
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of gel ageing and electrode corrosion on the performance of direct laser writing carbonization-enabled hydrogel-based moist-electric generators†
Xuewei Pia,
Yanbo Yao*b,
Dini Qina and
Tao Liu *a
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Soochow 215123, P. R. China. E-mail: tliu@suda.edu.cn
bSchool of Materials and Packaging Engineering, Fujian Polytechnic Normal University, Fuqing 350300, P. R. China. E-mail: yaoyb@fpnu.edu.cn
First published on 3rd June 2025
Abstract
The combined use of hydrogel active materials and porous and asymmetric electrodes enables the development of high-performance and high-power output moist-electric generators (MEGs). Herein, we report a direct laser writing carbonization (DLWc)-enabled approach for cost-effectively manufacturing the porous carbon electrode that can be easily integrated and assembled with the hydrogel active material for the scalable fabrication of MEGs. With the hydrogel active material composed of poly(vinyl alcohol) (PVA), poly(acrylic acid) (PAA), phytic acid (PA), glycerol and water, the best performing DLWc-enabled PVA/PAA/PA hydrogel-based MEG exhibited an open-circuit voltage of ∼0.8 V and a short-circuit current density of ∼110 μA cm−2 and delivered stable operation for more than 300 h in an ambient environment. In-depth studies further revealed the effect of time-ageing treatment of the hydrogel active material on its ionic conductivity and water uptake ability and the power performance of the finally assembled MEGs. Lastly, the combined investigations using microscopy, spectroscopy and electrochemical corrosion tests allowed us to unambiguously identify the critical roles of the corrosion reaction occurring at the metallic electrodes in enhancing the voltage and current performance of the hydrogel-based MEG.
Introduction
To address the challenge of climate change while fostering sustainable development, there is an increasing demand for sustainable energy sources and developing green energy technologies that are able to convert environmental energy into electricity.1–4 Among the different approaches, the emerging technology of moist-electric generators(MEGs),5–8 which relies on the ubiquitous water vapor to generate electricity, has received great attention owing to their simplicity in device structure, ease of manufacturing, and excellent performance in applications such as self-powered electronic skin,9 touchless interactive panel,10 wireless wearable electronics,11,12 multifunctional flexible sensing systems,13,14 and information encryption and display.15Similar to a battery or a supercapacitor, the basic structure of a MEG7,8 is composed of an active material and a pair of electrodes. The active material is able to absorb moisture, create ionic species and generate potential difference and electric current, and the electrodes are responsible for connecting with the external devices to deliver the required electric power. The general operation principle of a MEG relies on the concentration gradient of ionic species within the active material, which causes the directional migration of the charge carriers to give the desired voltage and current output.8
Notably, different active materials or their combinations have been pursued for creating the concentration gradient of hydrolysable/ionizable functional groups and thereby the corresponding ionic species. Among the different types of active materials that are used for developing MEGs, such as carbon-based materials,16–20 biomaterials,21–23 and metal oxides,11,24 ionically conductive hydrogels13,14,25–31 have recently gained much popularity owing to their strong hygroscopic capacity, tailorable compositions, ease of preparation, and excellent formability. Besides the active materials, asymmetric electrode structures can be used to introduce non-uniform moisture distribution in MEGs to create the desired concentration gradient for the ionic species.32,33 The typical construction of the asymmetric electrodes in a MEG comprises a porous electrode and a normal metallic electrode, which has been demonstrated to be beneficial for increasing the concentration and the concentration gradient for the charge carriers and thereby significantly improving the MEG performance.34–36 On the one hand, the use of the porous electrode enables easy access of the active material to the external moisture for increasing the concentration of charge carriers. On the other hand, the electric double-layer formed at the interface between the porous electrode and the active material creates additional voltage contribution to further enhance the MEG performance.2,7,15,36
Research has previously indicated that the combined use of the hydrogel active material and the porous and asymmetric electrode allows for the development of high-performance and high-power output MEGs. Given the advancement, there is still a need for robustly and cost-effectively manufacturing the porous electrode that can be easily integrated and assembled with the hydrogel active material for the scalable fabrication of MEGs. To this end, in the present study, we report a direct laser writing carbonization (DLWc)-enabled approach for fabricating high-performance hydrogel-based MEGs. In contrast to the other porous electrode manufacturing processes, the DLWc of the polyimide (PI) film enables the formation of porous carbon structures in situ supported by the compliant and foldable PI film at room temperature and ambient environment.37,38 This endows mechanical stability and integrity to the DLWc-processed carbon electrode and makes its integration/assembly much easier. In addition, the equipment and base materials used in a DLWc process are all readily available and can be purchased from different suppliers, which places the DLWc-enabled technology in a much better position for large-scale production and commercialization. Although the DLWc has been successfully applied for fabricating various functional devices including supercapacitors,40 piezoresistive sensors,41,42 solar-driven water evaporators,39 and humidity sensors,43 its advantage in the rapid preparation of porous electrodes for fabricating moisture-powered MEGs has not yet been demonstrated. As schematically shown in Fig. 1, our DLWc-enabled MEG (coined as DLWc-Hgel-MEG) has the typical 3-layer construction: The bottom layer is a normal metallic electrode; the top layer is the DLWc-processed porous carbon electrode; and in between the top and the bottom layer as well as in the pores of the porous electrode is the hydrogel active material. The hydrogel active material in our DLWc-Hgel-MEG was prepared by referring to the previous studies as disclosed by Byun et al.,44 Yu et al.45 and Akbar et al.46 The skeleton of the gel is an interpenetrating polymer network (IPN) composed of poly(vinyl alcohol) (PVA) and poly(acrylic acid) (PAA), which is mixed with a ternary solvent system composed of phytic acid (PA), glycerol and water. In this formulation, the role of PA is for increasing the ionic conductivity of the active material and, therefore, the power performance of the MEG. As usual, the presence of glycerol improves the anti-freezing capability of the active material to make the MEG operable at low temperatures. With the assistance of DLWc for rapidly fabricating the porous electrode, the best-performing DLWc-enabled PVA/PAA/PA hydrogel-based MEG showed an open-circuit voltage of ∼0.8 V and a short-circuit current density of ∼110 μA cm−2, and exhibited stable operation for more than 300 h in an ambient environment. Through a serial or parallel connection of multiple units, the practical use of DLWc-Hgel-MEGs for lighting LEDs and driving hydrometers is demonstrated.
