
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical dearomatization of 2-naphthols for C–O bond formation†
Han Byeol Kim‡
ab,
Dong Kyun Han‡a,
Jae Kyun Leec and
Seo-Jung Han *ab
*ab
aDepartment of Chemistry, Sogang University, 35 Baekbeom Ro, Seoul 04107, Republic of Korea. E-mail: sjhan@sogang.ac.kr
bCenter for Nano Materials, Sogang University, 35 Baekbeom Ro, Seoul 04107, Republic of Korea
cNeuro-Medicine Center, Korea Institute of Science and Technology, 5 Hwarang-ro 14-gil, Seongbuk-gu, Seoul 02792, Republic of Korea
First published on 15th May 2025
Abstract
We reported a metal-free electrochemical oxidative C–O homocoupling of 2-naphthols, followed by subsequent alkoxylation under mild conditions. This strategy offered an eco-friendly and cost-effective electrochemical approach using undivided cells. Additionally, the reaction exhibited broad tolerance to various substituted 2-naphthols and diverse alcohols, affording the corresponding naphthalenones in moderate to good yields.
Naphthols and their derivatives are readily available chemical feedstocks that play crucial roles as valuable intermediates in synthetic chemistry.1 In particular, the dearomatization of naphthol derivatives provides access to naphthalenone derivatives, which are frequently found in pharmaceuticals2 and natural products.3 Therefore, the development of efficient strategies for the dearomatization of naphthol derivatives has attracted significant interest from the synthetic community.4 Although these achievements have been remarkable, most of the reaction conditions require oxidants or metal/organocatalysts. Recently, electrochemical dearomatizations of phenols and naphthols have been reported.5,6 In 2023, Kalek and co-workers5 developed electrochemical dearomatizing spirolactonization and spiroetherification methods for naphthols (Scheme 1a). In addition, electrochemical dearomative methoxylation reactions of naphthols or phenols were reported by the same research group in 2024 (Scheme 1b). However, C–O homocoupling products were not generated under these reaction conditions. Although the Liu group6 reported electrochemical C–O homocoupling of 2-naphthols by oxidative dearomatization, the reaction required metal catalysts and high temperature (Scheme 1c). Herein, we discovered mild and metal-free electrochemical oxidative C–O homocoupling of 2-naphthols. Interestingly, the electrochemical alkoxylation of the C–O homocoupling product generated the alkoxylated naphthalenones (Scheme 1d).
 | ||
| Scheme 1 Electrochemical dearomative oxidation reactions of naphthols. | ||
1-Methyl-2-naphthol 1a was used as a model substrate to optimize the reaction conditions (Table 1, see the ESI† for full optimization table). The reaction was conducted in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of MeCN and MeOH as the solvent, using an undivided cell under constant current conditions. The C–O homocoupled product 2a was obtained in 44% yield at a constant current of 10 mA using a graphite cathode and a platinum anode (entry 1). The yield diminished when a platinum cathode and a graphite anode were employed (entry 2). We found that the reaction fared well with a platinum cathode and a platinum anode, providing the desired product 2a in 74% yield (entry 3). Adjusting the current either to 5 mA or 15 mA, instead of 10 mA decreased yield of product 2a (entries 4 and 5). An electrolyte screening revealed that the C–O homocoupling proceeded well when electrolytes containing bromide anions were used (entries 6–9). Among the bases screened, the addition of NaHCO3 as a base additive led to an improved yield of the C–O homocoupled product 2a (entries 10–12; see the ESI† for details). The desired product 2a was not observed in the absence of an electric current (entry 13). The structure of 2a was unambiguously confirmed by a X-ray diffraction analysis.
