
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvenient one-pot synthesis of 1-(4-(4-(2-[18F]fluoroethoxy)phenyl)piperazin-1-yl)ethanone ([18F]FEt-PPZ) for imaging tumors expressing sigma-1 receptors†
Somnath Karad,
Avik Chakrabortyad,
Lakshminarayanan. Na,
Rajesh C.a,
Pradip Chaudhuric,
Mukti Kanta Raya,
Kakoli Bosec,
Sharmila Banerjeecd,
Sandip Basuad and
Madhava B. Mallia *bd
*bd
aRadiation Medicine Centre, BARC, Parel, Mumbai-400012, India
bRadiopharmaceutical Division, BARC, Trombay, Mumbai-400085, India. E-mail: mallia@barc.gov.in; Fax: +91 22 2550 5151; Tel: +91 22 25590746
cAdvance Centre for Treatment, Research and Education in Cancer, Kharghar, Mumbai-410210, India
dHomi Bhabha National Institute, Anushaktinagar, Mumbai-400094, India
First published on 10th July 2025
Abstract
The overexpression of the sigma-1 receptor (σ1R) in a variety of cancers makes it a potential target for developing anticancer drugs or diagnostic/therapeutic radiopharmaceuticals. Molecules with piperidine or piperazine substructures have shown high binding affinity towards σ1R. Docking and MD simulation studies on commercially available piperidine/piperazine-containing compounds identified 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone (PPZ) as a potential ligand for targeting σ1R. Docking studies on its fluorinated analogue, 1-(4-(4-(2-fluoroethoxy)phenyl)piperazin-1-yl)ethanone (FEt-PPZ), showed good affinity towards σ1R, comparable to that of a clinically evaluated radiopharmaceuticals such as [18F]fluspidine. The root mean square deviation (RMSD) value of 2 A° in the MD simulations over a period of 50 ns indicated the excellent stability of the FEt-PPZ-σ1R receptor–ligand complex. Based on these results, the radiosynthesis of [18F]FEt-PPZ was carried out to evaluate its potential to target σ1R. The radiotracer was prepared through a one-pot, two-step method using a GE PETtrace cyclotron and PETtracer synthetic module with an activity yield of 32% ± 5.8% (n = 6). The radiochemical purity (RCP) of [18F]FEt-PPZ was >95%, as assessed through radioTLC and radio-HPLC. In vitro cell binding studies on [18F]FEt-PPZ in U-87 MG and B16F10 cells showed maximum uptakes of 13.28% ± 1.04% and 10.74% ± 0.82%, respectively, at 60 min post-incubation. Inhibition studies carried out in the presence of an excess non-radioactive compound (FEt-PPZ) showed a reduction of ∼55% in radiotracer uptake in glioma cells and ∼48% in melanoma cells, indicating its specificity towards σ1R. Bio-distribution and small animal PET/CT imaging studies on melanoma-tumor-bearing C57BL6 mice showed a significant tumor uptake (10.55% ± 1.3% ID per g organ) at 60 min post-injection and an hepatic and renal mode of excretion. Preclinical studies on [18F]FEt-PPZ developed using an in silico approach demonstrated its potential to target σ1R, thus warranting further investigation.
Introduction
Sigma-1 receptor (σ1R) overexpression is associated with different pathological conditions, such as dementia,1 Alzheimer's disease, amyotrophic lateral sclerosis,2,3 retinal degeneration4 and various types of cancers.5–7 The overexpression of σ1R in various cancers, such as breast cancer, melanoma, non-small cell lung carcinoma, esophageal squamous cell carcinoma, and glioma, makes it a potential target to develop anticancer drugs. Sigma receptors, discovered in 1976, were initially considered a subtype of opioid receptors (μ, δ and κ) owing to their affinity toward opioid ligands.8 However, with the greater understanding of their unique characteristics, they are now considered a separate category. Sigma receptors are divided into sigma-1 (σ1) and sigma-2 (σ2) subtypes. σ1R is a 25 kDa trans-membrane protein consisting of 223 amino acids, present at the mitochondrion-associated endoplasmic reticulum membrane.9 Initially, it was believed that σ1R has two trans-membrane regions and two ligand-binding domains. However, later reports on the crystal structure of the receptor showed that it had a single trans-membrane region and a single ligand-binding site. The ligand-binding site was found to be lipophilic and decorated with amino acids, including Val 84, Trp 89, Met 93, Leu 95, Tyr 103, Leu 105, Phe 107, Ile 124, Trp 164, Glu 172, and Leu 182.10 Among these, Glu 172, a highly conserved amino acid across different species, plays an important role in ligand-binding through electrostatic interaction.10The development of targeted radiopharmaceuticals for imaging or therapy is a key research focus because of their selective localization at disease sites and as they can help minimize radiation exposure to the other critical organs/tissues.11,12 Several σ1R-targeting PET-radiotracers, including [11C]SA4503,13 [18F]FMSA4503,14 [18F]FPS,15 [18F]FCT-146,16 [18F]Fluspidine,17 [18F]IAM6067,18 18F-labelled benzamide derivatives,19 [11C]HCC0923 (ref. 20) and [11C]HCC0929,20 have been reported for the scintigraphic imaging of the changes in σ1R distribution and density in tissues to obtain insights into their role in disease progression. Imaging the distribution of σ1Rs can aid a better understanding of the tumor physiology and their pathophysiological function, which can assist in the development of novel drugs for therapy. The σ1Rs are widely distributed in the central nervous system (CNS), where they are responsible for memory, pain modulation, motor functions and emotions. Although several of these σ1R radiopharmaceuticals have been evaluated clinically, they are not in routine clinical practice due to their inherent limitations, leaving scope for exploring new radiotracers for mapping σ1R.
Among the σ1R-targeting PET radiopharmaceuticals, the preparation of carbon-11 radiopharmaceuticals is much more challenging than fluorine-18 radiopharmaceuticals owing to the short half-life and complex chemistry of the former. In this context, efforts have been made to develop fluorine-18 radiopharmaceuticals following relatively simple chemistry and are continuing. At present SN2 and SNAr are two of the most widely used reaction mechanisms for radiolabeling with fluorine-18. This radiolabeling requires suitably modified aliphatic and aromatic precursors, often prepared through multistep, sometimes complex, organic syntheses.21,22 Apart from these methods, recent advances in this field include photocatalyzed23–25 and click chemistry-based26,27 radiolabeling procedures for the successful incorporation of fluorine-18 into the desired molecules. Despite the difficulties involved, radiolabeling with fluorine-18 is much more convenient than the preparation of short-lived carbon-11 radiopharmaceuticals. In this context, there is scope to look for potential fluorine-18 radiotracers for imaging σ1R that could be prepared from readily available commercial sources, rather than from precursor ligands prepared through multistep organic syntheses.
Recent years have witnessed the increased applications of various computational tools for designing drugs.28,29 These have helped to minimize the cost as well as the time involved in the conventional drug discovery methods through the actual synthesis of various ligands and their evaluation. In silico methods, like docking, and molecular dynamic simulation, can provide important insights into molecule–protein interactions, the binding energies, and the stability of the resulting small-molecule/protein–ligand complexes. Based on the available literature, we aimed to find a new molecule that could be easily radiolabeled with fluorine-18 and used as a marker for σ1R-expressing tumors.
