DOI:
10.1039/D5RA02745D
(Paper)
RSC Adv., 2025,
15, 19954-19965
Rational design and synthesis of potent active antimicrobial peptides based on American oyster defensin analogue A3†
Received
19th April 2025
, Accepted 3rd June 2025
First published on 12th June 2025
Abstract
The rise of drug-resistant microbes is increasingly recognized as a significant global health challenge. Microbial drug resistance diminishes the efficacy of existing treatment options, underscoring the urgent necessity for the development of novel antibiotic candidates to effectively address infections. Antimicrobial peptides, owing to their distinctive antibacterial mechanisms, are considered a promising alternative to conventional antibiotics. In this work, we modified the structure of the American oyster defensin (AOD) analogue A3 to obtain four novel antimicrobial peptides (D-A3, A3-C4, A3-C5, A3-C6). These synthesized peptides exhibited broad-spectrum antibacterial activity. Notably, it was demonstrated that A3-C4, A3-C5, and A3-C6 showed enhanced glutathione (GSH) stability compared to A3, while D-A3 exhibited superior protease stability. Importantly, none of the peptides displayed hemolytic toxicity. Mechanistic investigations suggested that the synthesized peptides exert their anti-bacterial effects primarily through membrane disruption. D-A3 and A3-C6 were peptides with the best antibacterial activity and enzymatic stability among the synthesized derived peptides. These studies of D-A3 and A3-C6 will contribute to the development of new candidate drugs for the treatment of microbial infections.
1. Introduction
Severe infections caused by microorganisms have posed significant challenges to humanity for centuries. Prior to the advent of antibiotics, infections such as pneumonia, plague, and syphilis were associated with high mortality rates, leading to substantial human casualties and economic losses.1–3 The discovery and subsequent utilization of antibiotics, exemplified by penicillin, have been instrumental in saving innumerable lives over the past century.4,5 The rapid advancement of technology has facilitated the successive development of numerous antibiotics, empowering healthcare professionals to effectively combat infections. Today, antibiotics are employed across various sectors, including healthcare, food production, and aquaculture.6–8
The extensive and pervasive utilization of antibiotics has resulted in a multitude of consequences. Primarily, prolonged antibiotic use contributes to the escalation of antimicrobial resistance (AMR), which progressively diminishes the efficacy of these agents.9 Additionally, antibiotic resistance can render certain antibiotics entirely ineffective against specific pathogens, necessitating a return to previously discontinued medications that were abandoned due to toxicity or adverse side effects.10 The presence of severe infections caused by resistant bacteria not only extends treatment durations and complicates patient treatment but also exacerbates both the economic and physical burdens on affected individuals.11 Currently, the swift emergence of highly resistant bacterial strains has attracted considerable attention.12 Predictions indicates that by the mid-21st century, antibiotic resistance could result in over 10 million fatalities annually. Should this trend persist without intervention, humans may revert to a period where microbial infections are largely unmanageable.13 Consequently, microbial resistance has emerged as a significant global health threat, thereby incentivizing researchers to develop new anti-infective therapeutics.14
In recent years, antimicrobial peptides (AMPs) have been recognized as significant therapeutic agents in combating infections. Several antimicrobial peptides, including Polymyxin, Teicoplanin, Bacitracin, and Vancomycin (often regarded as the “last line of defense” against infections), have been approved for commercial use.15,16 AMPs are ubiquitously found across various organisms and serve as the primary defense mechanism in the innate immune system, with a broad spectrum of antibacterial activity and low toxicity.17 Because they have a positively charged surface, most AMPs exert their bactericidal effects by disrupting bacterial cell membranes. This mode of action is distinct from that of conventional antibiotics, rendering AMPs less susceptible to the development of drug resistance.18–20 Due to their superior antibacterial efficacy and minimal toxic side effects, AMPs hold promises as potent agents for the clinical management of drug-resistant bacterial infections.21,22
American oyster defensin (AOD) is an AMP consisting of 38 amino acids and three disulfide bonds, showing significant antibacterial efficacy against both Gram-positive (G+) and Gram-negative (G−) bacteria. Nonetheless, its intricate structure results in elevated synthesis costs, thereby restricting its practical application.23 To develop AOD as a potential antibiotic, Seo et al. engineered and synthesized five arginine-rich cyclic 9-peptide according to AOD. Among these analogues, the A3 exhibited superior activity with minimum inhibitory concentration (MIC) values ranging from 0.4 to 6.5 μg mL−1 against B. subtilis, S. epidermidis, S. mutans, A.hydrophila, E. coli, P. aeruginosa, and S. enterica. Subsequent validation indicated that the A3 peptide had low hemolytic toxicity towards human red blood cells and minimal cytotoxicity against human dermal fibroblast (HDF) cells, rendering it a promising candidate for novel antibiotic development.24 Despite the considerable potential of the A3 as an AMP, several challenges impede the application of this candidate molecule. Primarily, as a cyclic peptide, its disulfide bond is prone to degradation in reducing environments within the body, which may result in diminished or lost activity. Potential strategy to address this issue involves substituting disulfide bond with triazole ring, synthesized through click chemistry reactions, thereby mitigating the negative effects associated with disulfide bond. Additionally, the rapid degradation of peptides by proteases in the body can lead to their inactivation. To prevent such enzymatic degradation, peptides can be synthesized using D-amino acids. The specificity of proteases prevents them from degrading peptides composed of D-amino acids.25–28
In this work, we improved the stability of the A3 through structural modifications, including substitution of all L-amino acids with D-amino acids or the replacement of the disulfide bond with a triazole ring. Furthermore, we synthesized the D-A3 (consisting of D-amino acids) and three triazole-containing peptides (A3-C4, A3-C5, A3-C6). We further examined the antibacterial activity, mechanism of action, stability, and toxicity of these peptides to acquire lead peptides with best antibacterial activity, enzymatic stability and serum stability for the development of novel antibiotic candidates.
2. Materials and methods
2.1. Materials
2.1.1. Test strain. Gram-positive bacteria: S. aureus, S. aureus ATCC-25923, B. subtilis CMCC-63501, B. cereus. Gram-negative bacteria: E. coli ATCC-25922, P. aeruginosa ATCC-27853, S. enterica, S. para-typhi B. The above bacteria were provided by the Medical Laboratory of Baotou Medical College.
2.1.2. Laboratory animals and breeding materials. The AB strain zebrafish was preserved and raised by the zebrafish model animal platform of the translational medicine platform of Baotou Medical College. The feeding method of zebrafish was as follows: 12 hours of light/12 hours of darkness alternating, water temperature 28 ± 1 °C, pH 7 ± 1.0, conductivity 485 s cm−1, and feeding the harvest shrimp twice a day.
2.1.3. Reagents and instruments. Fmoc-protected amino acids were obtained from GL Biochem (Shanghai)Ltd, Rink Amide AM resin was sourced from Tianjin Nankai Hecheng, and all other reagents were of analytically pure grade and commercially available. The CHA-S constant temperature oscillator was provided by Changzhou Guohua Electric Appliance Co., Ltd, the THZ-92A air bath constant temperature oscillator was from Boxun, the FD-1-50 vacuum freeze dryer was supplied by Beijing BIOCOOL, the Shimadzu high-performance liquid chromatography LC-20 was manufactured by Shimadzu Corporation, the SIL-16 autosampler for high-performance liquid chromatography was utilized, and the Thermo Scientific Ultimate 3000 preparative analytical chromatograph was employed. The J-815 circular dichroism spectrometer was obtained from Jasco (Shanghai) Trading Co., Ltd, the Lichen clean bench (SW-CJ-1FD) was used, the HWS-150 electric constant temperature incubator was from Beijing Kewei Yongxing Instrument Co., Ltd, the YXQ-LS-50A vertical pressure steam sterilizer was sourced from Shanghai Boxun Industrial Co., Ltd, the 0.5 McFarland turbidity tube was provided by the Medical Laboratory Technology Laboratory of Baotou Medical College, the Titan mini centrifuge (TIMI-10K) was used, the 1510 microplate reader was from Thermo Scientific, the Leica stereomicroscope (Stereozoom S9I) was utilized, and columns including Welch XB-C18 (4.6 mm × 250 mm, 5 μm) from Yuexu Technology (Shanghai) Co., Ltd, and Hypersil-GOLDTM preparative column (10 × 250 mm, 5 μm) from Thermo Scientific were employed.
2.2. Synthesis of A3 and its derived peptides
2.2.1. Synthesis of A3 and D-A3. The A3 peptide sequence is: NH2-CRRWRRRRC-OH, the structure of A3 is shown in Fig. S1.† We synthesized the A3 peptide and the D-A3 peptide, which consists entirely of D-amino acids, using 300 mg of Rink amide resin (substitution degree of 0.37 mmol g−1) and solid-phase peptide synthesis (SPPS) based on N-9-fluorenylmethoxycarbonyl (Fmoc) protection. The amino acid![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCTU:DIEA molar ratio for coupling was 4 eq.:3.8 eq.:8 eq. After swelling the resin and removing the protecting group, amino acids were coupled, with each amino acid requiring two coupling steps (45 min each) to ensure complete reaction. The coupling and deprotection steps were repeated, connecting all amino acids in sequence. The Kaiser test was used for deprotection and coupling verification after each step. After all amino acids were coupled, the protecting group of the last amino acid was removed. The resin was washed, and then 10 mL of cleavage reagent (TFA:H2O:TIPS = 95%:2.5%:2.5%) was added for cleavage, with a reaction time of 3 hours. After concentrating by nitrogen bubbling and precipitating via centrifugation with ice-cold methyl tert-butyl ether, the yellow crude peptide was obtained.
:HCTU:DIEA molar ratio for coupling was 4 eq.:3.8 eq.:8 eq. After swelling the resin and removing the protecting group, amino acids were coupled, with each amino acid requiring two coupling steps (45 min each) to ensure complete reaction. The coupling and deprotection steps were repeated, connecting all amino acids in sequence. The Kaiser test was used for deprotection and coupling verification after each step. After all amino acids were coupled, the protecting group of the last amino acid was removed. The resin was washed, and then 10 mL of cleavage reagent (TFA:H2O:TIPS = 95%:2.5%:2.5%) was added for cleavage, with a reaction time of 3 hours. After concentrating by nitrogen bubbling and precipitating via centrifugation with ice-cold methyl tert-butyl ether, the yellow crude peptide was obtained.Oxidation of peptides was carried out using dimethyl sulfoxide (DMSO) as the oxidant. Specifically, 3 mg of crude peptide was dissolved in 0.9 mL of phosphate buffer solution (PBS, pH 7.4), and then mixed with 100 μL of DMSO. The mixture was placed on a magnetic stirrer and allowed to react at room temperature. The progress of the reaction was monitored using HPLC. After 90 minutes of reaction, the cysteine residues on both sides of the A3 peptide were oxidized to form a cyclic peptide with a disulfide bond. Subsequently, the linear peptides and cyclic peptides were separated and purified using semi-preparative RP-HPLC. The peptides were then analyzed using ESI-MS.
2.2.2. Synthesis of A3-C4, A3-C5 and A3-C6. During the substitution of Cys, we replaced the C-terminal Cys of the A3 peptide with L-propargylglycine, while substituting the N-terminal Cys with (2S)-2-amino-4-azidobutanoic acid, (2S)-2-amino-5-azidovaleric acid, and (2S)-2-amino-6-azidohexanoic acid. The synthesis method was the same as that for the A3 peptide. When coupling non-natural amino acids (such as (S)-2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-azidobutanoic acid and Fmoc-L-propargylglycine), the molar ratio of amino acid:HATU:DIEPA used was 2 eq.:1.9 eq.:4 eq. After completing all amino acid couplings, the protecting groups were removed, and the resin was washed and cleaved. Nitrogen was then bubbled through to concentrate and remove excess solvent, followed by precipitation with ice-cold methyl tert-butyl ether and centrifugation to obtain the yellow crude peptide.The click chemistry reaction was catalyzed using copper sulfate. Specifically, 0.5 mM (approximately 7 mg) of linear A3-C4 crude peptide was dissolved in 0.9 mL of pure water. In a 2 mL centrifuge tube, 4 mg of anhydrous copper sulfate (CuSO4) and 11.8 mg of sodium ascorbate were weighed and dissolved in 2 mL of pure water. Initially, the solution appeared dark brown, but upon vigorous shaking, it rapidly turned orange-yellow. At this point, 100 μL of the mixed solution was promptly added to the peptide solution using a pipette, and the mixture was stirred and allowed to react at room temperature. The reaction progress was monitored using HPLC. After reacting for 90 minutes, the linear and cyclic peptides were separated and purified using semi-preparative RP-HPLC. The purified peptides were then obtained through freeze-drying. Both the purified linear A3-C4, linear A3-C5, linear A3-C6 and A3-C4, A3-C5, A3-C6 were analyzed using ESI-MS.
2.3. Circular dichroism
The secondary structural characteristics of A3 and A3-derived peptides (D-A3, A3-C4, A3-C5, A3-C6) in aqueous solution (pH 5.60) were tested using a circular dichroism spectrometer. The testing method involved preparing 1 mg mL−1 peptide solutions of the five peptides using pure water. A quartz cuvette with a 1 mm path length was used, with a scanning bandwidth of 1 nm, a scanning wavelength ranging from 300 nm to 170 nm, and a scanning speed of 200 nm min−1. Each sample's CD spectrum was measured three times, and the average value was taken.
2.4. Antibacterial activity of A3 and derived peptides
The minimum inhibitory concentrations (MIC) of A3 and its four derivative peptides against S. aureus, S. aureus ATCC-25923, B. subtilis CMCC-63501, B.cereus, E. coli ATCC-25922, P. aeruginosa ATCC-27853, S. enterica, and S. para-typhi B were determined using the agar well diffusion method.29 Bacterial suspensions at a concentration of 1 × 108 CFU mL−1 were prepared from logarithmic phase bacteria and used to inoculate nutrient agar plates containing 5% bacteria. Sterile 3 mm punchers were used to create uniform wells in the agar. The peptide solution was prepared at a maximum concentration of 1 mg mL−1 in 0.01% acetic acid aqueous solution, followed by two-fold serial dilutions. Subsequently, 10 μL of each peptide solution at different concentrations was carefully added to the wells, with 0.01% acetic acid aqueous solution serving as the negative control. Ten microliters of each peptide solution at different concentrations were carefully added to the wells, and 0.01% acetic acid water was used as a negative control. Three replicates were performed for each bacterial strain. The plates were incubated at 37 °C for 24 hours. After incubation, the plates were examined for the presence of zones of inhibition, and the corresponding peptide concentrations were recorded as the MIC. After the incubation period, check whether there are inhibition zones on the plates, and take the concentration corresponding to the appearance of the smallest inhibition zone as the MIC.
2.5. Antibacterial mechanism of A3 and its derived peptides
2.5.1. SEM. The bacteria in the logarithmic phase were prepared to 5 mL suspension, with a concentration of 1 × 108 CFU mL−1. The bacteria were collected by centrifuging at 5000 rpm for 8 minutes. The bacteria were treated with an antimicrobial peptide at a concentration of 1 × MIC. Bacteria treated with an equal volume of PBS were used as a blank group. After fully mixing, the drug and the bacteria were incubated at 37 °C for 2 hours. The mixture was centrifuged at 5000 rpm for 8 minutes, then the supernatant was carefully discarded. To the sediment, 2.5% glutaraldehyde was added and fixed overnight at 4 °C. Subsequently, the gradient dehydration was carried out for the fixed bacteria, using 50%, 70%, 90%, 95%, and 100% ethanol, with each dehydration step lasting for 10 minutes. After drying, a small amount of the sample was taken onto a conductive adhesive, then the sample was sprayed with gold. Finally, the sample was observed using SEM.
2.5.2. TEM. The bacteria in the logarithmic phase were prepared to 5 mL suspension, with a concentration of 1 × 108 CFU mL−1. The bacteria were collected by centrifuging at 5000 rpm for 8 minutes. The bacteria were treated with an antimicrobial peptide at a concentration of 1 × MIC. The bacteria treated with an equal volume of PBS were used as a blank group. After fully mixing, the drug and the bacteria were incubated at 37 °C for 2 hours. The mixture was centrifuged at 5000 rpm for 8 minutes, and then the supernatant was carefully discarded. To the sediment, add 2.5% glutaraldehyde. The sediment was fixed overnight at 4 °C. Subsequently, the gradient dehydration was carried out for the fixed bacteria, using 50%, 70%, 90%, 95%, and 100% ethanol, with each dehydration step lasting for 10 minutes. After drying, the sample was dispersed in ethanol and dropped onto an ultra-thin carbon film. Finally, excess sample was removed, and the remaining sample was observed using TEM.