:1 mixture of MeCN and MeOH as the solvent, using an undivided cell under constant current conditions. The C–O homocoupled product 2a was obtained in 44% yield at a constant current of 10 mA using a graphite cathode and a platinum anode (entry 1). The yield diminished when a platinum cathode and a graphite anode were employed (entry 2). We found that the reaction fared well with a platinum cathode and a platinum anode, providing the desired product 2a in 74% yield (entry 3). Adjusting the current either to 5 mA or 15 mA, instead of 10 mA decreased yield of product 2a (entries 4 and 5). An electrolyte screening revealed that the C–O homocoupling proceeded well when electrolytes containing bromide anions were used (entries 6–9). Among the bases screened, the addition of NaHCO3 as a base additive led to an improved yield of the C–O homocoupled product 2a (entries 10–12; see the ESI† for details). The desired product 2a was not observed in the absence of an electric current (entry 13). The structure of 2a was unambiguously confirmed by a X-ray diffraction analysis.
|
|||||
|---|---|---|---|---|---|
| Entry | Electrode (+)/(−) | Electrolyte | Icell (mA) | Additive (equiv.) | Yieldb (%) |
| a Reaction conditions: 1-methyl-2-naphthol 1a (0.32 mmol, 1.00 equiv.) and electrolyte (0.32 mmol, 1.00 equiv.) were dissolved in MeCN:MeOH (1:1, 0.05 M) and subjected to electrochemistry in an undivided cell under constant current conditions.b Determined by high-performance liquid chromatography (HPLC) using 3-nitrophenol as an internal standard.c Not observed. |
|||||
| 1 | C/Pt | n-Bu4NBF4 | 10 | — | 44 |
| 2 | Pt/C | n-Bu4NBF4 | 10 | — | 18 |
| 3 | Pt/Pt | n-Bu4NBF4 | 10 | — | 74 |
| 4 | Pt/Pt | n-Bu4NBF4 | 5 | — | 35 |
| 5 | Pt/Pt | n-Bu4NBF4 | 15 | — | 40 |
| 6 | Pt/Pt | n-Bu4NPF6 | 10 | — | 38 |
| 7 | Pt/Pt | LiBr | 10 | — | 74 |
| 8 | Pt/Pt | NaBr | 10 | — | 75 |
| 9 | Pt/Pt | n-Bu4Br | 10 | — | 79 |
| 10 | Pt/Pt | n-Bu4Br | 10 | NaHCO3 (0.5) | 80 |
| 11 | Pt/Pt | n-Bu4Br | 10 | NaHCO3 (1.5) | 96 |
| 12 | Pt/Pt | n-Bu4Br | 10 | NaHCO3 (3.0) | 75 |
| 13 | Pt/Pt | n-Bu4Br | — | NaHCO3 (1.5) | –c |

With the optimized conditions in hand, we explored the substrate scope of the electrochemical oxidative C–O homocoupling reactions (Scheme 2). Naphthols with linear alkyl substitutions at C(1) generated the corresponding products in good yields (2a–2c). Cyclopropylmethyl and cyclohexyl substitutions at the C(1) position of the 2-naphthols generated the desired products in lower yields (2d and 2e). In addition, allyl substitutions at the C(1) position of 2-naphthols were tolerated under our reaction conditions, producing the corresponding products in 35% and 42% yields, respectively (2f and 2g). We also investigated the substrate scope of the substitutions at the C(6) and C(7) positions of 1-methyl-2-naphthols. 1-Methyl-2-naphthols bearing ethyl, bromo, and phenyl substituents on C(6) afforded the corresponding products in moderate to good yields (2h, 2i, and 2j). The electronically variable aryl groups in C(6) were compatible with the reaction conditions (2k–2m). In addition, 1-methyl-2-naphthols bearing a methoxy substituent at the C(7) position provided 2n in 51% yield. Phenyl- and pyridine-substituted 1-naphthols were well tolerated, furnishing the corresponding products in good yields (2o and 2p).