 | ||
| Fig. 1 Schematic of the fabrication process of the DLWc-processed porous electrode (top) and the PVA/PAA/PA hydrogel active material (bottom), and their assembly for making the DLWc-enabled hydrogel-based MEG device-DLWc-Hgel-MEG. | ||
Beyond the fabrication and performance demonstration of DLWc-Hgel-MEGs, in the present study, we focus on two outstanding issues on the hydrogel-based MEG: the ageing effect of the hydrogel active material and the role of the normal metallic electrode. Research47–50 has previously provided evidence that ion aggregation, macro/microphase separation and polymer crystallization can change the mechanical and electrical properties of polymer electrolytes upon time-ageing and/or thermal cycling treatment. Given the complicated and the mixed compositions of the hydrogel active materials, there may exist similar ageing issues in the hydrogel-based MEG, which could affect the moisture absorption and desorption behaviours and the charge transport capability of the active material and ultimately influence the power performance of the MEG. To date, however, this ageing issue has not received sufficient attention from the MEG research community. To address this issue, in the present study, we investigated the effect of time-ageing of the solution and the gel of the PVA/PAA/PA active material on its ionic conductivity and water uptake ability as well as the power performance of the finally assembled MEG. It was found that time-ageing indeed plays an important role in dictating the current performance of the hydrogel-based MEG. As mentioned previously, the key for the operation of a MEG lies in creating the concentration gradient for the ionic species within the active materials. Nevertheless, a recent study51 has suggested that the corrosion reaction at the metallic electrode also plays a critical role in improving the performance of a MEG. By varying the type of metallic electrode of the DLWc-Hgel-MEG selected from copper, aluminium, silver, platinum, gold and carbon, we provided strong evidence to corroborate this conclusion. Namely, the corrosion reaction at the metallic electrode is critical in dictating the current performance of the hydrogel-based MEG. This work not only provides a simple method for the robust and cost-effective manufacturing of hydrogel-based MEGs, but also points the possible strategies for their performance optimization and reliable long-term operation capability.
Results and discussion
Structural characterization of the DLWc-processed porous carbon electrode and PVA/PAA/PA hydrogel
Previous studies,37–43 both experimental and theoretical modelling, have comprehensively investigated the effect of laser processing conditions such as extent of focus of the laser beam, laser power, laser irradiation time and laser scanning speed on the structures and properties of DLWc-processed carbons. In this work, we simply relied on Raman spectroscopy and SEM to reveal the chemical and microstructures of the DLWc-processed electrode, which was used for fabricating the MEG and processed under the specified conditions (see the Experimental section). Fig. 2a shows the Raman spectrum of the DLWc-processed porous carbon electrode, which, similar to the previous work, displays the characteristic peaks of amorphous carbon at 1350 cm−1 and 1587 cm−1, respectively, corresponding to the D-band assigned to the disordered/defective carbon structure and the G-band ascribed to the stretching vibration of the sp2 carbon network in graphitic materials. The inset of Fig. 2a is an optical picture of the DLWc-processed porous carbon electrode (9 × 9 square lattice). The SEM images shown in Fig. 2b and c, respectively, reveal the morphology of the DLWc-processed porous carbon electrode before and after the filling of the PVA/PAA/PA hydrogel active material (10 min of heating time). Similar to our previous studies,37 the as-prepared DLWc-processed carbon at each laser-irradiated lattice point (Fig. 2b) forms a crater-like structure, which exhibits a rich multi-scale porous structure that can be filled by the PVA/PAA/PA hydrogel (Fig. 2c) through capillary-driven imbibition of the PVA/AA/PA solution and the subsequent heating-induced polymerization, as described in the experimental section. Due to the large surface-to-volume ratio of the crater-like porous carbon structure created by DLWc, we expect that, by using the DLWc-processed porous electrode, water molecules in the ambient environment can easily get access to the hydrophilic sites of the PVA/PAA/PA hydrogel active material, which can be beneficial for enhancing the current performance of the finally assembled MEG. | ||
| Fig. 2 (a) Raman spectrum of the DLWc-processed porous carbon electrode. The inset shows the optical picture of the electrode, where the scale of the ruler is 1 mm. (b and c) SEM images of the DLWc-processed porous carbon electrodes before and after filling of the PVA/PAA/PA hydrogel active material. (d and e) Raman spectra for tracking the polymerization of AA in the solution of PVA/AA/PA initiated by UV exposure (solution aged for 0.5 day) and direct heating for different time durations. (f) Effect of time duration of UV exposure and direct heating time on the extent of polymerization of AA. | ||
The polymerization of AA in the solution of PVA/AA/PA initiated by UV exposure or direct heating was examined by Raman spectroscopy. Fig. 2d and e, respectively, show the Raman spectra of the PVA/AA/PA solution exposed to UV light (solution aged for 0.5 day; Fig. S1† shows the results for the sample aged for 2.5 days) and subjected to direct heating for different time durations. In both cases, we can see that the intensity of the peak at 1640 cm−1, which is assigned to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of the AA monomer, gradually decreases with the increasing UV exposure or heating time, an indication of the successful polymerization of AA to form a PAA polymer.52 With the Raman peak at 3440 cm−1 (–OH vibration) as an internal standard, we quantify the extent of polymerization of AA in the solution of PVA/AA/PA with the quantity 1 − [A1640/A3440]t/[A1640/A3440]t=0 (Fig. 2f), where t is the time duration of UV exposure or direct heating. As can be seen, upon heating for more than 10 min or subjecting to UV exposure for 30 s, the conversion of AA levels off, an indication of the complete polymerization of AA. Moreover, as far as the UV exposure is sufficiently long (>30 s), the time-ageing of the PVA/AA/PA solution (0.5 day vs. 2.5 days) has a negligible influence on the extent of polymerization of AA.
C of the AA monomer, gradually decreases with the increasing UV exposure or heating time, an indication of the successful polymerization of AA to form a PAA polymer.52 With the Raman peak at 3440 cm−1 (–OH vibration) as an internal standard, we quantify the extent of polymerization of AA in the solution of PVA/AA/PA with the quantity 1 − [A1640/A3440]t/[A1640/A3440]t=0 (Fig. 2f), where t is the time duration of UV exposure or direct heating. As can be seen, upon heating for more than 10 min or subjecting to UV exposure for 30 s, the conversion of AA levels off, an indication of the complete polymerization of AA. Moreover, as far as the UV exposure is sufficiently long (>30 s), the time-ageing of the PVA/AA/PA solution (0.5 day vs. 2.5 days) has a negligible influence on the extent of polymerization of AA.
