
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of PdO/Sr3Fe2O7−δ/α-Al2O3 catalysts for methane combustion†
Yeon-Bin Choi‡§
a,
Tae Wook Kang‡a,
Seo Young Kima,
Seon Tae Kima,
Byungseo Bae b,
Do Yun Kim*c,
Jin-Su Kwak*c and
Sun Woog Kim*a
b,
Do Yun Kim*c,
Jin-Su Kwak*c and
Sun Woog Kim*a
aElectronic Convergence Materials Division, Optic & Electronic Component Materials Center, Korea Institute of Ceramic Engineering and Technology, Jinju 52851, Republic of Korea. E-mail: skim80@kicet.re.kr
bAdvanced Resources Team, Yeongwol Industrial Promotion Agency, 21–28 Palgoe 1 Nonggongdanji, Yeongwolgun 26240, Republic of Korea
cHD Korea Shipbuilding & Offshore Engineering Co., Ltd, 477 Bundangsuseo-ro, Bundang-gu, Seongnam-si, Gyeonggi-do 13591, Republic of Korea. E-mail: doyun.kim@ksoe.co.kr
First published on 23rd June 2025
Abstract
Catalysts, consisting of palladium supported on Ruddlesden–Popper (RP) type Sr3Fe2O7−δ perovskite oxides, were synthesized using co-precipitation and impregnation methods for efficient methane combustion at low temperatures. Achieving effective and stable methane combustion over Sr3Fe2O7−δ perovskite-based catalysts necessitates the suppression of the transformation of the RP-type Sr3Fe2O7−δ perovskite structure supporting active Pd species and the thermal reaction process. In this study, we demonstrated that the use of α-Al2O3 as a supporting material in PdO/Sr3Fe2O7−δ/α-Al2O3 catalysts significantly improved the surface oxygen species and oxygen vacancy density as well as the structural stability of the composite oxide catalyst. Among the catalysts prepared, the 13 wt% PdO/16 wt% Sr3Fe2O7−δ/α-Al2O3 catalyst exhibited the highest catalytic activity for complete methane combustion at a low temperature of 300 °C.
Introduction
Methane (CH4) represents one of the cleanest fossil fuel sources and holds great potential in achieving carbon neutrality.1–3 It serves as the primary component in natural gas, shale gas, and coal-bed gas, with broad applications as an alternative to crude oil in various sectors such as ships, motor vehicles, power stations, and more.4–7 However, the detrimental impact of unburnt CH4 emissions is a pressing concern due to CH4's significantly higher global warming potential (at least seventy-two times greater than CO2), contributing to the ongoing greenhouse effect.8,9 Thus, the widespread adoption of LNG-powered ships and LNG power plants is essential to curb methane emissions.10–12Catalytic combustion has emerged as an effective method for mitigating these emissions by enabling CH4 oxidation at low temperatures, producing only water vapor and carbon dioxide with minimal NOx and CO emissions.13–20 Nonetheless, CH4, with its stable tetrahedral structure, is challenging to activate and oxidize under mild conditions. While Pd-supported CeO2–ZrO2 catalysts have been commonly used for methane combustion, they are sensitive to water vapor, which can hinder combustion and catalytic performance.21–25 Moreover, CeO2-based catalysts contain rare earth elements, making them economically challenging to produce.26 In addition, it has been reported that the complete combustion of methane for the catalyst systems loaded with Pd occurred at a high temperature over 400 °C under the conditions of high methane concentration over 1 vol% and low Pd loading amount under 4 wt%, as summarized in Table S1.† In order to realize the complete combustion of methane and further lower the temperature under these harsh conditions, it is necessary to develop a novel catalyst loaded with Pd.
To address these challenges, we aim to develop high-activity catalysts that facilitate CH4 combustion at low temperatures, reduce the activation energy for oxygen migration, and enhance water resistance.27,28 Among perovskite catalysts, Sr–Fe-based perovskite-type oxides, following the Mars–van Krevelen (MvK)-type mechanism,29–35 are promising for CH4 oxidation at moderate temperatures. Additionally, Ruddlesden–Popper (RP)-type layered perovskite oxides, such as Sr3Fe2O7−δ, offer unique electron and ion transport properties.36–39 The RP-type structure of Sr3Fe2O7−δ exhibits high oxygen migration capability,40–46 making it a suitable candidate for low-temperature methane combustion. The oxygen vacancy defects formed through the electron transition of Fe4+ → Fe3+ can enhance the oxygen mobility in the crystal lattice, thereby contributing to increased methane decomposition efficiency of the catalysts.47–49 However, to serve as an effective catalyst, Sr3Fe2O7−δ needs structural stability, water resistance, and high oxygen storage and release ability. While γ-Al2O3 is a common support for methane combustion catalysts due to its high specific surface area, it promotes unwanted reactions with water and lowers catalytic activity.50–52 In contrast, α-Al2O3 has weaker interactions with Pd species, high thermal stability, superior water resistance, and a lower specific surface area.53 In view of the above, this study focuses on using RP-type Sr3Fe2O7−δ perovskite oxide as a catalyst promoter for low-temperature methane combustion, supported by α-Al2O3 to enhance structural stability without forming Pd–O–Al bonds. The investigation includes the influence of the α-Al2O3 support on the catalyst and the methane oxidation performance of the resulting catalyst, considering oxygen storage and release ability, perovskite structure effects, and the role of the α-Al2O3 support.
Experimental
Material
Strontium nitrate (Sr(NO3)2, ≥98%) were guaranteed reagents from Daejung Chemicals & Metals Co. Ltd. Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O, ≥98%) was purchased from SIGMA-ALDRICH. 1 N-Potassium hydroxide standard solution (5–6%) was from Daejung Chemicals & Metals Co. Ltd. γ-Al2O3 powder (99.997%) were from Alfa Aesar. The physical properties, BET surface area (m2 g−1), and particle size (μm) of γ-Al2O3 were about 210, and 1–5, respectively. The core elements for oxidation of CH4 commercially Pd catalysts (<20% loading) were used from conversion of Palladium (II) nitrate hydrate (Pd(NO3)2, 37–42%, SIGMA-ALDRICH) to PdO by impregnation method.Synthesis of Sr3Fe2O7−δ-based complex oxide catalysts
The Sr3Fe2O7−δ/α-Al2O3 catalyst was prepared using a co-precipitation process. γ-Al2O3 powder (0.4 g, 99.997%, Alfa Aesar) was suspended in 1.0 mol L−1 Sr(NO3)2 aq. and 1.0 mol L−1 Fe(NO3)3 aq. in the stoichiometric ratio. The loading amount of Sr3Fe2O7−δ was adjusted to be 16wt% by using 0.46 mL of 1.0 mol L−1 Sr(NO3)2 aq. and 0.31 mL of 1.0 mol L−1 Fe(NO3)3 aq., respectively. After stirring at room temperature for 30 minutes, the pH of the solution was adjusted to 13.6 by adding 5 vol% KOH aqueous solution drop by drop, followed by further stirring at room temperature for 6 hours. The resulting product was collected by filtration, dried at 80 °C for 12 hours, and calcined at 1000 °C for 12 hours under ambient atmosphere to obtain the 16wt% Sr3Fe2O7−δ/α-Al2O3 catalysts. For comparison, Sr3Fe2O7 catalyst also synthesized as with the previous method.PdO was loaded onto 16wt%Sr3Fe2O7−δ/α-Al2O3 via the impregnation method. The 16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst was impregnated with PdO by suspending the catalyst powder (0.4 g) in 1.0 mol L−1 Pd(NO3)2 aq. (Sigma Aldrich), followed by adding 30 mL of deionized water. PdO loading ranged from 0 to 20 wt%. The mixture was stirred at room temperature for 6 hours, and then the solvent was evaporated at 180 °C. The residue was ground and calcined at 400 °C for 4 hours under atmospheric pressure to produce the corresponding αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (0 ≤ α ≤ 20). For comparison, 10wt%PdO/Sr3Fe2O7−δ and 10wt%PdO/α-Al2O3 catalysts were also synthesized, in which 10wt%PdO/Sr3Fe2O7−δ and 10wt%PdO/α-Al2O3 catalyst (using α-Al2O3 powder, 98%, DAEJUNG) were calcined at 400 °C for 4 hours.
