
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntibiotic-conjugated antimicrobial peptides for enhanced bacterial inhibition†
Xingrao Penga,
Yong Luoab,
Tianzhi Xua,
Zihan Chena,
Peiyao Chenc,
Cong Hud and
Shuang Liu *a
*a
aSchool of Materials Science and Engineering, Wuhan University of Technology, 122 Luoshi Road, Wuhan, Hubei 430070, China. E-mail: shuangliu@whut.edu.cn
bState Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University, Changsha 410082, China
cKey Laboratory of Fermentation Engineering (Ministry of Education), National “111” Center for Cellular Regulation and Molecular Pharmaceutics, Hubei Key Laboratory of Industrial Microbiology, School of Life and Health Sciences, Hubei University of Technology, Wuhan, Hubei 430068, China
dGuangxi Key Laboratory of Automatic Detecting Technology and Instruments, Guilin University of Electronic Technology, Guilin 541004, China
First published on 11th June 2025
Abstract
Herein, we report a pair of paenipeptin C′-based antimicrobial linear lipopeptides that significantly enhance bacterial inhibition through conjugation to antibiotics. When co-incubated with Escherichia coli or Staphylococcus aureus, these peptides induce bacterial death. The antimicrobial peptides target negatively charged bacterial membranes via electrostatic interactions, subsequently disrupting membrane integrity through aggregation and insertion, leading to membrane rupture and cytoplasmic leakage, as evidenced by bacterial morphology studies. Lipopeptides with longer alkyl chains penetrate deeper into the membrane structure, demonstrating stronger antibacterial effects. Additionally, the conjugated antibiotics may enhance bactericidal activity by inhibiting intracellular DNA gyrase. Therapeutic efficacy was further validated in a murine infected wound model. This work not only develops a class of broad-spectrum antimicrobial lipopeptides but also provides a novel strategy for developing antibiotic-conjugated antimicrobial peptides to enhance multiple antibacterial inhibition with minimal side effects.
Introduction
Bacterial infection threatens the health of humans, which may cause fever, tissue necrosis, functional impairment, and even damage to nerve and organ function.1–7 Traditionally, bacterial infections are usually treated with antibiotics, chemical agents which either kill or prevent the growth of microbes,8 including penicillin, aminopenicillins, tetracyclines, and cephalosporins. However, increasing antibiotic resistance has led to increased treatment failure, resulting in an urgent global health crisis.9–13 This situation underscores the urgent need for the development of novel and safe antimicrobial agents.14Antimicrobial peptides (AMPs), a class of natural low-molecular-weight peptides produced by organisms as part of the innate immune system,15,16 have attracted attention in anti-infective research due to their broad-spectrum antimicrobial activity, low propensity for acquired resistance, and membrane-targeting mechanisms.17–19 Paenipeptin C′, a synthetic linear antimicrobial peptide, was rationally designed through structural optimization of a natural lipopeptide template,20 originating from natural lipopeptide complexes (comprising both cyclic and linear structures) produced by the Paenibacillus sp. OSY-N strain. In 2017, Huang et al.21 first chemically synthesized a linear paenipeptin derivative and demonstrated that its antimicrobial activity was independent of macrocyclic structural elements. Paenipeptin C′ exerts antimicrobial effects through concentration-dependent membrane depolarization and potassium efflux. Its unique mechanism is further highlighted by its ability to disrupt biofilms and bind to bacterial surface components,22,23 such as lipopolysaccharides in Gram-negative bacteria and lipoteichoic acids in Gram-positive bacteria. However, monotherapy with membrane-targeting AMPs carries the risk of resistance development via bacterial membrane modification, necessitating synergistic strategies to enhance efficacy and mitigate resistance.24–27
Inspired by the unique antibacterial mechanism and superior antimicrobial activity of the antimicrobial peptide paenipeptin C′, we designed analogues of paenipeptin C′ as antimicrobial peptides 1 (lipid C6-Phe–Phe–Pae) and 2 (lipid C18-Pae) (Fig. 1a). We added lipid chains into the peptides since we believed that lipid chains can enhance the interaction with bacterial membrane to enhance the uptake.28–30 The diphenylalanine provides π–π interaction and β-sheet self-assembly, giving us the chance to study the influence of self-assembly on antibacterial efficacy. We also coupled ciprofloxacin (CPFx), a fluoroquinolone antibiotic, with the peptides 1 and 2 to produce CPFx-1 and CPFx-2, which can be hydrolyzed by esterase in bacteria and release CPFx to enhance the antibacterial efficacy.31–33 The conjugation of antimicrobial peptides improves the water solubility of the antibiotic, which also improves the uptake and the bacterial inhibiting efficacy. In vitro evaluations demonstrated that CPFx-2 exhibits potent broad-spectrum antimicrobial activity against E. coli and S. aureus through distinct mechanisms of action, while maintaining negligible cytotoxicity toward normal cells and tissues. In vivo antibacterial assessments confirmed the efficacy of CPFx-2 in promoting wound healing in a murine model of E. coli-infected wounds (Fig. 1).