Materials and method
General
All the chemicals used in the experiments were purchased from commercial sources and used as received without further purification. The chemicals 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone, ethylene ditosylate, 1-bromo 2-fluoro ethane, anhydrous acetonitrile, and DMSO were purchased from M/s. Sigma Aldrich (Bangalore, India). The σ2-receptor specific ligand, 1-[4-(6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolin-2-yl)butyl]-3-methyl-1,3-dihydro-1,3-benzimidazol-2-one (CM-389), used for the inhibition studies was purchased from M/s. MedchemExpress, USA. Alumina, sodium bicarbonate, HPLC grade water and acetonitrile were purchased from Merck, India. The QMA cartridge and Plus Light C18 column were purchased from Waters Corporation (MA, USA). IMDM and FBS medium were procured from Gibco, Thermo Fischer. Different modules of Schrodinger software were used for carrying out the computational studies. Energy minimizations of the protein and ligand were done using Protein Preparation Wizard and Ligprep, respectively. The docking studies were carried out using the Glide module and molecular dynamic simulation was done using Desmond. [18F]-Fluoride was produced in the GE PETtrace cyclotron (GE, USA) and the radiosynthesis was done in the GE TRACERlab module (Configured for 2-[18F]FDG production) (GE, USA). Silica gel-based instant thin layer chromatography (ITLC) paper was purchased from M/s. Varian, USA. RadioTLC scanner (Bioscan, GmbH) and HPLC (Knauer, Germany) equipped with both diode array detector as well as radioactive detector (Raytest, Germany) were used to analyze the purity of the products. Radioactivity measurements were performed using a well-type NaI(Tl) scintillation gamma counter with an open energy window (Para Electronics, India). The 1H/13C-NMR spectra were recorded on a 300 MHz Varian VXR 500S spectrophotometer (USA). 19F-NMR spectroscopy was performed using a Bruker Avance Neo (400 MHz) instrument. Mass spectra were recorded on a Waters Xevo G2-XS QTOF high-resolution mass spectrometer (USA), using electron spray ionization (ESI). PET scans were performed using a tri-modality gamma imaging system (Triumph@, Gamma Medica Ideas, Northridge, CA, USA).Computational studies
The crystal structures of σ1R (PDB id: 5HK1) and σ2R (PDB id: 7MFI) were obtained from the Protein Data Bank.30 Energy minimization was carried out using the protein preparation wizard with the default setting using the OPLS3 force field. The active site was identified and assigned from the literature and a grid box sized 10 × 10 × 10 Å was generated around the active site using the Receptor Grid Generation module. Energy minimization of the ligand was carried out using Ligprep. Docking was performed using Ligand Docking (in the Glide module) in extra precision mode. The MD simulations were done with the protein–ligand complex using Desmond. Here, σ1R consisted of a trans-membrane domain and a cytosolic domain. The N-terminal helix (amino acid no. 10–30) remained fixed at the endoplasmic reticulum trans-membrane region and the rest of the protein remained in the cytosol. For the MD simulation, a system was built by making an orthorhombic box around the protein–ligand complex. A dipalmitoyl phosphatidylcholine membrane was constructed and the N-terminal helical region (trans-membrane region) was inserted into the membrane, and the rest of the system was solvated with water (PI3P water model). Energy minimization was carried out to relax the whole system. The energy-minimized system was subjected to 50 ns MD simulation at 298 K and 1 bar pressure.Radiosynthesis of [18F]FEt-PPZ
The radiosynthesis was carried out following a one-pot, two-step procedure, as described below.Synthesis of the reference compound FEt-PPZ
The 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone (100 mg, 0.45 mmol) in acetonitrile (10 mL) was mixed with excess potassium carbonate (170 mg, 1.2 mmol) and 1-bromo-2-fluoroethane (62 mg, 0.49 mmol). The reaction mixture was refluxed for 12 h under continuous stirring. After cooling the reaction mixture, the solvent was removed under vacuum. The crude mixture was dissolved in water and extracted with chloroform (3 × 10 mL). The combined chloroform extracts were washed with saline and dried over anhydrous magnesium sulfate. The FEt-PPZ compound obtained after removing the solvent was used in the later experiments without further purification. 1H-NMR (CDCl3, δ ppm) 2.13 (s, 3H), 3.03 (t, 2H, J = 3Hz), 3.06 (t, 2H, J = 3Hz), 3.61 (t, 2H, J = 3Hz), 3.76 (t, 2H, J = 3Hz), 4.13–4.21 (dt, 2H), 4.68–4.79 (dt, 2H), 6.89 (m, 4H) [Fig. S1 in ESI†]; 13C-NMR (CDCl3, δ ppm) 21.32, 41.48, 46.36, 50.66, 51.07, 67.57, 67.73, 81.33, 82.69, 115.56, 118.79, 145.77, 153.12, 168.96 [Fig. S2 in ESI†]; F-NMR (CDCl3, δ ppm) −223 [Fig. S3 in ESI†], HRMS (ESI+) m/z: Calcd for C14H19FN2O2 (M + H)+: 267.1431, found: 267.1533 [Fig. S4 in ESI†].Quality control
HPLC
The radiochemical purity of [18F]FEt-PPZ was assessed by HPLC with a C18-reversed phase column equipped with a radioactivity detector. Here, about 10 μL of the test solution (∼0.3 MBq) was injected into the column and elution was monitored by observing the radioactivity profile. Aqueous 0.05 M trifluoro acetic acid (solvent A) and acetonitrile (solvent B) were used as mobile phases. Both solvents were filtered through a 0.22 μ filter. The elution started with 95% A and 5% B from 0 to 5 min. A linear gradient started at 5 min and the composition reached 50% A and 50% B at 15 min. At 25 min, the composition reached 5% A and 95% B. Afterwards, the solvent composition was reversed and at 30 min it reached its initial condition. The flow rate was maintained at 1 mL min−1.ITLC
The radiochemical purity of [18F]FEt-PPZ was determined by ITLC using acetonitrile:water (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) as the solvent system. About 5 μL of the test solution was placed ∼1.5 cm from the bottom of a 11.5 cm long ITLC strip and it was developed in the solvent system mentioned above. In this solvent system, free [18F]fluoride remained at the point of the spot (Rf = 0) and [18F]fluoroethyltosylate will move to the solvent front (Rf = 1). The developed strip was analyzed using a TLC scanner and from the peak area measurements, the %RCP of [18F]FEt-PPZ was calculated.
:5) as the solvent system. About 5 μL of the test solution was placed ∼1.5 cm from the bottom of a 11.5 cm long ITLC strip and it was developed in the solvent system mentioned above. In this solvent system, free [18F]fluoride remained at the point of the spot (Rf = 0) and [18F]fluoroethyltosylate will move to the solvent front (Rf = 1). The developed strip was analyzed using a TLC scanner and from the peak area measurements, the %RCP of [18F]FEt-PPZ was calculated.
Saline and serum stability studies
The in vitro stability of the radiotracer was evaluated in saline and in human serum at different time points. The human serum used for this study was isolated from blood voluntarily gifted by Mr Somnath Kar, Radiation Medicine Centre (first author of this article). The radiotracer (3.7 MBq) was incubated in saline (1 mL) at room temperature for 0, 1, 2, and 4 h, at the end of which the radiochemical purity was evaluated by radioITLC following the method described previously. For the serum stability studies, the radiotracer (3.7 MBq) was incubated in human serum (2 mL) at 37 °C. At 0, 1, 2, and 4 h, about 100 μL aliquots of the sample were withdrawn and an equal volume of acetonitrile was added to precipitate the serum proteins. Subsequently, the mixture was centrifuged to precipitate the proteins and the supernatant was analyzed by radioITLC to evaluate the stability.Octanol–water partition coefficient (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) PO/W)
PO/W)
For determination of the octanol–water partition coefficient, [18F]FEt-PPZ (1.8 MBq, 100 μL) was added to water (1.9 mL) in a 15 mL test tube. To this, n-octanol (2 mL) was added and the mixture was thoroughly mixed in a vortex mixture for 2 min. Subsequently, the two layers were separated by centrifugation at 3000 rpm for 3 min. An about 100 μL aliquot was taken from each phase and the associated activity was recorded with a γ-counter. The measurements were done in triplicate. The partition coefficient (LogPO/W) value was calculated as the logarithm of the ratio of the count in the n-octanol phase to the count in the aqueous phase.