 | ||
| Scheme 2 Substrate scope of substituted-2-naphthol for homocoupling reactions.a,b a Reaction conditions: 1-substituted-2-naphthol 1a (0.16 mmol, 1.00 equiv.), n-Bu4NBr (0.16 mmol, 1.00 equiv.) and NaHCO3 (0.24 mmol, 1.50 equiv.) were dissolved in MeCN:MeOH (1:1, 0.05 M) and subjected to electrochemistry in an undivided cell under constant current conditions. b The yield is that of the isolated product. | ||
We observed trace amounts of 1-methoxy-1-methyl-naphthalenone 3a were generated during the electrochemical dearomative C–O homocoupling reaction. Notably, 1-methoxy-1-methyl-naphthalenone 3a was detected only in the final stages of the reaction, whereas 3a was not observed at the beginning. Thus, we hypothesized that 3a could be produced from C–O homocoupling product 2a. We then attempted to optimize the reaction conditions for the methoxylation of 2a to obtain 1-methoxy-1-methyl-naphthalenone 3a (Table 2, see more details in ESI†). The yield of methoxylated naphthalenone 3a increased as the reaction current was decreased (entries 1–3). The addition of Na2HPO4 as a base additive produced methoxy 3a in a yield similar to that obtained with NaHCO3 (entries 3 and 4).7 However, the addition of excess Na2HPO4 resulted in a lower yield of 3a (entries 4–6). Altering the anode to either graphite or nickel was detrimental to the yield (entries 7 and 8). A survey of the different electrolytes revealed that n-Bu4NBF4 was the optimal electrolyte (entries 9 and 10). Desired product 3a was not detected in the absence of an electric current (entry 11).
|
|||||
|---|---|---|---|---|---|
| Entry | Electrode (+)/(−) | Electrolyte | Icell (mA) | Additive (equiv.) | Yieldb (%) |
| a Reaction conditions: compound 2a (0.32 mmol, 1.00 equiv.) and the electrolyte (0.32 mmol, 1.00 equiv.) were dissolved in MeCN:MeOH (1:1, 0.05 M) and subjected to electrochemistry in an undivided cell under constant current conditions.b Determined by 1H NMR using BHT as an internal standard.c Isolated yield.d Not observed. |
|||||
| 1 | C/Pt | n-Bu4NPF6 | 10 | NaHCO3 (0.5) | 60b |
| 2 | C/Pt | n-Bu4NPF6 | 5 | NaHCO3 (0.5) | 64b |
| 3 | C/Pt | n-Bu4NPF6 | 3 | NaHCO3 (0.5) | 66b(51c) |
| 4 | C/Pt | n-Bu4NPF6 | 3 | Na2HPO4 (0.5) | 66b(52c) |
| 5 | C/Pt | n-Bu4NPF6 | 3 | Na2HPO4 (1.0) | 61c |
| 6 | C/Pt | n-Bu4NPF6 | 3 | Na2HPO4 (1.5) | 45b |
| 7 | C/C | n-Bu4NPF6 | 3 | Na2HPO4 (1.0) | 47c |
| 8 | C/Ni | n-Bu4NPF6 | 3 | Na2HPO4 (1.0) | 57c |
| 9 | C/Pt | n-Bu4NBF4 | 3 | Na2HPO4 (1.0) | 69c |
| 10 | C/Pt | NaBF4 | 3 | Na2HPO4 (1.0) | 59c |
| 11 | C/Pt | n-Bu4NBF4 | — | Na2HPO4 (1.0) | –d |

We investigated the substrate scope of alcohols to obtain alkoxylated 2a (Scheme 3). In addition to the simple methoxy group, ethoxy, isopropoxy, and butoxy-substituted naphthalenones (3a–3d) were generated in moderate yields under our reaction conditions. In addition, cyclobutyloxy- and cyclohexyloxy-substituted naphthalenones were generated in 42% and 37% yields, respectively. The use of allyl and homoallyl alcohols afforded the corresponding products in moderate yields (3g–3i). The silyl ether functional group was tolerated under our reaction conditions, producing 3j in 35% yield. Additionally, the use of geraniol as a substrate generated 3k in 39% yield.
 | ||
| Scheme 3 Substrate scope of diverse alkoxylation reactions.a,b a Reaction conditions: 1-methyl-1-((1-methylnaphthalen-2-yl)oxy)naphthalen-2(1H)-one 2a (0.16 mmol, 1.00 equiv.), n-Bu4NBF4 (0.16 mmol, 1.00 equiv.) and Na2HPO4 (0.16 mmol, 1.00 equiv.) were dissolved in MeCN:MeOH (1:1, 0.05 M) and subjected to electrochemistry in an undivided cell under constant current conditions. b The yield is that of the isolated product. | ||
Next, the substrate scope of the C(6) and C(7) substituents was explored in the methoxylation reactions (Scheme 4). The substrate bearing an ethyl substituent at C(6) afforded 4a in 45% yield. In addition to the simple phenyl substituent, aryl substituents with electron-donating or electron-withdrawing groups fared well under the reaction conditions, affording the corresponding products in good yields (4b–4e). In addition, a bromo substituent on C(6) was tolerated under the reaction conditions furnishing 4f in 49% yield. Substrates containing methoxy and aryl substituents on C(7) produced the corresponding products in moderate yields (4g–4i).