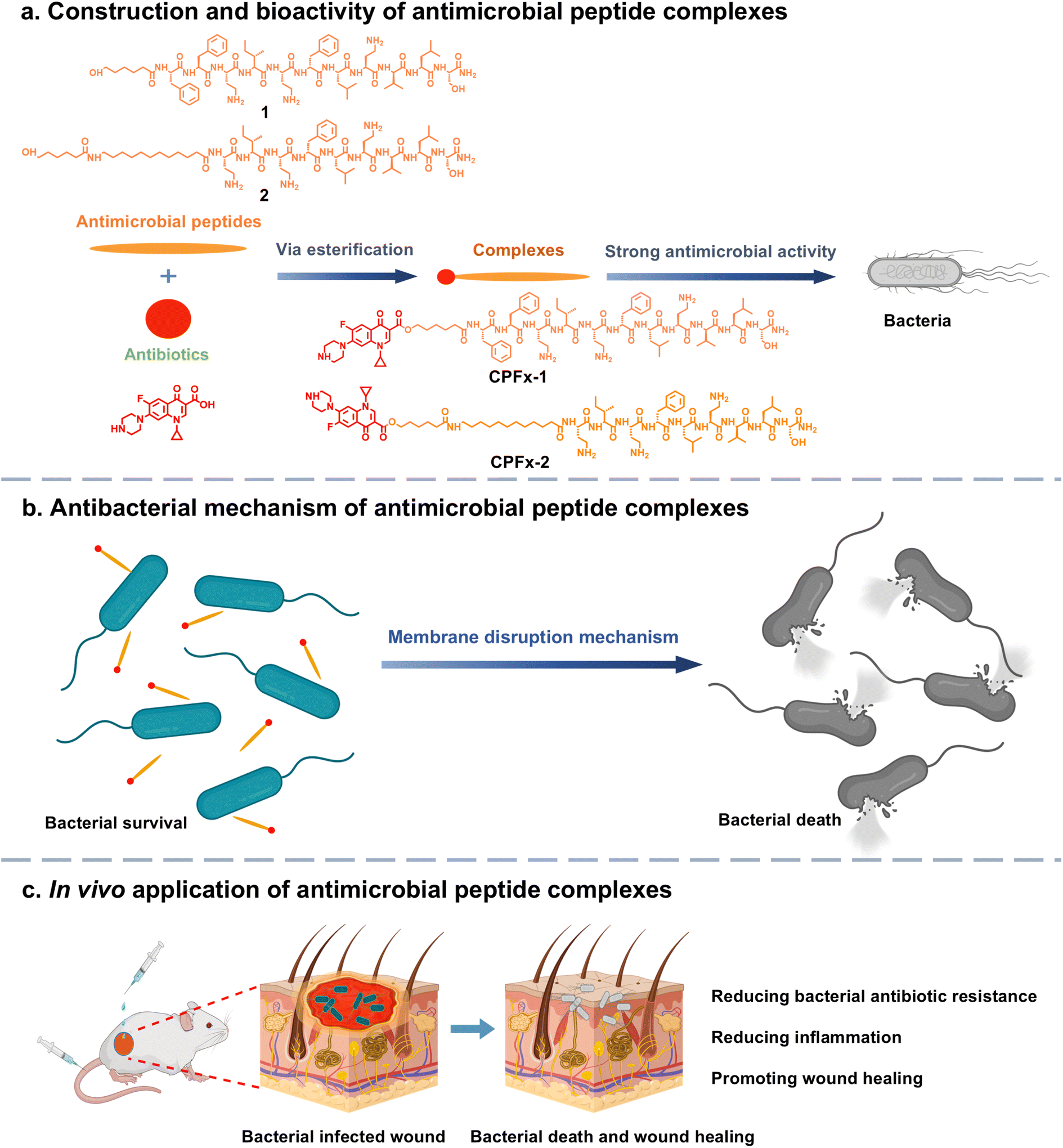 | ||
| Fig. 1 Schematic illustration of the construction and antibacterial of the four antimicrobial peptides. (a) The construction of antimicrobial peptides 1, 2 and their CPFx-conjugated analogues CPFx-1, CPFx-2. (b) Antibacterial mechanism of the complex through membrane targeting and disruption in bacteria. The positively charged complex first binds to the negatively charged bacterial surface via electrostatic interactions, followed by the insertion of its hydrophobic regions into the cell membrane, which leads to membrane rupture and leakage of cellular contents. (c) In both topical administration to infected murine wounds and intravenous injection via the tail vein, the complex demonstrated excellent antibacterial efficacy and enhanced wound-healing capacity. | ||
The current dual-mode antibacterial strategies mainly include: dual-antibiotic combination therapy,34 enzyme inhibitors combined with antibiotics,35 single-drug dual-target action,36 and nanomaterials combined with antibiotics,37 among others. Compared to the combined use of antimicrobial peptides and small-molecule antibiotics, our strategy involves conjugating antibiotics to antimicrobial peptides via ester bonds. This approach achieves precise membrane targeting through the electrostatic interactions between antimicrobial peptides and bacterial cell membranes. Additionally, the controlled hydrolysis of antibiotics is enabled by bacterial endogenous esterases, and it overcomes the issue of disparate drug distribution observed in traditional combination therapies.38,39
Results and discussion
Molecular synthesis and characterization
We used solid phase peptide synthesis (SPPS) to make the peptides (Scheme S2†). Briefly, after reacting CPFx and 6-hydroxyhexanoate to produce ester (Scheme S1†), we synthesized the designed peptides via SPPS by using Fmoc-protected amino acids. After purification by high performance liquid chromatography (HPLC), 1H NMR and MS (Fig. S1–S8†) were used to confirm the purity and identity of the precursors.Self-assembly evaluation
We first evaluated the stability of the four antimicrobial peptides by monitoring their absorbance over 24 hours in 10% fetal bovine serum (FBS) or in phosphate-buffered saline (PBS, pH 7.4) at 37 °C (Fig. 2a and S9†). The absorbance reflected instability factors of the material, such as hydrolysis (resulting in the generation of small molecules) and aggregation, both of which can increase the material's absorbance.40 The absorbance changes remained below 0.0975 after 24 hour incubation in either FBS or PBS (Fig. 2a and S9†), demonstrating their remarkable stability. This stability was evidenced by their resistance to degradation and aggregation. The resistance to degradation indicates the remarkable chemical stability, and the resistance to aggregation indicates the stable assembly. | ||
| Fig. 2 (a) Absorption spectra of antimicrobial peptides (50 μM) in 10% FBS at different time points. (b) Transmission electron microscopy (TEM) images of antimicrobial peptide (200 μM). (c) Dynamic light scattering (DLS) profiles of antimicrobial peptides (200 μM). | ||
We next assessed the self-assembly properties of the four antimicrobial peptides by measuring their critical micelle concentration (CMC) through light transmittance measurements across a concentration gradient (15.625–2000 μM) in PBS (pH 7.4). Fig. S10† demonstrates that peptides 1 and 2 showed no significant changes in light transmittance with increasing concentration at low concentrations, but exhibited decreased transmittance at higher concentrations, indicating their certain self-assembly capability at elevated concentrations. In contrast, CPFx-1 and CPFx-2 displayed concentration-dependent turbidity increases (reduced transmittance) at relatively lower concentrations, confirming supramolecular assembly formation. This behavior aligned with light scattering signatures of micellization, demonstrating that hydrophobic CPFx enhanced self-assembly. Notably, peptide 1 displayed a CMC of 959.36 μM, peptide 2 displayed a CMC of 976.07 μM, CPFx-1 displayed a CMC of 332.70 μM, while CPFx-2 displayed a CMC of 194.99 μM (Fig. S11†). The lower CMC of CPFx-2 confirmed a stronger self-assembly ability, resulting in the fact that the hydrophobic interactions induced by the C12 alkyl chain provided stronger self-assembly driving force than the π–π interactions induced by diphenylalanine.41