Catalyst characterization
The crystalline phase of the catalysts was identified by X-ray powder diffraction (XRD, Bruker D8 Advance) using Cu-Kα radiation (40 kV, 40 mA) in the 2θ range of 10–80° with a step size of 0.02° and a scan speed of 0.2°·min−1. The chemical composition of the samples was determined by inductively coupled plasma optical emission spectrometer (ICP-OES, Varian 720-ES). The crystallographic parameters were refined via Rietveld refinement (RIETAN-FP). In the refinement, the crystallographic parameters of Sr3Fe2O7 were obtained from the inorganic crystal structure data (Sr3Fe2O7: ICSD #74422) and were used as the starting model for refinement. The crystallite size was calculated from the measured XRD peak data using Scherrer's equation. The microstructure of the samples is observed by Cs-corrector transmission electron microscope (Cs-TEM, FEI ThemisZ) with energy-dispersive X-ray spectroscopy (EDS). The distribution of elements in the catalyst was measured by Cs-TEM with a back-scattered electron detector. The Brunauer–Emmett–Teller (BET) specific surface areas were measured at −196 °C through N2 adsorption (TriStar 3020, Shimadzu). The acidity and basicity were evaluated using the temperature-programmed desorption method using ammonia and carbon dioxide (NH3-TPD/CO2-TPD, Micromeritics AutoChem II 2920 Chemisorption Analyzer). After preheating the sample (0.1 g) at 500 °C for 1 h under a flow of helium gas (50 mL min−1), the sample was exposed to ammonia (10 vol% NH3/90 vol% He, 50 mL min−1) and carbon dioxide (10 vol% CO2/90 vol% He, 50 mL min−1) at 50 °C for 30 min, followed by purging at the same temperature for 15 min under helium flow (50 mL min−1). Subsequently, the temperature was increased at a rate of 10 °C min−1 under helium flow at a rate of 30 mL min−1 (BELCAT-B, Microtrac BEL). H2-temperature programmed reduction (H2-TPR) was conducted under 5 vol% H2/95 vol% Ar gas flow (50 mL min−1) at a heating rate of 5 °C min−1 (Micromeritics Auto Chem II 2920 Chemisorption Analyzer). Subsequently, the oxygen storage capacity (OSC) was investigated using the reduced sample after preheating under 5 vol% O2/95 vol% H2 gas flow (50 mL min−1) at a heating rate of 5 °C min−1, by means of the pulse-injection method at 500 °C (Micromeritics Auto Chem II 2920 Chemisorption Analyzer). X-ray photoelectron spectroscopy (XPS, AXIS SUPRA) data were acquired using Mg-Kα radiation (1253.6 eV); the spectra were fitted using the Shirley background and the Gaussian–Lorentzian lineshape analysis.Methane combustion measurement
The activity for methane oxidation was measured in a fixed-bed flow reactor consisting of a 10 mm-diameter quartz glass tube. The mixed gas comprised 0.2 vol% CH4 in air at a total flow rate of 60 mL min−1 (mass hourly space velocity = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 L kg−1 h−1) and was passed over 0.3 g of the catalyst; the gas was supplied to the catalyst bed using a mass flow controller. Prior to the measurements, the catalysts were pretreated at 400 °C for 2 h under a flow of argon (100 mL min−1). The concentration of methane gas at the outlet was analyzed using a non-dispersive infrared (NDIR) gas analyzer (DA-770, GASDNA Co., Ltd). The reaction temperature was increased from 150 to 500 °C in 25 °C intervals, and the methane conversion at each point was obtained after stabilization for 15 min.
000 L kg−1 h−1) and was passed over 0.3 g of the catalyst; the gas was supplied to the catalyst bed using a mass flow controller. Prior to the measurements, the catalysts were pretreated at 400 °C for 2 h under a flow of argon (100 mL min−1). The concentration of methane gas at the outlet was analyzed using a non-dispersive infrared (NDIR) gas analyzer (DA-770, GASDNA Co., Ltd). The reaction temperature was increased from 150 to 500 °C in 25 °C intervals, and the methane conversion at each point was obtained after stabilization for 15 min.
Results and discussion
Crystal structure of Sr–Fe perovskite-based catalysts
The crystal structure of the Sr3Fe2O7 powders was determined through X-ray diffraction (XRD) analysis using Rietveld refinement. The detailed crystallographic data and structure refinement parameters obtained by Rietveld refinement analysis are summarized in Tables 1 and S2.† The analysis used crystal structure data obtained from the inorganic crystal structure database (Sr3Fe2O7: ICSD #74422) as a reference. Fig. 1(a) and (b) displays the XRD patterns and crystal structure of the Sr3Fe2O7 powders synthesized in this study.| Sr3Fe2O7 | SrFeO2.75 | |
|---|---|---|
| Crystal system | Tetragonal | Orthorhombic |
| Space group | I4/mmm | Cmmm |
| a (nm) | 3.86239 | 9.75109 |
| b (nm) | 3.86239 | 9.81500 |
| c (nm) | 20.15442 | 2.91472 |
| Mass fractions of the phases | 0.9603 | 0.0397 |
| Rwp (%) | 6.379 | |
| Rp (%) | 3.343 | |
| S (%) | 5.2009 | |
| d1 | 0.0867 | |
| d2 | 0.0531 | |
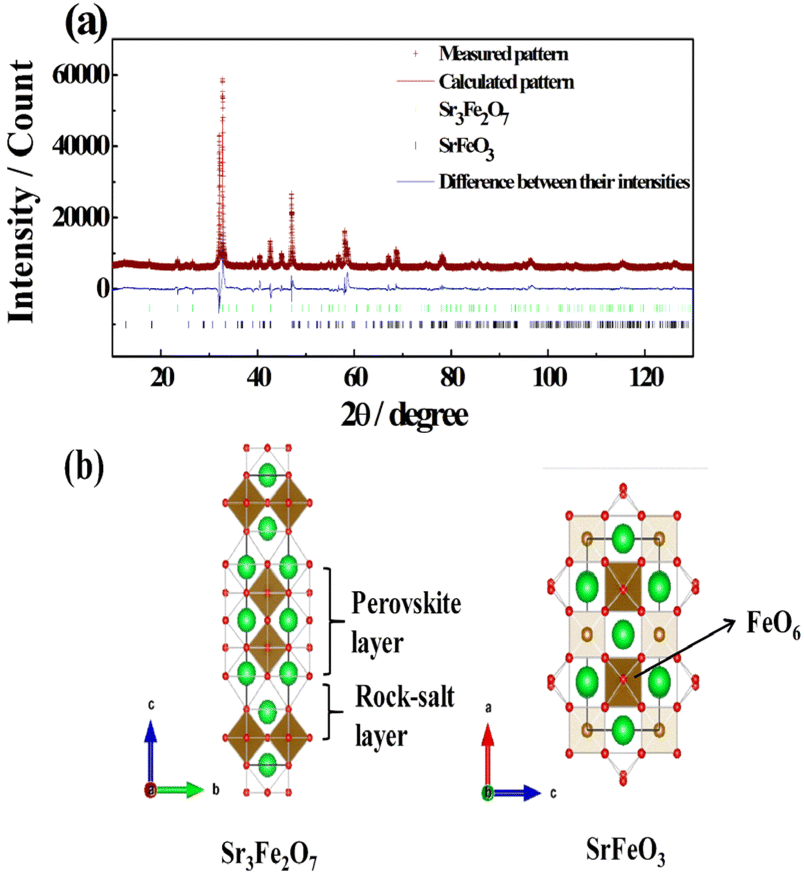 | ||
| Fig. 1 (a) Rietveld refinement of X-ray powder diffraction data for Sr3Fe2O7; white symbol: measured pattern, red solid line: calculated pattern, blue solid line: difference between the intensities and (b) crystal structure of Sr3Fe2O7 and SrFeO2.75. The circles correspond to Sr cations (green) and oxygen anions (red), which are located at the corners of an FeO6 octahedron. | ||