2.5.3. Crystal violet staining. Bacterial colonies in the logarithmic growth phase were collected to prepare a 5 mL bacterial suspension with a concentration of 1 × 108 CFU mL−1. A 200 μL aliquot of this suspension was transferred into 20 mL of LB liquid medium for subsequent experimentation. In a 96-well plate, 100 μL of physiological saline was added to serve as the blank control. For the experimental groups, solutions of A3, D-A3, and A3-C6 were introduced to achieve final concentrations of ½ × MIC, 1 × MIC, 2 × MIC, and 4 × MIC, respectively. Subsequently, 100 μL of the prepared bacterial suspension was added to both the blank control and experimental groups, followed by incubation at 37 °C for 24 hours. After incubation, the 96-well plate was removed from the 37 °C shaker and centrifuged at 3000 rpm for 5 minutes. The supernatant was discarded, and the plate was washed three times with PBS before being air-dried at room temperature. Subsequently, 200 μL of a 10% methanol solution was added to fix the biofilms for 15 minutes, followed by three additional washes with PBS. The biofilms were then stained with 0.1% crystal violet for 10 minutes, washed three times with PBS, and subsequently treated with 200 μL of 95% ethanol. The plate was subjected to gentle agitation at ambient temperature for a duration of 10 minutes, after which the optical density (OD) at a wavelength of 575 nm was recorded.
2.6. Stability of A3 and its derived peptides
2.6.1. Stability of A3 and derived peptides under reducing condition. Stability testing of A3 peptide and its derivatives was conducted using reduced GSH. GSH was dissolved in PBS (pH 7.4) to prepare a 100 mM stock solution. The A3 peptide and its derivatives were also respectively dissolved in PBS to prepare a 1 mM stock solution. The reaction between GSH and the peptide (at a molar ratio of 4:1) was carried out at room temperature. The reaction was monitored using HPLC at 0 min, 1 min, 5 min, 10 min, and 20 min. The reaction was then quenched by adding 50 μL of 2 M HCl to 10 μL reaction mixture. The residual peptide at each time point was monitored by HPLC. The degradation curve was plotted based on the residual amount.
2.6.2. Study on protease stability of A3 and its derived peptides. The trypsin at a concentration of 0.1 ng mL−1 and peptide solutions at a concentration of 1 mg mL−1 were respectively prepared, using PBS at pH 7.4. 900 μL of trypsin solution was transferred into a 1.5 mL EP tube. Then, 100 μL of each peptide solution was added to the EP tube containing the trypsin solution. The trypsin degradation reaction was conducted at 37 °C. During the experiment, 100 μL of the reaction solution was collected from mixture at 0 min, 1 min, 15 min, 30 min, 60 min, and 120 min. The reaction was terminated by adding 30 μL of 1 M HCl. The reaction process was monitored using HPLC.
2.6.3. Serum stability of A3 and its derivative peptides. Bacterial colonies (S. aureus ATCC-25923) in the logarithmic growth phase were isolated to prepare a 5 mL bacterial suspension with a concentration of 1 × 108 CFU mL−1 for subsequent experimentation. Frozen fetal bovine serum was thawed and subsequently mixed with A3, D-A3, and A3-C6 solutions to achieve the serum concentrations of 12.5% and 50.0%, respectively. Following thorough mixing, the mixture was incubated at 37 °C on a shaker for 2 h. The MIC was then determined using the two-fold dilution method. The bacterial suspension was diluted with the culture medium at a ratio of 1:100. In a 96-well plate, 180 μL of the bacterial culture medium was added to the second well, and 100 μL was added to wells 3 through 11. A 20 μL aliquot of the drug solution, pre-incubated with fetal bovine serum, was introduced into the first well, followed by successive two-fold gradient dilutions up to the 10th well. The 11th well served as the negative control. The 96-well plate was incubated on a shaker for 18–24 hours. Finally, the optical density at 600 nm (OD600) was measured using an enzyme-linked immunosorbent assay reader.
2.7. Toxicity of A3 and its derived peptides
2.7.1. Hemolytic toxicity test of A3 and derived peptides. Hemolytic toxicity experiments were conducted using rabbit red blood cells.30 Blood was drawn from the rabbit's marginal ear vein. The collected blood was then transferred to a 1.5 mL centrifuge tube and centrifuged at 3000 rpm for 10 minutes. After discarding the supernatant, the red blood cells were washed three times with physiological saline. Different concentrations (500 μg mL−1, 250 μg mL−1, 125 μg mL−1, 62.5 μg mL−1, 31.2 μg mL−1, 15.6 μg mL−1, 7.8 μg mL−1, 3.9 μg mL−1, 1.8 μg mL−1 and 0.9 μg mL−1) of A3 and derivative peptide solutions were respectively prepared using physiological saline. In a 0.2 mL centrifuge tube, 200 μL of peptide solution was mixed with 4 μL of red blood cells, and the mixture was carefully pipetted to ensure uniformity. Pure water was used as the positive control group, while the blank control group was treated with physiological saline. The mixtures were incubated at 37 °C for 1 hour, then centrifuged at 3000 rpm for 5 minutes. The supernatant was aspirated into a 96-well plate. The absorbance of supernatant at 540 nm was monitored using a microplate reader. The hemolysis rate was calculated using the following formula:
| Hemolysis rate (%) = (Aexperimental group − Ablank group)/(Apositive group − Ablank group) × 100% |
2.7.2. Toxicity of A3 and its derived peptides to zebrafish. The in vivo toxicity experiment was conducted using wild-type zebrafish (TO/WT).31 The methodology was as follows: In the evening before the experiment, male and female zebrafish (1:1 ratio) were placed in a mating tank and separated by a divider. On the second day, after the appearance of light, the divider was removed to allow zebrafish mating. Following spawning, dead embryos were discarded, and healthy zebrafish embryos were collected and placed in a shaking incubator at 28 °C overnight. Well-developed embryos were then selected and placed into a 6-well plate, with 20 embryos per well. Peptides were added to the wells at concentrations of 0 μg mL−1, 1.56 μg mL−1, 3.12 μg mL−1, 6.25 μg mL−1, 12.5 μg mL−1, 25.0 μg mL−1, and 50.0 μg mL−1. The number of hatched zebrafish was recorded using a stereo microscope every 24 hours. The body length of zebrafish was measured using a stereo microscope every 24 hours. This process was repeated for 5 days.
3. Results
3.1. Synthesis of peptides
3.1.1. Synthesis of D-A3. The A3, a cyclic peptide characterized by the presence of disulfide bond, demonstrated significant antibacterial efficacy against both Gram-positive and Gram-negative bacteria. In this investigation, D-A3 was synthesized via the Fmoc solid-phase peptide synthesis (SPPS) technique. The HCTU/DIEA condensation system was utilized to sequentially couple amino acids, commencing from the C-terminus of the peptide. Cleavage was achieved using trifluoroacetic acid (TFA), water, and triisopropylsilylacetylene (TIPS), followed by deposition with ice-cold methyl tert-butyl ether and nitrogen bubbling to yield the crude linear peptide. This crude linear peptide was dissolved in phosphate-buffered saline (PBS), and oxidation was initiated by the addition of 10% dimethyl sulfoxide (DMSO). The oxidation reaction was monitored using high-performance liquid chromatography (HPLC), as depicted in Fig. 1. Partial oxidation of the linear D-A3 was observed in the crude peptide. Upon the addition of DMSO, the peak of the linear peptide progressively diminished, while the peak of the cyclic peptide progressively increased. The oxidation reaction reached completion within 90 minutes. Subsequently, the peptides were separated and purified using semi-preparative liquid chromatography. The separated peptide was subsequently subjected to lyophilization utilizing a freeze dryer to obtain pure D-A3 (yield: 7.33%, retention time: 18.649). Finally, electrospray ionization mass spectrometry (ESI-MS) data suggested that D-A3 was successfully synthesized.
