, I. V. Kozhevnikov and E. V. Gusevskaya, J. Mol. Catal. A:Chem., 2006, 259, 99 CrossRef CAS.
- E. Rafiee, M. Joshaghani, F. Tork, A. Fakhri and S. Eavani, J. Mol. Catal. A:Chem., 2008, 283, 1 CrossRef CAS.
- J. T. Li, X. T. Meng, B. Bai and M. Xuan, Ultrason. Sonochem., 2010, 17, 14 CrossRef CAS PubMed.
- B. Yang, W. Hu and S. Zhang, RSC Adv., 2018, 8, 2267 RSC.
- Y. Lin, M. Cai, Z. Fang and H. Zhao, RSC Adv., 2016, 6, 85186 RSC.
- Z.-H. Ren, M.-N. Zhao and Z.-H. Guan, RSC Adv., 2016, 6, 16516 RSC.
- L. Ai, I. Y. Ajibola and B. Li, RSC Adv., 2021, 11, 36305 RSC.
- T. Maity, S. Bhunia, S. Das and S. Koner, RSC Adv., 2016, 6, 33380 RSC.
- J. Wang, Y. Cheng, J. Xiang and A. Wu, Synlett, 2019, 30, 743 CrossRef CAS.
- A. K. Sharma, H. Joshi and A. K. Singh, RSC Adv., 2020, 10, 6452 RSC.
- D. Liang, M. Wang, Y. Dong, Y. Guo and Q. Liu, RSC Adv., 2014, 4, 6564 RSC.
- M. Hosseini-Sarvari and Z. Razmi, RSC Adv., 2014, 4, 44105 RSC.
- F. M. Moghaddam, G. Tavakoli and A. Aliabadi, RSC Adv., 2015, 5, 59142–59153 RSC.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719–13725 CrossRef CAS PubMed.
- C. S. Pires, D. H. de Oliveira, M. R. B. Pontel, J. C. Kazmierczak, R. Cargnelutti, D. Alves, R. G. Jacob and R. F. Schumacher, Beilstein J. Org. Chem., 2018, 14, 2789 CrossRef CAS PubMed.
- R. Gondru, B. Janardhan and B. Rajitha, Res. Chem. Intermed., 2015, 11, 8099–8810 Search PubMed.
- J.-H. Xia, Q. Chen, J.-W. Yuan, W.-S. Shi, L.-R. Yang and Y.-M. Xiao, RSC Adv., 2023, 13, 26948 RSC.
- J.-C. Liou, S. S. Badsara, Y.-T. Huang and C.-F. Lee, RSC Adv., 2014, 4, 41237 RSC.
- W.-L. Xing, D.-G. Liu and M.-C. Fu, RSC Adv., 2021, 11, 4593 RSC.
- N. V. Orlov, I. V. Chistyakov, L. L. Khemchyan, V. P. Ananikov, I. P. Beletskaya and Z. A. Starikova, J. Org. Chem., 2014, 79, 12111–12121 CrossRef CAS PubMed.
- O. Vyhivskyi, D. N. Laikov, A. V. Finko, D. A. Skvortsov, I. V. Zhirkina, V. A. Tafeenko, N. V. Zyk, A. G. Majouga and E. K. Beloglazkina, J. Org. Chem., 2020, 85, 3160–3173 CrossRef CAS PubMed.
- Y. Li, H. Wang, X. Li, T. Chen and D. Zhao, Tetrahedron, 2010, 66, 8583–8586 CrossRef CAS.
- S. Roy, T. Chatterjee, B. Banerjee, N. Salam, A. Bhaumik and S. M. Islam, RSC Adv., 2014, 4, 46075–46083 RSC.
- M. Matsumura, H. Kumagai, Y. Murata, N. Kakusawa and S. Yasuike, J. Organomet. Chem., 2016, 807, 11–16 CrossRef CAS.
- K. H. V. Reddy, G. Satish, K. Ramesh, K. Karnakar and Y. Nageswar, Chem. Lett., 2012, 41, 585–587 CrossRef CAS.
- Y. Murata, S. Tsuchida, R. Nezaki, Y. Kitamura, M. Matsumura and S. Yasuike, RSC Adv., 2022, 12, 14502 RSC.
- L. Liu, D. Zhou, J. Dong, Y. Zhou, S.-F. Yin and L.-B. Han, J. Org. Chem., 2018, 83, 4190–4196 CrossRef CAS PubMed.
- R. Zhao, X. Huang, M. Wang, S. Hu, Y. Gao, P. Xu and Y. Zhao, J. Org. Chem., 2020, 85, 8185–8195 CrossRef CAS PubMed.
- J. Yuan, W.-P. To, Z.-Y. Zhang, C.-D. Yue, S. Meng, J. Chen, Y. Liu, G.-A. Yu and C.-M. Che, Org. Lett., 2018, 20, 7816–7820 CrossRef CAS PubMed.
- I. Kim, M. Min, D. Kang, K. Kim and S. Hong, Org. Lett., 2017, 19, 1394–1397 CrossRef CAS PubMed.
- D. Liu, J.-Q. Chen, X.-Z. Wang and P.-F. Xu, Adv. Synth. Catal., 2017, 359, 2773–2777 CrossRef CAS.
- P. Zhou, B. Hu, L. Li, K. Rao, J. Yang and F. Yu, J. Org. Chem., 2017, 82, 13268–13276 CrossRef CAS PubMed.
- P. Xu, Z. Wu, N. Zhou and C. Zhu, Org. Lett., 2016, 18, 1143–1145 CrossRef CAS PubMed.
- X. Chen, H. Wu, R. Yu, H. Zhu and Z. Wang, J. Org. Chem., 2021, 86, 8987–8996 CrossRef CAS PubMed.
- R. Isshiki, K. Muto and J. Yamaguchi, Org. Lett., 2018, 20, 1150–1153 CrossRef CAS PubMed.
- T. Wang, S. Sang, L. Liu, H. Qiao, Y. Gao and Y. Zhao, J. Org. Chem., 2014, 79, 608–617 CrossRef CAS PubMed.
- R. G. de Noronha, P. J. Costa, C. C. Romao, M. J. Calhorda and A. C. Fernandes, Organometallics, 2009, 28, 6206–6212 CrossRef CAS.
- Y. He, H. Wu and F. D. Toste, Chem. Sci., 2015, 6, 1194–1198 RSC.
- L. Niu, S. Wang, J. Liu, H. Yi, X.-A. Liang, T. Liu and A. Lei, Chem. Commun., 2018, 54, 1659 RSC.
- Y. Jian, M. Chen, B. Huang, W. Jia, C. Yang and W. Xia, Org. Lett., 2018, 20, 5370–5374 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*