Effect of time ageing on the water uptake and ionic conductance of the PVA/PAA/PA hydrogel
The gel samples obtained by UV exposure of the PVA/AA/PA solution (aged for 0.5 day, 2.5 days and 120 days) were immediately immersed in DI water for 24 h for recording their swelling behaviour to examine the effect of solution ageing on the water uptake capability of the PVA/PAA/PA hydrogel active material. Besides the solution ageing effect, we performed the same swelling experiments to investigate the gel ageing effect. In such a test, the gel sample prepared by UV exposure was aged at room temperature for 24 h before its immersion in DI water. With the condition of UV-exposure for 70 s as representative examples, Fig. 3a and b show the optical pictures, before and after water immersion treatment, of the gel samples, respectively, prepared from the PVA/AA/PA solution aged for 0.5 day and 2.5 days without gel ageing treatment. Fig. 3c and d show similar results for the gel samples subjected to both solution and gel ageing treatment. As can be seen, under the same UV exposure conditions, the samples without gel ageing (Fig. 3a and b) show a significantly higher swelling ratio than the samples subjected to 24 h of gel ageing (Fig. 3c and d). Moreover, the swelling ratio for the sample prepared from the solution aged for 0.5 day (Fig. 3a) is much higher than that aged for 2.5 days (Fig. 3b). A similar ageing effect was also observed for the PVA/PAA/PA hydrogel samples prepared by UV exposure at different time durations, as summarized in Fig. S2a–e.† It is noted that the gel samples prepared by UV exposure for less than 30 s from the PVA/AA/PA solution aged for 0.5 day and without gel aging treatment completely dissolved in water after the immersion treatment (Fig. S2a†). The results shown in Fig. 3a–d and S2† apparently indicate that the solution ageing and gel ageing treatment have a significant effect on the swelling behavior, and therefore, the water uptake capability for the PVA/PAA/PA hydrogel active material. To further explore this point, we summarize in Fig. 3e the water uptake capability ([mass after immersion − mass before immersion]/mass before immersion) for all the gel samples subjected to the swelling tests. Similar to the qualitative observations from the optical pictures shown in Fig. 3a–d and S2,† we can conclude from Fig. 3e that the gel aging treatment as well as longer solution ageing time can significantly reduce the water uptake capability of the PVA/PAA/PA hydrogel. In addition, as far as the UV-exposure time is long enough (>30 s), its influence on the water uptake capability of the PVA/PAA/PA hydrogel is insignificant. It is presumed that the build-up of the hydrogen bonding network and the crystallization of PVA with time facilitate the increase in the physical crosslinks in the PVA/PAA/PA hydrogel, which is responsible for the experimentally observed time-ageing effect, as demonstrated in Fig. 3 and S2.† To confirm the crystallization of PVA in the PVA/PAA/PA hydrogel, we performed X-ray diffraction measurements on the samples prepared from the solution aged for 0.5 and 2.5 days upon 30 s UV irradiation with and without gel ageing treatment. As indicated by the results shown in Fig. S3,† all samples clearly show the relatively broad diffraction peaks at 2θ ≈ 12°, 22° and 42°, which indicates the presence of small-sized PVA crystallites, in agreement with the previous study.53 | ||
| Fig. 3 (a–d) Optical images showing the effect of solution and gel ageing on water immersion (24 h)-induced swelling of the PVA/PAA/PA hydrogel prepared via UV exposure for 70 s (left: before immersion; right: immersion for 24 h; the scale of the ruler is 1 mm). (a) Solution ageing for 0.5 day without gel ageing. (b) Solution ageing for 2.5 days without gel ageing. (c) Solution ageing for 0.5 day and gel ageing for 24 h. (d) Solution ageing for 2.5 days and gel ageing for 24 h. (e) Summary of the effect of solution and gel ageing on the water uptake capability of the PVA/PAA/PA hydrogel prepared via UV exposure for different time durations. | ||
In addition to the swelling and the water uptake capability, the ionic conducting performance of the PVA/PAA/PA hydrogel is also subjected to the influence of time-ageing, as evidenced by the impedance spectra of the gel samples prepared at varied UV-exposure durations and time-ageing conditions (Fig. S4–S6†). As a representative example, Fig. 4a shows the complex plane impedance spectra of the PVA/PAA/PA hydrogel sample prepared from the PVA/AA/PA solution aged for 0.5 day without gel ageing treatment. The inset in Fig. 4a is the equivalent circuit designed for reproducing the experimentally measured impedance spectra for PVA/PAA/PA hydrogels. In the circuit, the parallel combination of the constant-phase element (CPE) and the corresponding resistor R reflects the distributed capacitive and resistive impedance contribution due to the association/dissociation of the acidic groups in the hydrogel. The presence of two different CPE/R elements (CPE1/R1 and CPE2/R2) in the circuit is for the consideration of the different association/dissociation constants for PA (phytic acid) and PAA (polyacrylic acid) in generating H+. The serial resistor R3 in the circuit takes account of the resistance contribution by Ni electrodes and the connecting wires. Using the commercial software Zview® and the designed equivalent circuit, we can fit the experimentally measured impedance spectra for all the PVA/PAA/PA hydrogel samples prepared at different conditions (Fig. 4a and S4–S6†). The fitting parameters are summarized in Table S1.† With R1 + R2 as an overall measure of the H+ transport capability in the hydrogel samples, in Fig. 4b, we compare the results of R1 + R2 for the PVA/PAA/PA hydrogel samples prepared from the PVA/AA/PA solution aged for 0.5 day, 2.5 days and 120 days without gel ageing treatment. As can be seen from Fig. 4b, regardless of the solution ageing time, the value of R1 + R2 proportionally increases with the increase in the UV-exposure time. This behaviour reflects that, with the increase in the UV-exposure time, the extent of AA polymerization correspondingly increases to result in the viscosity increase of the gel system, and therefore, it leads to a higher value of R1 + R2. Moreover, at the same UV-exposure duration, the value of R1 + R2 for the gel sample prepared from the solution aged for a longer time is consistently higher. This observation agrees with the ageing effect on the swelling and water uptake capability of the PVA/PAA/PA hydrogel. That is, the ageing results in an increase in the physical crosslinks, which create additional barriers to retard the H+ transport capability. With the additional gel aging treatment, the physical crosslinks can be further increased owing to the build-up of the hydrogen bonding network and the crystallization of PVA (Fig. S3†), which lead to an even higher value of R1 + R2, as shown in Fig. 4c.
 | ||
| Fig. 4 (a) Complex plane impedance diagram of the PVA/PAA/PA hydrogel prepared from the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for different time durations (no gel ageing treatment). The scattered data points are the experimental results, and the continuous lines are the corresponding equivalent circuit (inset) fitting results; the effect of (b) UV-exposure time and solution ageing time and (c) gel aging time on the ionic transport capability of the PVA/PAA/PA hydrogels, as measured by R1 + R2. | ||
As shown above, the water uptake and ionic conductance of the PVA/PAA/PA hydrogel strongly depend on the gel processing conditions; in particular, the solution ageing time and the gel ageing time. As a result, the ageing treatment also plays an important role in dictating the performance of the PVA/PAA/PAA hydrogel-based MEG, as discussed next.