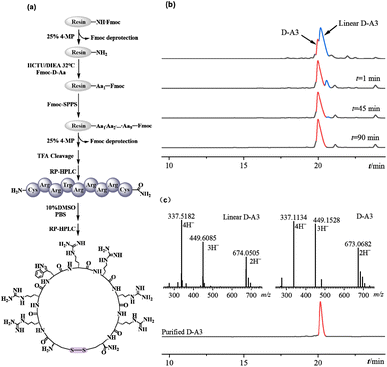 |
| | Fig. 1 Synthesis and characterization of D-A3. (a) Solid phase synthesis of D-A3; (b) the oxidation reaction of D-A3; (c) purification chromatogram and mass spectrum of D-A3. | |
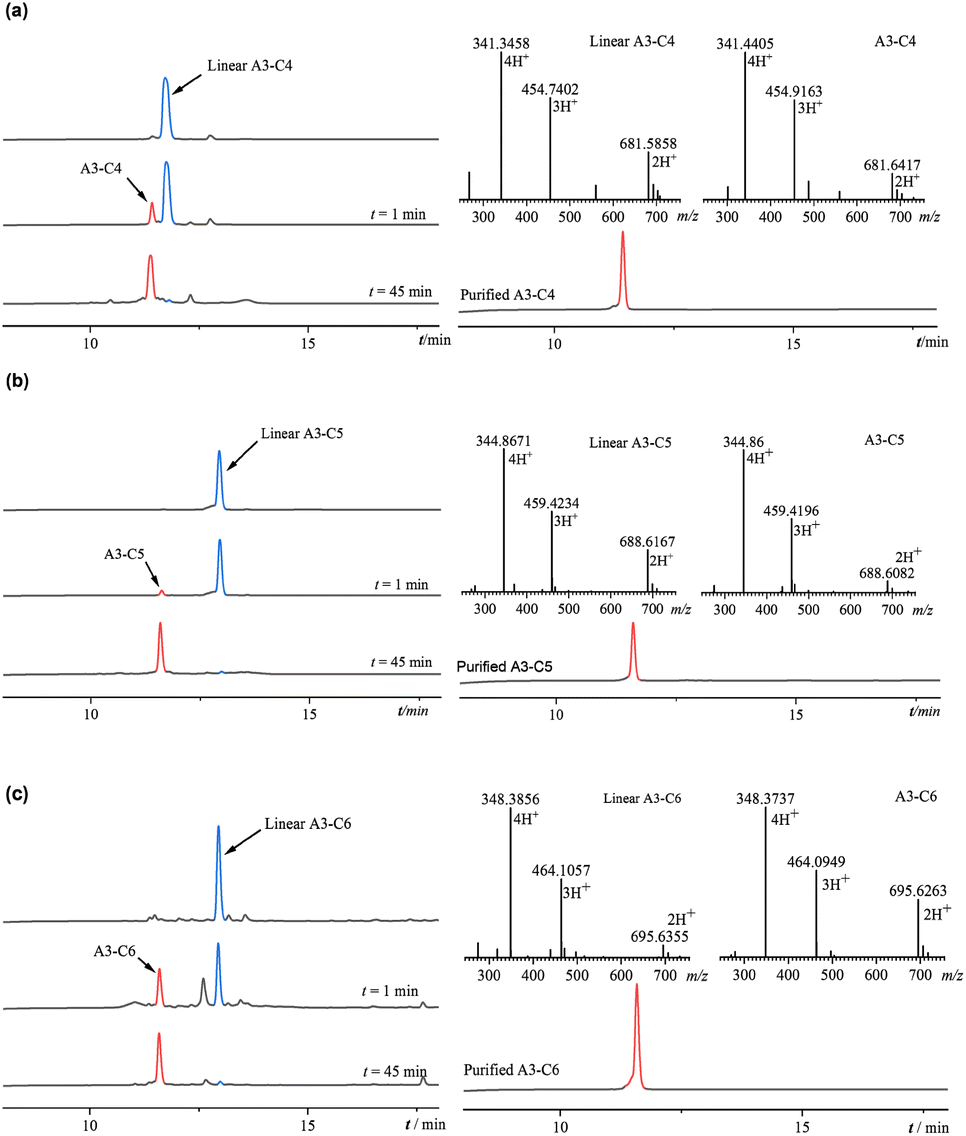
3.1.2. Synthesis of triazole-containing peptides. Cyclic peptides containing disulfide bonds exhibit instability in the reducing environment of the body, where they are prone to reduction by agents such as glutathione, resulting in a loss of activity. This study sought to enhance the stability of A3 by substituting its disulfide bond with triazole ring and to examine the impact of varying linker lengths (n = 4, 5, 6) on antibacterial activity following substitution. Three derivative peptides, (A3-C4, A3-C5, and A3-C6), were synthesized with the synthetic route depicted in Fig. 2. The cysteine residues at both termini of the peptides were replaced with propargylglycine and azido amino acids, each featuring different linker lengths. The resulting crude linear peptide was dissolved in water, and the reaction was initiated by adding a mixed solution of copper sulfate and sodium ascorbate. In the presence of monovalent copper ions, the alkyne and azide groups underwent a click chemistry reaction, forming a stable 1,4-triazole ring structure. As illustrated in Fig. 3, the reduction of the blue linear peptide peak and the emergence of a cyclic peptide product peak were observed in 1 minute. The click chemical reactions involving the three peptides were completed within 45 min. The resultant reaction products underwent purification via semi-preparative HPLC, followed by lyophilization (A3-C4 yield: 7.73%, retention time: 11.387; A3-C5 yield: 10.0%, retention time: 11.390; A3-C6 yield: 6.36%, retention time: 10.876). Subsequently, the molecular masses of A3-C4, A3-C5, and A3-C6 were respectively verified through ESI-MS. The molecular mass of A3-C4, A3-C5, and A3-C6 remained unchanged after the click chemical reaction. The retention time of A3-C4, A3-C5, and A3-C6 showed significant difference compared to the corresponding linear peptides. The above mass spectral and chromatographic data indicated that the successful synthesis of the anticipated triazole-containing peptides.
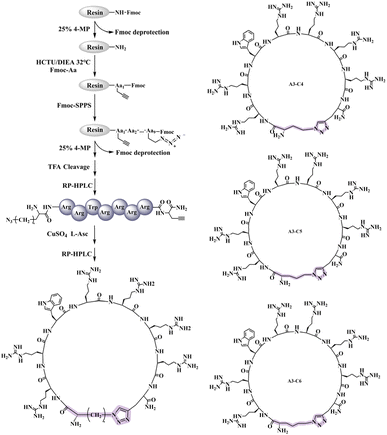 |
| | Fig. 2 Solid phase synthesis of triazole-containing peptides (A3-C4, A3-C5, A3-C6). | |
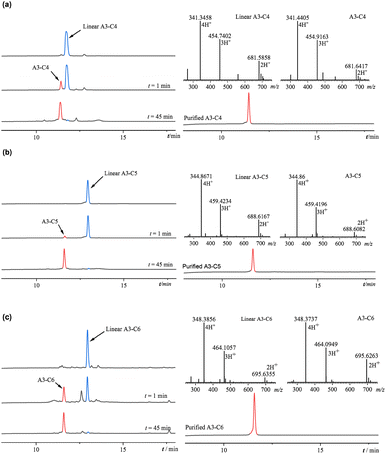 |
| | Fig. 3 (a) The click chemistry reaction of linear A3-C4; purification chromatogram and mass spectrum of A3-C4; (b) the click chemistry reaction of linear A3-C5; purification chromato-gram and mass spectrum of A3-C5; (c) the click chemistry reaction of linear A3-C6; purification chromato-gram and mass spectrum of A3-C6. | |
3.2. Secondary structure of A3 and its derived peptides
Circular dichroism (CD) spectrum serves as a robust technique for assessing the secondary structure of peptides. In this study, we analyzed the secondary structures of A3 and its derivative peptides (D-A3, A3-C4, A3-C5, A3-C6) in an aqueous environment at pH 5.6. As depicted in Fig. 4, the CD spectrum of A3 in water displayed a negative absorption peak near 205 nm, indicative of a disordered structure. Conversely, D-A3 exhibited a positive absorption peak at 205 nm, which is symmetrical to the absorption peak of the A3, suggesting a mirror-image relationship. This observation aligned with the CD characteristics typical of D-peptide. The symmetrical nature of the peaks for both peptides implied structural similarity, thereby confirming the successful synthesis of the D-peptide. Furthermore, we explored the impact of introducing a triazole ring into the A3 structure using CD spectrum. The CD spectra of the triazole-containing peptides showed a positive absorption peak around 190 nm and a negative absorption peak around 210 nm. The positions of absorption peaks were consistent with the characteristic absorption peak of β-sheet structure documented by Greenfield,29 suggesting that the three triazole-containing peptides had β-sheet conformation. CD spectrum revealed that the A3 underwent structural alterations after disulfide bond substitution, shifting from a disordered conformation to a β-sheet structure.