Quantitative real-time PCR analysis
The transcription levels of σ1R expression in the target organ and the cells used for the in vitro studies were analyzed by performing an RT-PCR study.31 Total RNA was extracted from the mice brain tissues, human malignant glioma cells (U-87 MG) and murine melanoma cells (B16F10) using TRIzol reagent (ThermoFisher) following the manufacturer's protocol. The procedure involved homogenizing the samples, followed by phase separation and RNA precipitation. Subsequently, RNA (∼1 μg) was reverse-transcribed into complementary DNA (cDNA) using a reverse transcriptase enzyme (Revert AidTM First strand cDNA synthesis Kit, MBI Fermentas, USA). The cDNA obtained from both the brain tissue and cell samples were subjected to analysis through RT-PCR employing a σ1R specific primer set (Table 1).| Gene name | Primer sequence (5′–3′) | Product size (bp) |
|---|---|---|
| Rat | ||
| σ1R: forward | 5′TGGATGGGCGCCATGT3′ | 68 |
| σ1R: reverse | 5′AAGGCGGTGCCGAAGAG3′ | |
RT-PCR was conducted using the FastStart Essential DNA Green Master reaction mix (SYBR Green CAT # 06402712001, Roche) on a LightCycler® 96 System (Roche, Switzerland). To ascertain the relative expression of the σ1R gene at the transcriptional level in the cells and tissues, normalization was performed using β-actin as an internal control, employing the 2−ΔΔct method.
In vitro cell binding and inhibition studies
Human malignant glioma cells U-87 MG (ATCC®, HTB-14™) and melanoma cancer cells B16F10 (ATCC®CRL-6475™) expressing σ1R were used for the in vitro cell binding study using a protocol described earlier.32 Cells were grown in 6-well tissue culture dishes using IMDM medium supplemented with 10% FBS in 5% CO2 at 37 °C, under humid conditions. The U-87 MG and B16F10 cells (1 × 106 cells) were incubated with ∼0.19 MBq of [18F]FEt-PPZ in cell binding buffer (IMDM with 0.2% BSA) for different time intervals (5, 15, 30, 45 and 60 min). The incubation was followed by centrifugation at 4000 rpm for 2 min and washing twice with ice-cold 0.05 M PBS (pH 7.4). The activity associated with the cell pellet and supernatant was then analyzed using a well-type NaI(Tl) scintillation gamma counter. The competitive inhibition study of [18F]FEt-PPZ was performed in the presence of excess cold FEt-PPZ and a specific σ2R ligand, CM-398, in separate experiments. Approximately the same number of cells (U-87 MG or B16F10) were incubated with 100-fold excess of non-radioactive FEt-PPZ (σ1R ligand) and CM-398 (σ2R ligand) for 1 h.33 Subsequently, a radiotracer was added and a similar procedure, mentioned earlier, was followed to determine the % cell binding at 60 min.In vivo biodistribution studies and small animal PET imaging
All the animal experiments were carried out after obtaining the approval of the Institutional Animal Ethics Committee (Approval No: BAEC/37/18, Dated: 07/04/22) following the principles of good laboratory practice (GLP). The biodistribution of the radiotracer was carried out in 3 to 4 weeks old female C57BL6 mice injected with B16F10 cells (1 × 106 cells per mouse) in the shoulder. The xenografts were allowed to grow for 1–2 weeks until the tumor reached the required size (∼1–1.5 cm3 volume).34 The mice were injected with freshly prepared [18F]FEt-PPZ (∼5 MBq per animal) through the tail vein and were then divided into three groups and kept for different time points (30, 60 and 120 min). At the end of the respective time points, the animals in the respective groups were sacrificed, and various organs/tissues were excised, weighed and the radioactivity associated with each organ/tissue was estimated using a flat-bed-type NaI(Tl) gamma counter. The biodistribution data were presented as percentages of the injected dose per gram (%ID per g) of the organ/tissue along with the relevant tumor/organ ratio of the accumulated activity for the vital organs.A PET-CT imaging study was performed in B16F10 xenograft tumor-bearing C57BL6 mouse to evaluate the localization and tumor uptake of [18F]FEt-PPZ using a tri-modality gamma imaging system. The animals were anaesthetized by isoflurane and the radiotracer (∼5 MBq) was injected through the tail vein. The scans were carried out at 30, 60, and 120 min post-injection. The PET scan was accomplished in 15 min followed by a CT scan. Image reconstruction was done by Maximum Likelihood Expectation Maximization (MLEM), an iterative algorithm. Reconstructed PET/CT images were analyzed using PMOD software, version 3.2 (PMOD Technologies Ltd, Zurich, Switzerland). The standard uptake value (SUV) was determined by drawing the ROI over the uptake in the tumor. The SUV was calculated using the following formula:
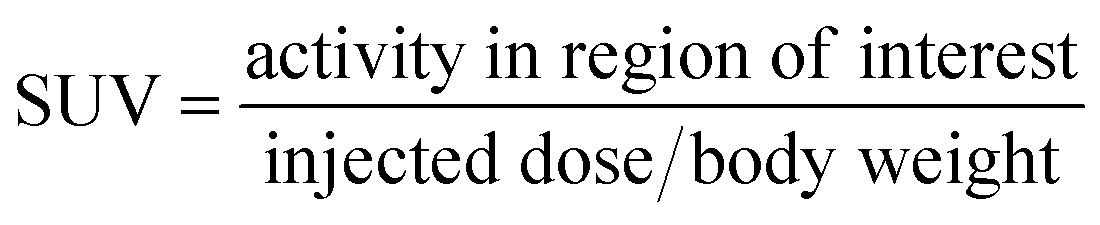
Statistical analysis
To ensure reproducibility of the results, measurements were made in triplicate (n = 3). Whenever possible, the results were expressed as the mean ± SD, which is the average with the spread of the standard deviation (SD). One-way ANOVA was conducted on the data using Sigma Stat 3.5 software, with the significance threshold set at p < 0.05.Result and discussion