To characterize secondary structures, we performed circular dichroism (CD) spectroscopy in PBS (pH 7.4) (Fig. S12, ESI†). Peptides 1 and CPFx-1 exhibited a strong negative peak at 200 nm accompanied by a weak negative band near 230 nm, indicating the potential coexistence of α-helical, β-sheet secondary structures and disordered regions in their conformations.42,43 Peptides 2 and CPFx-2 exhibited spectra with a negative peak at 200 nm and a positive peak at 190 nm, indicative of random coil conformations. We next used transmission electron microscopy (TEM) to verify the morphology of the peptides. The TEM images of all the four peptides revealed well-defined micelles (Fig. 2b). The micelles of 1 exhibited a diameter of 67.6 ± 8.9 nm, 2 exhibited a diameter of 70.7 ± 14.4 nm, CPFx-1 exhibited a diameter of 78.9 ± 7.4 nm and CPFx-2 exhibited a diameter of 103.1 ± 21.0 nm. The results showed that longer alkyl chains correlated with larger nanoparticle sizes, consistent with enhanced hydrophobic-driven assembly, which agrees with the CMC measurement results. The dynamic diameter measured (Fig. 2c) by dynamic light scattering (DLS) also validated the aforementioned results. Peptide 1 exhibited a dynamic diameter of 85.7 ± 17.0 nm, 2 exhibited a dynamic diameter of 116.1 ± 22.9 nm, CPFx-1 exhibited a dynamic diameter of 125.6 ± 19.1 nm and CPFx-2 exhibited a dynamic diameter of 127.4 ± 19.1 nm, consistent with their respective self-assembly capabilities. The results confirmed that the CPFx conjugation and incorporation of a long alkyl chain provide the most assembly driving force.
In vitro antimicrobial activity evaluation
After investigating the self-assembly capabilities of the four antimicrobial peptides, we evaluated their antibacterial efficacy against Gram-negative E. coli and Gram-positive S. aureus by quantifying bacterial viability.44 As shown in Fig. 3a, after 4 h of co-incubation with E. coli, CPFx-2 exhibited potent antibacterial activity even at a concentration as low as 6.25 μM, achieving a bacterial survival rate of 32.2%. Increasing the peptide concentration (0–50 μM) enhanced the antimicrobial effects of all four antimicrobial peptides, demonstrating a concentration-dependent bactericidal pattern against E. coli. After extending the incubation time to 12 h (Fig. 3b), CPFx-1 and CPFx-2 at concentrations of 6.25 μM further reduced E. coli survival to 20.6%, confirming their time-dependent antibacterial activity. For S. aureus, CPFx-2 displayed superior performance, with survival rates decreasing to 24.6% after 12 h incubation at a concentration of 50 μM (Fig. 3d). This highlights CPFx-2's broad-spectrum antimicrobial activity, effectively targeting both Gram-negative and Gram-positive pathogens. Notably, the conjugation of CPFx significantly enhanced the peptide's bactericidal potency, likely due to synergistic mechanisms between the peptide's membrane-disruptive action and ciprofloxacin's intracellular DNA gyrase inhibition.45,46 | ||
| Fig. 3 Changes in survival rate of E. coli after co-incubation with antimicrobial peptides at different concentrations for (a) 4 h and (b) 12 h. Changes in survival rate of S. aureus after co-incubation with antimicrobial peptides at different concentrations for (c) 4 h and (d) 12 h (n = 3, mean ± SD, ***p < 0.001, ****p < 0.0001). | ||
To further assess the in vitro antimicrobial performance of the peptides, we conducted an agar plate coating assay (Fig. 4a) against E. coli and S. aureus at a uniform peptide concentration of 50 μM. Compared with the control group (treated with PBS), all tested peptides (1, 2, CPFx-1 and CPFx-2) significantly reduced bacterial colony counts for both strains. Notably, CPFx-conjugated peptides demonstrated superior antimicrobial potency, with CPFx-2 nearly achieving complete eradication of both pathogens, as evidenced by the virtual absence of visible colonies. These results substantiated that the synergistic modification strategy combining CPFx conjugation and long-chain lipid integration enables a magnitude of enhancement in the antimicrobial performance relative to the parent peptide scaffolds.
 | ||
| Fig. 4 (a) LB agar plates showing viable bacterial counts of E. coli and S. aureus after treatment with antimicrobial peptides (50 μM) for 6 h, compared to a blank control group (PBS, pH 7.4). (b) Inhibition zone diameters of antimicrobial peptides against E. coli and S. aureus after 12 h of incubation. (c) Minimum Inhibitory Concentration (MIC) values of antimicrobial peptides against E. coli and S. aureus (n = 3, mean ± SD). | ||
We employed agar well diffusion assay to evaluate the antimicrobial activity of the four antimicrobial peptides.47 As shown in Fig. 4b, CPFx-2 demonstrated significantly larger inhibition zones compared to all other peptides in both E. coli and S. aureus cultures on LB agar plates at a standardized concentration of 50 μM. All the four peptide variants generated distinct bactericidal halos in LB agar plates (Fig. S13, ESI†), with sharply defined peripheries, confirming their broad-spectrum antimicrobial properties and rapid bactericidal action. Notably, CPFx-2 exhibited expanded clearance zones, indicative of superior antibacterial efficacy against both Gram-negative (E. coli) and Gram-positive (S. aureus) pathogens.