Effect of PVA/PAA/PA hydrogel aging on the electricity generation performance of the DLWc-Hgel-MEG
By using the PVA/PAA/PA hydrogels prepared by UV-exposure and aged under different conditions as the active material, the DLWc processed porous carbon filled with the thermally initiated PVA/PAA/PA hydrogel as the top electrode, and the commonly adopted copper as the bottom metallic electrode, we assembled the DLWc-Hgel-MEG to evaluate its open-circuit voltage-Uoc and short-circuit current-Isc at room temperature. As a representative example, Fig. 5a shows the change in Isc and Uoc with respect to the environmental humidity (step-wise controlled from 20% RH to 85% RH) for the MEG assembled with the PVA/PAA/PA hydrogel prepared from the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment. As can be seen, while the humidity is step-wise changed from 20% RH to 85% RH, the Isc of the MEG accordingly manifests a step-wise increasing behaviour. In contrast to Isc, the Uoc of the device shows a continuous decreasing trend with the increase in humidity. At 85% RH, the stabilized current output Isc of the MEG is ∼42 μA. Although the magnitude of Isc and Uoc may vary from one MEG to another, their general behaviours with the change in humidity are all similar to those given in Fig. 5a. In Fig. 5b, we compare the results of Isc and Uoc for the MEG assembled from the PVA/PAA/PA hydrogels that were prepared from the PVA/AA/PA solution aged for 0.5 day and 2.5 days and subjected to UV-exposure for different time durations without gel ageing treatment. The values of Isc and Uoc were taken as the device was stabilized at 85% RH for 11 h. As can be seen from Fig. 5b, the open-circuit voltage Uoc shows no dependence on both the UV-exposure time and the solution ageing time. Nevertheless, the short-circuit current Isc apparently decreases with the increase in the UV-exposure time. This is particularly true for the MEG prepared from the PVA/PAA/PA hydrogel with a shorter solution ageing time. Moreover, as compared to the MEG prepared from the gel with a longer solution ageing time (2.5 days and 120 days), the shorter solution ageing time (0.5 day) gives a slightly better current performance. By maintaining the UV-exposure time at 10 s, we further investigated the effect of gel ageing treatment on the current performance of the MEG, as shown in Fig. 5c. Evidently, when the environmental humidity is high (>60% RH), the gel ageing treatment for 24 h can significantly decrease the current performance of the DLWc-Hgel-MEG. In contrast to the current performance, the solution and the gel ageing treatment do not show significant influence on the voltage output of the DLWc-Hgel-MEG, as shown in Fig. 5d. On the basis of the earlier results of the effect of UV-exposure time, solution ageing time and gel ageing time on the water uptake and ionic conductance of the PVA/PAA/PA hydrogel, we presume that the deterioration of the current performance of the DLWc-Hgel-MEG is attributed to the retardation of the H+ transport capability caused by the increase in the viscosity and the physical crosslinks of the active gel material upon prolonged UV exposure, solution ageing and gel ageing treatment. | ||
| Fig. 5 (a) Output of the short-circuit current Isc and the open-circuit voltage Uoc for a representative DLWc-Hgel-MEG device tested at room temperature in a step-wise controlled humidity environment from 20% RH to 85% RH. The device is assembled with the PVA/PAA/PA hydrogel prepared from the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment. (b) Effect of solution ageing time and UV-exposure time on the output of Isc and Uoc for the DLWc-Hgel-MEG devices assembled from the PVA/PAA/PA hydrogels without gel ageing treatment tested at 85 RH%. Effect of (c) solution ageing (0.5 day vs. 2.5 days vs. 120 days) and (d) gel ageing treatment (0 h vs. 24 h) on the output of Isc and Uoc for the DLWc-Hgel-MEG devices assembled from the PVA/PAA/PA hydrogels tested at different humidity levels. | ||
Role of metallic electrode on the electricity generation mechanism of the DLWc-Hgel-MEG
According to the recent study,51 besides the concentration gradient of the ionic species within the active materials, the corrosion reaction at the metallic electrode plays a critical role in dictating the electricity generation performance for a MEG. To examine this point, we assembled multiple DLWc-Hgel-MEGs by using different types of metals (Cu, Al, Ag, Pt and Au) as the bottom electrode while keeping the same formation/preparation for the top porous carbon electrode and the PVA/PAA/PA hydrogel active material (prepared with the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment). For comparison, we also assembled a MEG with symmetric construction. Namely, the top and the bottom electrodes are all DLWc-processed porous carbon filled with the thermally initiated PVA/PAA/PA hydrogel. Fig. 6a and b, respectively, compare the open-circuit voltage Uoc and the short-circuit current density Jsc for these devices tested at different humidity levels. As can be seen, at a given humidity level, the output of Uoc and Jsc both strongly depends on the types of metallic electrodes used in assembling the MEG. Following the order of carbon ∼ Pt ∼ Au ≪ Ag < Cu < Al, the Uoc of the DLWc-Hgel-MEG device accordingly increases from ∼0 V to the highest value of ∼1.2 V. A similar trend was also observed for the output of Jsc, except that the MEG assembled with the Cu electrode shows an even higher value of Jsc than that with the Al electrode, when the humidity exceeds 60% RH. At the end of the above-mentioned testing, we disassembled the MEG and examined the contents of metallic elements in the PVA/PAA/PA hydrogel active material for each device by EDS. As shown in Fig. 6c, for the MEG, respectively, assembled with Al, Cu and Ag electrodes, we can correspondingly identify the presence of Al, Cu, and Ag in the PVA/PAA/PA hydrogel active material of the device. But this is not the case for the MEG assembled with Pt and Au electrode. In accordance with the EDS results, we identified the apparent changes in the surface morphology of the metallic electrodes of Al, Cu and Ag but not for the electrodes of Pt and Au, as shown in Fig. 6d, in which the surface morphology of the metallic electrode before assembling and after testing of the MEG is compared. | ||
| Fig. 6 (a and b) Dependence of the open-circuit voltage Uoc and short-circuit current density Jsc on the relative humidity for the DLWc-Hgel-MEG devices assembled with different types of metallic electrodes. (c) After 48 h of power generation testing, the EDS mapped metallic elements for the PVA/PAA/PA hydrogel active material in the MEG device assembled with different types of metallic electrodes. (d) SEM imaging for revealing the surface morphology change of the metallic electrodes of the MEG devices before assembling (top row) and after (bottom row) 48 h of power generation testing. | ||
The results shown in Fig. 6 strongly suggest that the corrosion reaction of the metallic electrodes play an important role in dictating the voltage and current performance of the DLWc-Hgel-MEG. Similar to a primary battery, the Al, Cu, and Ag electrodes in the correspondingly assembled MEG can be corroded in the acidic environment of the PVA/PAA/PA hydrogel active material to facilitate the high outputs of Uoc and Jsc. In contrast, the inert electrodes (Pt, Au and carbon) cannot be corroded under the present testing condition, and therefore, it leads to the correspondingly assembled MEG with relatively low performance for electricity generation. To further corroborate this point, we performed cyclic voltammetry and Tafel tests54 to examine the corrosion behavior of different types of metallic electrodes in the solution of PVA/AA/PA. The cyclic voltammetry testing results in a 3-electrode configuration (Hg/Hg2SO4 as the reference electrode and Pt as the counter electrode), as well as a 2-electrode configuration (DLWc-processed carbon as the counter electrode), as exhibited in Fig. 7a and S7,† respectively. The redox peaks of the Cu and Ag electrodes can be clearly identified in both cases, indicating the occurrence of Cu and Ag oxidation in the correspondingly assembled DLWc-Hgel-MEG. The correspondingly measured Tafel testing results (electrode potential vs. Hg/Hg2SO4 against the current density) are shown in Fig. 7b, from which we can obtain, for each tested metallic electrode, the corrosion potential Ucorr as well as the corrosion current density Jcorr determined by extrapolating the linear portion of the anodic branch (oxidation of the metallic electrode), as indicated in Fig. 7b. In Fig. 7c, we plot the corrosion potential Ucorr of the metallic electrodes against the open-circuit voltage Uoc for the correspondingly assembled MEG tested at different humidity levels. A strong correction between Ucorr and Uoc can be evidently identified. The similar results for the corrosion current density Jcorr and the short-circuit current density Jsc are shown in Fig. 7d. A positive correlation can also be identified between these two quantities. The correlation between Ucorr and Uoc as well as Jcorr and Jsc provide strong evidence to show that the corrosion reaction at the metallic electrodes is important for the enhanced voltage and current performance of the DLWc-Hgel-MEG. Besides this critical factor, certainly, the commonly advocated mechanism(s) for the operation of MEG, namely, the contribution by the directional migration of the charge carriers due to the concentration gradient of ionic species within the active material as well as the electric double-layer capacitance formed at the interface between the porous top electrode and the hydrogel active material may play a secondary role in controlling the performance of MEG.