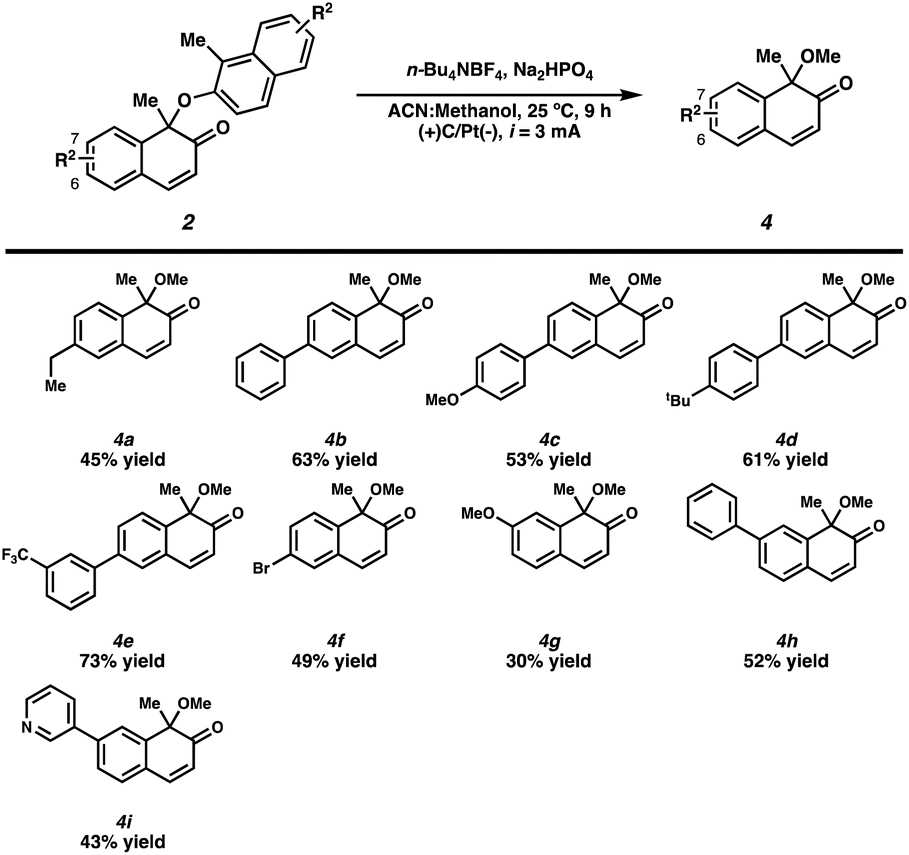 | ||
| Scheme 4 Substrate scope of methoxylation reactions.a,b a Reaction conditions: substituted-2-naphthol dimer 2 (0.16 mmol, 1.00 equiv.), n-Bu4NBF4 (0.16 mmol, 1.00 equiv.) and Na2HPO4 (0.16 mmol, 1.00 equiv.) were dissolved in MeCN:MeOH (1:1, 0.05 M) and subjected to electrochemistry in an undivided cell under constant current conditions. b The yield is that of the isolated product. | ||
To demonstrate the synthetic utility of our reaction, we performed a gram-scale synthesis, generating the desired product 2a in 75% yield, although a current of 20 mA was required for full conversion. Unfortunately, scaling up the alkoxylation of dimer 2a to the gram scale resulted in poor conversion under the optimized conditions. Moreover, reduction of ketone 3a with DIBAL produced alcohol 5 in 61% yield with high diastereoselectivity.8 Treatment of α,β-unsaturated ketone 3a with H2O2 afforded epoxide 6 in 73% yield with high diastereoselectivity.9 In addition, bromination of α,β-unsaturated ketone 3a with bromine provided 7, and a subsequent Suzuki coupling reaction with 4-fluorophenylboronic acid generated 8 in 89% yield.10 Alkene 9 was synthesized in 78% yield via the Wittig reaction of ketone 3a with methyltriphenylphosphonium bromide (Scheme 5).11
 | ||
| Scheme 5 Gram scale and diversification. | ||
1-Methylnaphthalen-2-ol 1a exhibited an oxidation peak at 1.86 V (vs. Ag/AgCl) (Fig. 1a). 1-Methyl-1-((1-methylnaphthalen-2-yl)oxy)naphthalen-2(1H)-one 2a showed a slightly higher oxidation peak at 1.93 V (Fig. 1b). Upon the addition of a base to the solution of 2a, the oxidation potential decreased to 1.76 V (vs. Ag/AgCl) (Fig. 1c), indicating that the presence of base facilitated the oxidation of 2a.