The peptides 1 and 2, without CPFx conjugation, exhibited a minimum inhibitory concentration (MIC) of 12.5 μM against E. coli (Fig. 4c), which was significantly higher than the MIC values (1.56 μM) of antibiotic-conjugated variants (CPFx-1 and CPFx-2). This demonstrated the enhanced antibacterial efficacy of CPFx-conjugated peptides against Gram-negative pathogens. Furthermore, CPFx-1 and CPFx-2 displayed substantially lower MIC against S. aureus (6.25 μM, respectively), indicating potent broad-spectrum antimicrobial activity. Notably, the superior performance of CPFx-2 (MIC = 3.125 μM) was likely attributed to its extended alkyl chains, which enhanced hydrophobic interactions with the phospholipid bilayer,48–50 thereby promoting membrane penetration and disruptive capacity. We compared the antibacterial activity data of antimicrobial peptides from other studies with CPFx-2 (Table S1†), and the results demonstrated that CPFx-2 exhibits superior antimicrobial activity. Furthermore, we assessed the cell viability of human normal bronchial epithelial cells (BEAS-2B). Even at 50 μM (a concentration significantly exceeding the MIC), CPFx-2-treated mammalian cells retained a survival rate >90% (Fig. S14†), with no statistically significant difference in LDH release (Fig. S15,† p > 0.05). These findings demonstrate the low cytotoxic effects of the antimicrobial peptides.
Antibacterial mechanisms of antimicrobial peptides
To elucidate the antimicrobial mechanisms of the peptides, we first characterized morphological changes of E. coli by using scanning electron microscopy (SEM) (Fig. 5a). Untreated cells exhibited smooth surfaces with intact membrane structures, while peptide-treated bacteria displayed varying degrees of membrane damage. Treatment with peptide 1 induced localized depressions on the bacterial surface, whereas peptide 2 caused pronounced wrinkling, accompanied by pits and pores. CPFx-conjugated peptides exacerbated membrane damage: CPFx-1 induced severe structural disruption, and CPFx-2 nearly obliterated bacterial structure, resulting in membrane fragmentation, cytoplasmic leakage, and structural collapse. These results confirmed that CPFx-2 induced bacteriolysis via membrane disruption, revealing synergistic mechanisms of CPFx conjugation and alkyl chain interactions. | ||
| Fig. 5 (a) Scanning electron microscopy (SEM) images of E. coli after co-incubation with antimicrobial peptides (50 μM) for 6 hours. (b) Zeta potential of CPFx, antimicrobial peptides, and bacterial (n = 3, mean ± SD). (c) NPN uptake of E. coli treated with antimicrobial peptides. Enhanced NPN uptake was reflected in an increase in NPN fluorescence intensity (n = 3, mean ± SD). | ||
We next measured the zeta potential of all four antimicrobial peptides (Fig. 5b). The results demonstrated that all the peptides exhibited positive surface charges in aqueous solution, attributable to their amine-rich side chains, while CPFx alone displayed a negative charge. Bacterial surfaces carried inherent negative charges due to anionic membrane components (E. coli: lipopolysaccharides; S. aureus: teichoic acids).51–53 This electrostatic profile explains why free CPFx fails to target bacterial membranes due to charge repulsion, whereas CPFx-conjugated peptides bind to negatively charged bacterial surfaces via electrostatic interactions, neutralizing surface charges and disrupting membrane integrity. Furthermore, peptide-mediated membrane targeting facilitates intracellular accumulation of CPFx, enhancing its DNA-damaging effects and enabling synergistic antibacterial action.
We also used N-phenyl-1-naphthylamine (NPN) to assess bacterial membrane damage, as NPN penetrates into hydrophobic regions of bacterial when the outer membrane is compromised, leading to a significant fluorescence increase.54–56 As shown in Fig. 5c, NPN fluorescence intensity increased significantly with rising peptide concentrations (PBS controls), indicating enhanced outer membrane permeability. Higher peptide concentrations likely promote membrane insertion and pore formation, enabling greater NPN influx and fluorescence signal amplification. At 50 μM, the hierarchy of membrane permeabilization efficacy (CPFx-2 > CPFx-1 > 2 > 1) agreed with SEM observations, further confirming that the peptides disrupt bacterial membranes through permeability alterations to exert their antibacterial effects.