 | ||
| Fig. 7 (a) Cyclic voltammograms for different types of metallic electrodes in the solution of PVA/AA/PA (Hg/Hg2SO4 as the reference electrode and Pt as the auxiliary electrode). (b) Tafel plots to show the corrosion behaviour of different types of metallic electrodes in the solution of PVA/AA/PA. The arrows indicate the method for determining the corrosion potential (vs. Hg/Hg2SO4 reference electrode) and the corrosion current density. (c) Correlation between the corrosion potential Ucorr (vs. Hg/Hg2SO4 reference electrode) of the metallic electrodes against the open-circuit voltage Uoc for the correspondingly assembled MEG devices tested at different humidity levels. (d) Correlation between the corrosion current density Jcorr of the metallic electrodes against the short-circuit current density Jsc for the correspondingly assembled MEG devices tested at different humidity levels. | ||
Load driving, application demonstration and long-term stability of DLWc-Hgel-MEG
With the device assembled by using the PVA/PAA/PA hydrogel prepared from the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment as an example, we evaluated the power transfer capability to a resistor load of the DLWc-Hgel-MEG in an ambient environment (temperature = 25 °C, humidity = 55 ± 3% RH). Fig. S9† shows the relationship between the load resistance and the MEG current and voltage output to the resistor. Fig. 8a summaries the correspondingly calculated power density for the tested MEG. Under the resistance (9.8 kΩ) matching condition, it manifested a peak power density of 26 μW cm−2, which is comparable to the best-performing MEG, as recently reported in the literature, such as 35 μW cm−2 for the PVA/PA/Gly-based MEG,55 32.6 μW cm−2 for the CS/SWNTs/PVA/CNF aerogel-based MEG,56 8.5 μW cm−2 for the electrospun hierarchical porous carbon nanofiber (HPCNF)-based device57 and 10.8 μW cm−2 for the silver electrode-based MEG.51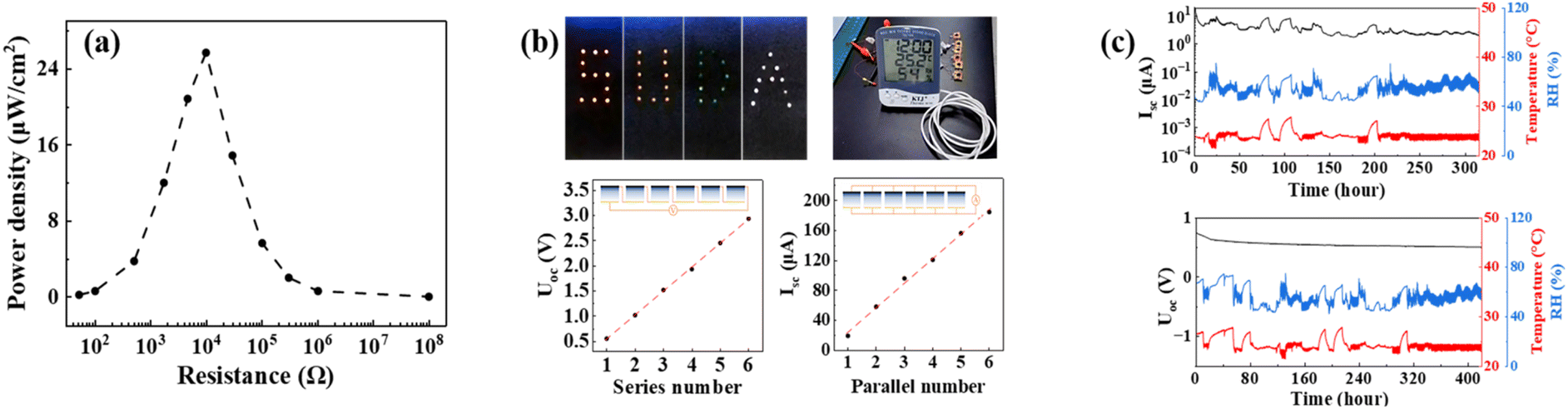 | ||
| Fig. 8 (a) Behaviour of the power output to a resistor load for the selected DLWc-Hgel-MEG device assembled from the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment tested in an ambient environment (temperature = 25 °C, humidity = 55 ± 3% RH). (b) Demonstration of the serial and parallel connections of multiple units of the DLWc-Hgel-MEG device for boosting the output of Isc and Uoc, and their use for lighting LEDs and powering the commercial hydrometer. (c) Long-term stability test in an ambient environment for a selected DLWc-Hgel-MEG device (prepared with the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment). | ||
The relatively low power capability of a single unit is a common issue for a MEG presently developed, which limits their practical use to some extent. In order to solve this problem, a common strategy is to integrate multiple units of MEG in serial/parallel connections. As shown in Fig. 8b, the current (voltage) output of the DLWc-Hgel-MEG can be proportionally enhanced by parallel (serial) connections of multiple units of DLWc-Hgel-MEG. With such an approach, in the same figure, the practical use of the DLWc-Hgel-MEG for lighting LEDs and for powering the commercial hydrometer has been demonstrated. To test the long-term operation performance of the DLWc-Hgel-MEG, we continuously monitored at room temperature in an open-lab environment the output of Isc and Uoc of a selected MEG prepared with the PVA/AA/PA solution aged for 0.5 day and subjected to UV-exposure for 10 s without gel ageing treatment. As shown in Fig. 8c, during the period of half a month operation, this MEG is able to continuously and stably deliver the required voltage and current output, which indicates the good long-term stability of our DLWc-Hgel-MEG. To date, the performance evaluation of the DLWc-Hgel-MEG was carried out at room temperature. Beyond the room temperature, we also tested a selected MEG prepared with the PVA/AA/PA solution aged for 0.5 day and UV-exposure for 10 s without gel ageing treatment at 85% ± 5% RH in the temperature range of 0–70 °C. The output of Isc and Uoc with respect to temperature is shown in Fig. S10.† As can be seen, the value of Isc increases from ∼10 μA at 0 °C to more than 100 μA at 70 °C. In contrast, the value of Uoc shows only a minor decrease from 0.55 V at 0 °C to 0.50 V at 70 °C. The dramatic increase in the current output with temperature is attributed to the significant mobility enhancement of the ionic charges, due to temperature-induced viscosity drop of the PVA/PAA/PA hydrogel active material.