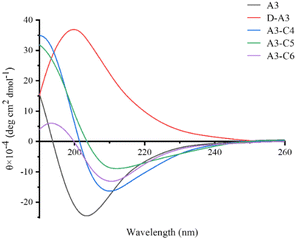 |
| | Fig. 4 CD spectra of A3 and derived peptides. | |
3.3. Antibacterial activity of A3 and its derived peptides
The antimicrobial efficacy of A3 and its derivative peptides was assessed utilizing the radial diffusion method at 1 mg mL−1. In the qualitative antibacterial assay (Fig. 5), we evaluated the antibacterial efficacy of A3 peptide and its derivatives against S. aureus, B. subtilis, B.cereus, E. coli, P. aeruginosa, S. enterica and S. paratyphi B, respectively. All five peptides exhibited antibacterial activity against bacteria, as evidenced by the formation of clear inhibition zones. Notably, D-A3 exhibited the highest activity, characterized by the largest inhibition zone, whereas the inhibition zones of the three triazole-containing peptides were marginally smaller than that of A3, among the 7 types of bacteria mentioned above.
 |
| | Fig. 5 Qualitative antibacterial test of A3 and its derived peptides against Gram-negative bacteria and Gram-positive bacteria. | |
In addition, we assessed the minimum inhibitory concentration (MIC) of the A3 and its derivative peptides utilizing the agar well diffusion method. The MIC was determined by evaluating five peptides against four Gram-positive bacteria (B. subtilis,B. cereus, and two strains of S. aureus) and four Gram-negative bacteria (E. coli, P. aeruginosa, and two strains of S. enterica) at various concentrations, using a twofold dilution method (Table 1). Our findings indicated that A3 had significant bactericidal activity against both Gram-positive and Gram-negative bacteria, corroborating previous research by Seo. All derivative peptides exhibited broad-spectrum antibacterial activity, with MIC values ranging from 0.9 to 250 μg mL−1 for the strains tested. A3 and D-A3 showed the same antibacterial activity against S. aureus ATCC-25923 and B. subtilis CMCC-63501. For S. aureus, B. cereus, S. enterica, and S. para-typhi B, the antibacterial efficacy of D-A3 was twice higher than that of A3. Notably, the MIC value of D-A3 was 3.9 μg mL−1 for E. coli; this value for A3 was only 15.62 μg mL−1. The antibacterial efficacy of D-A3 against P. aeruginosa (15.6 μg mL−1) was markedly superior to that of A3 (125 μg mL−1), demonstrating an eight-fold enhancement in activity. The triazole-containing peptides preserved their broad-spectrum antibacterial properties, exhibiting MIC values ranging from 7.8 to 250 μg mL−1. Nevertheless, their antibacterial potency against certain bacterial strains was reduced compared to A3, with decreases ranging from 2 to 8 folds. For P. aeruginosa ATCC-27853 and S. enterica, A3-C6 exhibited equivalent antibacterial activity to A3 (125 μg mL−1). Among the three triazole-containing compounds, similar antibacterial activity was observed for S. aureus ATCC-25923 and S. paratyphi B, with MIC values of 125 μg mL−1 and 15.6 μg mL−1, respectively. In the case of S. aureus and E. coli ATCC-25922, the antibacterial efficacy of A3-C4 and A3-C6 was equivalent and surpassed that of A3-C5. A3-C6 was demonstrated to have the highest antimicrobial activity among the triazole-containing peptides. A3-C4, A3-C5 and A3-C6 prepared from different azido amino acid precursors, showed different antibacterial activities. The activity of A3-C6 prepared from azido amino acid precursors with longer linker was stronger than that of peptides prepared from azido amino acid precursors with shorter linker. The antibacterial activity of triazole-containing peptides showed that the linker length of azido amino acids precursors would affect the antibacterial activity by changing the size of cyclic peptide ring. These results suggest that the activity of antimicrobial peptides can be regulated by the length of linker of azido amino acid precursors. Overall, the antibacterial activity of D-A3 was comparable to or exceeded that of A3.
Table 1 Minimum inhibitory concentration (MIC) of A3 and derived peptides
| Microbe |
Gram stain |
MIC (μg ml−1) |
| A3 |
D-A3 |
A3-C4 |
A3-C5 |
A3-C6 |
| S. aureus |
+ |
31.2 |
15.6 |
62.5 |
125.0 |
62.5 |
| S. aureus ATCC-25923 |
+ |
1.9 |
1.9 |
7.8 |
7.8 |
7.8 |
| B. subtilis CMCC-63501 |
+ |
1.9 |
1.9 |
31.2 |
31.2 |
15.6 |
| B. cereus |
+ |
31.2 |
15.6 |
125.0 |
125.0 |
62.5 |
| E. coli ATCC-25922 |
− |
15.62 |
3.9 |
125 |
250 |
125 |
| P. aeruginosa ATCC-27853 |
− |
125.0 |
15.6 |
250.0 |
250.0 |
125.0 |
| S. enterica |
− |
125.0 |
62.5 |
250.0 |
250.0 |
125.0 |
| S. para-typhi B |
− |
1.9 |
0.9 |
15.6 |
15.6 |
15.6 |
CD data suggested that the triazole-containing peptides adopted a β-sheet conformation in aqueous solution, whereas the A3 remained in a disordered structure. The D-A3 maintained excellent antibacterial activity against certain bacteria due to its structural similarity. The enhanced antibacterial efficacy of D-A3 against specific bacteria, such as E. coli ATCC-25922 and P. aeruginosa ATCC-27853, compared to A3, may result from the improved compatibility of the D-peptide with specific bacterial targets, thereby augmenting antibacterial effectiveness. Modifying A3 by replacing disulfide bonds with a triazole ring could alter its structure, leading to diminished antibacterial activity for the three triazole-containing peptides. In comparing the antibacterial activities of the three triazole-containing peptides, it was observed that A3-C6 demonstrated slightly superior antibacterial activity compared to A3-C4 and A3-C5. It probably is that the extended linker of A3-C6 enabled its structure to more closely mimic the structure of the A3, thereby enhancing its antibacterial activity.