In vivo anti-infectious efficacy of antimicrobial peptides
Encouraged by the exceptional antimicrobial activity of the antimicrobial peptides, we evaluated their in vivo therapeutic potential for E. coli-infected wounds by using a murine model.57 We administered topical treatment to the infected wounds with the peptides at a concentration of 200 μM or PBS as a control, and recorded the wound healing progress every three days via photographic documentation (Fig. 6a). On day 1 of treatment, all treatment groups exhibited uniformly sized wounds with visible redness, swelling, and inflammation at the edges, typical of early-stage infection. By day 3, peptides 1, 2, and CPFx-1 showed persistent redness and inflammation, similar to the control group, while the CPFx-2-treated group demonstrated significant improvement in the wound environment, including reduced wound area and scab formation at the edges, indicating its superior anti-inflammatory and pro-healing effects in the early treatment phase. By day 6, the control group showed no improvement, whereas the antimicrobial peptide-treated groups exhibited reduced redness and inflammation, with varying degrees of scab formation and reduced wound area, particularly in the CPFx-2-treated group, which showed the most significant improvement. By day 9, the wounds in the CPFx-2-treated group were almost completely healed, with fully regenerated epidermis and the smallest wound area compared to other antimicrobial peptide-treated groups, confirming its optimal therapeutic efficacy. To further evaluate the systemic therapeutic effects of antimicrobial peptides on infected wounds (Fig. 6b), we administered the drugs via tail vein injection to ensure systemic circulation, and the results were consistent with those of topical treatment. On day 3 of treatment, the infected wounds in the CPFx-2-treated group exhibited significantly reduced redness, swelling, and inflammatory responses, accompanied by a noticeable decrease in wound area. By day 6, the CPFx-2-treated group demonstrated the most substantial reduction in wound area, optimal scab formation, and the most pronounced healing progress. On day 9, all four antimicrobial peptide-treated groups showed markedly smaller wound areas compared to the PBS control group, with complete scab formation observed in all cases. Notably, the CPFx-2-treated group exhibited near-complete wound closure, with the smallest residual wound area among all antimicrobial peptide-treated groups, confirming the superior efficacy of CPFx-2 in tail vein injection therapy. The results indicated that all four antimicrobial peptides exhibited certain antibacterial effects by both topical treatment and intravenous injection, significantly reducing wound redness and inflammation while promoting wound healing. Among them, CPFx-2 showed the best antibacterial efficacy, demonstrating significant antibacterial and pro-healing effects early in the treatment, confirming that the CPFx conjugation strategy significantly enhanced the antibacterial activity of antimicrobial peptides.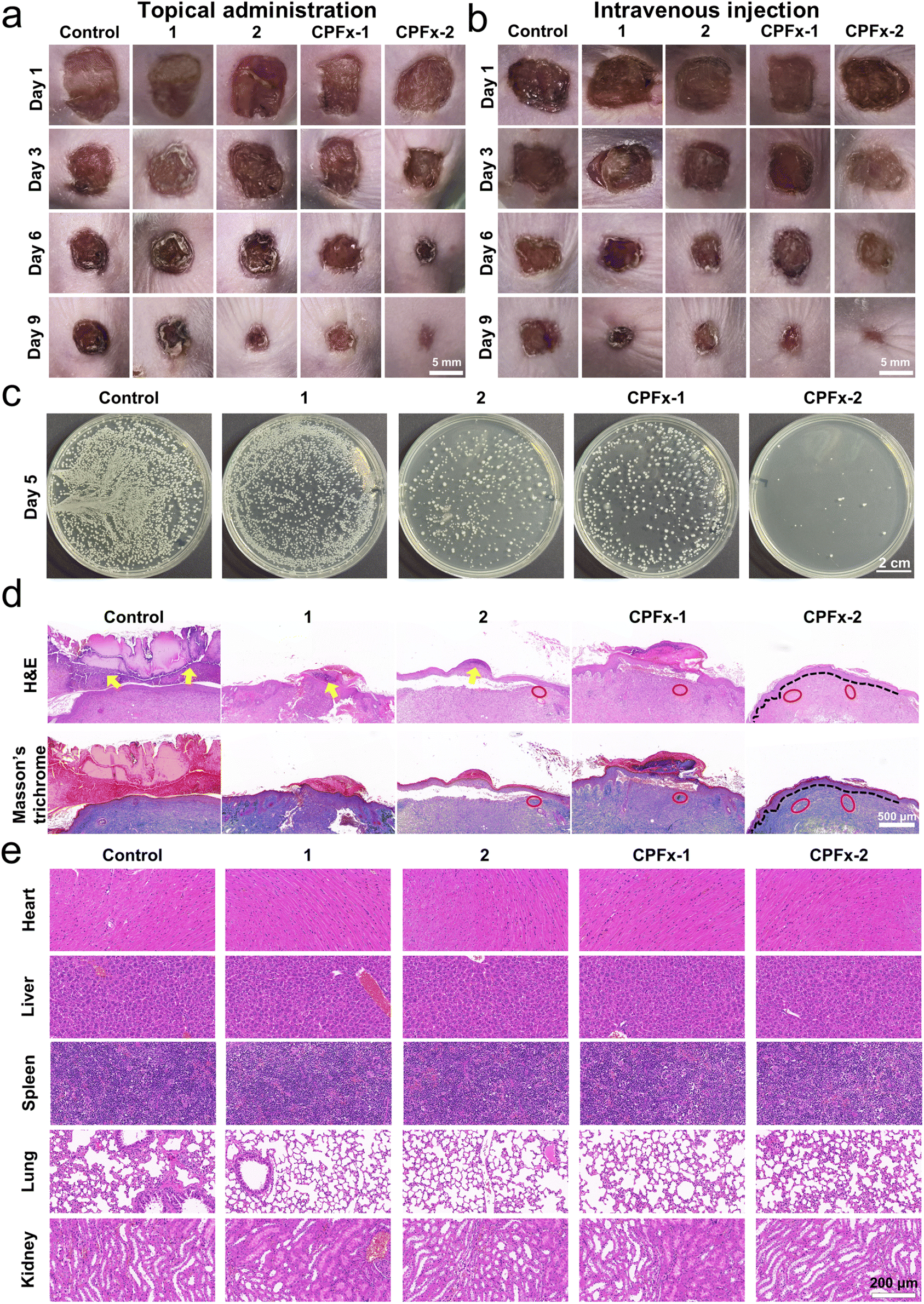 | ||
| Fig. 6 Photographs of infected wounds treated with (a) topical administration and (b) intravenous injection on day 1, 3, 6, and 9. (c) Plate coating photographs of infected wounds on day 5 after topical administration. (d) Histological analysis of wound tissue sections stained with H&E and Masson's trichrome on day 7 after topical administration (yellow arrows: inflammatory cells; red circles: collagen fibers; black dashed lines: epidermis–dermis boundary). (e) H&E-stained images of heart, liver, spleen, lung, and kidney from the E. coli-infected wound model on day 7 post-topical administration. | ||
To assess the antibacterial effects of the peptides at the mid-treatment stage, we performed agar plate coating assays of infected wounds on day 5 (Fig. 6c). The control group exhibited dense and numerous bacterial colonies, indicating normal growth and proliferation of E. coli, while the antimicrobial peptide-treated groups showed significantly reduced and sparser bacterial colonies. Notably, the CPFx-2 group exhibited almost no bacterial colonies on the agar plates, indicating that all peptides exhibited certain antibacterial activity against E. coli at the mid-treatment stage, with CPFx-2 showing the most significant efficacy, nearly completely inhibiting the growth and proliferation of E. coli. This further validated the conclusions from the murine wound infection model experiments.