Conclusions
By using the direct laser writing carbonization (DLWc) method, in combination with the interpenetrating polymer network (IPN) composed of poly(vinyl alcohol) (PVA) and poly(acrylic acid) (PAA) as well as the ternary solvents of phytic acid (PA), glycerol and water as hydrogel active materials, we demonstrated a cost-effective and scalable method for fabricating hydrogel-based MEGs. The DLWc-enabled PVA/PAA/PA hydrogel-based MEG exhibited excellent voltage, current and power performance as well as long-term stability. Beyond the fabrication and performance demonstration, we clarified two outstanding issues on the hydrogel-based MEG: the ageing treatment for the hydrogel active material plays an important role in dictating its ionic conductivity and water uptake ability as well as the power performance of the finally assembled MEG. The corrosion reaction at the metallic electrode in a hydrogel-based MEG is critical for enhancing the voltage and current performance of a hydrogel-based MEG. This work is valuable for the scalable manufacturing and performance optimization of MEGs, which will be beneficial for the development of green power sources in the future.Experimental section
Materials
Polyvinyl alcohol (PVA, degree of hydrolysis: 99%, Mw ∼145![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1), phytic acid (PA, 50 wt% in water), glycerol (99%, extra pure) and diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. Acrylic acid (AA, >97%, GC) was purchased from Shanghai Macklin Biochemical Technology Co. Ltd. Ammonium persulfate (APS, AR) was purchased from Chinasun Specialty Products Co. Ltd. Nitric acid (concentration of 68 wt%) was supplied by Sinopharm Chemical Reagent Co. Ltd, China. A polyimide (PI) film (Kapton®) with a thickness of 125 μm was purchased from Dupont, USA. All chemicals were used as received without further purification.
000 g mol−1), phytic acid (PA, 50 wt% in water), glycerol (99%, extra pure) and diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO) were purchased from Shanghai Aladdin Biochemical Technology Co. Ltd. Acrylic acid (AA, >97%, GC) was purchased from Shanghai Macklin Biochemical Technology Co. Ltd. Ammonium persulfate (APS, AR) was purchased from Chinasun Specialty Products Co. Ltd. Nitric acid (concentration of 68 wt%) was supplied by Sinopharm Chemical Reagent Co. Ltd, China. A polyimide (PI) film (Kapton®) with a thickness of 125 μm was purchased from Dupont, USA. All chemicals were used as received without further purification.
Preparation of porous carbon electrodes by the DLWc method
A laser engraving and cutting machine equipped with a CO2 laser of wavelength 10.6 μm (SCE4030, Wuhan Sunic Photoelectricity Equipment Manufacture Co. Ltd) was used to apply DLWc operation on a piece of PI film of size 10 mm × 10 mm × 125 μm, which was placed on a stainless-steel plate. In the DLWc process, a predefined lattice pattern (9 × 9 square lattice) was processed by laser drilling with a laser power set at 5 W. At each lattice point, the laser irradiation was maintained for 0.15 seconds. Subsequent to the DLWc process, the self-supported porous carbon electrode was further treated in a 6 M HNO3 solution at 95 °C for 2.5 hours for improving its water wettability.Preparation of PVA/AA/PA solution and its use for making hydrogel active materials and the assembly of DLWc-Hgel-MEGs
The solution of PVA/AA/PA was first prepared, which was later used for preparing the PVA/PAA/PA hydrogel active material in a DLWc-Hgel-MEG by either photo-initiation or thermal-initiation process. In brief, a solution of glycerol in water was first prepared by mixing 0.9 g deionized water and 1.1 g glycerol. To this solution, 0.1 g PVA were added and stirred at 95 °C until the complete dissolution of PVA. Upon cooling down the previously prepared PVA solution, 0.9 g AA and 1.9 g PA were then added and the mixture was stirred at room temperature for 15 minutes to obtain a homogeneous solution of PVA/AA/PA in a glycerol/water mixture.To prepare the hydrogel active material that is used for filling the pores of the porous electrode, 5.5 mg APS was added to the previously prepared PVA/AA/PA solution, an aliquot of which was then dropped onto the DLWc-processed carbon electrode to fill the pores via capillary action. Subsequently, the electrode imbibed with the PVA/AA/PA solution was heated at 75 °C for 10 min to thermally initiate the polymerization of AA and eventually result in the desired PVA/PAA/PA hydrogel active material. A similar method was also used for preparing the thermally initiated PVA/PAA/PA hydrogel samples for evaluating their AC impedance and swelling behaviour.
To prepare the hydrogel active material that is sandwiched between the top DLWc-processed porous carbon electrode and the bottom normal metallic electrode, 5.5 mg TPO was added to the previously prepared PVA/AA/PA solution. Before its use, this solution was aged at 4 °C in a refrigerator for 0.5, 2.5 days or 120 days for investigating the effect of solution time-ageing on the PVA/PAA/PA hydrogel active material. An aliquot of the aged PVA/AA/PA solution was filled into a pre-assembled box-mold with PMMA as the frame and the normal metallic electrode as the bottom and subjected to UV exposure (365 nm light source, H-CM-DGY, Howsuper Technology Co. Ltd) for 10 s/30 s/50 s/70 s to initiate the polymerization of AA and eventually result in the desired PVA/PAA/PA hydrogel active material. The thickness of the hydrogel active material was controlled at 1.8 mm by manipulating the amount of PVA/AA/PA solution filled in the box-mold. Finally, such prepared PVA/PAA/PA hydrogel active material was bonded with the previously prepared DLWc-processed carbon electrode by gluing the PI support of the electrode with the PMMA frame with epoxy adhesive to give a single unit of the DLWc-Hgel-MEG. The selection of 1.8 mm was based on an evaluation on the performance of the DLWc-Hgel-MEG under fixed testing conditions but with varying thickness of the hydrogel active material from 0.3 to 5.1 mm (Fig. S8†). A similar method was also used for preparing the photo-initiated PVA/PAA/PA hydrogel samples for evaluating their AC impedance and swelling behaviour.