3.4. Antibacterial mechanism of A3 and its derived peptides
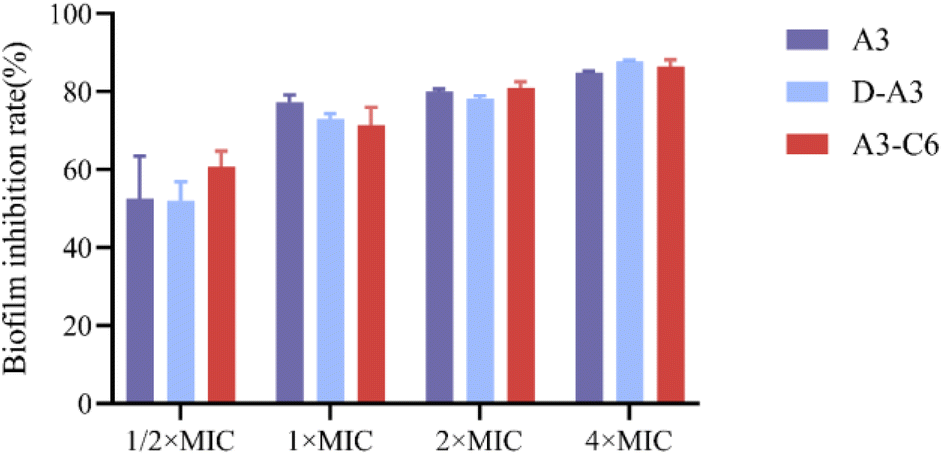
To elucidate the mode of action of A3 and its derivative peptides on bacteria, crystal violet staining was performed. Crystal violet is an alkaline dye that can bind to bacterial intracellular DNA or extracellular polysaccharides to form dark blue crystals. Crystal violet is used for bacterial DNA or biofilm staining. In order to understand the effect of A3 and its derived peptides on bacterial biofilm, the absorption of crystal violet by S. aureus ATCC-25923 (treated with different concentrations of peptides) was measured. According to the absorption of crystal violet in different samples (Fig. S13†), the inhibition rate of different concentrations of A3 and its derived peptides on biofilm was calculated by using the inhibition rate calculation.32 As illustrated in Fig. 6, the inhibitory effect of peptides on biofilm formation is concentration dependent. At a concentration of 1/2 MIC, A3, D-A3 and A3-C6 inhibited biofilm formation of S. aureus ATCC-25923 by about 50% or more, comparing to the untreated group. At a concentration of 2 MIC, A3, D-A3 and A3-C6 inhibited bio-film formation of S. aureus ATCC-25923 by about 80%. Seo et al. reported the action mode of A3 was that it interacted with intracellular components such as DNA or DNA amplification reactions24. These results suggest that an additional action mode of A3 and its derivative peptides is that they can inhibit the biofilm formation of bacteria.
 |
| | Fig. 6 Biofilm inhibition rate of S. aureus ATCC-25923 in the presence of different concentrations of A3, D-A3 and A3-C6. | |
To confirm the effect of A3 and its derivative peptides on bacteria, scanning electron microscopy (SEM) was used. The study focused on three bacterial strains: S. enterica, S. aureus ATCC-25923, and P. aeruginosa ATCC-27853. Initially, SEM was utilized to observe morphological alterations in S. enterica following peptide treatment, with phosphate-buffered saline (PBS)-treated bacteria serving as a negative control. As illustrated in Fig. 7a–f, the results demonstrated that PBS-treated S. enterica maintained a rod-shaped appearance with uniform size, intact morphology, and a smooth surface, exhibiting no signs of depression, shrinkage, or damage. Conversely, treatment with A3, D-A3, A3-C4, A3-C5, and A3-C6 resulted in S. enterica exhibiting irregular morphologies and varying degrees of surface depression compared to the PBS group. Notably, the D-A3 treatment group displayed the most pronounced shrinkage, while S. enterica treated with A3-C4, A3-C5, and A3-C6 exhibited similar degrees of morphological shrinkage. The D-A3 and A3-C6 have already been demonstrated to have significant antibacterial activity against P. aeruginosa ATCC-27853. SEM was employed to examine the effects of these peptides on P. aeruginosa ATCC-27853. As depicted in Fig. S13,† the PBS-treated P. aeruginosa ATCC-27853 exhibited a relatively smooth surface with no significant damage. In contrast, peptide-treated bacteria showed degrees of membrane damage, including shrinkage and, in some cases, rupture. Subsequently, another potential antimicrobial peptide A3-C6 was utilized to treat S. aureus ATCC-25923. The morphological alterations of S. aureus ATCC-25923 were assessed using SEM. As illustrated in Fig. S14,† the S. aureus ATCC-25923 in the control group maintained a spherical shape with an intact external morphology and a smooth surface.
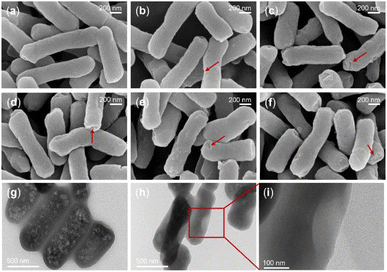 |
| | Fig. 7 SEM images of S. enterica treated with A3 and derived peptides or PBS, and TEM images of P. aeruginosa ATCC-27853 treated with D-A3. (a) SEM image of the PBS treatment group; (b) SEM image of the A3 treatment group; (c) SEM image of the D-A3 treatment group; (d) SEM image of the A3-C4 treatment group; (e) SEM image of the A3-C5 treatment group; (f) SEM image of the A3-C6 treatment group; (g) TEM image of the PBS treatment for P. aeruginosa ATCC-27853 at 500 nm; (h) TEM image of D-A3 treatment for P. aeruginosa ATCC-27853 at 500 nm; (i) TEM image of D-A3 treatment for P. aeruginosa ATCC-27853 at 100 nm. | |
We utilized transmission electron microscopy (TEM) to investigate the impact of the D-A3, which had exceptional antibacterial activity, on P. aeruginosa ATCC-27853. As illustrated in Fig. 7g, in the absence of the peptide, P. aeruginosa ATCC-27853 retained an intact morphological structure with a consistent distribution of intracellular materials. Conversely, following peptide treatment, the bacteria exhibited pronounced alterations, including ruptured or collapsed surfaces, uneven intracellular material distribution, potential vacuolation, and compromised cell membrane integrity.
3.5. Stability of A3 and its derived peptides
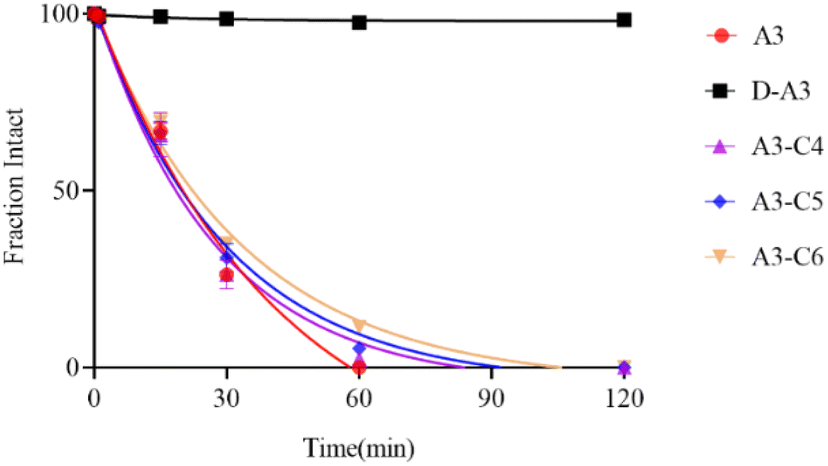
3.5.1. Stability of A3 and its derived peptides under reducing conditions. Glutathione (GSH), a significant reducing agent within the body, is ubiquitously present and plays a vital role in essential biochemical processes, including the tricarboxylic acid cycle, glucose metabolism, and redox reactions. Cyclic peptides containing disulfide bonds are prone to reduction by agents such as GSH, resulting in diminished potency. Consequently, we examined the stability of A3 and its derivative peptides under reducing conditions. The stability profiles of A3 and its derivative peptides were assessed using HPLC in the presence of 4 mM GSH at various time intervals. Upon exposure to GSH, A3 and D-A3 were nearly completely reduced within 1 minute, in Fig. 8. In contrast, under the sa. me conditions, the peak of A3-C4, A3-C5, and A3-C6 remained unchanged after 30 minutes. According to the test results, the A3 and D-A3 displayed limited stability in the presence of 4 mM GSH, characterized by a rapid reduction of the disulfide bond. In contrast, the three triazole-containing peptides exhibited comparable stability against GSH treatment, suggesting enhanced stability under reducing conditions relative to the A3 and D-A3.
 |
| | Fig. 8 GSH stability of A3 and derived peptides. | |
3.5.2. Protease stability of A3 and its derived peptides. To assess the stability of A3-derived peptides in the presence of proteases, we performed an in vitro enzymatic stability assay on A3 and its derivatives using trypsin. As depicted in Fig. 9, the results of protease stability revealed that A3 was completely degraded after trypsin treatment for 60 minutes. In contrast, A3-C4, A3-C5, and A3-C6 were degraded within 85–110 minutes, respectively exhibiting half-lives of 18.4 minutes, 20.0 minutes, and 22.3 minutes. The three triazole-containing peptides were demonstrated comparable stability against trypsin, which was marginally superior to that of A3. This enhanced stability of triazole-containing peptides was likely due to the formation of a β-sheet structure in solution, leading to a reduced degradation rate during enzymatic breakdown. Notably, the D-A3 showed no peak of degradation within 120 minutes, indicating a substantially extended degradation time. This suggests that D-amino acids effectively inhibited trypsin-induced peptide degradation. Consequently, D-A3 exhibited excellent protease stability, positioning it as a promising candidate for oral bioactivity.