We performed histological analysis on the murine models (Fig. 6d). The control group exhibited abundant inflammatory cells, necrotic epidermis, poor wound healing, and disorganized tissue structure. In contrast, 1 and 2-treated groups showed reduced inflammatory cells, indicating alleviated inflammation, with partial collagen deposition and improved wound healing. The groups with CPFx-conjugated antimicrobial peptide treatments showed significantly reduced inflammatory cells and increased collagen deposition in the infected wounds. Notably, the CPFx-2 group exhibited clear dermal–epidermal boundaries, fully regenerated epidermis, continuous and uniformly thick stratum corneum, and densely arranged collagen fibers, indicating that CPFx-2 significantly inhibited E. coli infection of the wounds and promoted collagen deposition, demonstrating remarkable wound-healing capabilities.
To evaluate the biosafety of antimicrobial peptides in vivo, we recorded murine body weights during the treatment, and performed H&E staining of major organs (heart, liver, spleen, lung, and kidney), and assessed hemocompatibility. Local and systemic administration of antimicrobial peptides did not result in weight loss (Fig. S16, ESI†), and H&E staining of major organ sections post-treatment revealed no damage (Fig. 6e). Additionally, all the peptides caused no hemolysis of murine red blood cells (Fig. S17, ESI†), indicating that all the peptides exhibited excellent biosafety in vivo.
Conclusion
Based on the structural optimization of the linear lipopeptide paenipeptin C′, we designed and developed two high-performance antimicrobial peptides: peptide 1 (lipid C6-Phe–Phe–Pae, containing diphenylalanine and a C6 lipid chain) and peptide 2 (lipid C18-Pae, featuring a C18 lipid chain). To further enhance their antibacterial activity, we conjugated these peptides with CPFx via ester bonds, yielding CPFx-1 and CPFx-2. CPFx-2 exhibited the most favorable self-assembly properties, demonstrating the lowest critical micelle concentration (CMC) and forming the largest nanoparticles, as confirmed by transmission electron microscopy (TEM) and dynamic light scattering (DLS). In vitro antibacterial assays revealed that both CPFx-1 and CPFx-2 displayed strong inhibitory effects against E. coli and S. aureus. Notably, CPFx-2, with its longer lipid chain, exhibited superior antibacterial activity. In a murine wound infection model, CPFx-2 not only significantly reduced local inflammation but also accelerated wound healing, confirming that CPFx conjugation and extended lipid chains synergistically enhance antimicrobial efficacy. Mechanistic studies demonstrated that the antimicrobial peptides first target negatively charged bacterial membranes via electrostatic interactions, followed by aggregation and membrane insertion, which disrupt membrane integrity, increases permeability, and ultimately leads to membrane rupture and cytoplasmic leakage. CPFx-2 exhibited the strongest bactericidal effect, attributed to its longer alkyl chain, which facilitates deeper membrane interaction and structural disruption. Additionally, intracellular esterase-mediated hydrolysis may release CPFx, enabling its DNA gyrase inhibition mechanism, thereby establishing a dual-action antibacterial strategy.In summary, we designed and synthesized CPFx-conjugated antimicrobial peptides to synergistically enhance bactericidal activity against E. coli and S. aureus. We performed comprehensive in vitro characterization, antibacterial activity assays, and mechanistic investigations, along with validation of therapeutic efficacy in a murine E. coli-infected wound model. Our study presents a CPFx-antimicrobial peptide conjugate drug design with clear synergistic mechanisms. Although we have verified the excellent stability of CPFx-conjugated antimicrobial peptides under simulated physiological conditions, the ester bonds in CPFx conjugates may be hydrolyzed prematurely due to multiple factors including pH, esterase activity, and reactive oxygen species, which could affect the efficacy of the drug. In the future, we will conduct more comprehensive pharmacokinetic and stability tests to verify the translational potential of CPFx-antimicrobial peptide conjugate drugs.
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Central China Normal University and approved by the Animal Ethics Committee of Central China Normal University Administrative Panel on Laboratory Animal Care (permit number CCNU-IACUC-2023-001).Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Xingrao Peng: data curation, formal analysis, methodology and writing – original draft; Yong Luo: data curation, formal analysis, visualization and methodology; Tianzhi Xu: formal analysis; Zihan Chen: formal analysis; Peiyao Chen: formal analysis; Cong Hu: funding acquisition; Shuang Liu: conceptualization, resources, supervision, funding acquisition and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Hubei Provincial Natural Science Foundation of China (Grants 2023AFB659), Open Project Program of State Key Laboratory of Chemo/Biosensing and Chemometrics, Hunan University ((2023)0758), Open Project Program of Guangxi Key Laboratory of Automatic Detecting Technology and Instruments, Guilin University of Electronic Technology (YQ23205).References
- R. A. Fisher, B. Gollan and S. Helaine, Nat. Rev. Microbiol., 2017, 15, 453–464 CrossRef CAS PubMed.
- R. E. Hancock, A. Nijnik and D. J. Philpott, Nat. Rev. Microbiol., 2012, 10, 243–254 CrossRef CAS PubMed.
- W. Zhan, G. Gao, Z. Liu, X. Liu, L. Xu, M. Wang, H. D. Xu, R. Tang, J. Cao, X. Sun and G. Liang, Adv. Healthcare Mater., 2023, 12, e2203283 CrossRef PubMed.