Characterization and MEG performance evaluation
Optical microscopy (OM, MJ43, Microshot Technology Co. Ltd) and scanning electron microscopy (SEM, S-4700, Hitachi) were used for examining the morphology and structure of the DLWc-processed carbon electrode as well as the morphology of the normal metallic electrode before and after the assembly and operation of the DLWc-Hgel-MEG. Energy-dispersive spectroscopy (EDS, EVO18, Carl Zeiss AG) was performed to analyse the contents of metallic elements in the PVA/PAA/PA hydrogel active material after the operation of the DLWc-Hgel-MEG. The chemical structures of the PVA/PAA/PA hydrogel and the DLWc-processed carbon were characterized by Raman spectroscopy (LabRAM Soleil, HORIBA, 50× objective, 532 nm excitation laser). X-ray diffraction measurement was performed using an XRD diffractometer (Bruker) to acquire the crystallization information of PVA in the selected PVA/PAA/PA hydrogel samples.An electrochemical workstation impedance analyzer (RST5260F, Suzhou Risetest Electronic Co. Ltd) was used for performing the Tafel test by linear sweeping voltammetry (LSV) at a rate of 1 mV s−1 to examine the corrosion behavior of the metallic electrode (copper, aluminium, silver, platinum, gold to carbon) in the solution of PVA/AA/PAA. A three-electrode configuration was used for this purpose, in which a Pt wire and Hg/Hg2SO4 were used as the counter and reference electrodes, respectively. The same instrument was also applied to acquire the AC impedance spectra in the frequency range of 0.01 Hz to 1 MHz for the PVA/PAA/PA hydrogel prepared under different conditions for evaluating their ionic conductivity. A pair of Ni blocking electrodes were used for such a test. With a Pt wire and Hg/Hg2SO4 as the counter and reference electrodes, respectively, a three-electrode testing configuration was used for performing the cyclic voltammetry test on the six different types of electrodes in the PVA/AA/PA solution. With DLWc-processed carbon as the counter electrode, the CV test was also performed by using a two-electrode configuration for the six different types of electrodes in the PVA/AA/PA solution. In both cases, the scanning speed was set at 100 mV s−1. The open-circuit voltage (VOC) and the short-circuit current (ISC) of the DLWc-Hgel-MEG were evaluated using a Keithley 3706A system switch/multimeter. During the measurement, the device was set in a humidity-controlled testing chamber, the relative humidity (RH) of which was controlled by using different types of saturated salt solution. The RH value of the testing chamber was monitored using a hygrometer (USB-TH, Shandong Renke Control Technology Co., Ltd). All electrical tests were performed at room temperature unless otherwise stated. For the selected DLWc-Hgel-MEG, a programmable temperature and humidity chamber (2 K-150 GB, Qinzhuo Environmental Testing Equipment Co. Ltd) and a biochemical incubator (BPC-70F, Shanghai Yiheng Scientific Instruments Co., Ltd) were used for evaluating the temperature-dependent electricity generation performance.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Xuewei Pi: methodology, formal analysis, investigation, writing original draft. Yanbo Yao: conceptualization, methodology, writing – review & editing, funding acquisition. Dini Qin: investigation, resources. Tao Liu: conceptualization, methodology, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Natural Science Foundation of Fujian Province (2024J01970), State and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions, Jiangsu Key Laboratory of Advanced Functional Polymer Materials and Jiangsu Engineering Laboratory of Novel Functional Polymeric Materials.References
- O. H. Ando Junior, A. L. O. Maran and N. C. Henao, Renewable Sustainable Energy Rev., 2018, 91, 376–393 CrossRef.
- Z. Zhang, X. Li, J. Yin, Y. Xu, W. Fei, M. Xue, Q. Wang, J. Zhou and W. Guo, Nat. Nanotechnol., 2018, 13, 1109–1119 CrossRef CAS PubMed.
- S. Maiti, S. K. Karan, J. K. Kim and B. B. Khatua, Adv. Energy Mater., 2019, 9, 1803027 CrossRef.
- X. Wang, F. Lin, X. Wang, S. Fang, J. Tan, W. Chu, R. Rong, J. Yin, Z. Zhang, Y. Liu and W. Guo, Chem. Soc. Rev., 2022, 51, 4902–4927 RSC.
- J. Bai, Y. Huang, H. Cheng and L. Qu, Nanoscale, 2019, 11, 23083–23091 RSC.
- H. Yan, Z. Liu and R. Qi, Nano Energy, 2022, 101, 107591 CrossRef CAS.
- Y. Cao, B. Xu, Z. Li and H. Fu, Adv. Funct. Mater., 2023, 33, 2301420 CrossRef CAS.
- T. Xu, X. Ding, H. Cheng, G. Han and L. Qu, Adv. Mater., 2024, 36, 2209661 CrossRef CAS.
- X. Zhang, M. Wang, Y. Wu, X. Chen, K. Wu, Q. Fu and H. Deng, Adv. Funct. Mater., 2023, 33, 2210027 CrossRef CAS.
- H. Cheng, Y. Huang, L. Qu, Q. Cheng, G. Shi and L. Jiang, Nano Energy, 2018, 45, 37–43 CrossRef CAS.
- D. Shen, M. Xiao, G. Zou, L. Liu, W. W. Duley and Y. N. Zhou, Adv. Mater., 2018, 30, e1705925 CrossRef PubMed.
- Y. Li, S. Tian, X. Chen, Y. Liao, F. Jiang, J. Ye, Y. He, Y. Gui, Z. Lian, G. Liu, J. Dai, L. Li, J. Chen, S. Liu, R. Zhu, Y. Lu and M. Gao, J. Mater. Chem. A, 2024, 12, 33039–33052 RSC.
- L. Li, Z. Chen, M. Hao, S. Wang, F. Sun, Z. Zhao and T. Zhang, Nano Lett., 2019, 19, 5544–5552 CrossRef CAS PubMed.
- L. Li, M. Hao, X. Yang, F. Sun, Y. Bai, H. Ding, S. Wang and T. Zhang, Nano Energy, 2020, 72, 104663 CrossRef CAS.
- Y. Zhang, Z. Yu, H. Qu, S. Guo, J. Yang, S. Zhang, L. Yang, S. Cheng, J. Wang and S. C. Tan, Adv. Mater., 2024, 36, 2208081 CrossRef CAS PubMed.
- F. Zhao, H. Cheng, Z. Zhang, L. Jiang and L. Qu, Adv. Mater., 2015, 27, 4351–4357 CrossRef CAS PubMed.
- Y. Liang, F. Zhao, Z. Cheng, Y. Deng, Y. Xiao, H. Cheng, P. Zhang, Y. Huang, H. Shao and L. Qu, Energy Environ. Sci., 2018, 11, 1730–1735 RSC.
- K. Gao, J. Sun, X. Lin, Y. Li, X. Sun, N. Chen and L. Qu, J. Mater. Chem. A, 2021, 9, 24488–24494 RSC.
- Y. Huang, H. Cheng, G. Shi and L. Qu, ACS Appl. Mater. Interfaces, 2017, 9, 38170–38175 CrossRef CAS.
- Z. Jin, Q. Cao, H. Gong, B. Chen, Y. Jiang, Y. Su, J. Zhou and Y. Li, J. Power Sources, 2024, 608, 234264 CrossRef CAS.