 |
| | Fig. 9 Trypsin degradation curves of A3 and derived peptides. | |
3.5.3. Serum stability of A3 and its derivative peptides. To investigate the stability of A3, D-A3 and A3-C6 in serum, we separately co-incubated A3 and its derivative peptides with fetal bovine serum. Subsequently, we observed the effect of different amounts of serum on the MIC values of peptides against S. aureus ATCC-25923, using broth dilution method. As shown in Table 2, after A3 and its derivative peptides co-incubated with 12.5% or 50.0% fetal bovine serum for 2 h, the MIC value of A3 was 1-fold or 2-fold decrease, respectively. D-A3 showed strong stability in different serum concentrations, with no change in MIC, indicating its excellent stability in serum. The result suggests that replacing all L amino acids in A3 with D amino acids can significantly improve its serum stability. After 12.5% or 50.0% serum treatment, MIC respectively decreased 1-fold or 2-fold for A3-C6. The decrease in MIC value of A3-C6 was the same as that of A3, indicating that A3-C6 and A3 had the same ability to resist serum.
Table 2 Serum stability of A3 and its derivative peptides
| |
MIC (μg mL−1) |
| Control |
12.5% serum |
50% serum |
| A3 |
1.9 |
3.9 |
7.8 |
| D-A3 |
1.9 |
1.9 |
1.9 |
| A3-C6 |
7.8 |
15.6 |
31.2 |
3.6. Toxicity of A3 and its derived peptides
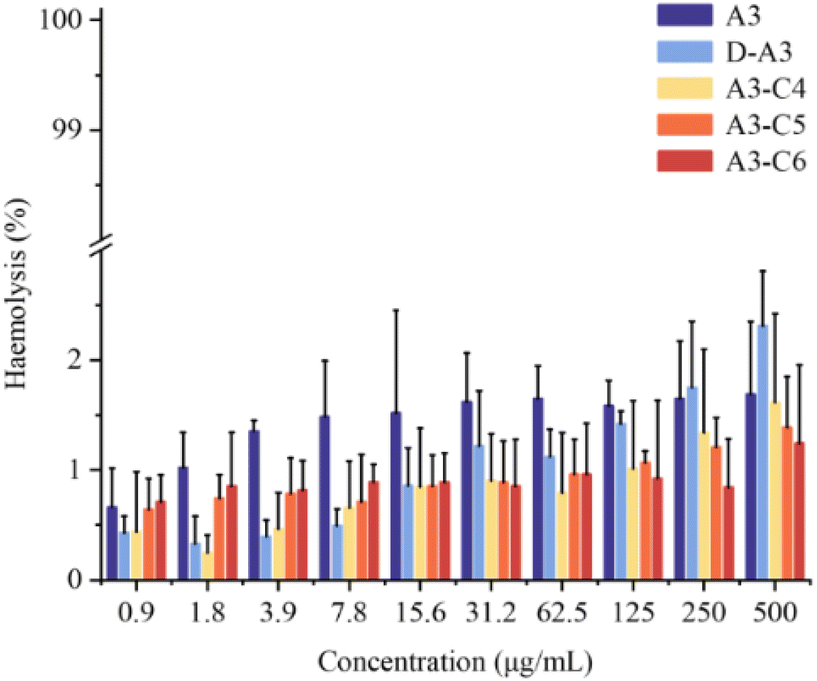
3.6.1. Hemolytic toxicity of A3 and its derived peptides. To evaluate the toxicity of A3-derived peptides, we performed an in vitro toxicity assessment for A3 and its derivatives using a red blood cell hemolysis assay. A3 and four derived peptides were incubated with rabbit red blood cells at 37 °C. After 1 hour, the optical density (OD) values of each group were measured using a microplate reader. Pure water served as a positive control, while physiological saline was employed as a negative control to determine the hemolysis rate. In the assay, the hemolysis rate for the positive control group with pure water was 100%, whereas the negative control with physiological saline exhibited a hemolysis rate of 0%. The hemolysis rate of five peptides (at concentrations ranging from 0.9 to 500 μg mL−1) remained below 5% (Fig. 10). Notably, even at 60–500 times MIC the hemolysis rates of derived peptides did not exceed 2.5%. These results indicate that the four derived peptides possess minimal hemolytic toxicity towards rabbit red blood cells.
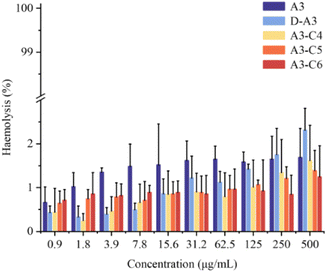 |
| | Fig. 10 Hemolytic activity of A3 and derived peptides. | |
3.6.2. Toxicity of A3 and its derived peptides to zebrafish. In this study, A3 and its derivatives had favorable water solubility, rendering them appropriate for assessing in vivo toxicity, using zebrafish. Fig. 11 illustrates the impact of various treatment groups on the zebrafish embryo hatching rate. The hatching rate of zebrafish embryos exposed to A3 decreased progressively with increasing drug concentrations during the 48-hour exposure period. At 50 μg mL−1, the hatching rate reached approximately 75% by 48 hours. Notably, complete hatching (100%) was observed across all groups after 72 hours (Fig. 11). In comparison to A3, the D-A3 exhibited a similar pattern of reduced hatching rates, when increasing its concentration. The 48-hour hatching rate of D-A3 was marginally lower than that of A3. At a concentration of 12.5 μg mL−1, where D-A3 predominantly manifests its antibacterial properties, the 48-hour hatching rates for both A3 and D-A3 were comparable. The hatching rate of A3-C4 at all tested concentrations, attained 100% after 72 hours. Conversely, A3-C5 exhibited failed hatchings at 48 hours at concentrations of 25 μg mL−1 and 50 μg mL−1. After 72 hours, the hatching rates of A3-C5 at these concentrations were 8% and 55%, respectively, suggesting a significant impact on zebrafish embryo hatching. Consequently, subsequent experiments concentrated on other derivative peptides. The overall 48-hour hatching rate of A3-C6 was comparable to that of A3, indicating a relatively low impact of A3-C6 on zebrafish embryo hatching. Follow-up observations were conducted to evaluate whether drug administration influenced the growth and development of zebrafish. Four days post-treatment, the body length of the zebrafish was measured, with results presented in Fig. 11f. At a concentration of 6 μg mL−1, there was slight decrease in zebrafish body length, after D-A3 treatment. No significant differences in body length were found between triazole-containing peptides and either group containing A3 or negative group. This suggests that three derivative peptides exhibit low toxicity for the growth and development of zebrafish.
 |
| | Fig. 11 Effects of A3 and derived peptides on the hatching rate of zebrafish embryo at 48 hours (a)–(e) and the development of zebrafish after 5 days (f). Compared with the control group (0 μg mL−1), *p < 0.05, **p < 0.01, ****p < 0.0001. | |
4. Conclusions
The rapid emergence of drug-resistant bacteria has heightened due to the misuse of traditional antibiotics. In response, researchers are actively seeking novel antibiotics to address these infections. AMPs have emerged as a promising alternative due to their distinctive antibacterial mechanisms. The structures of antimicrobial peptides, constrained by disulfide bonds, endow them with diverse spatial conformations, enabling them to perform a wide range of functions. Consequently, AMPs represent a potentially valuable class of therapeutic agents. However, despite the fact that many AMPs have been discovered, they are vulnerable to rapid degradation by reducing agents and various proteases. This instability has resulted in a limited number of peptide drugs successfully advancing from laboratory research to clinical application, thereby posing a significant challenge to the development of peptide-based antibiotics.