Previous studies have shown piperidine, piperazine, and aromatic rings, etc. as common scaffolds in potential σ1R-targeting molecules.35 In an attempt to identify potential molecules to target σ1R, we carried out molecular docking studies of commercially available compounds with piperidine, piperazine and benzene scaffolds. Since the aim was to prepare a fluorine-18-labeled PET radiotracer through [18F]fluoroethylation, the presence of suitable structural features (–OH, –NH groups) was an important consideration while shortlisting commercial compounds. To validate the accuracy of the docking methodology, initial docking was performed using PD14448, a co-crystallized ligand, which successfully reproduced the expected top binding configuration. Subsequently, the shortlisted commercial compounds with piperidine, piperazine and benzene scaffolds were docked against the σ1R receptor (PDB id: 5HK1) [T1 in ESI†]. We found that 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone (CAS No. 67914-60-7) had the best G-score (Glide score) of −5.6 kcal mol−1. However, it is reasonable to assume that the docking score may change, favorably or otherwise, upon radiolabeling the compound through [18F]fluoroethylation. Therefore, another round of docking studies with the fluoroethylated analogues of all the shortlisted commercial compounds was carried out [see T1 in the ESI†]. The results indicated that 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone (PPZ) [Fig. 1(a)] and its fluoroethylated analogue, 1-(4-(4-(2-fluoroethoxy)phenyl)piperazin-1-yl)ethanone (FEt-PPZ) [Fig. 1(b)], showed the best docking scores. In fact, FEt-PPZ was found to have a better affinity for σ1R (−9.4 kcal mol−1) than its precursor compound PPZ (−5.6 kcal mol−1). This compound was, therefore, selected for the further studies.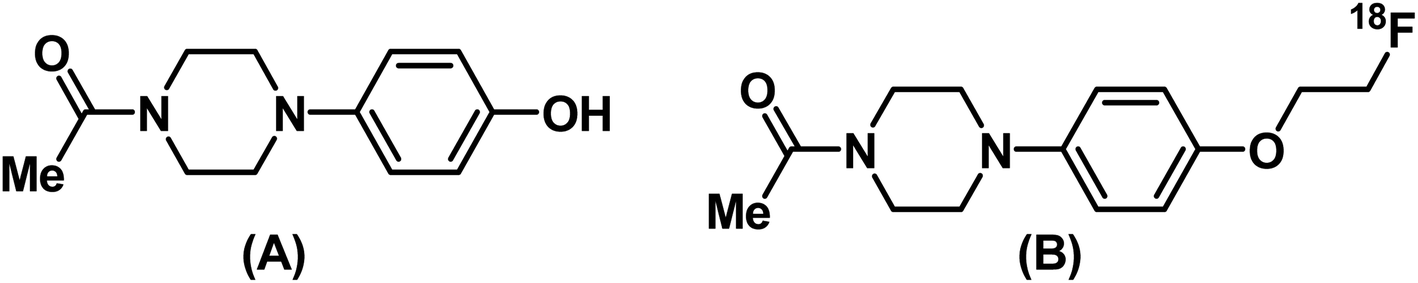 | ||
| Fig. 1 Structures of (A) 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone (PPZ) and (B) 1-(4-(4-(2-[18F]fluoroethoxy)phenyl)piperazin-1-yl)ethanone ([18F]FEt-PPZ). | ||
For comparing with FEt-PPZ, the G-scores of other reported PET radiopharmaceuticals targeting σ1R were also determined. Their G-scores ranged from −11.4 to −9.2 kcal mol−1 [Table 2] and it could be noted that the G-score for FEt-PPZ lay within this range. While the affinity of FEt-PPZ towards σ1R was comparable with the other reported PET radiopharmaceuticals, it is pertinent to note that PPZ is a commercially available compound, whereas the precursors to the reported PET radiopharmaceuticals needed difficult multistep syntheses. As the σ1R and σ2R receptors are closely related, docking studies were also performed for FEt-PPZ as well as the other reported PET radiopharmaceuticals with σ2R [Table 2]. The G-score for FEt-PPZ against σ2R was found to be −6.49 kcal mol−1, which indicated its relatively higher affinity towards σ1R compared to σ2R. Similar observations were noted with the other reported PET radiopharmaceuticals.
| PET radiopharmaceutical | σ1R G-score (kcal mol−1) | σ2R G-score (kcal mol−1) |
|---|---|---|
| [18F]FMSA4503 (ref. 14) | −11.75 | −9.5 |
| [11C]HCC0929 (ref. 20) | −11.5 | −6.7 |
| [18F]Benzamide19 | −11.0 | −7.38 |
| [11C]HCC0923 (ref. 20) | −10.6 | −6.9 |
| [18F]FCT146 (ref. 16) | −10.44 | −7.08 |
| [18F]FPS15 | −10.28 | −8.9 |
| [18F]FEt-PPZ | −9.4 | −6.49 |
| [18F]Fluspidine17 | −9.2 | −8.74 |
The 2D interaction diagram (Fig. 2) showed that compound FEt-PPZ established a variety of intermolecular interactions within the receptor's binding pocket to achieve structural stability. Specifically, the electrostatic interaction between Glu172 and the cationic amine of the ligand, a pivotal interaction in ligand binding at the active site of σ1R, was consistent with the prior literature.20 Additionally, the formation of a pi–cation interaction between the cationic amine and Phe107 further enhanced the ligand's stability. It was observed that the ligand was engaged in multiple hydrophobic interactions with key amino acids, such as Tyr103, Leu105, Ile124, Ile178, Thr181, and Leu182, contributing to the overall binding profile. Comprehensive MD simulation spanning 50 ns revealed that the protein–ligand complex maintained a stable trajectory throughout, confirming the ligand's enduring presence within the binding pocket. The computational analysis affirmed the fluoro-ethylated compound's favourable binding energy and underscored the sustained stability of its complex with the σ1R.
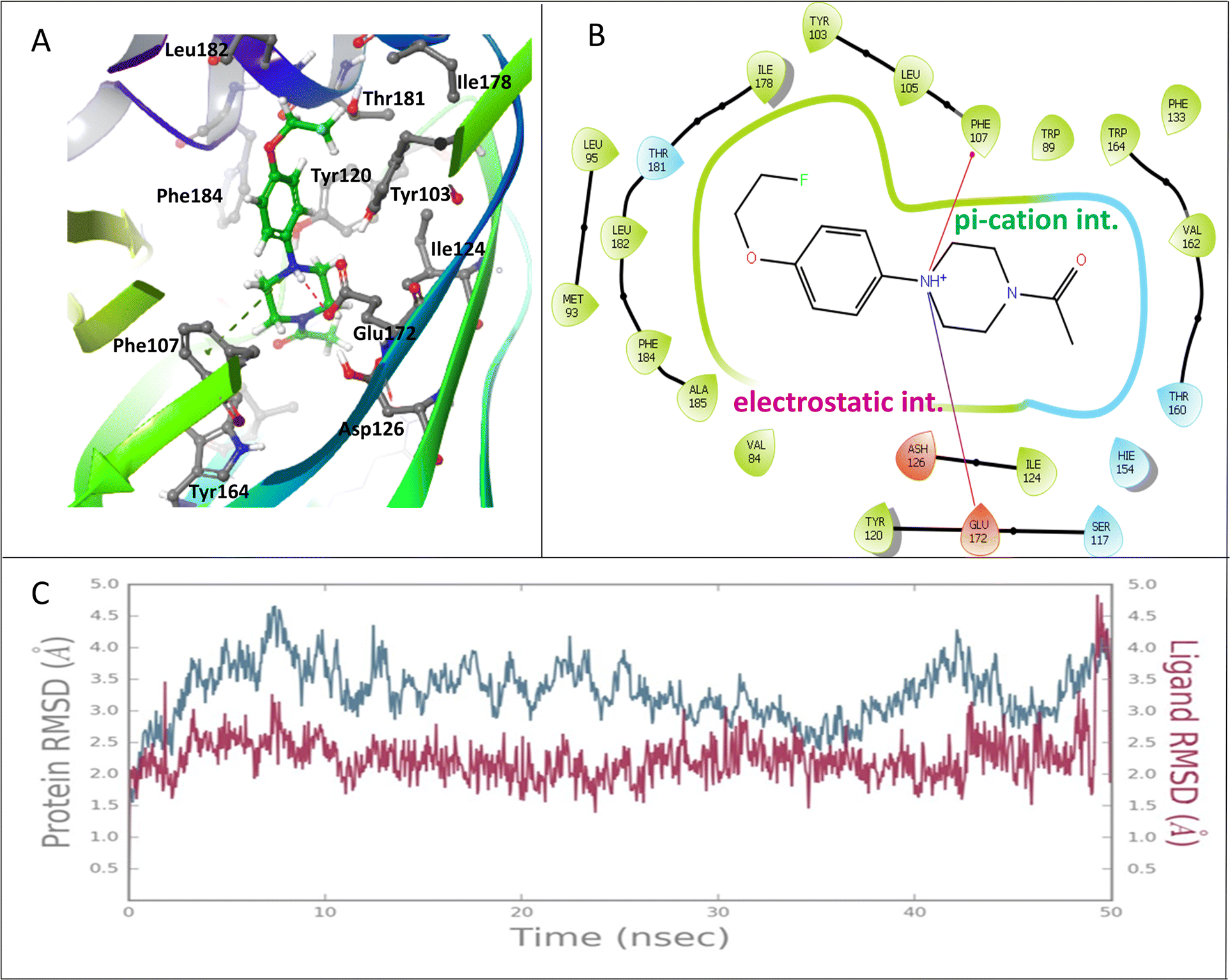 | ||
| Fig. 2 Results of the docking and MD simulation studies between FEt-PPZ and σ1R (PDB ID: 5HK1). (A) Mode of interaction between the protein and the ligand. (B) 2D interaction diagram showing the interactions between FEt-PPZ and σ1R. (C) MD simulation study showing the RMSD values of the protein and ligand over a period of 50 ns. | ||
Radiofluoroethylation is a well-known method to introduce radioactive fluorine (18F) into a molecule containing suitable functional groups.36 Fig. 3 shows the scheme for the radiosynthesis of [18F]FEt-PPZ under optimized conditions [T2 in ESI†]. The final product was obtained by an SPE-based purification process using neutral alumina and two Light C18 cartridges. The radiochemical purity (RCP) of the product was determined using radioITLC and radioHPLC (Fig. 4). The RCP of [18F]FEt-PPZ was found to be more than 99%. In the radioHPLC, a single peak was observed at around 15 min. In the radioITLC, the radiolabeled compound showed an Rf of 0.7. The activity yield of [18F]FEt-PPZ was 32% ± 5.8% (n = 6).