The XRD data matched well with the lattice constants of Sr3Fe2O7 (ICSD #74422), albeit with some SrFeO2.75 as an impurity phase. Rietveld refinement yielded favorable weighted profile R-factors, confirming the phase purity of the samples. At room temperature, Sr3Fe2O7 was found to crystallize in a tetragonal system (space group I4/mmm) with lattice parameters a = 3.86239 nm, b = 3.86239 nm, and c = 20.15442 nm (see Table 1). The mass fraction of Sr3Fe2O7 in the synthesized powder was determined to be 96%, making it the predominant phase. The Sr3Fe2O7 oxide exhibited the RP-type structure composed of (SrFeO3)2(SrO). A second perovskite-type phase, SrFeO2.75, was also present. As a promoter, the synthesized Sr3Fe2O7 was used to create a composite oxide catalyst for low-temperature methane combustion. For this purpose, a composite oxide catalyst, composed of 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3, was synthesized by varying the composition. The composition of the samples was analyzed by ICP-OES and the results are summarized in Table S3.† It was confirmed that all samples were synthesized in each stoichiometric ratio. XRD patterns of the synthesized catalysts were compared with those of 10wt%PdO/Sr3Fe2O7−δ, 10wt%PdO/α-Al2O3, and 16wt% Sr3Fe2O7−δ/α-Al2O3 as shown in Fig. 2. The XRD pattern of the 10wt%PdO/Sr3Fe2O7−δ catalyst showed PdO and the orthorhombic SrFeO2.75 (SrFeO3−δ) perovskite phase. Supporting PdO on Sr3Fe2O7 led to the transformation of the RP-type Sr3Fe2O7 structure into the simple perovskite-type SrFeO3−δ, likely due to the migration of oxide ions from Sr3Fe2O7 to the Pd active sites.
 | ||
| Fig. 2 XRD pattern of (a) 10wt%PdO/SrFeO3−δ, (b) 10wt%PdO/α-Al2O3, (c) 16wt%Sr3Fe2O7−δ/α-Al2O3, and (d) 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts. | ||
In contrast, the XRD pattern of the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst showed PdO, tetragonal Sr3Fe2O7 (Sr3Fe2O7−δ) perovskite phase with space group I4/mmm, α-Al2O3 and γ-Al2O3 phase, in which phase ratio of the α-Al2O3 and γ-Al2O3 phase was 84.4:15.6% form the Rietveld refinement. In the XRD patterns of the 16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst, a small amount of γ-Al2O3 phase was also obtained and the phase ratio of γ-Al2O3 phase was 17.3%. This confirmed the preservation of the Sr3Fe2O7 perovskite phase when supported on α-Al2O3, indicating improved structural stability. In addition, α-Al2O3 contributes to the enhancement of the dispersity of the PdO on catalyst promoter. The crystallite size of the PdO of 10wt%PdO/SrFeO3−δ, 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3, and 10wt%PdO/α-Al2O3 was calculated using Scherrer's equation. As shown in Table 2, the structural effects were supported by differences in PdO crystallite sizes, with the 10wt%PdO/SrFeO3−δ catalyst exhibiting larger crystallites compared to the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. This difference indicates that the bulk migration of the oxide ions from Sr3Fe2O7 to the Pd active site in the 10wt%PdO/SrFeO3−δ catalyst without the α-Al2O3 support can facilitate oxidation and aggregation of the Pd active centers, which increases the size of the PdO crystallites. In addition, this result was also supported by the PdO dispersion on the respective catalysts. The PdO dispersion of 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 (7.69%) was determined to be higher than that of 10wt%PdO/SrFeO3−δ (7.58%), as shown in Table S4.† In comparison, the 10wt%PdO/α-Al2O3 catalyst without the Sr3Fe2O7 promoter had the smallest PdO crystallite size (3.71 nm) because there was no source of oxide ions for migration to the PdO active species.
| Sample | PdO crystallite size/nm | Specific surface area/m2 g−1 |
|---|---|---|
| γ-Al2O3 | — | 148.0 |
| α-Al2O3 | — | 14.6 |
| Sr3Fe2O7 | — | 30.5 |
| 10wt%PdO/SrFeO3−δ | 19.1 | 21.6 |
| 10wt%PdO/α-Al2O3 | 3.7 | 2.5 |
| 16wt%Sr3Fe2O7−δ/α-Al2O3 | — | 80.9 |
| 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 | 9.2 | 70.2 |
To determine an amount of PdO on the surface of the catalysts, we carried out XPS analysis for 10wt%PdO/SrFeO3−δ and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3, and calculated the Pd2+/(Pd2+ + Pd0) ratio using peak areas of Pd 3d core-level (see in Fig. S3 and Table S4†). The Pd 3d XPS spectra comprise two spin–orbit multiplets corresponding to the 3d3/2 and 3d5/2 core levels, and each peak of the 3d3/2 and 3d5/2 core levels can be divided into two Gaussian peaks, where peak at high binding energy corresponds to the Pd2+ and the other peak at low binding energy corresponds to the Pd0. The Pd2+/(Pd2+ + Pd0) ratio of 10wt%PdO/SrFeO3−δ (42.49%) was higher than that of 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 (40.31%). This result indicates that a larger amount of PdO was loaded onto the surface of 10wt%PdO/SrFeO3−δ compared to the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 and it also supports that bulk migration of the oxide ions from the promoter to the Pd active sites was more extensively occurred at the sample of 10wt%PdO/SrFeO3−δ. In addition, the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst (70.19 m2 g−1) exhibited a higher specific surface area than that of 10wt%PdO/SrFeO3−δ catalyst (21.58 m2 g−1) without supporting Al2O3 as shown in Table 2. On the other hand, the specific surface area of 10wt%PdO/α-Al2O3 catalyst (2.51 m2 g−1) was lower than that of 10wt%PdO/SrFeO3−δ catalyst. This result indicates that a high specific surface area of the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst may be due to the presence of the γ-Al2O3 phase, which usually contributes to the enhancement of the specific surface area of catalyst materials.
To investigate the effect of the α-Al2O3 support on the thermal and structural stability of the catalysts, the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 and 10wt%PdO/SrFeO3−δ catalysts were evaluated at various temperatures, 200–600 °C. The catalyst samples were re-heated at each temperature for 1 h and their crystal structure was identified (see in Fig. S1†). In the case of the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3, the Sr3Fe2O7−δ perovskite structure was well maintained in all samples re-heated at 200–600 °C. On the other hand, the SrFeO3−δ perovskite structure for 10wt%PdO/SrFeO3−δ was transformed to a composite phase of SrFe12O19 and Sr2Fe2O5 after heat treatment at 200 °C. It indicates that α-Al2O3 is significantly effective in inhibiting the crystal structure change for these catalysts. Moreover, when Sr3Fe2O7 was supported on α-Al2O3, the XRD peak of the Sr3Fe2O7 perovskite phase shifted to the lower angle side (refer to Fig. S7†), which indicates that the lattice of the Sr3Fe2O7 perovskite structure was expanded by the formation of oxygen vacancies in the Sr3Fe2O7 lattice. The release of oxide ions from Sr3Fe2O7 can be associated with transformation of the γ-Al2O3 monoclinic phase, the raw material used for the support, into the hexagonal α-Al2O3 phase with a volume reduction of ∼10% after calcination at 1000 °C during synthesis of the catalyst.50 This result confirms that in the case of the 16wt% Sr3Fe2O7−δ/α-Al2O3 and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts, the Sr3Fe2O7−δ promoter with oxygen vacancies was formed without any change in the RP-type Sr3Fe2O7 perovskite structure when Sr3Fe2O7 was supported on α-Al2O3.
Surface and bulk oxide property of Sr–Fe perovskite-based catalysts
To investigate the surface acidic and basic property of the prepared Sr–Fe perovskite-based catalysts, NH3- and CO2-TPD measurements were evaluated as shown in Fig. 3(a) and (b). The 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst displayed narrow desorption peaks at around 100, 230, and 490 °C in the NH3-TPD profile, signifying weak, medium-strength, and strongly acidic sites, including Lewis acid sites. In comparison, the 10wt%PdO/SrFeO3−δ, 10wt%PdO/α-Al2O3, and α-Al2O3 catalysts exhibited broad and narrow peaks mainly associated with weak acid sites and Lewis acid sites. In the CO2-TPD profiles, the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst showed a pronounced area peak at around 110 °C, while the other catalysts showed weak CO2 adsorption. This result indicates that the 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3 has the superior basic sites compared to that of the other catalysts. The number of acidic and basic sites on each catalyst was determined from peak areas in the NH3- and CO2-TPD profiles. As shown in Table 3, the number of acidic sites on the catalyst was dramatically increased when PdO was supported on α-Al2O3, which is due to the increased acidic sites of Pd2+ cations on the catalyst surface. The highest number of basic sites was obtained for the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst (220.0 μmol g−1), which is higher than that of 10wt%PdO/SrFeO3−δ (148 μmol g−1). This result indicates that the concentration of surface oxygen species which act as a Lewis basic site on the 16wt%Sr3Fe2O7−δ/α-Al2O3 is higher than that on the SrFeO3−δ bulk, which may be due to the surface area of 16wt%Sr3Fe2O7−δ/α-Al2O3 was higher than that of SrFeO3−δ bulk. | ||
| Fig. 3 (a) NH3-TPD and (b) CO2-TPD profiles of α-Al2O3, 10wt%PdO/α-Al2O3, 10wt%PdO/SrFeO3−δ, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3, (c) H2-TPR profile of α-Al2O3, Sr3Fe2O7, 16wt%Sr3Fe2O7−δ/α-Al2O3, 10wt%PdO/α-Al2O3, 10wt%PdO/SrFeO3−δ, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. (d) O2-TPD profile of Sr3Fe2O7, 16wt%Sr3Fe2O7−δ/α-Al2O3, 10wt%PdO/SrFeO3−δ, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. | ||
| Sample | Number of acidic site/μmol g−1 | Number of basic site/μmol g−1 | Reduction temperature/°C | H2 consumption amount/mmol g−1 | OSC/μmol O2·g−1 | O2 desorption site/μmol g−1 | Oxygen vacancy density/×1017 sites m−2 |
|---|---|---|---|---|---|---|---|
| α-Al2O3 | 27.5 | 53.8 | — | 0 | 0 | — | — |
| Sr3Fe2O7 | — | — | 450, 495 | 1.237(SrFeOx) | 4.899 | 44 | 1.59 |
| 16wt%Sr3Fe2O7−δ/α-Al2O3 | — | — | 345 | 0.111(SrFeOx) | 1.752 | 84 | 3.95 |
| 10wt%PdO/α-Al2O3 | 41.0 | 15.5 | 62 | 0.258(PdO) | 0.703 | — | — |
| 10wt%PdO/SrFeO3−δ | 109.5 | 148.2 | 73, 325, 440, 495 | 0.587(PdO) | 35.174 | 75 | 3.21 |
| 2.119(SrFeOx) | |||||||
| 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3 | 169.0 | 220.0 | 56, 83, 193, 420, 490 | 0.104(PdO) | 35.695 | 357 | 15.44 |
| 0.677(SrFeOx) |
To investigate the oxygen migration properties of the prepared Sr–Fe perovskite-based catalysts, H2-TPR and O2 pulse injection measurements were performed (Fig. 3(c), S2† and Table 3). The α-Al2O3 support showed no H2 reduction activity or oxygen storage capacity. The H2-TPR profile of the Sr3Fe2O7 perovskite bulk oxide exhibited H2 reduction peaks at high temperatures of around 450 and 495 °C by bulk oxygen species. In contrast, the 16wt%Sr3Fe2O7−δ/α-Al2O3 showed a lower temperature H2 reduction peak at 345 °C by surface oxygen reduction. It is because that the higher specific surface area and the more surface oxygen species, the lower the activation energy required for oxygen release induced to facilitate H2 reduction even at low temperatures. The 16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst exhibited lower H2 consumption amount and oxygen storage capacity (OSC) than the Sr3Fe2O7 perovskite bulk due to the small absolute amount of oxygen species released from the lattice of Sr3Fe2O7−δ. When assessing supported PdO species, the PdO reduction peak of 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst was exhibited at the lowest temperature of 56 °C compared to 10wt%PdO/α-Al2O3 (62 °C) and 10wt%PdO/SrFeO3−δ (73 °C). The H2 consumption amount of PdO reduction peak were determined as follows; 10wt%PdO/SrFeO3−δ (587 μmol g−1) > 10wt%PdO/α-Al2O3 (258 μmol g−1) > 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 (104 μmol g−1). The reduction peak of SrFeOx oxide in the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst occurred at lower temperatures (83, 193, 420 and 490 °C) than that in the 10wt%PdO/SrFeO3−δ catalyst (325, 440 and 495 °C). The H2 consumption amount of SrFeOx oxide reduction peak were determined as follows; 10wt%PdO/SrFeO3−δ (2119 μmol g−1) > 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 (677 μmol g−1). As the PdO active species was supported, the activation energy for oxygen release from SrFeOx oxide was reduced, and then, H2 consumption amount and OSC of PdO supported catalysts tended to increase compared to catalyst without PdO supported. On the other hand, although H2 consumption amount was low, the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst, which has high concentration of surface oxygen species, showed more low-temperature activity for H2 reduction than the 10wt%PdO/SrFeO3−δ catalyst, which has mainly bulk oxygen species. O2-TPD analysis was performed to confirm the formation of oxygen vacancies on the surface of prepared catalysts and the results are shown in Fig. 3(d) and Table 3. The 16wt%Sr3Fe2O7−δ/α-Al2O3 showed a higher O2 desorption area than that of the Sr3Fe2O7 perovskite bulk oxide at low temperature of around 100 °C. The O2 desorption at low temperature can be induced from physically adsorbed oxygen on surface oxygen vacancy. The 16wt%Sr3Fe2O7−δ/α-Al2O3 showed a higher oxygen vacancy density which estimated based on the amount of ad-O2 derived from the O2-TPD than that of the Sr3Fe2O7 bulk (Table 3). This result indicates that the surface oxygen vacancy of the 16wt%Sr3Fe2O7−δ/α-Al2O3 is a higher than that of the Sr3Fe2O7 perovskite. This is because that the lattice of the Sr3Fe2O7 perovskite structure of 16wt%Sr3Fe2O7−δ/α-Al2O3 was expanded by the formation of oxygen vacancies as the oxygen ions are released from Sr3Fe2O7 lattice associated with the transformation of the γ-Al2O3 monoclinic phase into the α-Al2O3 hexagonal phase. The O2 desorption area of the catalysts was effectively increased when PdO was supported on the surface of the catalysts and the O2 desorption area. and the oxygen vacancy density of 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 was superior to 10wt%PdO/SrFeO3−δ. These results indicate that the amount of oxygen ions released from the surface oxygen and migrating to the Pd active sites is higher for 16wt%Sr3Fe2O7−δ/α-Al2O3 due to the high oxygen ion conductivity and the oxygen vacancy density compared to the Sr3Fe2O7 bulk. This usually results in the high methane gas decomposition performance of catalysts.
Catalytic activity for methane combustion over Sr–Fe perovskite-based catalyst
The catalytic activity of catalysts with different compositions for methane combustion was evaluated. Fig. 4 illustrates the temperature dependence of methane combustion over the various prepared catalysts, including 10wt%PdO/SrFeO3−δ, 10wt%PdO/α-Al2O3, 16wt%Sr3Fe2O7−δ/α-Al2O3, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. | ||
| Fig. 4 Temperature-dependence of methane oxidation over 10wt%PdO/SrFeO3−δ, 10wt%PdO/α-Al2O3, 16wt%Sr3Fe2O7−δ/α-Al2O3, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts. | ||
The 10wt%PdO/α-Al2O3 catalyst enabled complete methane combustion at 400 °C. Upon the addition of the Sr3Fe2O7−δ co-catalyst to the 10wt%PdO/α-Al2O3 catalyst, the temperature for complete methane combustion decreased to 325 °C. This result indicates that catalytic activity for methane combustion at low temperatures can be enhanced by adding the Sr3Fe2O7−δ co-catalyst, which increase surface oxygen species and oxygen vacancy density of the catalyst, thus promoting methane oxidation at low-temperature. To confirm the promoting effect of the PdO active sites and α-Al2O3 support on catalytic methane combustion, we compared the catalytic activity of the 16wt%Sr3Fe2O7−δ/α-Al2O3 and 10wt%PdO/SrFeO3−δ catalysts with that of the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst. The 16wt%Sr3Fe2O7−δ/α-Al2O3 showed limited catalytic activity for methane combustion. Since methane oxidation primarily occurs on the PdO active sites, the 10wt%PdO/SrFeO3−δ catalysts achieved complete methane combustion at 425 °C, approximately 100 °C higher than the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst. This difference arises from the fact that the 10wt%PdO/SrFeO3−δ catalysts exhibit lower surface oxygen species and oxygen vacancy density compared to the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts, attributed to the high activation energy for oxygen migration in SrFeO3−δ compared to the Sr3Fe2O7−δ perovskite oxide. Among the prepared catalysts with different compositions, the highest catalytic activity was achieved with the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst sample, where a methane conversion of 100% was attained at 325 °C. Nevertheless, when compared to cerium oxide (CeO2)-based catalysts with higher oxygen storage capacity than Sr3Fe2O7−δ perovskite oxide,51–55,57 the catalytic methane combustion activity of the 10wt%PdO/16wt%Ce0.92Si0.08O2−δ/γ-Al2O3 catalysts56 was still higher than that of the 10wt% PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts under low temperature below 300 °C as shown in Fig. S6.†
For a more detailed investigation of the methane decomposition mechanism of the Sr3Fe2O7−δ perovskite oxide-based catalyst, XPS analysis of Fe 2p and O 1s before and after the reaction was performed,58–60 as presented in Fig. S4, S5, and Table S4.† An increase in the ratio of Fe3+/Fe and a decrease in the ratio of Fe4+/Fe can be observed in both SrFeO3−δ and Sr3Fe2O7−δ before and after methane combustion reaction. This implies that the reduction of FeO2 to Fe2O3 occurs during the methane decomposition reaction, activating the redox property of the catalyst due to the valence change of Fe3+/4+. In result of O 1s analysis, the ratio of surface oxygen species (Oads/O) (53.67%) was higher than the ratio of lattice oxygen species (Olatt/O) (46.67%) in before reaction of 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. On the other hand, the ratio of surface oxygen species (Oads/O) (33.25%) was lower than the ratio of lattice oxygen species (Olatt/O) (66.75%) in before reaction of 10wt%PdO/SrFeO3−δ. And then, no the fraction of surface oxygen species was measured in XPS spectra of O 1s of 10wt%PdO/α-Al2O3. From this result, it can be confirmed that the highest concentration of surface oxygen species is distributed on 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 compared to 10wt%PdO/SrFeO3−δ and 10wt%PdO/α-Al2O3. Additionally, a relative decrease in the ratio of surface oxygen species (Oads/O) can be observed before and after the reaction with increase of the ratio of lattice oxygen species (Olatt/O) in SrFeOx promoted catalysts. This indicates that the surface oxygen species of the catalyst actively participate in the reaction activity during the methane combustion reaction. Moreover, the high Fe3+/Fe and Oads/O ratios after the reaction indicate that the mobility of surface oxygen associated with the reduction of FeO2 to Fe2O3 was higher for 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 than for 10wt%PdO/SrFeO3−δ.
For a more detailed discussion of the effects of catalyst composition on the catalytic activity for low-temperature methane combustion, we calculated the turnover frequency (TOF) to density of surface PdO active site, oxygen vacancies and specific reaction rates for methane conversion at 275 °C. Table 4 presents the methane conversion, specific reaction rate (r275°C) and TOF at 275 °C to the density of PdO active site (PdO) and oxygen vacancies for the 10wt%PdO/α-Al2O3, 10wt%PdO/SrFeO3−δ, and 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts. The 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst exhibited the highest reaction rate (0.619 mmol s−1 g−1) compared to that of the 10wt%PdO/SrFeO3−δ (0.132 mmol s−1 g−1) and 10wt%PdO/α-Al2O3 (0.75 mmol s−1 g−1) catalysts, which is consistent with the catalytic methane conversion activities as shown in Fig. 4. In particular, the TOF derived from normalization of the rate constant with the surface density of PdO active sites was found to increase as the SrFeOx oxide supported to 10wt%PdO/α-Al2O3, reaching 7.955 × 103 h−1 for the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. When methane is decomposition at the PdO active site, even under the same PdO loading conditions, the TOF value increases as the SrFeOx oxide is loaded. This is because the oxygen species released by SrFeOx increases the oxidizing properties of the PdO active species. Moreover, the TOF derived from normalization of the rate constant with the surface density of oxygen vacancy in the 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 was superior to 10wt%PdO/SrFeO3−δ, reaching 36.10 × 103 h−1 for the most activity, which is consistent with the catalytic methane conversion activities. This observation confirms that by supporting the Sr3Fe2O7−δ co-catalyst, the catalytic activity for low-temperature methane combustion can be facilitated by improving the density of oxygen vacancy. Therefore, it can be concluded that, despite having identical PdO loadings, these factors are the primary contributors to the superior methane oxidation capacity of the PdO active species in 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3 compared to those in the 10wt%PdO/SrFeO3−δ catalysts.
| Sample | Methane conversion at 275 °C/% | r275°C/mmol−1 g−1 | TOF × 103/h−1 (PdO) | TOF × 103/h−1 (oxygen vacancies) |
|---|---|---|---|---|
| 10wt%PdO/α-Al2O3 | 3.50 | 0.075 | 0.002 | — |
| 10wt%PdO/SrFeO3−δ | 9.85 | 0.132 | 0.103 | 7.28 |
| 10wt%PdO/16wt% Sr3Fe2O7−δ/α-Al2O3 | 49.85 | 0.619 | 7.955 | 36.10 |
Effect of PdO loading in Sr–Fe perovskite-based catalyst
To investigate the impact of PdO loading on the catalytic activity for methane combustion, we synthesized a series of αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (1 ≤ α ≤ 20) and evaluated their catalytic performance. The X-ray diffraction (XRD) peaks in Fig. 5(a) were accurately indexed to PdO, the Sr3Fe2O6 phase, and α-Al2O3, with no impurity phases detected. Notably, the intensity of the PdO peak increased with higher PdO loading, leading to an increased calculated crystallite size of PdO (refer to Table 5). Conversely, the specific surface area of the αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (1 ≤ α ≤ 20) decreased as PdO loading increased (see Table 5). This decrease can be attributed to the aggregation of PdO particles on the catalyst's surface. High-resolution TEM images obtained via Cs-TEM revealed Pd(200) crystal lattices with a d-spacing of 0.22 nm in 13wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (see Fig. S8(b)†). The particle size of PdO was approximately 6–7 nm, which is smaller than crystallite size of PdO calculated from the XRD analysis results. These differences may be due to the aggregation of PdO in high concentration PdO supported αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts. Additionally, EDS images (Fig. S8(a)†) confirm the homogeneous distribution of Pd, Sr, Fe, and Al elements, affirming the uniformity of the as-prepared catalyst samples. While Al elements are widely distributed, Sr, Fe, and Pd are locally and uniformly distributed. These results indicate that PdO and Sr3Fe2O7−δ are effectively supported on surface of α-Al2O3, with PdO expected to exist either in the form of PdO/Sr3Fe2O7−δ or independently on the surface of Al2O3.61–63 | ||
| Fig. 5 (a) XRD pattern of αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts with different PdO loadings (α = 1, 3, 5, 10, 13, 15, 20). (b) O2-TPD profile of αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts with different PdO loadings (α = 5, 10, 13, 15, 20). | ||
| Sample | PdO crystallite size/nm | Specific surface area/m2 g−1 |
|---|---|---|
| γ-Al2O3 | — | 148.0 |
| α-Al2O3 | — | 14.6 |
| Sr3Fe2O7 | — | 30.5 |
| 10wt%PdO/SrFeO3−δ | 19.1 | 21.6 |
| 10wt%PdO/α-Al2O3 | 3.7 | 2.5 |
| 16wt%Sr3Fe2O7−δ/α-Al2O3 | — | 80.9 |
| 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 | 9.2 | 70.2 |
Fig. 5(b) presents the O2-TPD profile recorded on the αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (5 ≤ α ≤ 20). When PdO was supported up to 13wt%, the oxygen desorption area continued to increase, and the desorption temperature decreased to around 100 °C of low temperature. The highest oxygen desorption area was obtained at the sample of α = 13 wt%. The increase of the oxygen desorption area can be explained by the production of oxygen vacancies on the crystal surface and within the lattice to maintain charge compensation, because Fe4+ is reduced to Fe3+ during CH4 gas decomposition reaction. However, in range of α > 13wt%, the amount of oxygen desorption decreased, and the desorption temperature increased. It can be inferred that the formation of oxygen vacancies can be suppressed by excessive aggregation of Pd metal in range of α > 13wt% caused to reduce the property of electron donor. From this O2-TPD analysis, the density of oxygen vacancy was estimated based on the desorption amount of O2, as shown in Table 5. Among the prepared catalysts, 13wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts showed the highest oxygen vacancy density of 31.71 × 1017 site m−2. We assessed the catalytic activity for methane combustion over the αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 (1 ≤ α ≤ 20) catalysts, and the results are presented in Fig. 6(a). The temperature required for complete methane combustion decreased to 300 °C with an increase in PdO loading to α = 13. However, at higher PdO loadings (α ≥ 15), the temperature of complete methane combustion rose to 325 °C, and catalytic activity decreased due to PdO aggregation and a reduction in the catalysts' surface area. These findings confirmed that the optimal PdO loading for methane combustion is 13 wt%. To investigate the effect on the catalytic activity for methane combustion in more detail, we calculated the TOF and specific reaction rates for the methane conversion reaction, varying the loading amount of PdO and the density of oxygen vacancies. In Fig. 6(b), the correlations of the specific reaction rate (r225°C) and the turnover frequency (TOF) to the density of PdO active sites at 275 °C for oxidation of methane for the various catalysts is presented. To isolate the effect of PdO loading, we normalized the TOF and the specific reaction rate at 225 °C with surface density and mass of PdO active site. Additionally, it was determined that the specific reaction rate and TOF also continuously increased as the oxygen vacancy density increased (Fig. 6(c)). The highest oxygen vacancy density and the fastest methane combustion rate were observed when the PdO loading amount was 13 wt%. The 13wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst showed methane conversion of 100% at the lowest temperature of 300 °C. It can be determined that the highest oxygen vacancy density and surface oxygen species in the catalyst promoted methane combustion activity in combination with PdO active species. The catalyst recyclability test for methane combustion was performed using the 13wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst and the results are shown in Fig. S9(a).† Although the methane combustion activity decreased slightly at some temperature with each iteration of methane combustion analysis, they are showed the same temperature of initial and complete methane combustion. From the XRD patterns of the samples after cycling test, no impurity phase corresponding to the decomposition of the Sr3Fe2O7−δ catalyst was observed (Fig. S9(b)†). Additionally, the methane combustion activity of αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts with low PdO loading amount (α = 1, 3) under a methane concentration of 2.5% and mass hourly space velocity of 120000 L kg−1 h−1 are shown in Fig. S9(c).† 1wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst did not show the complete methane combustion activity under 600 °C. On the other hand, 3wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalyst showed the complete methane combustion activity at 400 °C with T50 of around 280 °C and T90 of 350 °C. In similar methane concentration and PdO loading amount, the novel catalyst based on Sr3Fe2O7−δ perovskite oxide prepared in this study showed superior methane combustion activity under low temperature compared to the reported catalyst.64,65
 | ||
| Fig. 6 (a) Temperature-dependence of methane oxidation over the α wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts with different PdO loadings (α = 1, 3, 5, 10, 13, 15, 20). (b) Correlations of the specific reaction rate (r225°C) and the turnover frequency (TOF) to the density of PdO active sites at 225 °C for oxidation of methane over the αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (α = 5, 10, 13, 15, 20). (c) Correlations of the specific reaction rate and TOF to the density of oxygen vacancy estimated based on the amount of O2 derived from the O2-TPD analysis of the αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 catalysts (α = 5, 10, 13, 15, 20). | ||
Conclusions
We prepared PdO/Sr3Fe2O7−δ/α-Al2O3 composite oxide catalysts using a co-precipitation and impregnation method and assessed their catalytic performance in low-temperature methane combustion. Sr3Fe2O7 perovskite oxide is known for its exceptional oxygen storage and release capabilities. However, to utilize it effectively in low-temperature methane oxidation, we needed to enhance its structural and thermal stability. This was essential to prevent the transformation of the Sr3Fe2O7 perovskite structure, as it can change to SrFeO3−δ when supporting PdO active species on Sr3Fe2O7 oxide and calcining at 400 °C for 4 hours. In this study, we successfully synthesized αwt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 composite oxide catalysts with Sr3Fe2O7−δ as a co-catalyst. These catalysts formed only oxygen vacancies without distorting the crystal structure of Sr3Fe2O7. By employing an α-Al2O3 support, we improved the thermal stability of the catalysts. The structural integrity remained intact even after heat treatment at 600 °C. Notably, the 16wt%Sr3Fe2O7−δ/α-Al2O3 co-catalyst, without PdO active species, displayed no methane oxidation activity. In comparison, the 10wt%PdO/SrFeO3−δ catalyst, lacking α-Al2O3 support, exhibited lower methane oxidation activity compared to 10wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3. This comparison solidified the position of the composite oxide catalyst, comprising PdO/Sr3Fe2O7−δ/α-Al2O3, as the superior choice. It offered higher oxygen vacancy density and surface oxygen species, making it the most suitable catalyst for methane oxidation among various compositions. Finally, after optimizing the PdO loading, the 13wt%PdO/16wt%Sr3Fe2O7−δ/α-Al2O3 composite oxide catalyst achieved complete methane combustion at the remarkably low temperature of 300 °C, demonstrating the highest methane oxidation activity. Also, this study underscores the potential of this novel catalyst, where Sr3Fe2O7−δ perovskite oxide plays a pivotal role as a promoter in methane combustion at low temperature even with low PdO loading amount of 3 wt%.Data availability
The datasets used and/or analysed during the current study for the article cannot be made available to recommended repositories due to legal confidentiality requirements for research data management plans (DMPs) of Funder guidelines.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the Ministry of Trade, Industry and Energy, Korea, under the “World Class Plus Program (R&D, P177000004)” supervised by the Korea Institute for Advancement of Technology (KIAT). This research was supported by Korea Institute of Marine Science & Technology Promotion (KIMST) Funded by the Ministry of Oceans and Fisheries, Korea (RS-2023-00256331).References
- H. Lin, Y. Liu, J. Deng, L. Jing and H. Dai, Catalysts, 2023, 13(2), 427 CrossRef CAS.
- Y. Gao, M. Jiang, L. Yang, Z. Li, F. Tian and Y. He, Front. Chem., 2022, 10, 959422 CrossRef CAS PubMed.
- L. He, Y. Fan, J. Bellettre, J. Yue and L. Luo, Front. Chem., 2020, 119, 109589 CAS.
- V. A. Likhanov and O. P. Lopatin, Therm. Eng., 2017, 64, 935–944 CrossRef CAS.
- T. Y. Kim, S. B. Jo, C. H. Lee, S. H. Kang, J. W. Kim, S. C. Lee and J. C. Kim, Korean J. Chem. Eng., 2020, 37, 1690–1698 CrossRef CAS.
- Y. S. Yu, X. W. Zhang, J. Liu, Y. Lee and X. S. Li, Energy Environ. Sci., 2021, 14, 5611–5668 RSC.
- G. Zhao, X. Pan, Z. Zhang, Y. Liu and Y. Lu, J. Catal., 2020, 384, 122–135 CrossRef CAS.
- S. J. Yeo, J. S. Kim and W. J. Lee, J. Cleaner Prod., 2021, 30, 129955 Search PubMed.
- I. Tvedten, S. Bauer and J. Clean, Energy Res. Soc. Sci., 2022, 86, 102423 CrossRef.
- B. Wang, S. A. Suazo, Y. P. Torres and E. Nikolla, Catal. Today, 2017, 285, 147–158 CrossRef CAS.
- K. Murata, D. Kosuge, J. Ohyama, Y. Mahara, Y. Yamamoto, S. Arai and A. Satsuma, ACS Catal., 2019, 10, 1381–1387 CrossRef.
- M. Cargnello, J. D. Jaén, J. H. Garrido, K. Bakhmutsky, T. Montini, J. C. Gámez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- F. F. Tao, J. J. Shan, L. Nguyen, Z. Wang, S. Zhang, L. Zhang, Z. Wu, W. Huang, S. Zeng and P. Hu, Nat. Commun., 2015, 6, 7798 CrossRef CAS PubMed.
- G. Ercolino, P. Stelmachowski, G. Grzybek, A. Kotarba and S. Specchia, Appl. Catal., B, 2017, 206, 712–725 CrossRef CAS.
- T. M. Onn, S. Zhang, L. Arroyo-Ramirez, Y. C. Chung, G. W. Graham, X. Pan and R. J. Gorte, ACS Catal., 2015, 5, 5696–5701 CrossRef CAS.
- X. Zou, Z. Rui and H. Ji, ACS Catal., 2017, 7, 1615–1625 CrossRef CAS.
- J. H. Park, J. H. Cho, Y. J. Kim, E. S. Kim, H. S. Han and C. H. Shin, Appl. Catal., B, 2014, 160–161, 135–143 CrossRef CAS.
- H. Peng, C. Rao, N. Zhang, X. Wang, W. Liu, W. Mao, L. Han, P. Zhang and S. Dai, Angew. Chem., Int. Ed., 2018, 57, 8953–8957 CrossRef CAS PubMed.
- Q. Dai, Q. Zhu, Y. Lou and X. Wang, J. Catal., 2018, 357, 29–40 CrossRef.
- A. R. Supandi, N. Nunotani and N. Imanaka, J. Asian Ceram. Soc., 2020, 8, 745 CrossRef.
- P. G. Choi, N. Nunotani and N. Imanaka, J. Asian Ceram. Soc., 2018, 6, 368–373 CrossRef.
- Y. B. Choi, N. Nunotani and N. Imanaka, Mater. Lett., 2020, 278(1–4), 128392 CrossRef CAS.
- Y. B. Choi, N. Nunotani, K. Morita and N. Imanaka, J. Asian Ceram. Soc., 2022, 10, 178 CrossRef.
- Y. B. Choi, N. Nunotani, K. Morita and N. Imanaka, Catalysts, 2022, 12, 69 CrossRef CAS.
- M. Jeong, N. Nunotani and N. Imanaka, Bull. Chem. Soc. Jpn., 2018, 91, 158 CrossRef CAS.
- Y. Ding, Y. Jia, M. Jiang, Y. Guo, L. Wang, Q. Ke, M. N. Ha, S. Dai and W. Zhan, Chem. Eng. J., 2021, 416, 129150 CrossRef CAS.
- M. Alyani and K. J. Smith, Ind. Eng. Chem. Res., 2016, 55, 8309–8318 CrossRef CAS.
- D. Ciuparu, M. R. Lyubovsky, E. Altman, L. D. Pfefferle and A. Datye, Catal. Rev.:Sci. Eng., 2002, 44, 593–649 CrossRef CAS.
- W. R. Schwartz, D. Ciuparu and L. D. Pfefferle, J. Phys. Chem. C, 2012, 116, 8587–8593 CrossRef CAS.
- R. Gholami, M. Alyani and K. J. Smith, Catalysts, 2015, 5, 561–594 CrossRef CAS.