In this study, we engineered and synthesized four novel derivative peptides based on the A3 by incorporating D-amino acids or replacing the disulfide bond with a triazole ring, with the objective of enhancing their stability and biological activity under reducing conditions. Initially, we synthesized D-A3, A3-C4, A3-C5, and A3-C6 utilizing Fmoc SPPS. The successful synthesis of the target peptides was verified via liquid-phase reaction monitoring and mass spectrometry. Subsequently, we assessed the antibacterial activity of the synthesized peptides using the pore diffusion method. The antibacterial experimental data revealed that the derivative peptides exhibited broad-spectrum antibacterial activity. Notably, D-A3 displayed superior antibacterial efficacy compared to A3, particularly against specific bacteria such as E. coli ATCC-25922 and P. aeruginosa ATCC-27853. However, the antibacterial activity of the triazole-containing peptides showed a slight decrease compared to A3. Through investigation of the antibacterial mechanism of the peptides, we found an additional mechanism that they inhibit biofilm formation. Stability assays demonstrated that the triazole-containing peptides exhibited substantial stability under reducing conditions, whereas D-A3 showed significant resistance to trypsin or serum degradation. Toxicity assessments revealed that D-A3, A3-C4, A3-C5, and A3-C6 exhibited minimal hemolytic toxicity (<5%). Furthermore, zebrafish toxicity assays indicated that D-A3 and A3 had comparable toxicity levels, while A3-C6 had the least impact on zebrafish embryo hatching. In summary, our results suggest that the A3 derivative peptides exert antibacterial effects through membrane disruption. Notably, the D-A3 and A3-C6 demonstrate promising antibacterial activity, enhanced enzymatic stability, and low toxicity, thereby presenting themselves as potential candidates for the development of efficient AMPs for bacterial infection treatment.
Ethical statement
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Baotou Medical College and approved by the Animal Ethics Committee of Baotou Medical College.
Data availability
All supporting data can be founding the ESI file.†
Author contributions
Methodology, conceptualization, data curation, writing original draft, Jiawei Zhao. Methodology, data curation, Xin Zhang. Methodology, data curation, Yu Liu and Zhixing Geng. Methodology, Jili Dai. Supervision and review, Ye Guo. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge Meiying Zhang for her help in providing various bacteria required for the experiment. This research was funded by the National Natural Science Foundation of China, grant numbers 21967017, and the Natural Science Foundation of Inner Mongolia, grant number 2023MS02001, and 2022 Inner Mongolia Autonomous Region “Grassland Talents” Project Youth Innovation and Entrepreneurship Talent Project, grant numbers 2022CYYC1C88.
References
- N. Yadav and V. S. Chauhan, Adv. Colloid Interface Sci., 2024, 333, 103282 CrossRef CAS PubMed.
- A. Mercer, Pathog. Global Health, 2021, 115(3), 151–167 CrossRef CAS PubMed.
- R. M. Olson and D. M. Anderson, PLoS One, 2019, 14(5), e0217440 CrossRef CAS PubMed.
- S. Paul, S. Verma and Y.-C. Chen, ACS Infect. Dis., 2024, 10(4), 1034–1055 CrossRef CAS PubMed.
- X. Gong, Y. Han, T. Wang, G. Song, H. Chen, H. Tang, X. Huang, K. Deng, S. Wang and Y. Wang, Adv. Mater., 2025, 37(4), e2414357 Search PubMed.
- T. Pulingam, T. Parumasivam, A. M. Gazzali, A. M. Sulaiman, J. Y. Chee, M. Lakshmanan, C. F. Chin and K. Sudesh, Eur. J. Pharm. Sci., 2022, 170, 106103 CrossRef CAS PubMed.
- C. Xu, L. Kong, H. Gao, X. Cheng and X. Wang, Front. Microbiol., 2022, 13, 822689 CrossRef PubMed.
- K. Xu, X. Zhao, Y. Tan, J. Wu, Y. Cai, J. Zhou and X. Wang, Biomater. Adv., 2023, 155, 213684 CrossRef CAS PubMed.
- Y. Hou, T. Tan, Z. Guo, Y. Ji, J. Hu and Y. Zhang, Biomater. Sci., 2022, 10, 3831–3844 Search PubMed.
- Y. Zhu, W. Xu, W. Chen, B. Li, G. Li, H. Deng, L. Zhang, C. Shao and A. Shan, Sci. Adv., 2025, 11(6), eads3844 CrossRef CAS PubMed.
- X. Y. Luo, C. M. Hu, Q. Yin, X. M. Zhang, Z. Z. Liu, C. K. Zhou, J. G. Zhang, W. Chen and Y. J. Yang, Adv. Sci., 2024, 11(30), e2401793 CrossRef PubMed.
- B. Li, Y. Liu, P. Yan, X. Ouyang, Z. Ba, Y. Wang, T. Yang, Z. Yu, B. Ren, C. Zhong, H. Liu, Y. Zhang, S. Gou and J. Ni, Eur. J. Med. Chem., 2025, 283, 117149 CrossRef CAS PubMed.
- G. Subramaniam and M. Girish, Indian J. Pediatr., 2020, 87(11), 937–944 CrossRef PubMed.
- K. Botelho Sampaio de Oliveira, M. Lopes Leite, V. Albuquerque Cunha, N. Brito da Cunha and O. Luiz Franco, Drug Discovery Today, 2023, 28(8), 103629 CrossRef CAS PubMed.
- C. H. Chen and T. K. Lu, Antibiotics, 2020, 9(1), 24 CrossRef CAS PubMed.
- A. M. Turner, J. Y. H. Lee, C. L. Gorrie, B. P. Howden and G. P. Carter, Front. Microbiol., 2021, 12, 637656 CrossRef PubMed.
- N. Zhang, X. Gu, D. Song, P. Zhang, N. Zhang, W. Chen, S. Ji, Y. Qi and S. Ma, Bioorg. Chem., 2022, 119, 105583 CrossRef CAS PubMed.
- Y. Tian, Y. Hou, J. Tian, J. Zheng, Z. Xiao, J. Hu and Y. Zhang, J. Mater. Chem. B, 2024, 12(33), 8122–8132 RSC.
- M. Chen, N. Lin, X. Liu, X. Tang, Z. Wang and D. Zhang, Front. Immunol., 2023, 14, 1168517 CrossRef CAS PubMed.
- C. Bucataru and C. Ciobanasu, Microbiol. Res., 2024, 286, 127822 CrossRef CAS PubMed.
- C. Tian, N. Zhao, L. Yang, F. Lin, R. Cai, Y. Zhang, J. Peng and G. Guo, Front. Cell. Infect. Microbiol., 2024, 14, 1334378 CrossRef CAS PubMed.
- M. V. Arasu and N. A. Al-Dhabi, J. Infect. Public Health, 2023, 16(12), 2031–2037 CrossRef PubMed.
- J.-K. Seo, J. M. Crawford, K. L. Stone and E. J. Noga, Biochem. Biophys. Res. Commun., 2005, 338(4), 1998–2004 CrossRef CAS PubMed.
- J. K. Seo, D. G. Kim, J. E. Lee, K. S. Park, I. A. Lee, K. Y. Lee, Y. O. Kim and B. H. Nam, Mar. Drugs, 2021, 19(8), 451 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37(10), 1186–1197 CrossRef CAS PubMed.
- Z. Lai, X. Yuan, H. Chen, Y. Zhu, N. Dong and A. Shan, Biotechnol. Adv., 2022, 59, 107962 CrossRef CAS PubMed.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12(8), 1325–1351 RSC.
- A. M. White, S. J. de Veer, G. Wu, P. J. Harvey, K. Yap, G. J. King, J. E. Swedberg, C. K. Wang, R. H. P. Law, T. Durek and D. J. Craik, Angew Chem. Int. Ed. Engl., 2020, 59(28), 11273–11277 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1(6), 2876–2890 CrossRef CAS PubMed.
- N. Akram, M. Usman, S. Haider, M. S. Akhtar and K. Gul, Polymers, 2022, 14(17), 3701 CrossRef CAS PubMed.
- A. Aspatwar, M. M. Hammaren, M. Parikka and S. Parkkila, J. Visualized Exp., 2019, 25, 150 Search PubMed.
- Y. Liu, Y. Jiang, J. Zhu, J. Huang and H. Zhang, Carbohydr. Polym., 2019, 206, 412–419 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a