 | ||
| Fig. 3 Radiosyntheses of (A) [18F]FEt-PPZ and (B) FEt-PPZ. | ||
 | ||
| Fig. 4 HPLC elution profiles of (A) [18F]FEt-PPZ and (B) FEt-PPZ. (C) Saline and serum stabilities of the compound at up to 4 h. | ||
For structural characterization, FEt-PPZ was synthesized in a macroscopic scale. This was achieved through the O-alkylation of PPZ under basic conditions, as mentioned in the Experimental section. A group of two doublets of triplets integrating four protons between 4 and 5 ppm in the 1H-NMR spectrum represented the fluoroethyl group attached to PPZ [Fig. S1 in the ESI†]. The observed splitting pattern was consistent with the coupling of a fluorine atom with the protons of the ethyl chain. Similarly, the 13C-NMR spectrum was consistent with the expected structure of FEt-PPZ [Fig. S2 in the ESI†]. A peak at −223 ppm in the F-NMR indicated the presence of fluorine in the molecule [Fig. S3 in the ESI†]. The molecular ion peak at m/z 267.1533 in the HRMS further confirmed the formation of FEt-PPZ [Fig. S4 in the ESI†]. The HPLC elution profile of FEt-PPZ was compared with the radioactivity profile of [18F]FEt-PPZ. It could be noted that the UV spectrum of the reference compound, FEt-PPZ [Fig. 4(b)], displayed a single peak at 14.5 min, closely matching the retention time of [18F]FEt-PPZ [Fig. 4(a)] prepared at the trace level. The UV-elution profile [Fig. S5 in the ESI†] corresponding to [18F]FEt-PPZ did not show any absorption, indicating the absence of the precursor material in the SPE-purified product. The molar activity of the radiotracer was determined to be 8.89 GBq μg−1 [S7 in the ESI†].
[18F]FEt-PPZ was found to be stable in saline at room temperature as well as in serum at 37 °C at up to 4 h. The stability was ascertained by radioITLC as described in the previous section. A typical radioITLC profile of [18F]FEt-PPZ is shown in Fig. S6 in the ESI.† In both cases, the %RCP of [18F]FEt-PPZ was found to be more than 95% even at 4 h post-incubation. The calculated LogPO/W value was 0.72 ± 0.11 (n = 3), suggesting the lipophilic nature of the compound.
Before carrying out the in vitro cell binding studies with [18F]FEt-PPZ, the expressions of σ1R were analyzed in the mouse brain, U-87 MG cells and B16F10 cells. The receptor expression was analyzed by RT-PCR. The results obtained are shown in Fig. 5. The expression of σ1R was observed in both the cells as well as in the brain. The highest expression of σ1R was observed in the mouse brain tissue followed by the U-87 MG cells and B16F10 cells, where the expression levels of σ1R were comparable.
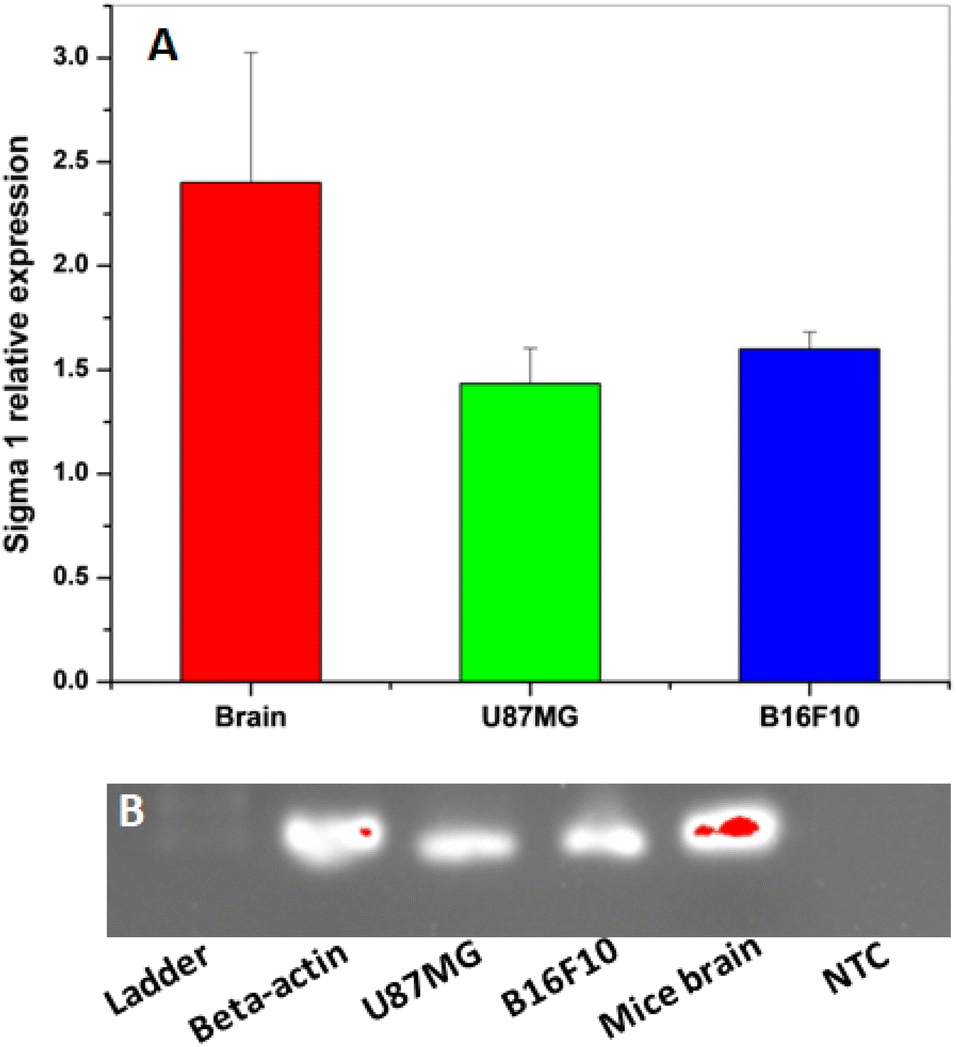 | ||
| Fig. 5 (A) Relative mRNA expression levels of σ1R gene in mouse brain tissue, glioma cells (U-87MG) and melanoma cells (B16F10). (B) Relative mRNA expression levels of σ1R gene analyzed by SYBR-Green Based Quantitative RT-PCT on cells (U-87MG and B16F10) and mouse brain tissue. The results expressed are mean ± SD (n = 3) when compared with the housekeeping gene (NTC: Non-Template Control; Ladder: DNA Marker 200bp). | ||