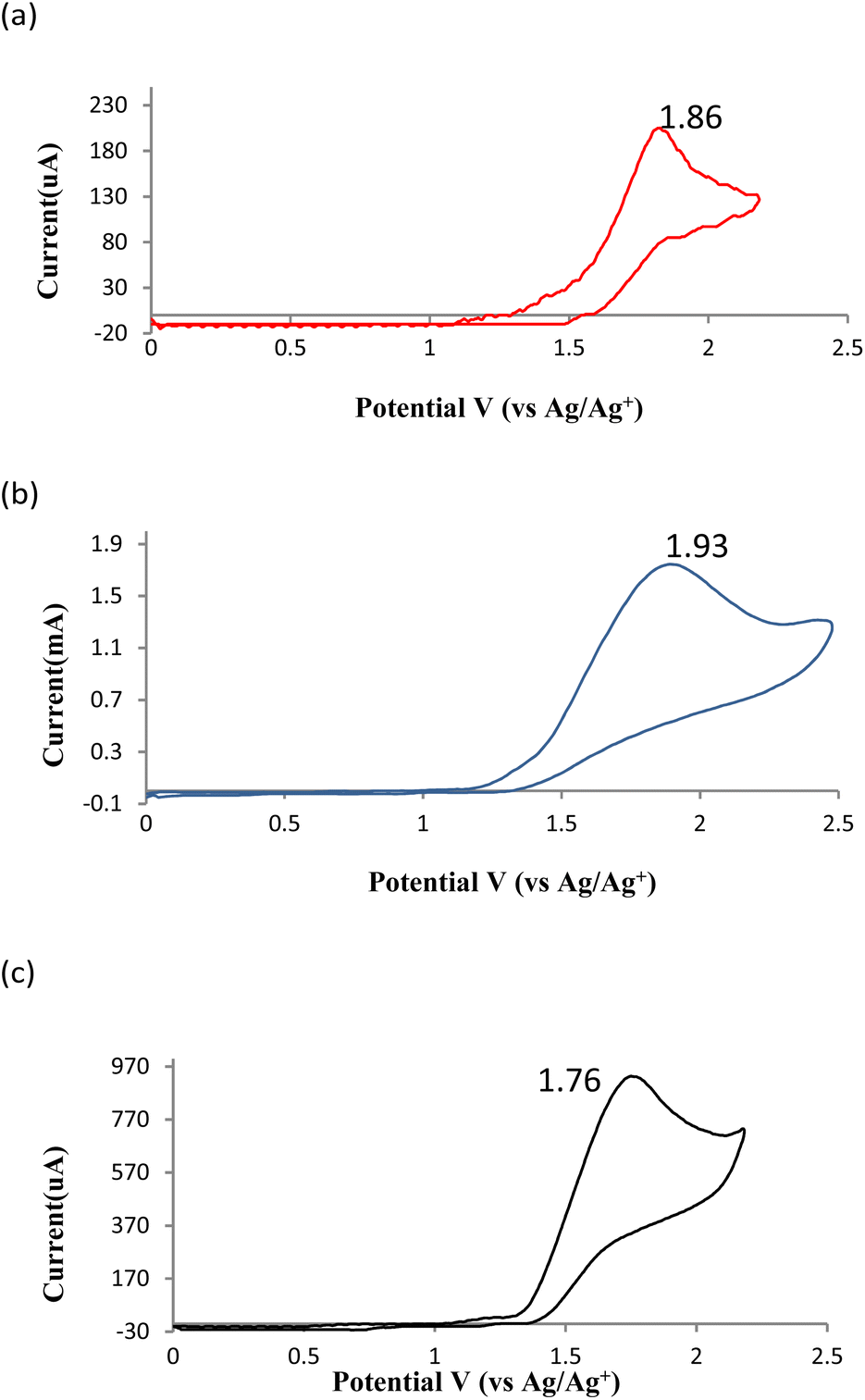 | ||
| Fig. 1 (a) 1a in 0.1 M nBu4NBF4 in ACN, (b) 2a in 0.1 M nBu4NBF4 in ACN, (c) 2a + Na2HPO4 in 0.1 M nBu4NBF4 in ACN. | ||
Conclusions
In summary, we have developed an efficient and sustainable electrochemical oxidative dearomatization of 2-naphthols, achieving C–O homocoupling and subsequent alkoxylation under mild and metal-free conditions. The optimized reaction conditions afforded the corresponding naphthalenone in moderate to good yields with broad functional group compatibility. Moreover, the utility of our products was demonstrated through gram scale and derivatization of the synthesized naphthalenones. Thus, this study provides environmentally friendly electrochemical oxidative approaches and shows potential for the synthesis of pharmaceuticals and natural product scaffolds.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 2a has been deposited at the CCDC under 2419735.Author contributions
Han Byeol Kim: data curation, investigation, methodology, and writing – original draft. Dong Kyun Han: data curation, investigation, methodology, and writing – original draft. Jae Kyun Lee: data curation. Seo-Jung Han: conceptualization, formal analysis, funding acquisition, investigation, methodology, project administration, resources, supervision, writing – original draft, and writing – review and editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This research was supported by Global – Learning & Academic research institution for Master's·PhD students, and