- K. Persson, L. D. Pfefferle, W. Schwartz, A. Ersson and S. G. Jaras, Appl. Catal., B, 2007, 74, 242–250 CrossRef CAS.
- S. J. Ramos, G. S. Dinali, C. Oliveira, G. C. Martins, C. G. Moreira, J. Siqueira and L. R. G. Guilherme, Curr. Pollut. Rep., 2016, 2, 28–50 CrossRef CAS.
- J. Wu, R. Ye, D. J. Xu, L. Wan, T. R. Reina, H. Sun, Y. Ni, Z. F. Zhou and X. Deng, Front. Chem., 2022, 10, 961355 CrossRef CAS PubMed.
- P. Zhang, Z. Lou, L. Gong, J. Xu, Q. Chen, M. J. Reece, H. Yan, Z. Dashevsky and F. Gao, J. Alloys Compd., 2023, 937, 168366 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- L. He, Y. L. Fan, J. Bellettre, J. Yue and L. G. Luo, Renewable Sustainable Energy Rev., 2020, 119, 109589 CrossRef CAS.
- L. He, Y. L. Fan, F. C. Guo, K. R. Yang, J. A. Heinlein, S. M. Bamonte and J. J. Fee, J. Am. Chem. Soc., 2020, 142, 17119–17130 CrossRef PubMed.
- F. Zasada, J. Janas, W. Piskorz, M. Gorczyńska and Z. Sojka, ACS Catal., 2017, 7, 2853–2867 CrossRef CAS.
- K. Beppu, S. Hosokawa, A. Demizu, Y. Oshino, K. Tamai, K. Kato, K. Wada, H. Asakura, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2018, 21, 11186–11193 CrossRef.
- L. Peng, Q. Li, L. Sun and H. Zhao, Catalysts, 2021, 11, 1400 CrossRef CAS.
- H. Falcon, J. A. Barbero, J. A. Alonso, M. J. Martınez-Lope and J. L. G. Fierro, Chem. Mater., 2002, 14, 2325–2333 CrossRef CAS.
- R. Bornovski, L. Huang, E. P. Komarala, J. M. Rondinelli and B. A. Rosen, ACS Appl. Mater. Interfaces, 2019, 11, 33850–33858 CrossRef CAS PubMed.
- T. Akbay, A. Staykov, J. Druce, H. Téllez, T. Ishihara and J. A. Kilner, J. Mater. Chem. A, 2016, 4, 13113–13124 RSC.
- D. Halwidl, W. Mayr-Schmölzer, M. Setvin, D. Fobes, J. Peng, Z. Mao, M. Schmid, F. Mittendorfer, J. Redinger and U. A. Diebold, J. Mater. Chem. A, 2018, 6, 5703–5713 RSC.
- W. Tan, D. Huan, W. Yang, N. Shi, W. Wang, R. Peng, X. Wu and Y. A. Lu, RSC Adv., 2018, 8, 26448–26460 RSC.
- B. Yang, X. Wu, B. K. Inusa, D. H. Kuo, P. Zhang, C. Li, T. Liu, D. Lu, Z. Yuan, J. Lin and X. Chen, Chem. Eng. J., 2025, 514, 163151 CrossRef CAS.
- X. Wu, T. L. Wan, B. Yang, D. H. Kuo, P. Zhang, M. Liu, S. N. Adawara, D. Lu, J. Lin and X. Chen, J. Mater. Chem. A, 2025, 13, 17976 RSC.
- Z. Su, B. Wu, C. Li, D. H. Kuo, P. Zhang, L. Chen, D. Lu, J. Lin, X. Chen and Z. Yuan, Chem. Eng. J., 2025, 510, 161621 CrossRef CAS.
- Y. Ling, F. Wang, R. A. Budiman, T. Nakamura and K. Amezawa, Phys. Chem. Chem. Phys., 2015, 17, 7489–7497 RSC.
- Y. Ling, F. Wang, Y. Okamoto, T. Nakamura and K. Amezawa, Solid State Ionics, 2016, 288, 298–302 CrossRef CAS.
- Z. Wang, W. Yang, S. P. Shafi, L. Bi, Z. Wang, R. Peng, C. Xia, W. Liu and Y. Lu, J. Mater. Chem. A, 2015, 3, 8405–8412 RSC.
- Y. Zhou, Z. Zhou, J. Sun, L. Liu, F. Luo, G. Xu, X. Cao and M. Xu, EcoMat, 2023, 5, 12347 CrossRef.
- K. Tamai, S. Hosokawa, H. Okamoto, H. Asakura, K. Teramura and T. Tanaka, ACS Appl. Mater. Interfaces, 2019, 11, 26985–26993 CrossRef CAS PubMed.
- A. Takamatsu, K. Tamai, S. Hosokawa, T. Tanaka, M. Ehara and R. Fukuda, ACS Appl. Mater. Interfaces, 2021, 13, 7216–7226 CrossRef CAS PubMed.
- K. Tamai, S. Hosokawa, K. Onishi, C. Watanabe, K. Kato, H. Asakura, K. Teramura and T. Tanaka, ACS Catal., 2020, 10, 2528–2537 CrossRef CAS.
- M. Jeong, N. Nunotani, N. Moriyama and N. Imanaka, J. Asian Ceram. Soc., 2016, 4, 259 CrossRef.
- B. Wu, B. Yang, X. Wu, D. H. Kuo, Z. Su, L. Chen, P. Zhang, M. T. Mosisa, D. Lu, Z. Yuan, J. Lin and X. Chen, ACS Appl. Mater. Interfaces, 2024, 16, 58764–58779 CrossRef CAS PubMed.
- Z. Su, B. Wu, D. H. Kuo, L. Chen, P. Zhang, B. Yang, D. Lu, J. Lin and X. Chen, J. Mater. Chem. A, 2024, 12, 28486 RSC.
- P. Zhang, L. Chen, D. H. Kuo, B. Wu, Z. Su, D. Lu, Q. Wu, J. Li, J. Lin and X. Chen, J. Mater. Chem. A, 2024, 12, 7163 RSC.
- X. Chen, T. Huang, D. H. Kuo, H. Sun, P. Li, O. A. Zelekew, A. B. Abdeta, Q. Wu, J. Zhang, Z. Yuan and J. Lin, Appl. Catal., B, 2021, 298, 120542 CrossRef CAS.
- X. Chen, H. Sun, D. H. Kuo, A. B. Abdeta, O. A. Zelekew, Y. Guo, J. Zhang, Z. Yuan and J. Lin, Appl. Catal., B, 2021, 287, 119992 CrossRef CAS.
- L. Chen, P. Zhang, D. H. Kuo, J. Jiang, B. Wu, Z. Su, O. A. Zelekew, J. Lin, D. Lu and X. Chen, J. Mater. Chem. A, 2024, 12, 9871–9885 RSC.
- Y. J. Zhang, J. R. Niu, G. Z. Wang, D. Li, H. X. Dai, H. He and W. G. Qiu, J. Rare Earths, 2006, 24, 1–5 Search PubMed.
- Z. X. Wu, J. G. Deng, Y. X. Liu, S. H. Xie, Y. Jiang, X. T. Zhao, J. Yang, H. Arandiyan, G. S. Guo and H. X. Dai, J. Catal., 2015, 332, 13–24 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02851e |
| ‡ These authors contributed equally to this work. |
| § Current address: Advanced Resources Team, Yeongwol Industrial Promotion Agency, 21–28 Palgoe 1 Nonggongdanji, Yeongwolgun 26240, Republic of Korea. |
| This journal is © The Royal Society of Chemistry 2025 |