When incubated, [18F]FEt-PPZ exhibited an affinity for both melanoma (B16F10) and glioma (U-87 MG) cells, with a stronger affinity observed towards the glioma cells. In both instances, the cellular uptake was found to increase over time, reaching its maximum value at 45 min after incubation and plateauing subsequently. The maximum uptakes of [18F]FEt-PPZ observed with the U-87 MG and B16F10 cells was 13.05 ± 1.96% and 10.82% ± 1.69%, respectively [Fig. 6]. The comparable %cell binding values of the radiotracer between the two cells corroborated well with the comparable expression levels of σ1R in them [Fig. 5]. Competitive inhibition studies using non-radioactive FEt-PPZ resulted in a significant reduction in tracer uptake, approximately 55% in U-87 MG cells and 48% in B16F10 cells, indicating specific binding to σ1R. The docking score (Table 2) clearly indicated the preference of [18F]FEt-PPZ for σ1R. To verify this finding experimentally, competitive inhibition studies of [18F]FEt-PPZ were carried out in the presence of excess CM-398, a σ2R receptor specific ligand. The results indicated that preincubation with excess CM-398 led only to a relatively lower inhibition (∼10%) in [18F]FEt-PPZ binding in both cell types [Fig. 6]. This observation was in concordance with the preferential affinity of [18F]FEt-PPZ towards σ1R compared to σ2R, as indicated by the docking studies.
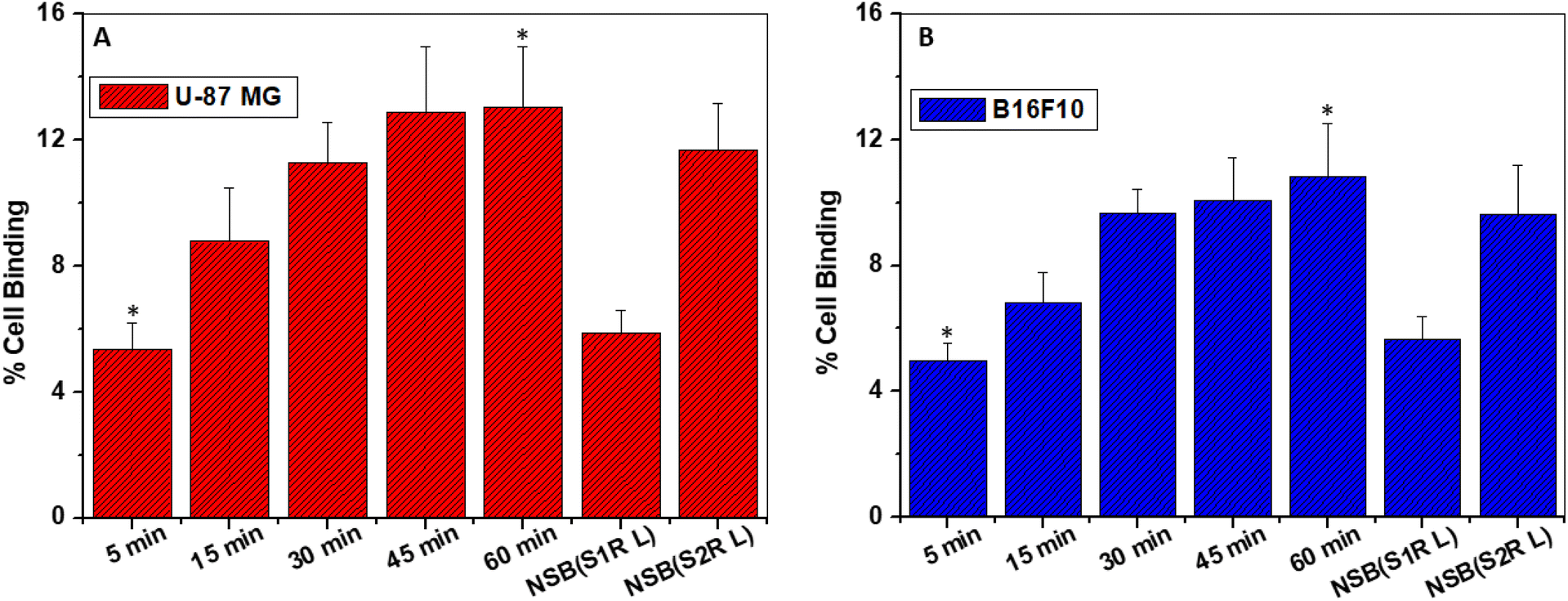 | ||
| Fig. 6 Results of the in vitro cell binding studies of [18F]FEt-PPZ on σ1R expressing (A) U-87 MG and (B) B16F10 cells. Cells were incubated with ∼0.19 MBq of [18F]FEt-PPZ for 5, 15, 30, 45 and 60 min time intervals at 37 °C. Non-specific cell binding (NSB) was assessed via competition assay, wherein cells were preincubated with excess cold FEt-PPZ (S1R L) and CM-398 (S2R L) (60 min, 37 °C), followed by incubation with [18F]FEt-PPZ for 60 min. All data points are expressed as mean ± SD (n = 3) (*P < 0.05). | ||
The distributions of [18F]FEt-PPZ in the melanoma-tumor-bearing mice at different time intervals (30, 60, and 120 min) post-injection are shown in Fig. 7(a). The radiotracer could be seen effectively penetrating the blood–brain barrier (BBB), showing a peak uptake of 6.16 ± 0.18% ID per g at 30 min post-injection (p.i), which subsequently decreased to 3.55% ± 0.26% ID per g at 120 min p.i. A relatively faster clearance of activity was found in the lung, heart, bone, and muscle. The radiotracer exhibited significant uptake and retention in the tumors at all time points. The highest uptake, 10.55% ± 1.3%, was noted at 60 min p.i. The tumor-to-blood (T/B) and tumor-to-muscle (T/M) ratio improved with time till 1 h post-injection [Fig. 7(b)], remaining nearly the same thereafter for the next 1 h. The maximum T/B and T/M ratios obtained were 1.54/0.15 and 4.68/0.16, respectively. Clearance of [18F]FEt-PPZ activity was found both through the kidney and hepatobiliary routes, as indicated by the moderate to high activity levels in the liver, spleen, intestine, and kidneys. In the animal PET/CT imaging [Fig. 7(c)], notable tumor uptake was observed, which was in concordance with the biodistribution data. The SUV values of the tumor at 30, 60 and 120 min were calculated and found to be 11.2, 9.8 and 7.1, respectively. A high SUV value suggested radiotracer uptake in the tumor. Significant activity was found in the abdomen region at each time point, supporting the biodistribution results regarding the renal and hepatic routes of excretion.