Postdocs (LAMP) Program of the National Research Foundation of Korea (NRF) grant funded by the Ministry of Education (No. RS-2024-00441954). Moreover, this study was supported by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (RS-2024-00396224). This research was also supported by the National Research Council of Science & Technology (NST) granted by the Korea government (MSIT) (No. CAP23011-100). Moreover, this work was supported by the Sogang University Research Grant of 2025 (202510001.01).References
- (a) J. A. M. Vargas, D. P. Day and A. C. B. Burtoloso, Eur. J. Org Chem., 2021, 5, 741–756 CrossRef; (b) J. Liu, J. Chen, T. Liu, J. Liu and Y. Zeng, Chin. J. Org. Chem., 2023, 43, 379–410 CrossRef CAS; (c) X. P. Hui, L. F. Niu, Y. C. Xin, R. L. Wang, F. Jiang and P. F. Xu, Synlett, 2010, 5, 765–768 CrossRef.
- (a) W. Mahabusarakam, C. Hematasin, S. Chakthong, S. Voravuthikunchai and I. Olawumi, Planta Med., 2010, 76, 345–349 CrossRef CAS PubMed; (b) S. R. M. Ibrahim, S. A. Fadil, H. A. Fadil, B. A. Eshamawi, S. G. A. Mohamed and G. A. Mohamed, Toxins, 2022, 14, 154 CrossRef CAS PubMed; (c) C. F. Schwender, S. Faber, C. Blaum and J. Schavel, J. Med. Chem., 1970, 13, 684 CrossRef CAS PubMed; (d) J. T. Randolph, C. A. Flentge, P. P. Huang, D. K. Hutchinson, L. L. Klein, H. B. Lim, R. Mondal, T. Reisch, D. A. Montgomery, W. W. Jiang, S. V. Masse, L. E. Herandez, R. F. Henry, Y. Liu, G. Koev, W. M. Kati, K. D. Stewart, D. W. A. Beno, A. Molla and D. J. Kempf, J. Med. Chem., 2009, 52, 3174–3183 CrossRef CAS.