- W. Zhan, L. Xu, Z. Liu, X. Liu, G. Gao, T. Xia, X. Cheng, X. Sun, F. G. Wu, Q. Yu and G. Liang, Angew. Chem., Int. Ed., 2023, 62, e202306427 CrossRef CAS PubMed.
- Y. Zhang, S. Ai, Z. Yu, L. Wang, H. Tao, B. Wang, D. Kong, Z. Yang and Y. Wang, Adv. Funct. Mater., 2024, 34, 2314607 CrossRef CAS.
- J. Li, R. Guan, A. Wuethrich, M. Yan, J. Cheng, G. Liu, J. Zhan, M. Trau and Y. Sun, Anal. Chem., 2024, 96, 4495–4504 CrossRef CAS PubMed.
- Q. Li, H. Ye, F. Zhao, Y. Li, Z. Zhang, Q. Yan and Y. Sun, Dalton Trans., 2024, 53, 3434–3444 RSC.
- B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC.
- S. Mölstad, S. Löfmark, K. Carlin, M. Erntell, O. Aspevall, L. Blad, H. Hanberger, K. Hedin, J. Hellman, C. Norman, G. Skoog, C. S. Lundborg, K. Tegmark Wisell, C. Åhrén and O. Cars, Bull. W. H. O., 2017, 95, 764–773 CrossRef PubMed.
- M. Talaat, B. Zayed, S. Tolba, E. Abdou, M. Gomaa, D. Itani, Y. Hutin and R. Hajjeh, Emerging Infect. Dis., 2022, 28, 717–724 CrossRef PubMed.
- Y. Xu, C. Li, J. An, X. Ma, J. Yang, L. Luo, Y. Deng, J. S. Kim and Y. Sun, Sci. China: Chem., 2023, 66, 155–163 CrossRef CAS.
- L. E. Manhart, W. M. Geisler, C. S. Bradshaw, J. S. Jensen and D. H. Martin, Emerging Infect. Dis., 2022, 28, 1–11 Search PubMed.
- C. Årdal, M. Gawad, E. Baraldi, M. Jahre and C. Edlund, Bull. W. H. O., 2025, 103, 51–56 CrossRef PubMed.
- Y. Xu, C. Li, X. Ma, W. Tuo, L. Tu, X. Li, Y. sun, P. J. Stang and Y. Sun, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2209904119 CrossRef CAS PubMed.
- M. E. Schafer, H. Browne, J. B. Goldberg and D. E. Greenberg, Acc. Chem. Res., 2021, 54, 2377–2385 CrossRef CAS PubMed.
- A. A. Bahar and D. Ren, Pharmaceuticals, 2013, 6, 1543–1575 CrossRef PubMed.
- M. Mahlapuu, J. Håkansson, L. Ringstad and C. Björn, Front. Cell. Infect. Microbiol., 2016, 6, 194 Search PubMed.
- J. Li, J. J. Koh, S. Liu, R. Lakshminarayanan, C. S. Verma and R. W. Beuerman, Front. Neurosci., 2017, 11, 73 Search PubMed.
- S. A. Sousa, J. R. Feliciano, T. Pita, C. F. Soeiro, B. L. Mendes, L. G. Alves and J. H. Leitão, Antibiotics, 2021, 10, 942 CrossRef CAS PubMed.
- S. H. Moon, X. Zhang, G. Zheng, D. G. Meeker, M. S. Smeltzer and E. Huang, J. Med. Chem., 2017, 60, 9630–9640 CrossRef CAS PubMed.
- E. Huang, X. Yang, L. Zhang, S. H. Moon and A. E. Yousef, FEMS Microbiol. Lett., 2017, 364, fnx049 Search PubMed.
- J. Talapko and I. Škrlec, Pharmaceuticals, 2020, 13, 229 CrossRef PubMed.
- S. H. Moon and E. Huang, BMC Microbiol., 2019, 19, 6 CrossRef PubMed.
- M. S. Zharkova, D. S. Orlov, O. Y. Golubeva, O. B. Chakchir, I. E. Eliseev, T. M. Grinchuk and O. V. Shamova, Front. Cell. Infect. Microbiol., 2019, 9, 128 CrossRef CAS PubMed.
- C. Santos, G. R. Rodrigues, L. F. Lima, M. C. G. Reis, N. B. Cunha, S. C. Dias and O. L. Franco, Front. Bioeng. Biotechnol., 2022, 10, 1051456 CrossRef PubMed.
- X. Wu, Z. Li, X. Li, Y. Tian, Y. Fan, C. Yu, B. Zhou, Y. Liu, R. Xiang and L. Yang, Drug Des., Dev. Ther., 2017, 11, 939–946 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- A. Scheeder, M. Brockhoff, E. N. Ward, G. S Kaminski Schierle, L. Mela and G. F. Kaminski, J. Am. Chem. Soc., 2023, 145, 28240–28250 CrossRef CAS PubMed.
- D. M. Correia, L. C. Fernandes, M. M. Fernandes, B. Hermenegildo, R. M. Meira, C. Ribeiro, S. Ribeiro, J. Reguera and S. L. Méndez, Adv. Funct. Mater., 2021, 31, 2104148 CrossRef.
- H. Zhou, M. D. Weir, J. M. Antonucci, G. E. Schumacher, X. Zhou and H. H. Xu, Int. J. Oral. Sci., 2014, 6, 77–86 CrossRef CAS PubMed.
- J. T. Mhlongo, A. Y. Waddad, F. Albericio and B. G. de la Torre, Adv. Sci., 2023, 10, e2300472 CrossRef PubMed.
- S. Chernysh, N. Gordya, D. Tulin and A. Yakovlev, Infect. Drug Resist., 2018, 11, 501–514 CrossRef CAS PubMed.