 | ||
| Fig. 7 (A) Biodistribution data of [18F]FEt-PPZ in syngeneic B16F10-tumor-bearing C57BL6 mice. The radio-conjugate [18F]FEt-PPZ (∼5 MBq per mouse) was injected through the tail vein (n = 3) for biodistribution studies; (B) variation of the T/B and T/M ratios in the B16F10-tumor-bearing C57BL6 mice over a period of 2 h. (C) Soft tissue coronal hybrid image (PET/CT) of the B16F10-tumor-bearing mouse over a period of 2 h. The mouse was injected with ∼5 MBq of [18F]FEt-PPZ, and the images were acquired at 30, 60, and 120 min post injection. Uptake of the radiotracer was observed in the tumor, brain, kidney and intestine. | ||
Conclusions
A PET radiotracer targeting σ1R was prepared by a one-pot synthesis from a commercially available molecule by adopting in silico methods. As indicated in the docking and molecular dynamics studies, the radiotracer showed significant binding to the σ1R-expressing melanoma (B16F10) and glioma (U-87 MG) cells. Inhibition studies of the radiotracer carried out in the presence of excess FEt-PPZ and CM-398, a ligand for σ2R, clearly indicated the preferential affinity of [18F]FEt-PPZ for σ1R over σ2R, as also predicted by the docking studies. The biodistribution and imaging studies in melanoma-tumor-bearing mice also showed significant uptake and retention of the radiotracer in the tumors. The work presented here constitutes the successful use of in silico methods for developing a diagnostic radiopharmaceutical against cancers expressing σ1 receptors. Future work will be directed towards reducing the observed non-target uptake of the radiotracer, which could be due to its lipophilic nature.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The author declare no competing interest.References
- D. A. Ryskamp, S. Korban, V. Zhemkov, N. Kraskovskaya and I. Bezprozvanny, Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases, Front. Neurosci., 2019, 13(862), 1–20, DOI:10.3389/fnins.2019.00862.
- J.-L. Jin, M. Fang, Y.-X. Zhao and X.-Y. Liu, Roles of Sigma-1 Receptors in Alzheimer's Disease, Int. J. Clin. Exp. Med., 2015, 8(4), 4808–4820 Search PubMed.
- S.-Y. A. Tsai, M. J. Pokrass, N. R. Klauer, H. Nohara and T.-P. Su, Sigma-1 Receptor Regulates Tau Phosphorylation and Axon Extension by Shaping P35 Turnover via Myristic Acid, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(21), 6742–6747, DOI:10.1073/pnas.1422001112.
- S. B. Smith, J. Wang, X. Cui, B. A. Mysona, J. Zhao and K. E. Bollinger, Sigma 1 Receptor: A Novel Therapeutic Target in Retinal Disease, Prog. Retin. Eye Res., 2018, 67, 130–149, DOI:10.1016/j.preteyeres.2018.07.003.
- D. Crottès, H. Guizouarn, P. Martin, F. Borgese and O. Soriani, The Sigma-1 Receptor: A Regulator of Cancer Cell Electrical Plasticity?, Front. Physiol., 2013, 4(175), 1–10, DOI:10.3389/fphys.2013.00175.
- F. J. Kim and C. M. Maher, Sigma1 Pharmacology in the Context of Cancer, Handb. Exp. Pharmacol., 2017, 244, 237–308, DOI:10.1007/164_2017_38.
- Y. Wu, X. Bai, X. Li, C. Zhu and Z. P. Wu, Overexpression of Sigma-1 Receptor in MCF-7 Cells Enhances Proliferation via the Classic Protein Kinase C Subtype Signaling Pathway, Oncol. Lett., 2018, 16(5), 6763–6769, DOI:10.3892/ol.2018.9448.
- W. R. Martin, C. G. Eades, J. A. Thompson, R. E. Huppler and P. E. Gilbert, The Effects of Morphine- and Nalorphine- like Drugs in the Nondependent and Morphine-Dependent Chronic Spinal Dog, J. Pharmacol. Exp. Ther., 1976, 197(3), 517–532 CrossRef CAS PubMed.
- T.-Y. Weng, S.-Y. A. Tsai and T.-P. Su, Roles of Sigma-1 Receptors on Mitochondrial Functions Relevant to Neurodegenerative Diseases, J. Biomed. Sci., 2017, 24(1), 74, DOI:10.1186/s12929-017-0380-6.
- H. R. Schmidt, S. Zheng, E. Gurpinar, A. Koehl, A. Manglik and A. C. Kruse, Crystal Structure of the Human Σ1 Receptor, Nature, 2016, 532(7600), 527–530, DOI:10.1038/nature17391.
- G. Sgouros, L. Bodei, M. R. McDevitt and J. R. Nedrow, Radiopharmaceutical Therapy in Cancer: Clinical Advances and Challenges, Nat. Rev. Drug Discovery, 2020, 19(9), 589–608, DOI:10.1038/s41573-020-0073-9.
- Druggability of Targets for Diagnostic Radiopharmaceuticals | ACS Pharmacology & Translational Science, https://www.pubs.acs.org/doi/10.1021/acsptsci.3c00081, accessed 2024-06-28 Search PubMed.
- K. Kawamura, K. Ishiwata, Y. Shimada, Y. Kimura, T. Kobayashi, K. Matsuno, Y. Homma and M. Senda, Preclinical Evaluation of [11C]SA4503: Radiation Dosimetry, in Vivo Selectivity and PET Imaging of Sigma1 Receptors in the Cat Brain, Ann. Nucl. Med., 2000, 14(4), 285–292, DOI:10.1007/BF02988211.
- K. Kawamura, H. Tsukada, K. Shiba, C. Tsuji, N. Harada, Y. Kimura and K. Ishiwata, Synthesis and Evaluation of Fluorine-18-Labeled SA4503 as a Selective Sigma1 Receptor Ligand for Positron Emission Tomography, Nucl. Med. Biol., 2007, 34(5), 571–577, DOI:10.1016/j.nucmedbio.2007.03.009.
- R. N. Waterhouse and T. L. Collier, In Vivo Evaluation of [18F]1-(3-Fluoropropyl)-4-(4-Cyanophenoxymethyl)Piperidine: A Selective Sigma-1 Receptor Radioligand for PET, Nucl. Med. Biol., 1997, 24(2), 127–134, DOI:10.1016/s0969-8051(96)00184-9.
- T. Hjørnevik, P. W. Cipriano, B. Shen, J. H. Park, P. Gulaka, D. Holley, H. Gandhi, D. Yoon, E. S. Mittra, G. Zaharchuk, S. S. Gambhir, C. R. McCurdy, F. T. Chin and S. Biswal, Biodistribution and Radiation Dosimetry of 18F-FTC-146 in Humans, J. Nucl. Med., 2017, 58(12), 2004–2009, DOI:10.2967/jnumed.117.192641.
- S. Fischer, C. Wiese, E. Große Maestrup, A. Hiller, W. Deuther-Conrad, M. Scheunemann, D. Schepmann, J. Steinbach, B. Wünsch and P. Brust, Molecular Imaging of σ Receptors: Synthesis and Evaluation of the Potent Σ1 Selective Radioligand [18F]Fluspidine, Eur. J. Nucl. Med. Mol. Imaging, 2011, 38(3), 540–551, DOI:10.1007/s00259-010-1658-z.