- X. Liu, H. Gao, J. E. Ward, X. Liu, B. Yin, T. Fu, J. Chen, D. R. Lovley and J. Yao, Nature, 2020, 578, 550–554 CrossRef CAS.
- X. Liu, T. Ueki, H. Gao, T. L. Woodard, K. P. Nevin, T. Fu, S. Fu, L. Sun, D. R. Lovley and J. Yao, Nat. Commun., 2022, 13, 4369 CrossRef CAS PubMed.
- J. Liu, L. Huang, W. He, X. Cai, Y. Wang, L. Zhou and Y. Yuan, Nano Energy, 2022, 102, 107709 CrossRef CAS.
- B. Shao, Z. Song, X. Chen, Y. Wu, Y. Li, C. Song, F. Yang, T. Song, Y. Wang, S.-T. Lee and B. Sun, ACS Nano, 2021, 15, 7472–7481 CrossRef CAS PubMed.
- S. Liu, R. Yang, T. Yang, Z. Luo, B. Su and X. Lin, J. Mater. Chem. A, 2022, 10, 20905–20913 RSC.
- J. Bai, Q. Liao, H. Yao, T. Guang, T. He, H. Cheng and L. Qu, Energy Environ. Sci., 2023, 16, 3088–3097 RSC.
- W. He, P. Li, H. Wang, Y. Hu, B. Lu, C. Weng, H. Cheng and L. Qu, ACS Nano, 2024, 18, 12096–12104 CrossRef CAS PubMed.
- H. Zhang, N. He, B. Wang, B. Ding, B. Jiang, D. Tang and L. Li, Adv. Mater., 2023, 35, 2300398 CrossRef CAS PubMed.
- Z. Huang, C. Li, W. Ying, N. Pan, X. Lei, J. Zhang, R. Wang and J. Wang, Nano Energy, 2024, 126, 109673 CrossRef CAS.
- X. Wen, Z. Sun, X. Xie, Q. Zhou, H. Liu, L. Wang, X. Qin and S. C. Tan, Adv. Funct. Mater., 2024, 34, 2311128 CrossRef CAS.
- Z. Sun, X. Wen, S. Guo, M. Zhou, L. Wang, X. Qin and S. C. Tan, Nano Energy, 2023, 116, 108748 CrossRef CAS.
- Z. Sun, L. Feng, C. Xiong, X. He, L. Wang, X. Qin and J. Yu, J. Mater. Chem. A, 2021, 9, 7085–7093 RSC.
- Y. Gao, X. Cai, Y. Zhao, W. Huang, J. Lv, J. Wang, H. Liang, Z. Hao, H. Tan and J. Cai, J. Mater. Chem. A, 2024, 12, 12216–12224 RSC.
- Z. Sun, X. Wen, L. Wang, J. Yu and X. Qin, Energy Environ. Sci., 2022, 15, 4584–4591 RSC.
- Z. Sun, L. Feng, X. Wen, L. Wang, X. Qin and J. Yu, Mater. Horiz., 2021, 8, 2303–2309 RSC.
- J.-C. Feng, S.-X. Li, Z.-P. Zhang, Y. An, Q.-S. Gao, Z. Sun and H. Xia, Nano Energy, 2024, 119, 109103 CrossRef CAS.
- X. Ruan, R. Wang, J. Luo, Y. Yao and T. Liu, Mater. Des., 2018, 160, 1168–1177 CrossRef CAS.
- R. Wang, X. Duan, J. Yao, X. Ruan, Y. Yao and T. Liu, J. Appl. Polym. Sci., 2020, 137, 48978 CrossRef CAS.
- C. Xu, F. Gao, J. Tang, Y. Yao and T. Liu, J. Mater. Chem. A, 2022, 10, 12692–12701 RSC.
- J. Lin, Z. Peng, Y. Liu, F. Ruiz-Zepeda, R. Ye, E. L. G. Samuel, M. J. Yacaman, B. I. Yakobson and J. M. Tour, Nat. Commun., 2014, 5, 5714 CrossRef CAS PubMed.
- R. Rahimi, M. Ochoa, W. Yu and B. Ziaie, ACS Appl. Mater. Interfaces, 2015, 7, 4463–4470 CrossRef CAS PubMed.
- S. Luo, P. T. Hoang and T. Liu, Carbon, 2016, 96, 522–531 CrossRef CAS.
- H. He, Y. Yao and T. Liu, Sens. Actuators, B, 2023, 393, 134194 CrossRef CAS.
- J. Byun, Y. M. Lee and C.-S. Cho, J. Appl. Polym. Sci., 1996, 61, 697–702 CrossRef CAS.
- J. Yu, C. Dang, H. Liu, M. Wang, X. Feng, C. Zhang, J. Kang and H. Qi, Macromol. Mater. Eng., 2020, 305, 2000475 CrossRef CAS.
- Z. A. Akbar, J.-W. Jeon and S.-Y. Jang, Energy Environ. Sci., 2020, 13, 2915–2923 RSC.
- B. Kumar, S. Koka, S. J. Rodrigues and M. Nookala, Solid State Ionics, 2003, 156, 163–170 CrossRef CAS.
- A. K. Łasińska, M. Marzantowicz, J. R. Dygas, F. Krok, Z. Florjańczyk, A. Tomaszewska, E. Zygadło-Monikowska, Z. Żukowska and U. Lafont, Electrochim. Acta, 2015, 169, 61–72 CrossRef.
- R. J. Sengwa, P. Dhatarwal and S. Choudhary, Solid State Ionics, 2018, 324, 247–259 CrossRef CAS.
- M. Marzantowicz, J. R. Dygas, F. Krok, Z. Florjańczyk, E. Zygadło-Monikowska and G. Lapienis, Solid State Ionics, 2011, 192, 137–142 CrossRef CAS.
- J. Mo, X. Wang, X. Lin, X. Feng, C. Qiu, S. Tao, P. Chen, K. Zhu and H. Qi, Chem. Eng. J., 2024, 491, 152055 CrossRef CAS.
- C. Murli and Y. Song, J. Phys. Chem. B, 2010, 114, 9744–9750 CrossRef CAS PubMed.
- S. Mandal and A. K. Dasmahapatra, J. Polym. Res., 2021, 28, 269 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, 2nd edn, Wiley, New York, 2001 Search PubMed.
- S. Yang, X. Tao, W. Chen, J. Mao, H. Luo, S. Lin, L. Zhang and J. Hao, Adv. Mater., 2022, 34, 2200693 CrossRef CAS PubMed.
- X. Zhang, Z. Dai, J. Chen, X. Chen, X. Lin, S. Yang, K. Wu, Q. Fu and H. Deng, Energy Environ. Sci., 2023, 16, 3600–3611 RSC.
- X. Lu, T. Yang, C. Fu, Z. Jiang, Y. Zhang, K. Shang, C. He, J. Zhou and Q. He, Adv. Energy Mater., 2022, 12, 2202634 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02872h |
| This journal is © The Royal Society of Chemistry 2025 |