- A. León-Buitimea, C. R. Garza-Cárdenas, M. F. Román-García, C. A. Ramírez-Díaz, M. Ulloa-Ramírez and J. R. Morones-Ramírez, Antibiotics, 2022, 11, 794 CrossRef PubMed.
- M. Unemo and K. Workowski, Lancet Infect. Dis., 2018, 18, 486–488 CrossRef PubMed.
- K. Yamamoto, T. Sato, A. Hao, K. Asao, R. Kaguchi, S. Kusaka, R. R. Ruddarraju, D. Kazamori, K. Seo, S. Takahashi, M. Horiuchi, S. Yokota, S. Lee and S. Ichikawa, Nat. Commun., 2024, 15, 5085 CrossRef CAS PubMed.
- S. Batson, C. Chiara, V. Majce, A. J. Lloyd, S. Gobec, D. Rea, V. Fülöp, C. W. Thoroughgood, K. J. Simmons, C. G. Dowson, C. W. G. Fishwick, L. P. S. Carvalho and D. I. Roper, Nat. Commun., 2017, 8, 1939 CrossRef PubMed.
- A. L. Buitimea, C. R. G. Cárdenas, M. F. R. García, C. A. R. Díaz, M. U. Ramírez and J. R. M. Ramírez, Antibiotics, 2022, 11, 794 CrossRef PubMed.
- G. Spoletini, M. Kennedy, L. Flint, T. Graham, C. Etherington, N. Shaw, P. Whitaker, M. Denton, I. Clifton and D. Peckham, Pulm. Pharmacol. Ther., 2018, 50, 82–87 CrossRef CAS PubMed.
- V. I. Band, D. A. Hufnagel, S. Jaggavarapu, E. X. Sherman, J. E. Wozniak, S. W. Satola, M. M. Farley, J. T. Jacob, E. M. Burd and W. S. Weiss, Nat. Microbiol., 2019, 4, 1627–1635 CrossRef CAS PubMed.
- S. Oh, H. Hur, Y. Kim, S. Shin, H. Woo, J. Choi and H. H. Lee, Nanomaterials, 2021, 11, 2887 CrossRef CAS PubMed.
- Q. Zhang, W. Tan, Z. Liu, Y. Zhang, W. S. Wei, S. Fraden and B. Xu, J. Am. Chem. Soc., 2024, 146, 12901–12906 CrossRef CAS PubMed.
- G. Hu, D. Wang, L. Sun, R. Su, M. Corazzin, X. Sun, L. Dou, M. Zhang, L. Zhao, L. Su and Y. Jin, Foods, 2022, 11, 2655 CrossRef CAS PubMed.
- T. Wang, X. Li, L. Chen, L. Li and S. Janaswamy, Front. Chem., 2020, 8, 662 CrossRef CAS PubMed.
- P. J. Petersen, N. V. Jacobus, W. J. Weiss, P. E. Sum and R. T. Testa, Antimicrob. Agents Chemother., 1999, 43, 738–744 CrossRef CAS PubMed.
- N. A. S. Neto, J. T. A. Oliveira, T. K. B. Aguiar, L. P. Bezerra, L. A. C. Branco, F. P. Mesquita, C. D. T. Freitas and P. F. N. Souza, Pathogens, 2022, 11, 995 CrossRef CAS PubMed.
- J. Puño-Sarmiento, E. M. Anderson, A. J. Park, C. M. Khursigara and D. E. Barnett Foster, Sci. Rep., 2020, 10, 10029 CrossRef PubMed.
- E. Andersson, V. Rydengård, A. Sonesson, M. Mörgelin, L. Björck and A. Schmidtchen, Eur. J. Biochem., 2004, 271, 1219–1226 CrossRef CAS PubMed.
- S. Liu, Q. Zhang, X. Peng, C. Hu, S. Wang and Y. Sun, Biomater. Sci., 2024, 12, 1274–1280 RSC.
- S. Liu, Q. Zhang, A. N. Shy, M. Yi, H. He, S. Lu and B. Xu, J. Am. Chem. Soc., 2021, 143, 15852–15862 CrossRef CAS PubMed.
- S. Liu, Q. Zhang, H. He, M. Yi, W. Tan, J. Guo and B. Xu, Angew. Chem., Int. Ed., 2022, 61, e202210568 CrossRef CAS PubMed.
- A. Sowa-Jasiłek, A. Zdybicka-Barabas, S. Stączek, B. Pawlikowska-Pawlęga, K. Grygorczuk-Płaneta, K. Skrzypiec, W. I. Gruszecki, P. Mak and M. Cytryńska, Int. J. Mol. Sci., 2020, 21, 1912 CrossRef PubMed.
- J. H. Kim, J. Lee, J. Park and Y. S. Gho, Semin. Cell Dev. Biol., 2015, 40, 97–104 CrossRef CAS PubMed.
- M. Gross, S. E. Cramton, F. Götz and A. Peschel, Infect. Immun., 2001, 69, 3423–3426 CrossRef CAS PubMed.
- R. Bucuresteanu, M. Ionita, V. Chihaia, A. Ficai, R. D. Trusca, C. I. Ilie, A. Kuncser, A. M. Holban, G. Mihaescu, G. Petcu, A. Nicolaev, R. M. Costescu, M. Husch, V. Parvulescu and L. M. Ditu, Int. J. Mol. Sci., 2022, 23, 6888 CrossRef CAS PubMed.
- I. M. Helander and T. Mattila-Sandholm, J. Appl. Microbiol., 2000, 88, 213–219 CrossRef CAS PubMed.
- M. Lelis, S. Tuckute, M. Urbonavicius, S. Varnagiris, S. Sakalauskaite and R. Daugelavicius, Inorganics, 2023, 11, 59 CrossRef CAS.
- L. Li, Y. Zhou, P. Li, Q. Xu, K. Li, H. Hu, W. Bing and Z. Zhang, Front. Bioeng. Biotechnol., 2022, 10, 1066306 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02932e |
| This journal is © The Royal Society of Chemistry 2025 |