- F.-X. Lepelletier, M. Vandesquille, M.-C. Asselin, C. Prenant, A. C. Robinson, D. M. A. Mann, M. Green, E. Barnett, S. D. Banister, M. Mottinelli, C. Mesangeau, C. R. McCurdy, I. B. Fricke, A. H. Jacobs, M. Kassiou and H. Boutin, Evaluation of 18F-IAM6067 as a Sigma-1 Receptor PET Tracer for Neurodegeneration in Vivo in Rodents and in Human Tissue, Theranostics, 2020, 10(18), 7938–7955, DOI:10.7150/thno.47585.
- D. Yang, A. Comeau, W. D. Bowen, R. H. Mach, B. D. Ross, H. Hong and M. E. Van Dort, Design and Investigation of a [18F]-Labeled Benzamide Derivative as a High Affinity Dual Sigma Receptor Subtype Radioligand for Prostate Tumor Imaging, Mol. Pharm., 2017, 14(3), 770–780, DOI:10.1021/acs.molpharmaceut.6b01020.
- Y. Lan, P. Bai, Z. Chen, R. Neelamegam, M. S. Placzek, H. Wang, S. A. Fiedler, J. Yang, G. Yuan, X. Qu, H. R. Schmidt, J. Song, M. D. Normandin, C. Ran and C. Wang, Novel Radioligands for Imaging Sigma-1 Receptor in Brain Using Positron Emission Tomography (PET), Acta Pharm. Sin. B, 2019, 9(6), 1204–1215, DOI:10.1016/j.apsb.2019.07.002.
- S. Preshlock, M. Tredwell and V. Gouverneur, 18 F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography, Chem. Rev., 2016, 116(2), 719–766, DOI:10.1021/acs.chemrev.5b00493.
- J. Rong and S. H. Liang Aliphatic [18F]Fluorination Chemistry for Positron Emission Tomography. in Fluorination, ed. Hu, J. and Umemoto, T., Springer Singapore, Singapore, 2020, pp. 1–14. DOI:10.1007/978-981-10-3896-9_38.
- W. Chen, Z. Huang, N. E. S. Tay, B. Giglio, M. Wang, H. Wang, Z. Wu, D. A. Nicewicz and Z. Li, Direct Arene C–H Fluorination with18 F− via Organic Photoredox Catalysis, Science, 2019, 364(6446), 1170–1174, DOI:10.1126/science.aav7019.
- N. E. S. Tay, W. Chen, A. Levens, V. A. Pistritto, Z. Huang, Z. Wu, Z. Li and D. A. Nicewicz, 19F- and 18F-Arene Deoxyfluorination via Organic Photoredox-Catalysed Polarity-Reversed Nucleophilic Aromatic Substitution, Nat. Catal., 2020, 3(9), 734–742, DOI:10.1038/s41929-020-0495-0.
- W. Chen, H. Wang, N. E. S. Tay, V. A. Pistritto, K.-P. Li, T. Zhang, Z. Wu, D. A. Nicewicz and Z. Li, Arene Radiofluorination Enabled by Photoredox-Mediated Halide Interconversion, Nat. Chem., 2022, 14(2), 216–223, DOI:10.1038/s41557-021-00835-7.
- C. Yang, K. Lu, J. Li, H. Wu and W. Chen, Rapid Construction of18 F-Triazolyl-Tetrazines through the Click Reaction, J. Org. Chem., 2024, 89(20), 14673–14678, DOI:10.1021/acs.joc.4c00574.
- Q. Zheng, H. Xu, H. Wang, W.-G. H. Du, N. Wang, H. Xiong, Y. Gu, L. Noodleman, K. B. Sharpless, G. Yang and P. Wu, Sulfur [18 F]Fluoride Exchange Click Chemistry Enabled Ultrafast Late-Stage Radiosynthesis, J. Am. Chem. Soc., 2021, 143(10), 3753–3763, DOI:10.1021/jacs.0c09306.
- F. D. Prieto-Martínez; E. López-López; K. Eurídice Juárez-Mercado and J. L. Medina-Franco Chapter 2 - Computational Drug Design Methods—Current and Future Perspectives. in In Silico Drug Design, ed. Roy, K., Academic Press, 2019, pp. 19–44. DOI:10.1016/B978-0-12-816125-8.00002-X.
- X. Lin, X. Li and X. Lin, A Review on Applications of Computational Methods in Drug Screening and Design, Molecules, 2020, 25(6), 1375, DOI:10.3390/molecules25061375.
- A. Alon, J. Lyu, J. M. Braz, T. A. Tummino, V. Craik, M. J. O’Meara, C. M. Webb, D. S. Radchenko, Y. S. Moroz, X.-P. Huang, Y. Liu, B. L. Roth, J. J. Irwin, A. I. Basbaum, B. K. Shoichet and A. C. Kruse, Structures of the Σ2 Receptor Enable Docking for Bioactive Ligand Discovery, Nature, 2021, 600(7890), 759–764, DOI:10.1038/s41586-021-04175-x.
- S. Patra, J. Dey, S. Kar and A. Chakraborty, Delivery of Chlorambucil to the Brain Using Surface Modified Solid Lipid Nanoparticles, ACS Appl. Bio Mater., 2024, 7(5), 3403–3413, DOI:10.1021/acsabm.4c00326.
- A. Chakraborty, A. Mitra, S. Sahu, M. Tawate, S. Lad, Kamaldeep, S. Rakshit, T. Upadhye Bannore, S. Gaikwad, G. Dhotre, M. K. Ray, A. Damle, S. Basu and S. Banerjee, Intricacies in the Preparation of Patient Doses of [177Lu]Lu-Rituximab and [177Lu]Lu-Trastuzumab Using Low Specific Activity [177Lu]LuCl3: Methodological Aspects, Mol. Imaging Biol., 2024, 26(1), 61–80, DOI:10.1007/s11307-023-01846-1.
- S. Intagliata, A. Sharma, T. I. King, C. Mesangeau, M. Seminerio, F. T. Chin, L. L. Wilson, R. R. Matsumoto, J. P. McLaughlin, B. A. Avery and C. R. McCurdy, Discovery of a Highly Selective Sigma-2 Receptor Ligand, 1-(4-(6,7-Dimethoxy-3,4-Dihydroisoquinolin-2(1H)-Yl)Butyl)-3-Methyl-1H-Benzo[d]Imidazole-2(3H)-One (CM398), with Drug-Like Properties and Antinociceptive Effects In Vivo, AAPS J, 2020, 22(5), 94, DOI:10.1208/s12248-020-00472-x.
- S. Patra, J. Dey, S. Kar, A. Chakraborty and M. Tawate, Methotrexate-Loaded Surface-Modified Solid Lipid Nanoparticles Targeting Cancer Expressing COX-2 Enzyme, Langmuir, 2024, 40, 14811–14822, DOI:10.1021/acs.langmuir.4c00638.
- L. D. Luca, L. Lombardo, S. Mirabile, A. Marrazzo, M. Dichiara, G. Cosentino, E. Amata and R. Gitto, Discovery and Computational Studies of Piperidine/Piperazine-Based Compounds Endowed with Sigma Receptor Affinity, RSC Med. Chem., 2023, 14(9), 1734–1742, 10.1039/D3MD00291H.
- Y. Qi, Y. Li, Y. Fang, H. Gao, B. Qiang, S. Wang and H. Zhang, Design, Synthesis, Biological Evaluation, and Molecular Docking of 2,4-Diaminopyrimidine Derivatives Targeting Focal Adhesion Kinase as Tumor Radiotracers, Mol. Pharm., 2021, 18(4), 1634–1642, DOI:10.1021/acs.molpharmaceut.0c01088.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02999f |
| This journal is © The Royal Society of Chemistry 2025 |