- (a) R. Shan, M. Standler and O. Sterner, J. Nat. Prod., 1997, 60, 804–805 CrossRef CAS; (b) J. Li, L. A. Li, H. P. Long, Y. P. Jian, Y. Xue, W. X. Wang, G. S. Tan, Z. C. Gong and J. K. Liu, Phytochemistry, 2021, 186, 112729 CrossRef CAS PubMed; (c) G. Chen, X. M. Pi and C. Y. Yu, Nat. Prod. Res., 2015, 29, 174–179 CrossRef CAS; (d) P. W. Jeffs and D. G. Lynn, J. Org. Chem., 1975, 40, 2958–2960 CrossRef CAS; (e) S. P. Roche and J. A. Porco Jr, Angew. Chem., Int. Ed., 2011, 50, 4068–4093 CrossRef CAS.
- (a) J. An and M. Bandini, Eur. J. Org Chem., 2020, 27, 4087–4097 CrossRef; (b) J. Zheng, S. B. Wang, C. Zheng and S. L You, J. Am. Chem. Soc., 2015, 137, 4880–4883 CrossRef CAS; (c) S. S. Zhang, J. Xue, Q. Gu, X. Jiang and S. L. You, Org. Biomol. Chem., 2021, 19, 8761–8771 RSC; (d) Q. X. Zhang, J. H. Xie, Q. Gu and S. L. You, Chem. Commun., 2023, 59, 3590–3593 RSC; (e) Y. Li, K. Feng, R. Zhao, C. Zhu and H. Xu, Synlett, 2024, 35, 1089–1100 CrossRef CAS; (f) Q. Yin, S. G. Wang, X. W. Liang, D. W. Gao, J. Zheng and S. L. You, Chem. Sci., 2015, 6, 4179–4183 RSC; (g) Z. L. Xia, C. Zheng, R. Q. Xu and S. L. You, Nat. Commun., 2019, 10, 3150 CrossRef; (h) D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Angew. Chem., Int. Ed., 2017, 56(12), 3242–3246 CrossRef CAS.
- (a) I. Tomczyk and M. Kalek, Chem. - Eur. J., 2024, 30, e202303916 CrossRef CAS; (b) S. S. Beigbaghlou, R. S. Yafele and M. kaleck, Synthesis, 2023, 55, 4173–4180 CrossRef.
- (a) R. Bag, N. P. Mishra, D. Saha and P. Banerjee, J. Org. Chem., 2024, 89, 2200–2211 CrossRef CAS; (b) J. L. Wan and J. M. Huang, Adv. Synth. Catal., 2023, 365, 1211–1216 CrossRef CAS; (c) C. Zhang, F. Bu, C. Zeng, D. Wang, L. Lu, H. Zhang and A. Lei, CCS Chem., 2022, 4, 1199–1207 CrossRef CAS; (d) F. Sprang, J. D. Herszman and S. R. Waldvogel, Green Chem., 2022, 24, 5116–5124 RSC; (e) T. Chen, S. Chen, S. Fu, S. Qin and B. Liu, Synlett, 2019, 30, 903–909 CrossRef CAS.
- Upon employing Na2HPO4 as an additive for the reaction of 1-methyl-2-naphthol 1a, both C–O homocoupled 2a and alkoxylated 3a were obtained in 34% and 17%, respectively. These results indicate that the direct synthesis of the alkoxylated compound from 1-methyl-2-naphthol using Na2HPO4 as an additive is challenging
 .
. - Q. Yin, S. G. Wang, X. W. Liang, D. W. Gao, J. Zheng and S. L. You, Chem. Sci., 2015, 6, 4179–4183 RSC.
- H. Egami, T. Rouno, T. Niwa, K. Masuda, K. Yamashita and Y. Hamashima, Angew. Chem., Int. Ed., 2020, 59(33), 14101–14105 CrossRef CAS PubMed.
- B. N. Norris, T. Pan and T. Y. Meyer, Org. Lett., 2010, 12(23), 5514–5517 CrossRef CAS PubMed.
- K. C. Nicolaou, Y. Tang, J. Wang, A. F. Stepan, A. Li and A. Montero, J. Am. Chem. Soc., 2007, 129(48), 14850–14851 CrossRef CAS.

Footnotes |
| † Electronic supplementary information (ESI) available: All experimental details, 1H, 13C NMR spectra, CV, and proposed mechanism. CCDC 2419735. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra02693h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |