
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable synthesis of Schiff base derivatives via an ionic liquid and a microwave-assisted approach: structural, biological, and computational evaluation†
Nilesh Bhusari‡
a,
Abhay Bagul‡b,
Vipin Kumar Mishrac,
Aisha Tufail d,
Digambar Gaikwad*b and
Amit Dubey*e
d,
Digambar Gaikwad*b and
Amit Dubey*e
aDepartment of Chemistry, Maulana Azad College of Arts, Science and Commerce, Chhatrapati Sambhajinagar 431004, Maharashtra, India
bDepartment of Forensic Chemistry, Government Institute of Forensic Sciences, Chhatrapati Sambhajinagar 431004, Maharashtra, India. E-mail: gaikwad.dd.dg@gmail.com
cChemistry Division, School of Advanced Sciences and Languages, VIT Bhopal University, Bhopal, India
dComputational Chemistry and Drug Discovery Division, Quanta Calculus, Greater Noida-201310, Uttar Pradesh, India
eCenter for Global Health Research, Saveetha Medical College and Hospital, Saveetha Institute of Medical and Technical Sciences, Chennai-600077, Tamil Nadu, India. E-mail: ameetbioinfo@gmail.com; amitdubey@saveetha.com
First published on 3rd July 2025
Abstract
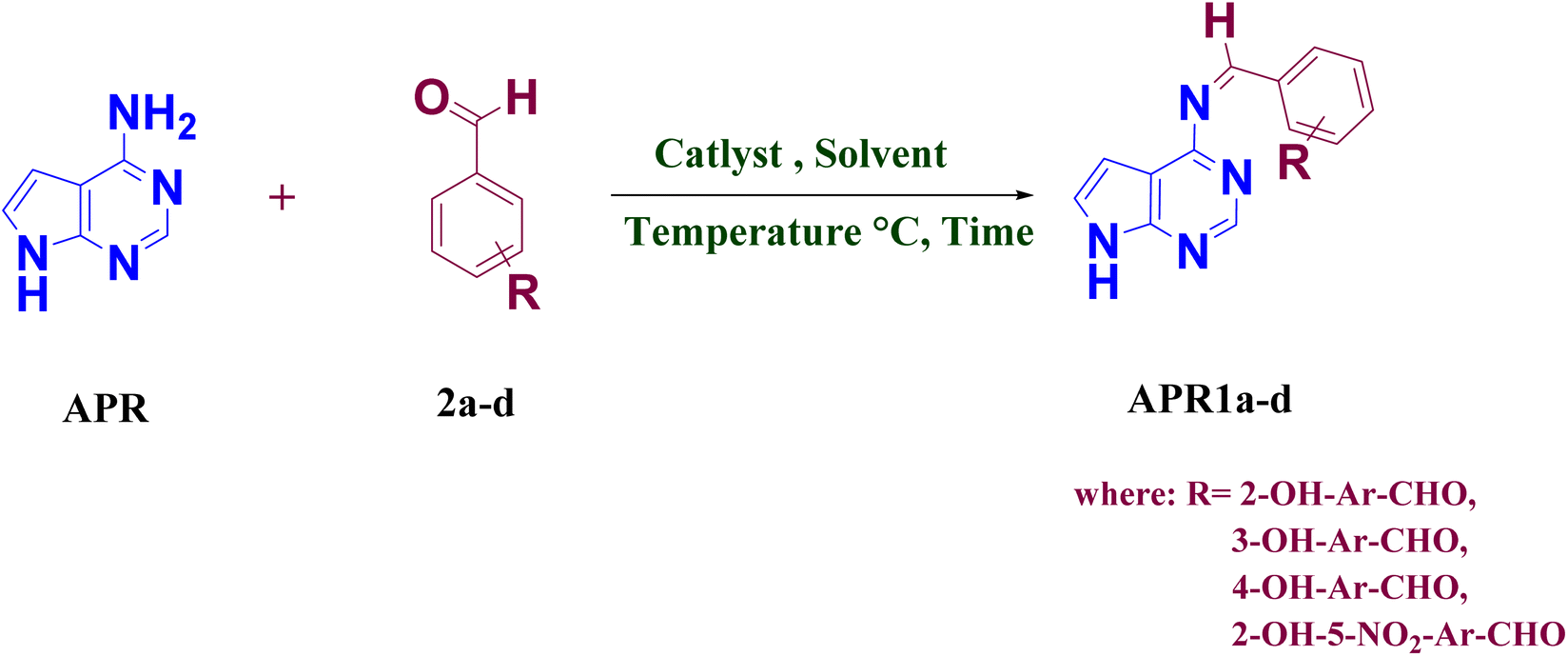
A sustainable and efficient microwave-assisted strategy was developed for the synthesis of novel 4-amino-pyrrolo [2,3-d]pyrimidine-based Schiff base derivatives (APR1a–d), utilizing the ionic liquid 1-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([HMIM][TFSI]) as both catalyst and solvent. This green protocol afforded high yields (82–94%) under mild conditions with excellent reusability of the ionic liquid. Structural confirmation was achieved via FT-IR, 1H and 13C NMR, and mass spectrometry. The antimicrobial potential of the synthesized compounds was assessed against a panel of Gram-positive and Gram-negative bacteria, as well as pathogenic yeast strains. Among them, APR1d exhibited the most potent antibacterial activity (up to 28 mm inhibition against B. subtilis) and broad-spectrum antifungal efficacy (up to 19 mm inhibition against C. albicans and S. cerevisiae). Cytotoxicity analysis via brine shrimp lethality assay indicated low toxicity, with LC50 values of 3.50 × 10−4 M (APR1b) and 8.50 × 10−4 M (APR1c). Density Functional Theory (DFT) analysis revealed that APR1d possessed the smallest HOMO–LUMO gap (0.0679 eV) and highest electrophilicity index (0.4288 eV), supporting its high reactivity. Molecular electrostatic potential maps and global reactivity descriptors (μ, η, S, ω, χ) further elucidated the electronic distribution and interaction potential. Molecular docking and MM/PBSA analyses confirmed APR1d's strong and stable binding to key microbial target proteins, surpassing standard drugs in binding affinity. These results underscore APR1d as a promising lead candidate with significant therapeutic potential, while highlighting the synergy of green chemistry, computational modeling, and biological validation in modern drug discovery.
1 Introduction
Over the years, the increasing challenge of infections caused by bacterial and fungal pathogens has heightened the demand for novel antibacterial and antifungal drugs, particularly due to the rise of drug-resistant strains.1–3 A significant number of currently available antibacterial and antifungal agents are derived from heterocyclic scaffolds, as these molecules possess broad pharmacological applications, making them indispensable in medicinal chemistry.4–8 The presence of heteroatoms within these structures imparts specific molecular characteristics that enable interactions with diverse biological targets.9–14 Among the wide range of heterocycles, naturally occurring pyrimidine compounds are well known for their broad spectrum of biological activities,15–17 including antibacterial,18–20 antifungal,21–23 anticancer,24–28 anti-inflammatory,29–31 antidepressant,30–34 antiallergic,35 antiviral,36–38 and antioxidant39–41 properties. Additionally, heterocyclic frameworks such as pyrrolopyrimidines have garnered considerable interest in drug discovery due to their potent biological effects.42,43 Another class of bioactive compounds, imines or azomethines (commonly known as Schiff bases), formed by the condensation of primary amines with aldehydes or ketones, also exhibit significant pharmacological potential.44–46 In light of these promising properties, we aimed to synthesize pyrimidine-linked Schiff bases by condensing 4-amino-pyrropyrimidine with benzaldehyde to generate novel Schiff base derivatives and assess their biological efficacy.The increasing complexity of organic molecules necessitates the development of innovative synthetic strategies that are efficient, selective, and environmentally sustainable.47,48 Traditional synthetic approaches often rely on volatile organic solvents, hazardous reagents, and high-energy input, which pose both environmental and operational challenges. Green chemistry has emerged as a solution, focusing on minimizing waste, reducing energy consumption, and improving overall process efficiency.49,50 In this context, eco-friendly pharmaceutical synthesis has gained traction,51,52 with microwave-assisted synthesis standing out as a powerful method due to its ability to shorten reaction times, improve yields, and lower energy requirements.53–55
Furthermore, the use of ionic liquids (ILs) in microwave-assisted reactions enhances product yield due to their unique physicochemical properties.56–59 ILs are non-volatile, non-flammable, highly solvating, recyclable, and thermally stable,60–63 making them viable alternatives to conventional volatile organic solvents64 or effective catalysts65 for organic transformations.66 The efficacy of many chemical reactions is influenced by the acidity or basicity of the medium, and task-specific ILs designed to serve as both solvents and catalysts play a crucial role in optimizing reaction conditions.67,68 In this study, we employed [HMIM] [TFSI], an acidic IL that functions as both a catalyst and solvent, offering the additional advantage of recyclability and reusability. The N-methyl imidazole ILs was tested for reaction optimization were shown in Fig. 1.
 | ||
| Fig. 1 Structure of imidazolium-based IL employed in the reaction optimization. | ||
In this study, we present an environmentally friendly synthetic approach for producing novel 4-amino-pyrrolo-pyrimidine linked Schiff base compounds (APR1a–d) using [HMIM][TFSI] IL as a dual catalyst and solvent under microwave irradiation. This method not only aligns with green chemistry principles but also enhances reaction efficiency. Key highlights of this research include the use of ILs for catalysis and solvent purposes, their recyclability, and microwave-assisted reaction conditions.
The synthesized Schiff base derivatives were thoroughly characterized using a combination of physicochemical techniques, elemental analysis, and spectral methods including FT-IR, 1H NMR, 13C NMR, and mass spectrometry. Their biological potential was evaluated through antimicrobial and cytotoxicity assays, which revealed significant activity against a range of bacterial and fungal strains. To further investigate their structural and electronic properties, Density Functional Theory (DFT) calculations were performed, providing insight into molecular geometry, frontier orbital energies, and global reactivity descriptors-key parameters in the assessment of drug-like behavior.
Molecular docking studies were also conducted to explore the binding interactions of the synthesized compounds with selected bacterial and fungal target proteins, offering mechanistic perspectives that complement the experimental antimicrobial results. The antibacterial activity was assessed in vitro against both Gram-positive (Staphylococcus aureus, Bacillus subtilis) and Gram-negative (Escherichia coli, Pseudomonas aeruginosa) bacterial strains using streptomycin as a reference standard. Similarly, antifungal efficacy was evaluated against yeast strains (Candida albicans and Saccharomyces cerevisiae) in comparison with fluconazole.
This study underscores the value of integrating green chemistry approaches in the development of pharmacologically relevant heterocycles, highlighting the role of ionic liquids and microwave-assisted synthesis in enhancing reaction efficiency, environmental compatibility, and the biological relevance of the resulting compounds.
2 Experimental
2.1. Materials
All chemicals utilized in the present study were of high purity, designated as AnalaR grade, encompassing organic compounds and solvents such as 4-amino pyrrolopyrimidine, benzaldehydes, 1-hexyl-3-methylimidazolium bis(trifluormethylsulfonyl)imide [HMIM][TFSI], solvents and others, all sourced from Sigma Aldrich, were employed without the need for additional purification.2.2. Apparatus
All reactions were conducted in oven-dried round-bottom flasks. Microwave-assisted reactions were carried out in a Biotage Microwave reactor. The progress of the reactions was monitored via Thin-layer chromatography (TLC) was performed using Merck silica gel 60F254 plates (0.25 mm thickness), with visualization under UV light at 254 nm, with MDC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol mobile phase 7:3(v/v) combination serves as the developing solvent system. To determine the uncorrected melting points of the synthesized compounds, open capillary tubes were employed. Fourier Transform Infrared (FT-IR) spectra in the 4000–500 cm−1 region were recorded on a Bruker FT-IR spectrophotometer using KBr pellets. The chemical elemental analysis for the determination of C, H, and N was conducted using the Carlo-Erba LA-118 microdosimeter. 1H NMR and 13C NMR spectra were obtained at room temperature in DMSO-d6, Tetramethylsilane (TMS) as the internal standard on a BRUKER AVANCE III HD NMR 400 MHz spectrometer. ESI- Mass spectra were obtained using the Waters Acquity TQD LC/MS/MS system. The HPLC analysis was conducted using a Shimadzu 1100 Series spectrometer equipped with a Agilent 1290 Infinity II Prep LC system, Shimadzu LC-20AD BLK with UV detection at 230 nm.
:methanol mobile phase 7:3(v/v) combination serves as the developing solvent system. To determine the uncorrected melting points of the synthesized compounds, open capillary tubes were employed. Fourier Transform Infrared (FT-IR) spectra in the 4000–500 cm−1 region were recorded on a Bruker FT-IR spectrophotometer using KBr pellets. The chemical elemental analysis for the determination of C, H, and N was conducted using the Carlo-Erba LA-118 microdosimeter. 1H NMR and 13C NMR spectra were obtained at room temperature in DMSO-d6, Tetramethylsilane (TMS) as the internal standard on a BRUKER AVANCE III HD NMR 400 MHz spectrometer. ESI- Mass spectra were obtained using the Waters Acquity TQD LC/MS/MS system. The HPLC analysis was conducted using a Shimadzu 1100 Series spectrometer equipped with a Agilent 1290 Infinity II Prep LC system, Shimadzu LC-20AD BLK with UV detection at 230 nm.
2.3. High performance liquid chromatography
The High-Performance Liquid Chromatography (HPLC) analysis was conducted using an Agilent 1290 Infinity II Prep LC system equipped with a SHIMADZU LC-20AD BLK pump. The separation was performed on an Ascentis® Supelco C18 column, measuring 250 mm in length and 4.6 mm in diameter, with a particle size of 5 μm. The mobile phase consisted of 65% acetonitrile in water, delivered at a constant flow rate of 1.5 mL min−1. Samples were injected at a volume of 10 μL, and detection was carried out using a diode array detector (DAD) set to a wavelength of 230 nm. HPLC analysis confirmed the purity of all tested compounds to be ≥95.0%, with representative chromatograms provided in the supplementary Section (refer Fig. S17, S18 and Tables S1–S3†).2.4. General procedure for the preparation of 4-amino-pyrrolo-pyrimidine linked Schiff base (APR1a–d)
The synthesis of 4-amino-pyrrolo-pyrimidine linked Schiff base compounds (APR1a–d) was achieved through a one-step condensation reaction between 4-amino-7H-pyrrolo [2,3-d] pyrimidine (APR) (1.0 mmol) and substituted benzaldehyde (2a–d) (1.0 mmol), utilizing [HMIM][TFSI] IL (3 volume) as both the catalyst and solvent (Scheme 1). The reaction mixture was subjected to microwave irradiation at 100–150 W for 10 minutes at 80 °C for 30 second time cycle. The reaction progress was monitored periodically using thin-layer chromatography (TLC) in MDC:methanol mobile phase 7:3(v/v). After completion of reaction, cool reaction mass at 25–30 °C. and quenched with water (10 volume). The precipitated solids was filtered and washed with water (1 volume × 2 times) to get the targeted wet crude compounds (APR1a–d). After that all wet crude compounds dried in a tray dryer for 8 hours at 50–55 °C to obtained dry crude compounds (APR1a–d). The obtained crude compounds was recrystallized using ethanol (4 volume) to produce a series of wet pure compounds (APR1a–d) which were dried in try dryer for 8 hours at 50–55 °C to obtained dry pure compounds (APR1a–d), as shown in Table 1 and Scheme 1. The ionic liquid-catalyzed, microwave-assisted green synthesis of 4-amino-pyrrolo-pyrimidine Schiff bases approach provided enhanced yields (91–94%) with reduced reaction times demonstrating its efficiency as a green synthetic strategy.
 | ||
| Scheme 1 Synthesis of substituted benzaldehyde-APRD derivatives. | ||
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Temperature (°C) | Time (min h−1) | Yieldb (%) |
| a Reaction conditions; 4-amino-7H-pyrrolo [2,3-d] pyrimidine (APR) (1.0 mmol), substituted benzaldehydes (2a–d) (1.0 mmol), solvent (5 volume), IL (3 volume), catalyst 5 mol%.b Isolated yield of pure products.c trace amounts of products were observed by TLC; MW = 80 °C. | |||||
| 1 | — | EtOH | RT | 10 h | No reaction |
| 2 | — | EtOH | 80 | 10 h | Tracec |
| 3 | AcOH | EtOH | RT | 10 h | No reaction |
| 4 | AcOH | EtOH | 80 | 4 h | 65–68% |
| 5 | AcOH | MeOH | 60 | 4 h | 61–65% |
| 6 | AcOH | DMF | 80 | 4 h | 52–56% |
| 7 | AcOH | Acetonitrile | 80 | 4 h | 55–60% |
| 8 | AcOH | EtOH | MW | 10 min | 70–73% |
| 9 | [HMIM][TFSI] | EtOH | MW | 10 min | 76–79% |
| 10 | [HMIM][TFSI] | [HMIM][TFSI] | MW | 10 min | 91–94% |
![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C
C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ), 1680 (CN–), 1302 (C–N), 762 (disubstituted benzene ring); 1H NMR (400 MHz in DMSO): δ 11.145 (Ar-NH), δ 10.828 (Ar-OH), δ 9.221 (Ar-CHNH), δ 7.438–7.702 (Ar-CH) ppm. 13C NMR [400 MHz, DMSO, δ(ppm)]: 157.26 (–CN imine carbon), 151.38 (pyrimidine C between 2 N atom), 148.50 (pyrimidine C to adjust N group), 131.49 (–CH), 120.02 (C1 phenyl ring), 119.96 (C3 phenyl ring), 116.02 (pyrrole C1), 104.04 (pyrrole C2), 101.71 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.12.CC), 1657 (CN–), 1343 (C–N), 783 (disubstituted benzene ring). 1H NMR (400 MHz in DMSO): δ 11.145 (Ar-NH), δ 10.828 (Ar-OH), δ 9.231 (Ar-CH = NH), δ 7.438–7.702 (Ar-CH), 13C NMR [400 MHz, DMSO, δ(ppm)]: 163.07 (–CN imine carbon), 149.33 (pyrimidine C between 2 N atom), 140.67 (pyrimidine C to adjust N group), 127.98 (–CH), 126.48(C1 phenyl ring), 123.02 (C3 phenyl ring), 120.01 (pyrrole C1), 117.44 (pyrrole C2), 102.02 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.22.CC), 1662 (CN–), 1347 (C–N aromatic amine), 723 (Disubstituted benzene ring). 1H NMR (400 MHz inn DMSO): δ 12.38 (Ar-NH), δ 12.258 (Ar-OH), δ 10.341 (Ar-CHNH), δ 6.974–8.408 (Ar-CH), 13C NMR [400 MHz, DMSO, δ(ppm)]: 160.02 (–CN imine carbon), 152.06 (pyrimidine C between 2 N atom), 142.42 (pyrimidine C to adjust N group), 130.53 (–CH), 126.78(C1 phenyl ring), 124.55 (C3 phenyl ring), 116.99 (pyrrole C1), 102.09 (pyrrole C2), 99.93 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.15.CC), 1677 (CN–), 1360 (C–N aromatic amine), 1360 (–N–O) symmetrical stretching, 1470 (N–O) asymmetrical stretching, 818 (trisubstituted benzene ring). 1H NMR (400 MHz in DMSO): δ 12.392 (Ar-NH), δ 10.593 (Ar-OH), δ 8.047 (Ar-CHNH), δ 7.333–7.586 (Ar-CH). 13C NMR [400 MHz, DMSO, δ(ppm)]: 163.02 (–CN imine carbon), 149.39 (pyrimidine C between 2 N atom), 140.07 (pyrimidine C to adjust N group), 127.38 (-CH = ), 126.40 (C1 phenyl ring), 123.02 (C3 phenyl ring), 120.01 (pyrrole C1), 117.40 (pyrrole C2), 102.02 (pyrrole C3). MS of C13H9N5O3: m/z: 283.24; (M + H)+: 283.99.
), 1680 (CN–), 1302 (C–N), 762 (disubstituted benzene ring); 1H NMR (400 MHz in DMSO): δ 11.145 (Ar-NH), δ 10.828 (Ar-OH), δ 9.221 (Ar-CHNH), δ 7.438–7.702 (Ar-CH) ppm. 13C NMR [400 MHz, DMSO, δ(ppm)]: 157.26 (–CN imine carbon), 151.38 (pyrimidine C between 2 N atom), 148.50 (pyrimidine C to adjust N group), 131.49 (–CH), 120.02 (C1 phenyl ring), 119.96 (C3 phenyl ring), 116.02 (pyrrole C1), 104.04 (pyrrole C2), 101.71 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.12.CC), 1657 (CN–), 1343 (C–N), 783 (disubstituted benzene ring). 1H NMR (400 MHz in DMSO): δ 11.145 (Ar-NH), δ 10.828 (Ar-OH), δ 9.231 (Ar-CH = NH), δ 7.438–7.702 (Ar-CH), 13C NMR [400 MHz, DMSO, δ(ppm)]: 163.07 (–CN imine carbon), 149.33 (pyrimidine C between 2 N atom), 140.67 (pyrimidine C to adjust N group), 127.98 (–CH), 126.48(C1 phenyl ring), 123.02 (C3 phenyl ring), 120.01 (pyrrole C1), 117.44 (pyrrole C2), 102.02 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.22.CC), 1662 (CN–), 1347 (C–N aromatic amine), 723 (Disubstituted benzene ring). 1H NMR (400 MHz inn DMSO): δ 12.38 (Ar-NH), δ 12.258 (Ar-OH), δ 10.341 (Ar-CHNH), δ 6.974–8.408 (Ar-CH), 13C NMR [400 MHz, DMSO, δ(ppm)]: 160.02 (–CN imine carbon), 152.06 (pyrimidine C between 2 N atom), 142.42 (pyrimidine C to adjust N group), 130.53 (–CH), 126.78(C1 phenyl ring), 124.55 (C3 phenyl ring), 116.99 (pyrrole C1), 102.09 (pyrrole C2), 99.93 (pyrrole C3). MS of C13H10N4O: m/z:238.24; (M + H)+:239.15.CC), 1677 (CN–), 1360 (C–N aromatic amine), 1360 (–N–O) symmetrical stretching, 1470 (N–O) asymmetrical stretching, 818 (trisubstituted benzene ring). 1H NMR (400 MHz in DMSO): δ 12.392 (Ar-NH), δ 10.593 (Ar-OH), δ 8.047 (Ar-CHNH), δ 7.333–7.586 (Ar-CH). 13C NMR [400 MHz, DMSO, δ(ppm)]: 163.02 (–CN imine carbon), 149.39 (pyrimidine C between 2 N atom), 140.07 (pyrimidine C to adjust N group), 127.38 (-CH = ), 126.40 (C1 phenyl ring), 123.02 (C3 phenyl ring), 120.01 (pyrrole C1), 117.40 (pyrrole C2), 102.02 (pyrrole C3). MS of C13H9N5O3: m/z: 283.24; (M + H)+: 283.99.2.5. Biological activities
In the pursuit of combating infectious diseases and discovering potent anti-infectious medications, a comprehensive evaluation was conducted on the synthesized compounds to assess their impact on the specified biological activities outlined below.2.5.1.1 Test microorganisms. In this study, two Gram-positive bacteria (Staphylococcus aureus MTCC 96 and Bacillus subtilis MTCC 121) and two Gram-negative bacteria (Escherichia coli MTCC 1652 and Pseudomonas aeruginosa MTCC 741) were employed to evaluate the antimicrobial activity of the synthesized compounds. All bacterial cultures were obtained from the Microbial Type Culture Collection (MTCC) at IMTECH-Chandigarh. The medium utilized for antimicrobial testing was Muller Hinton agar, which was autoclaved at 15 lbs in−2 for 15 minutes.
2.5.1.2 Antibacterial activity. The antimicrobial activity of the newly synthesized compounds was assessed using the agar wells diffusion technique.69 For the evaluation, the inoculum size was adjusted to approximately 108 colony-forming units (cfu mL−1) by suspending the culture in sterile distilled water. Petri dishes containing 20 mL of Muller Hilton agar medium were swabbed with a culture of the relevant microbial strains and allowed to absorb for 15 minutes. Using a sterile borer, wells (6 mm in diameter) were created, and 100 μL of a 4.0 mg mL−1 concentration solution of each compound, reconstituted in DMSO, was added to the preinoculated plates. All plates were then incubated at 37 °C for 24 hours, and the antimicrobial activity of the prepared compounds was determined by measuring the zone of inhibition around the wells. DMSO served as the negative control, while Streptomycin functioned as the positive control. This process was replicated on three plates for each organism.
2.5.1.3 Determination of minimum inhibitory concentration (MIC). The determination of the Minimum Inhibitory Concentration (MIC) for all the compounds was carried out using the modified agar well diffusion method.70,71 Various concentrations, spanning from 10 to 1000 μg mL−1, were prepared for the synthesized compounds from a stock solution of 4 mg mL−1 in DMSO. A 100 μL volume of each dilution was dispensed into wells (in triplicate) on agar plates that had already been seeded with 100 μL of a standardized inoculum (108 cfu mL−1) of the target microbial strain. Subsequently, these plates were incubated at 37 °C for 24 hours, and the inhibition zones were observed. Streptomycin antibiotic served as the positive control.
2.5.1.4 Antifungal activity. All the synthesized compounds underwent assessment for their antifungal efficacy against freshly cultured strains of C. albicans and Saccharomyces cerevisiae using the cup plate method, following a standard protocol.72 The culture media were sterilized by autoclaving for 15 minutes at a pressure of 10 pounds per square inch. Each microbial strain was cultured by spreading 20 mL of Mueller-Hinton agar media onto a Petri plate and allowing it to absorb for 15 minutes before continuing with the cultivation process. Wells with a diameter of 6 mm were then created on the agar plates using a sterile borer. Subsequently, 100 μl of each test compound (APR1a–d), dissolved in DMSO, was added to the previously inoculated plates. The plates were then maintained at a temperature of 37 °C for three days. The inhibition zones for each test compound were measured, with fluconazole serving as the positive control and DMSO as the negative control. The entire procedure was repeated three times, and efficacy of fluconazole was used as a reference point in this investigation.
2.6. Computational workflow overview
A comprehensive computational approach was employed to investigate the electronic structure, stability, reactivity, molecular interactions, and pharmacokinetic properties of the synthesized compounds. This study integrates Density Functional Theory (DFT) calculations, Molecular Electrostatic Potential (MESP) analysis, Molecular Docking, Molecular Dynamics (MD) Simulations, ADMET Profiling, and Pharmacophore Modeling to provide a holistic understanding of the compounds' physicochemical and biological properties.3 Results and discussion
3.1. Optimization of reaction conditions
The optimized reaction conditions were determined using both conventional and microwave irradiation methods by reacting 4-amino-7H-pyrrolo [2,3-d] pyrimidine (APR) with substituted benzaldehyde (2a–d). Table 1 presents the optimized conditions under various parameters.Initially, the reaction was conducted at room temperature for 10 hours using EtOH as the solvent, but no progress was observed (entry 1). When the reaction was carried out at 80 °C, only a trace amount of the product was detected by TLC (entry 2). Subsequently, a AcOH was added and stirred at room temperature for 10 hours, but no reaction occurred (entry 3). However, refluxing the reaction at 80 °C for 4 hours resulted in a 65–68% yield (entry 4).
To assess the effect of different solvents on product yield, the reaction was performed using MeOH, DMF, and acetonitrile while keeping other conditions constant, as presented in Table 1. The yields obtained were 61–65% with MeOH, 52–56% with DMF, and 55–60% with acetonitrile (entries 5, 6, and 7, respectively). Additionally, the reaction was conducted under microwave irradiation using a catalytic amount of AcOH in EtOH, yielding 70–73% of the product (entry 8). This was higher than the yield obtained through conventional heating, demonstrating the greater efficiency of microwave irradiation for synthesizing the desired compounds. Further optimization was performed using ionic liquids (ILs).
The catalytic efficiency of imidazolium-based ionic liquid [HMIM][TFSI] was evaluated using EtOH as the solvent under microwave irradiation at 80 °C for 10 minutes. The addition of imidazolium-based ionic liquids, [HMIM][TFSI], resulted in product yields of 76–79% (entry 9). Further, the reactions were examined using imidazolium-based ionic liquid [HMIM][TFSI] both as catalysts and solvents under microwave irradiation for 10 minutes at 80 °C. Under these conditions, [[HMIM][TFSI] resulted in product yields of 91–94%, respectively (entries 10). Overall, the results indicate that the tested ionic liquid, [HMIM][TFSI] exhibited the highest conversion rate under microwave irradiation, making it the most efficient catalytic system.
3.2. Synthesis and analysis of desired 4-amino pyrrolo-pyrimidine-linked Schiff base compounds APR1a–d
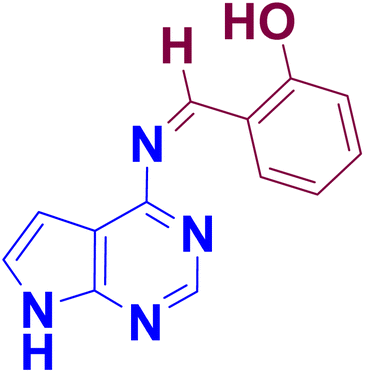
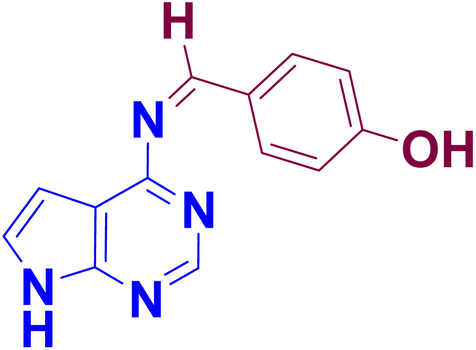
The synthesis of 4-amino pyrrolo-pyrimidine-linked Schiff base APR1a–d (Table 2) was carried out with the established optimized reaction conditions (Table 1), The 4-amino-7H-pyrrolo [2,3-d] pyrimidine (APR) was treated with substituted benzaldehydes (2a–d) in the presence of [HMIM][TFSI] serving as both a catalyst and solvent. The synthetic procedure is detailed in the experimental section. Furthermore, the structures of the synthesized compounds were confirmed through FT-IR, NMR, 13C NMR, MS and elemental analysis.




3.3. Recycling and reuse of ionic liquid
The recycling of [HMIM] [TFSI] IL was performed following the method with minor modification outlined in the literature.96 The IL was recovered from aqueous filtrate (mother liquor) obtained from multiple reaction sets, starting with an initial extraction using dichloromethane. The resulting solution was then filtered to remove any residual particles. Subsequently, the solvent was evaporated from the filtrate under vacuum below 45 °C, and the recycled IL was dried for 8 hours under vacuum before reuse.To evaluate the efficiency of [HMIM][TFSI] in the synthesis of 4-amino pyrrolo-pyrimidine-linked Schiff base (APR1a–d) compounds, reactions were conducted up to four times using the IL. After isolating the product, the remaining ionic liquid was subjected to the recycling process described above and reused in the subsequent cycle. This procedure was repeated for four cycles, with the results presented in Scheme 2.
 | ||
| Scheme 2 Recycling and reuse of [HMIM][TFSI] IL; yield (%): 1 = employing fresh IL (1st cycle), 2 = recycled IL (2nd cycle), 3 = recycled IL (3rd cycle), 4 = recycled IL (4th cycle). | ||
3.4. Plausible reaction mechanism
The plausible reaction mechanism for the synthesis of 4-amino-pyrrolo-pyrimidine-linked Schiff base (APR1a–d) compounds is illustrated in Scheme 3.97,98 | ||
| Scheme 3 Plausible reaction mechanism for the [HMIM][TFSI]-catalyzed, synthesis of 4-amino-pyrrolo-pyrimidine-linked Schiff base (APR1a–d) compounds. | ||
Step 1: electrophilic activation and carbene formation.
The substituted benzaldehydes (2a–d) undergo protonation at the carbonyl oxygen by the acidic C2–H proton of the 1-hexyl-3-methylimidazolium cation ([HMIM]+) in the ionic liquid [HMIM][TFSI]. This protonation enhances the electrophilicity of the carbonyl carbon. Simultaneously, deprotonation at the C2–H position of the imidazolium cation in [HMIM][TFSI], leading to the formation of a transient N-heterocyclic carbene (NHC), as illustrated in Step 1 of Scheme 3.
Step 2: NHC-mediated nucleophilic addition.
This NHC acts as a base/nucleophile, facilitating the nucleophilic attack of the amino nitrogen lone pair from 4-amino-7H-pyrrolo[2,3-d]pyrimidine (APR) on the activated carbonyl carbon of 2a–d. This step forms the tetrahedral carbinolamine intermediate.
Step 3: [HMIM][TFSI] ionic liquid regeneration.
The NHC is rapidly reprotonated (likely by protonated carbinolamine oxygen), regenerating the original [HMIM][TFSI] ionic liquid (Step 3). This step closes the catalytic cycle involving the NHC.
Step 4: dehydration.
The elimination of water from the carbinolamine intermediate leads to the formation of the target Schiff base derivatives (APR1a–d).
3.5. Physical and spectral analysis
To confirm the structural identification of synthesized compounds various instrumental analyses like FT-IR, NMR, 13C NMR, MS and elemental analysis were conducted.NN–) group,101,102 observed in the 1654–1673 cm−1 region, appeared as a distinct new band, confirming the successful formation of all synthesized compounds. This also indicates that the stretching vibrations of the aldehyde (CHO) and amino (NH2) groups in the APR compound are suppressed. The band observed at 3118–3314 cm−1, identified as aromatic (NH), suggests the presence of these compounds in their processed forms.103 The bands observed between 2912 and 3098 cm−1 are characteristic of aldehydic substances. In the infrared spectra of APR1a–d compounds, two distinct bands at 1582–1588 cm−1 and 1475–1509 cm−1 correspond to the CC group of the aromatic ring. Additionally, the FT-IR spectra of APR1a–d compounds show the aromatic (C–N) band at 1316–1335 cm−1, the di/trisubstituted benzene ring at 723–729 cm−1, and the monosubstituted benzene ring at 654–691 cm−1.N) carbon, exhibits a peak in the range of 157.26–163.07 ppm, characteristic of Schiff bases, confirming the successful synthesis of the compounds (APR1a–d). The carbon positioned between two nitrogen atoms in the pyrimidine ring resonates between 149.39–152.06 ppm, while the carbon adjacent to a nitrogen group appears within 140.07–148.50 ppm, reflecting electronic influences from the nitrogen atoms. The –CH = carbon resonates between 127.38–131.49 ppm, indicating its involvement in a conjugated system. The phenyl ring carbons (C1 and C3) display signals in the ranges of 120.02–126.78 ppm and 119.96–124.55 ppm, respectively, which are characteristic of aromatic carbon environments. Similarly, the pyrrole ring carbons (C1, C2, and C3) resonate within 116.02–120.01 ppm, 102.09–117.44 ppm, and 99.93–102.02 ppm, respectively, consistent with electron-rich heterocyclic systems.These 13C NMR chemical shifts provide valuable insights into the structural and electronic properties of the synthesized compounds.
Similarly, the MS spectra of APR1b, APR1c, and APR1d exhibit molecular ion peaks at m/z 239.22, m/z 239.15, and m/z 283.99, respectively. These values align with their theoretical molecular masses, further confirming the successful synthesis and structural integrity of the target compounds. Additionally, the consistent m/z values of the [M + H]+ ion peaks validate the formation of the target compounds, reinforcing the molecular structures. This strong agreement provides compelling evidence for the structural integrity of the synthesized compounds.
Overall, the ESI-MS data effectively validates the structural characterization of the synthesized compounds, confirming their molecular compositions and paving the way for further exploration of their chemical properties and potential applications.
3.6. Biological activities
Antibiotics, essential to modern medicine, are becoming increasingly ineffective due to rising antimicrobial resistance (AMR), posing a serious global health threat. The devastating impact of drug-resistant infections was underscored by a landmark 2019 analysis on the global burden of bacterial AMR, which reported 1.27 million direct deaths and 4.9 million associated deaths caused by resistant bacterial infections. Therefore, in the search for potential agents to combat bacterial antimicrobial resistance, along with the associated oxidative stress and inflammation, the following activities were evaluated. | ||
| Fig. 2 Antibacterial activity of the synthesised compounds and standard drug. | ||
Key observations.
(1) Potent activity against S. aureus: all synthesized compounds (APR1a–d) exhibited greater inhibition against S. aureus (21.5–25.0 mm) than streptomycin (15.5 mm), indicating strong efficacy against this Gram-positive bacterium.
(2) Strongest broad–spectrum activity – the compound APR1d showed the highest antibacterial activity across all tested strains, particularly against B. subtilis (28.0 mm), E. coli (20.0 mm), and P. aeruginosa (23.0 mm), surpassing streptomycin in effectiveness.
(3) Gram-negative activity (E. coli & P. aeruginosa): All compounds displayed moderate to strong inhibition against E. coli (15.0–20.0 mm) and P. aeruginosa (14.0–23.0 mm), with APR1d showing the highest activity, suggesting potential efficacy against Gram-negative bacteria.
(4) Weaker activity against B. subtilis: except for compound APR1d (28.0 mm), the other compounds showed low to moderate inhibition against B. subtilis (7.5–11.0 mm), with streptomycin (14.5 mm) displaying stronger inhibition than synthesized compounds (APR1a–d).
(5) By correlating antibacterial activity with HPLC purity data, we observed that higher-purity compounds (≥95% purity; see Fig. S17, S18 and Tables S1–S3†) exhibited enhanced biological performance, ensuring the reliability of our data.
Key takeaways:
• APR1d emerges as the most promising compound, exhibiting the highest activity across all tested bacterial strains.
• The synthesized compounds, especially APR1c and APR1d, demonstrate superior antibacterial efficacy compared to Streptomycin, suggesting their potential as new antimicrobial agents.
• The varied inhibition patterns indicate potential selectivity of the compounds toward different bacterial species, which could be useful in targeted antibiotic development.
3.7. Antifungal activity
Table S3† and Fig. 3 present the antifungal activity of the synthesized compounds (APR1a–d) and the standard drug fluconazole against Candida albicans and Saccharyomyces cerevisiae, measured by the zone of inhibition (mm). Linking these results with HPLC purity data revealed how purity affects antifungal performance. | ||
| Fig. 3 Antifungal activity of the synthesised compounds and standard drug. | ||
(1) Compound APR1d exhibits the highest activity: APR1d shows the strongest inhibition against both Candida albicans (19.0 mm) and Saccharomyces cerevisiae (19.0 mm), making it the most potent antifungal compound.
(2) Better performance than fluconazole: all synthesized compounds, except APR1c against Candida albicans (11.0 mm), show superior inhibition compared to fluconazole (10.5 mm and 14.0 mm), indicating enhanced antifungal efficacy.
(3) Strain-specific trends: APR1a (16.5 mm) is highly effective against Candida albicans, whereas APR1b (15.5 mm) and APR1c (14.5 mm) show moderate activity against Saccharomyces cerevisiae.
(4) Our analysis showed that compounds with ≥95% purity (HPLC; see Fig. S17, S18 and Tables S1–S3†) demonstrated stronger antibacterial effects, confirming data reliability.
(5) Key takeaways.
• APR1d is the most effective antifungal agent, displaying broad-spectrum activity against both fungal strains.
• The synthesized compounds show higher antifungal potency than fluconazole, making them promising candidates for further pharmaceutical development.
• Additional studies, including mechanism of action, cytotoxicity, and structure–activity relationship (SAR) analysis, are needed to confirm their therapeutic potential.
 | ||
| Fig. 4 Cytotoxic activity of the all compounds. | ||
Key observations.
(1) APR1c (LC50 > 8.50 × 10−4 M) shows the least cytotoxicity, as it has the highest LC50 value.
(2) APR1b (LC50 > 3.50 × 10−4 M) exhibits the highest cytotoxicity among the synthesized compounds, suggesting it may have potential bioactivity.
(3) APR1d (LC50 > 4.50 × 10−4 M) and APR1a (LC50 > 6.50 × 10−4 M) show intermediate cytotoxic effects.
(4) Vincristine sulphate (LC50 > 3.24 × 10−6 M), a known anticancer drug, exhibits the highest cytotoxicity, which is expected due to its strong bioactivity against cancer cells.
(5) All the compounds (APR1a–d) was obtained in ≥95% purity (HPLC, see Fig. S17, S18 and Tables S1–S3†)), ensuring reliability of biological data.
Key takeaways:
• The synthesized compounds APR1a–d demonstrate varying degrees of cytotoxicity, with APR1b being the most active.
• However, their toxicity levels are significantly lower than Vincristine Sulphate, suggesting that they might be safer but require further bioactivity screening.
• Further studies, including detailed mechanistic investigations, are necessary to determine their potential therapeutic applications.
3.8. Structure activity relationship (SAR)
The exploration of the structure–activity relationship (Scheme 4) proves instrumental in discerning the correlation between molecular structure and biological activity. All synthesized compounds showcase remarkable activity across various biological assessments. | ||
| Scheme 4 Structure activity relationship of synthesized compounds. | ||
• The presence of multiple nitrogen atoms in the fused bicyclic ring system enhances hydrogen bonding and potential interaction with biological targets such as enzymes and receptors.
• It can participate in key interactions with amino acid residues in biological receptors, possibly forming hydrogen bonds that stabilize ligand-receptor binding.
• The electron-withdrawing effect may influence the electronic distribution over the ring, potentially affecting binding affinity.
(4) The azomethine (–CN) group introduces planarity and electron delocalization in the molecule, which can enhance target binding affinity through π–π stacking interactions with aromatic amino acids (e.g., in enzyme active sites). The presence of an azomethine (–CN) group in a synthesised compound (APR1a–d) significantly influences biological activity, target interaction, and pharmacokinetic properties. It plays a role in Kinase inhibition (anticancer potential), DNA/protein interaction (cytotoxicity) and Metal complexation (antimicrobial activity).
(5) The presence of an electron-donating or withdrawing group near the imine affects its reactivity:
• Electron-withdrawing group (–NO2) in the compound APR1d can increase stability and reduce nucleophilic attack.
• Electron-donating groups (–OH) in synthesised compounds (APR1a–d) can enhance interactions with proteins.
3.9. Computational insights into stability, reactivity, and potential applications
The computational analysis of the studied structures provides valuable insights into their electronic properties, stability, and reactivity (Table S4†). By examining the total energy, binding energy, frontier molecular orbitals, and various reactivity descriptors, we can deduce the potential applications and stability of these compounds.3.9.1.1 Electronic properties and reactivity: understanding charge transfer and conductivity. The HOMO–LUMO gap (refer Table S4† and Fig. 5), a crucial parameter defining electronic transitions and molecular reactivity, is observed to be the smallest in structure APR1d (0.0679 eV). This lower band gap signifies higher electronic conductivity and greater reactivity, making APR1d an attractive candidate for optoelectronic and redox-related applications. The calculated chemical hardness (η) values further reinforce this trend, with APR1d showing the lowest hardness (0.0339 eV), indicating a highly polarizable system that can easily interact with its environment.
 | ||
| Fig. 5 HOMO–LUMO of synthesized compounds. | ||
Conversely, chemical softness (S), which measures a molecule's tendency to exchange electrons, is highest for APRD1d (14.7216 eV−1) (Table S4† and Fig. 5). This suggests that APR1d can efficiently participate in charge transfer processes, a desirable trait in molecular electronics and drug design. Additionally, the electronegativity (χ) and chemical potential (μ) values further confirm the high reactivity of APR1d, making it a promising candidate for further functionalization and practical applications.
3.9.1.2 Electrophilicity and dipole moment: implications for reactivity and applications. The electrophilicity index (ω), an essential indicator of a molecule's ability to accept electrons, is highest for APR1d (0.4288 eV) (Table S4 and Fig. S21†). This suggests that structure APR1d has the strongest potential to participate in electrophilic reactions, which could be beneficial in designing novel materials for energy storage or targeted drug interactions.
Dipole moment values also provide insights into the molecular polarity and interaction potential. Structure APR1a exhibits the highest dipole moment (1.9717 D) (Table S4†), indicating a higher degree of asymmetry in charge distribution, which can enhance intermolecular interactions, particularly in biological environments. In contrast, structure APR1c has the lowest dipole moment (0.7816 D) (Table S4†), suggesting a more symmetrical charge distribution, which may reduce solubility but enhance packing efficiency in solid-state applications.
4 Conclusion and future prospects: unlocking potential for advanced applications
The computational findings reveal that structure APR1d stands out as the most reactive and stable candidate, with superior electronic properties, high chemical softness, and strong electrophilic potential. These characteristics make it an excellent candidate for applications in molecular electronics, catalysis, and biomedical engineering.Future experimental validation and functionalization of APR1d could further enhance its properties, paving the way for its use in next-generation materials. Additionally, the high dipole moment of APR1d suggests potential for targeted drug delivery applications, where molecular polarity plays a crucial role in biological interactions.
These results provide a strong foundation for further investigations, bridging computational insights with real-world applications to unlock the full potential of these novel molecular structures.
4.1 Molecular electrostatic potential (MESP) analysis: charge distribution, reactivity, and potential applications
• Structure APR1a exhibits well-distributed electrostatic potential, with electron-rich (negative potential) regions localized around electronegative atoms, such as oxygen and nitrogen (Table S5†). This suggests a strong tendency for hydrogen bonding and nucleophilic interactions, making APR1a a promising candidate for biomolecular binding and drug–receptor interactions.
• Structure APR1b shows a slightly more uniform MESP distribution compared to APR1a, with a moderate electrostatic potential gradient. The presence of both donor and acceptor regions suggests balanced reactivity, potentially favoring selective molecular recognition in enzyme inhibition or drug–target interactions.
• Structure APR1c demonstrates less polarization in its MESP map, indicating a reduced dipole moment and weaker intermolecular interactions compared to 1a and 1b. However, its electron-deficient regions suggest that APR1c could act as an effective electrophile, participating in charge-transfer processes under specific conditions.
• Structure APR1d stands out with highly localized negative potential regions, indicating enhanced nucleophilic reactivity. This strong electrostatic variation suggests that APR1d is the most chemically active structure, aligning with its low band gap energy (0.0679 eV) and high electrophilicity index (0.4288 eV) observed in the DFT results (Table S4†). Such properties enhance its ability to interact with biomolecules, metal centers in catalysis, and redox-active environments.
These findings reinforce the potential of APR1d as the most reactive and bioactive candidate, while APR1a and APR1b exhibit strong hydrogen bonding capabilities, making them suitable for biological applications.
4.1.1.1 Correlation between MESP and electronic properties. The trends observed in MESP maps align with the HOMO–LUMO energy gap, chemical hardness, and dipole moment values from the computational studies:
• Lower band gap energy (as seen in APR1d) corresponds to higher charge transfer potential, which is reflected in its pronounced MESP variations.
• Higher chemical softness (S) enhances electronic adaptability, a key trait for catalytic, sensor, and drug development applications. The most chemically soft structure, APR1d (S = 14.7216 eV−1) (Table S4† and Fig. 8), also shows the most intense electrostatic variations in MESP, reinforcing its high reactivity.
• Dipole moment trends align with MESP asymmetry: APR1a (1.97 D) shows the most asymmetric charge distribution, favoring polar interactions, while APR1c (0.78 D) (Table S4† and Fig. S23†) has the most uniform MESP distribution, indicating lower polarity and potential applications in materials science.
5 Conclusion and future directions
The MESP analysis, coupled with electronic property evaluation, highlights APR1d as the most reactive structure, making it highly suitable for catalytic applications, charge-transfer processes, and bioactive compound development. Meanwhile, APR1a and APR1b demonstrate strong polar interactions, suggesting their potential in drug design and molecular recognition.Future studies should focus on experimental validation through spectroscopic and biological assays, further exploring the functionalization potential of these structures for advanced applications in materials science, catalysis, and medicinal chemistry.
5.1 Molecular docking analysis: binding affinity, hydrogen bonding, and steric interactions of compounds against E. coli and Candida albicans
The molecular docking results presented in Table S6† and visually represented in Fig. 6 and 7 provide compelling insights into the potential antibacterial and antifungal efficacy of the tested compounds against E. coli (PDB: 1hnj) and Candida albicans (PDB: 5v5z). The observed binding affinities, hydrogen bonding interactions, and steric interactions suggest promising bioactivity, reinforcing the therapeutic potential of these compounds. | ||
| Fig. 6 Molecular docking visualization of E. coli target protein (PDB: 1HNJ, white surface) in complex with top-performing candidate molecules. Binding poses of APR1a (red), APR1b (blue), APR1c (cyan), APR1d (yellow), and reference drug Fluconazole (green) are depicted in ball-and-stick representation. The overlay highlights distinct binding orientations and favorable interactions within the active site, underscoring the potential of the APR1a–d series as strong therapeutic leads for antimicrobial development. | ||
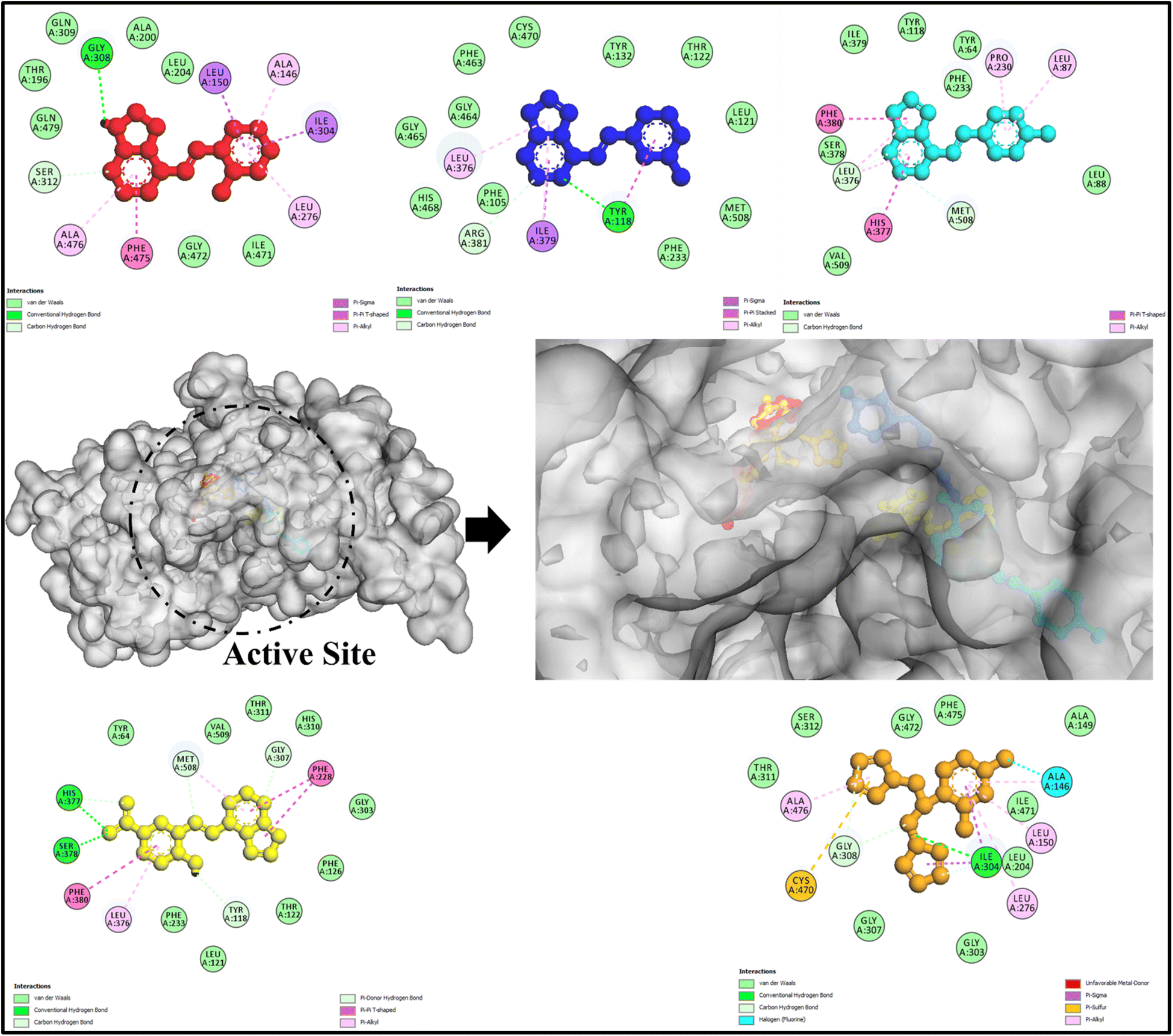 | ||
| Fig. 7 Molecular docking visualization of Candida albicans target protein (PDB: 5V5Z, grey surface) in complex with top-performing candidate molecules. The binding conformations of APR1a (red), APR1b (blue), APR1c (cyan), APR1d (yellow), and the reference drug streptomycin (orange) are shown in ball-and-stick models. The superimposed docked poses illustrate favorable orientation and occupancy within the active site cavity, highlighting the APR1a–d series' promising potential as next-generation antifungal agents through enhanced target engagement and binding complementarity. | ||
Similarly, APR1b demonstrated a notable binding energy of −7.3 kcal mol−1, interacting primarily with Asn247, which is crucial for its stability within the active site. Compound APR1a and APR1c exhibited moderate binding affinities of −7.0 and −7.1 kcal mol−1, respectively, indicating potential antibacterial activity but slightly lower than APR1b and APR1d. The lack of hydrogen bonding in APR1c suggests that steric interactions with Asn247 play a significant role in its stability within the binding pocket (Fig. 6).
In comparison, streptomycin, the control drug, displayed weaker binding affinity (−6.2 kcal mol−1) but showed multiple hydrogen bonding interactions with Asp150, Gly152, Met207, Gly209, and Asn210. This suggests that the tested compounds, particularly APR1d and APR1b, might offer stronger or comparable antibacterial activity while interacting with key active site residues.
5.1.1.1 Interaction with Candida albicans (PDB: 5v5z). The docking results against Candida albicans (refer Table S6† and Fig. 7). further reinforce the promising antifungal potential of the synthesized compounds. Notably, compound APR1d exhibited the highest binding affinity (−8.0 kcal mol−1), outperforming fluconazole, the standard antifungal drug (−7.8 kcal mol−1). This enhanced binding suggests that APR1d could serve as a strong antifungal candidate. The presence of hydrogen bonding with His377 and Ser378, along with steric interactions involving Gly307, His377, and Ser378, indicates a well-fitted binding conformation within the fungal target site.
Compounds APR1a and APR1b both exhibited significant binding affinities of −7.8 kcal mol−1, equaling fluconazole. However, APR1b formed hydrogen bonding interactions with Tyr118, a crucial residue for ligand stabilization, while APR1a lacked hydrogen bonding but still maintained a high affinity through steric interactions. Compound APR1c, with a binding energy of −7.5 kcal mol−1, formed interactions with His377 and Ser378, which are key contributors to ligand stabilization.
Fluconazole, while displaying strong hydrogen bonding with Ile304 and Thr311 and steric interactions with multiple residues (Leu204, Leu276, Ile304, Gly308, Thr311, Gly472), did not exhibit a significantly stronger binding affinity compared to the tested compounds. This suggests that the novel compounds, particularly APR1d, could provide comparable or improved antifungal efficacy.
6 Significance of findings
The docking studies indicate that the synthesized compounds, especially APR1d and APR1b, demonstrate superior binding affinities compared to conventional antibiotics and antifungal agents (Fig. S24†). The stronger binding interactions of these compounds suggest potential improvements in drug efficacy, selectivity, and possibly lower resistance development in pathogens. Furthermore, the presence of critical hydrogen bonds and steric interactions enhances the stability of these compounds within the active sites of bacterial and fungal targets, making them promising candidates for further biological evaluation.Future experimental validation, including in vitro antimicrobial assays, minimum inhibitory concentration (MIC) studies, and cytotoxicity evaluations, will be crucial in determining their real-world applicability. If these compounds retain their high efficacy in biological settings, they could serve as viable alternatives to existing antimicrobial agents, addressing the growing concern of antimicrobial resistance.
Overall, the molecular docking findings present highly promising antibacterial and antifungal potential for the tested compounds. The superior binding affinities of APR1d and APR1b, particularly against E. coli and Candida albicans (Fig. S24†), highlight their potential as novel antimicrobial agents. These results pave the way for further optimization and experimental validation, with the potential to develop next-generation antimicrobial therapeutics.
6.1. Molecular dynamic simulations analysis
The evaluation of the stability of protein-ligand complexes involved a comprehensive analysis using molecular dynamics simulation outlined in Tables S7–S9.† Various parameters, such as Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of Gyration (Rg), Solvent Accessible Surface Area (SASA), and hydrogen bond analysis, were employed throughout the simulation. To assess the stability of the system, Cα atom RMSD values were examined for both the Apo (Escherichia coli) and the specific ligand bound complexes, Escherichia coli with Streptomycin drug (used as control in current study) and synthesis drug (APR1d compound), as shown in Fig. S25B.† The Apo form of Escherichia coli consistently exhibited high RMSD values in compare to control and APR1d ligand bound complexes. Although the synthesis (APR1d ligand) complexes displayed stable RMSD values, maintaining a nearly constant value throughout the entire simulation period (Fig. S25B†). This result implies that the stability of these complexes is affected by the presence of ligands. RMSF has been used to analyze the flexibility, or fluctuations, of individual residues within Escherichia coli over time. The RMSF values of selected complexes and Apo showed some similarities (Fig. S25A†). It is noteworthy that the composite APR1d ligand complex has the lowest RMSF value, indicating higher stability compared to the other two systems (Apo and streptomycin bound Escherichia coli). Additionally, the radius of gyration (Rg), a measure of the tightness or spatial distribution of atoms in a molecule, was studied to analyze overall shape and conformational changes in biomolecules. In this study, Rg showed similarities in structural compactness between Apo and Streptomycin complexes (Fig. S25C†). However, APR1d -ligand bound complex have lesser Rg value, shows the higher compactness. The Ligand RMSD plot shown in Fig. 8 provides insights into the dynamic's behavior of ligands molecules within Escherichia coli. The Root Mean Square Deviation (RMSD) of the APR1d ligand is lower when compared to that of the control ligand. This indicates that the APR1d ligand exhibits less deviation from the desired structure, suggesting potentially tighter binding or a better fit within the binding site compared to the control ligand shown in Fig. S25D.†Hydrogen bonds play a crucial role in influencing the strength of interactions within the MD simulations, with a higher number of hydrogen bonds formed between ligands and proteins helping to form tighter binding and stable complexes In this study, ligand-protein complex stability was evaluated using a hydrogen bond analysis over the course of the simulation. Interestingly, the complexes control and APR1d ligand, which interacted with residues Met207, Asn210, Arg151 and Phe304, respectively, shown in Fig. 8. The residues involved in hydrogen bonding, as identified, correspond with the results obtained from the docking analysis. The trajectory was clustered into five groups, and the most highly populated cluster, comprising 60% of the total simulation, was chosen for investigating the interactions between the ligand and protein shown in Fig. 8.
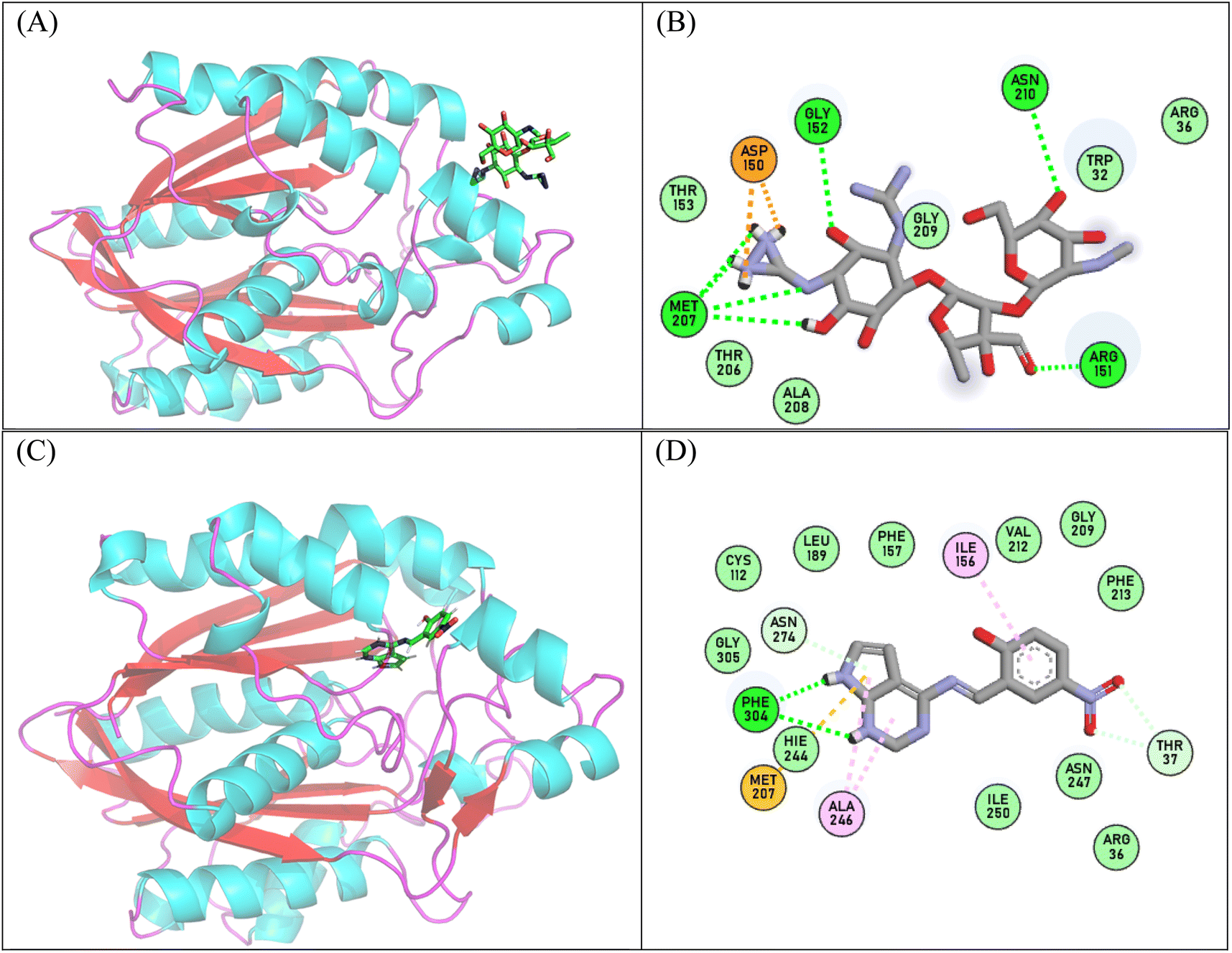 | ||
| Fig. 8 The representation structure of the most populated cluster of streptomycin and APR1d ligand bound E. coli complex during 100 ns molecular dynamics simulation (A) binding site of streptomycin (B) interaction of streptomycin with E. coli (C) binding site of APR1d ligand and (D) interaction of APR1d ligand with E. coli.The ligand (green) with stick and protein complex shown as cartoon. | ||
The AMBER18 software's Molecular Mechanics Poisson-Boltzmann Surface Area (MM-PBSA) module was utilized to compute the binding free energy between the ligand and the complex. This encompassed all non-bonded interactions, such as van der Waals forces, electrostatic interactions, polar solvation, and nonpolar salvations resulting in a total binding free energy of complexes. The binding free energy is higher for the synthesized drug (−28 kcal mol−1) compared to the control (−9 kcal mol−1) shown in Tables S7–S9.† This suggests that the synthesized drug has a stronger affinity for its target compared to the control compound.
Similar analyses were conducted for complexes involving a control ligand and a synthesized ligand with Candida albicans. Both RMSD and RMSF exhibited lower values in the complex with the synthesized ligand bound to Candida albicans shown in Fig. S26.† The synthesized ligand displayed significantly lower RMSD values, indicating its stability within the binding site of Candida albicans (shown in Fig. S26A†). Additionally, interactions between the ligands and Candida albicans were assessed. It was observed that the control ligand interacted with Ile447/Gly448, whereas the synthesized ligand interacted with His377/His378 residues of Candida albicans shown in Fig. 9. The binding stability of the ligand complex systems was reflected by the binding free energy, revealing that the synthesized ligand exhibited a more negative binding energy (−28 kcal mol−1) compared to the control ligand (−25 kcal mol−1) shown in Tables S8 andS9.†
 | ||
| Fig. 9 The representation structure of the most populated cluster of fluconazole and APR1d ligand bound C. Albicans complex during 100 ns molecular dynamics simulation (A) binding site of streptomycin (B) interaction of streptomycin with E. coli (C) binding site of APR1d ligand and (D) interaction of APR1d ligand with C. Albicans protein. The ligand (green) with stick and protein complex shown as cartoon. | ||
In conclusion, the comparative analysis between complexes involving a control ligand and a synthesized ligand with E. coli/C. albicans revealed several key findings. Firstly, both RMSD and RMSF values were lower in the complex with the synthesized ligand (APR1d drugs), indicating greater stability within the binding site of E. coli as well as C. albicans. Importantly, the binding free energy analysis underscored the enhanced binding stability of the synthesized ligand complex, as evidenced by its more negative binding energy compared to the control ligand. These findings collectively suggest the potential efficacy and specificity of the synthesized ligand in targeting E. coli/C. albicans, highlighting its promise for further investigation and development as a therapeutic agent.
6.1.1.1 Physicochemical properties. All four compounds exhibit similar molecular weights (∼238–283 g mol−1), indicating their potential for effective interaction with biological targets. The number of rotatable bonds (2–3) suggests good molecular flexibility, which can impact binding affinity. Compound APR1d, however, possesses a slightly higher number of heavy atoms (21) and hydrogen bond acceptors (6), which may contribute to increased molecular interactions.
Total polar surface area (TPSA) values ranged from 56.98 Å2 to 119.98 Å2, with compound APR1d showing the highest TPSA. Generally, TPSA values below 140 Å2 indicate favorable oral bioavailability, suggesting that all compounds can be efficiently absorbed. However, the fraction of sp3 hybridized carbons (Csp3) was 0.08 for 1a and 0.00 for the others, implying a predominantly planar and rigid structure, which may influence binding specificity.
6.1.1.2 Lipophilicity and water solubility. The lipophilicity values, expressed as the consensus Log P (Po/W), suggest that compounds APR1b and APR1c (log P = 2.02) are more hydrophobic than 1a (log P = 0.14) and APR1d (log P = 1.22). This indicates that APR1b and APR1c might have higher membrane permeability but could also pose solubility challenges.
Solubility analysis showed that APR1a is highly soluble (Log S = 0.01, ALI model), while the other compounds are classified as soluble to moderately soluble. This suggests that 1a may have an advantage in terms of dissolution and bioavailability, whereas APR1b, APR1c, and APR1d might require solubility-enhancing strategies.
6.1.1.3 Pharmacokinetics and drug metabolism. Gastrointestinal (GI) absorption was high for all four compounds, reinforcing their potential for oral administration. However, only APR1b and APR1c demonstrated blood–brain barrier (BBB) permeability, indicating their potential for central nervous system (CNS) applications. In contrast, APR1a and APR1d are unlikely to cross the BBB, making them more suitable for peripheral targets.
Interestingly, none of the compounds acted as substrates for P-glycoprotein (P-gp), suggesting that they are less prone to efflux mechanisms, which could enhance their intracellular retention. However, 1b and 1c were predicted to inhibit CYP1A2, which might lead to drug–drug interactions if co-administered with CYP1A2-metabolized drugs.
Skin permeability (log Kp) values ranged from −8.28 cm s−1 (APR1a) to −6.38 cm s−1 (APR1b and APR1c), suggesting limited transdermal absorption, making them less favorable for topical formulations.
6.1.1.4 Drug-likeness and medicinal chemistry evaluation. All compounds met Lipinski's rule of five without violations, indicating favorable oral bioavailability. However, only APR1b, APR1c, and APR1d passed the Ghose filter, while APR1a exhibited one violation. The bioavailability score remained consistent at 0.55 across all four compounds, reinforcing their likelihood of systemic availability.
In terms of medicinal chemistry, PAINS (Pan Assay Interference Compounds) alerts were absent, suggesting a low likelihood of non-specific interactions. However, the Brenk filter identified an imine alert in APR1a, APR1b, and APR1c, with APR1d showing additional alerts due to the presence of nitro and oxygen-nitrogen single-bond groups, which might affect toxicity or reactivity. The synthetic accessibility scores ranged from 2.50 to 3.43, indicating moderate ease of synthesis, with APR1c being the easiest to synthesize.
Overall, the analysis presents a promising outlook for these compounds, with each exhibiting unique strength. Compound APR1a stands out for its high solubility and minimal toxicity risks, making it an ideal candidate for further optimization. Compounds APR1b and APR1c show potential for CNS applications due to their BBB permeability but require careful consideration of CYP1A2 inhibition. Compound APR1d, with its higher TPSA and solubility profile, could be advantageous for systemic applications, though its medicinal chemistry alerts warrant further investigation.
Future studies, including in vitro and in vivo evaluations, will be crucial to validate these findings and further refine the lead candidates for pharmaceutical development.
6.1.1.5 Structural and molecular interaction insights: evaluating drug-like potential. The molecular interactions and structural characteristics of the four compounds (APR1a, APR1b, APR1c, and APR1d) reveal promising features that could influence their pharmacological potential. Key attributes such as hydrogen bonding capacity, hydrophobicity, and aromaticity (refer Table S11 and Fig. S28†) play a crucial role in determining their biological activity, stability, and drug-likeness.
6.1.1.6 Hydrogen bonding capacity: balancing stability and binding affinity. Hydrogen bonding is a critical factor in drug–target interactions, affecting solubility, bioavailability, and binding specificity. Compounds APR1a, APR1b, and APR1c share an identical profile with five hydrogen bond acceptors and three hydrogen bond donors, suggesting a uniform potential for hydrogen bond formation with biological targets. This consistency may lead to stable interactions with enzymes or receptors, contributing to their potential efficacy.
In contrast, compound APR1d exhibits a higher number of hydrogen bond acceptors (7), which could enhance its ability to form multiple interactions with polar residues in protein binding sites. This characteristic may improve its binding affinity and specificity, making APR1d a strong candidate for further optimization. However, excessive hydrogen bonding can sometimes reduce membrane permeability, which needs to be carefully evaluated in further studies.
6.1.1.7 Hydrophobicity and aromatic ring contribution: influencing drug permeability. Hydrophobicity is crucial for membrane permeability, as highly hydrophobic compounds tend to pass through cell membranes more efficiently. Compounds APR1a, APR1b, and APR1c exhibit two hydrophobic regions, suggesting a balanced profile that may allow for effective cellular uptake while maintaining adequate aqueous solubility. On the other hand, APR1d presents only one hydrophobic region, which could slightly impact its membrane permeability but might enhance solubility in aqueous environments.
All four compounds share a common aromatic ring count of six, indicating a stable planar structure that could facilitate π–π interactions with biological targets. Aromaticity often enhances binding affinity in drug–receptor interactions, which could contribute to the compounds' therapeutic potential.
6.1.1.8 Ionizability and drug stability: enhancing pharmacokinetic properties. None of the compounds exhibit negative or positive ionizable groups, which suggests that they maintain a neutral charge across physiological pH ranges. This characteristic is advantageous, as excessive ionization can lead to poor membrane permeability and reduced bioavailability. The absence of ionizable groups indicates that these compounds may have a stable pharmacokinetic profile, avoiding potential issues related to pH-dependent solubility or unwanted electrostatic interactions.
The structural and physicochemical analysis of these compounds highlights their promising potential for drug development. While APR1a, APR1b, and APR1c share a similar balance of hydrogen bonding and hydrophobicity, APR1d stands out with an increased hydrogen bond acceptor count, which could enhance its binding interactions. The uniform aromaticity across all compounds suggests strong target engagement, while the absence of ionizable groups supports stability across physiological conditions.
Future investigations, including in vitro binding assays and in vivo pharmacokinetic studies, will be essential to validate these findings and optimize the most promising candidates for therapeutic applications.
7 Conclusion
A green, microwave-assisted synthesis of 4-amino-pyrrolo[2,3-d]pyrimidine Schiff bases (APR1a–d) using reusable [HMIM][TFSI] ionic liquid achieved high yields (82–94%).APR1dexhibited exceptional antimicrobial activity (e.g., 28.0 mm zone against B. subtilis), outperforming fluconazole, and demonstrated low cytotoxicity (LC50 > 3.50 × 10−4 M). HPLC analysis confirmed ≥95% purity for all compounds (APR1a–d), validating the reliability of biological evaluations. DFT and docking studies revealed its high reactivity (HOMO–LUMO gap: 0.0679 eV) and strong target binding (ΔG = −28.8 kcal mol−1). With favorable ADMET properties and no Lipinski violations, APR1d emerges as a promising antimicrobial lead.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Nilesh Bhusari: synthesis, characterization, biological activities, writing original draft (synthesis, characterization, biological activities), data curation, investigation, conceptualization, methodology, validation, and funding Abhay Bagul: writing the original draft, writing—review and editing, data curation, investigation, visualization and validation. Vipin Kumar Mishra: molecular dynamics simulations, validation and formal analysis. Aisha Tufail: writing the original draft (in silico work), software (DFT and MESP), visualization, validation. Digamber D. Gaikwad: supervision, writing—review & editing, validation, data curation and formal analysis. Amit Dubey: supervision, data curation, investigation, conceptualization, writing the original draft (in silico work), software (pharmacophore modeling, DFT and MESP), visualization, methodology, writing – review & editing, validation and formal analysis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- H. Y. Li, W. Q. Yang, X. Z. Zhou, F. Shao, T. Shen, H. Y. Guan, J. Zheng and L. M. Zhang, Antibacterial and antifungal sesquiterpenoids: chemistry, resource, and activity, Biomolecules, 2022, 12(9), 1271 CrossRef CAS PubMed.
- R. H. Abd El-Aleam, R. F. George, H. H. Georgey and H. M. Abdel-Rahman, Bacterial virulence factors: a target for heterocyclic compounds to combat bacterial resistance, RSC Adv., 2021, 11(58), 36459–36482 RSC.
- K. C. Howard, E. K. Dennis, D. S. Watt and S. Garneau-Tsodikova, A comprehensive overview of the medicinal chemistry of antifungal drugs: perspectives and promise, Chem. Soc. Rev., 2020, 49(8), 2426–2480 RSC.
- M. B. Islam, M. I. Islam, N. Nath, T. B. Emran, M. R. Rahman, R. Sharma and M. M. Matin, Recent advances in pyridine scaffold: focus on chemistry, synthesis, and antibacterial activities, Biomed Res. Int., 2023, 2023, 9967591 CrossRef PubMed.
- K. U. Sadek, R. A. Mekheimer, M. Abd-Elmonem, F. A. Abo-Elsoud, A. M. Hayallah, S. M. Mostafa, M. H. Abdellattif, M. A. Abourehab, T. A. Farghaly and A. Elkamhawy, Recent developments in the synthesis of hybrid heterocycles: a promising approach to develop multi-target antibacterial agents, J. Mol. Struct., 2023, 1286, 135616 CrossRef CAS.
- R. Kumar and G. Singh, Substituted benzimidazoles as antibacterial and antifungal agents: a review, Pharmacophore, 2022, 13(2), 41–55 CrossRef.
- E. Kabir and M. Uzzaman, A review on biological and medicinal impact of heterocyclic compounds, Results Chem., 2022, 4, 100606 CrossRef CAS.
- K. Haider, M. Shafeeque, S. Yahya and M. S. Yar, A comprehensive review on pyrazoline based heterocyclic hybrids as potent anticancer agents, Eur. J. Med. Chem. Rep., 2022, 5, 100042 CAS.
- B. Kaur, G. Singh, V. Sharma and I. Singh, Sulphur containing heterocyclic compounds as anticancer agents, Anti-Cancer Agents Med. Chem., 2023, 23(8), 869–881 CrossRef CAS PubMed.
- K. Laxmikeshav, P. Kumari and N. Shankaraiah, Expedition of sulfur-containing heterocyclic derivatives as cytotoxic agents in medicinal chemistry: a decade update, Med. Res. Rev., 2022, 42(1), 513–575 CrossRef CAS PubMed.
- H. Sachdeva, S. Khaturia, M. Saquib, N. Khatik, A. R. Khandelwal, R. Meena and K. Sharma, Oxygen- and sulphur-containing heterocyclic compounds as potential anticancer agents, Appl. Biochem. Biotechnol., 2022, 194(12), 6438–6467 CrossRef CAS PubMed.
- E. Frank and G. Szöllősi, Nitrogen-containing Heterocycles as Significant Molecular Scaffolds for Medicinal and Other Applications, MDPI, 2021, p. 4617 Search PubMed.
- M. M. Heravi and V. Zadsirjan, Prescribed drugs containing nitrogen heterocycles: an overview, RSC Adv., 2020, 10(72), 44247–44311 RSC.
- N. N. Makhova, L. I. Belen’kii, G. A. Gazieva, I. L. Dalinger, L. S. Konstantinova, V. V. Kuznetsov, A. N. Kravchenko, M. M. Krayushkin, O. A. Rakitin and A. M. Starosotnikov, Progress in the chemistry of nitrogen-, oxygen- and sulfur-containing heterocyclic systems, Russ. Chem. Rev., 2020, 89(1), 55 CrossRef CAS.
- D. S. Williamson, G. P. Smith, G. K. Mikkelsen, T. Jensen, P. Acheson-Dossang and L. Badolo, et al., Design and synthesis of pyrrolo[2,3-d]pyrimidine-derived leucine-rich repeat kinase 2 (LRRK2) inhibitors using a checkpoint kinase 1 (CHK1)-derived crystallographic surrogate, J. Med. Chem., 2021, 64(14), 10312–10332 CrossRef CAS PubMed.
- S. Pathania and R. K. Rawal, Pyrrolopyrimidines: an update on recent advancements in their medicinal attributes, Eur. J. Med. Chem., 2018, 157, 503–526 CrossRef CAS PubMed.
- A. E. Van der Westhuyzen, L. V. Frolova, A. Kornienko and W. A. van Otterlo. The Rigidins: Isolation, bioactivity, and total synthesis—novel pyrrolo[2,3-d]pyrimidine analogues using multicomponent reactions, in The Alkaloids: Chemistry and Biology, ed. Knölker H.-J., Academic Press, 2018, pp. 191–220 Search PubMed.
- S. Kumar and B. Narasimhan, Therapeutic potential of heterocyclic pyrimidine scaffolds, Chem. Cent. J., 2018, 12, 38 CrossRef PubMed.
- K. Ahmed, M. I. Choudhary and R. S. Z. Saleem, Heterocyclic pyrimidine derivatives as promising antibacterial agents, Eur. J. Med. Chem., 2023, 259, 115701 CrossRef CAS PubMed.
- A. Bagul, M. Kumar, A. Tufail, N. Tufail, D. Gaikwad and A. Dubey, Synergistic exploration of antimicrobial potency, cytotoxicity, and molecular mechanisms: a tripartite investigation integrating in vitro, in vivo, and in silico approaches for pyrimidine-based metal(II) complexes, Appl. Organomet. Chem., 2024, 38(7) DOI:10.1002/aoc.7521.
- J. An, W. Lan, Q. Fei, P. Li and W. Wu, Synthesis, antifungal, and antibacterial activities of novel benzoylurea derivatives containing a pyrimidine moiety, Molecules, 2023, 28(18), 6498, DOI:10.3390/molecules28186498.
- S. X. Wu, J. Liu and B. X. Tan, et al., Design, synthesis, antibacterial, and antifungal activity evaluation of novel pyrimidine derivatives incorporating amide and 1,3,4-thiadiazole thioether moieties, Russ. J. Gen. Chem., 2024, 94, 3448–3455, DOI:10.1134/S1070363224120375.
- A. M. Abd-Elaziz, H. M. Aly and N. M. Saleh, et al., Synthesis and characterization of novel pyrimidine derivatives as a promising tool for antimicrobial agent and in-vitro cytotoxicity, J. Iran Chem. Soc., 2022, 19, 2279–2296, DOI:10.1007/s13738-021-02448-w.
- T. Miwa, T. Hitaka, H. Akimoto and H. Nomura, Novel pyrrolo[2,3-d]pyrimidine antifolates: synthesis and antitumor activities, J. Med. Chem., 1991, 34(2), 555–560, DOI:10.1021/jm00106a012.
- M. S. Madhurya, V. Thakur, S. Dastari and N. Shankaraiah, Pyrrolo[2,3-d]pyrimidines as potential kinase inhibitors in cancer drug discovery: a critical review, Bioorg. Chem., 2024, 153, 107867, DOI:10.1016/j.bioorg.2024.107867.
- A. D. Bagul, D. D. Gaikwad, A. Tufail, J. N. Sangshetti, M. G. Damale and A. Dubey, Pyrrolopyrimidinehydrazide-2-chlorobenzaldehyde metal complexes: synthesis, characterization, and extraordinary exploration through in vitro, in vivo cytotoxicity, and cutting-edge computational analyses, Appl. Organomet. Chem., 2024, 39(2) DOI:10.1002/aoc.7809.
- M. Albratty and H. A. Alhazmi, Novel pyridine and pyrimidine derivatives as promising anticancer agents: a review, Arab J. Chem., 2022, 15(6), 103846, DOI:10.1016/j.arabjc.2022.103846.
- B. Tylińska, B. Wiatrak, Ż. Czyżnikowska, A. Cieśla-Niechwiadowicz, E. Gębarowska and A. Janicka-Kłos, Novel pyrimidine derivatives as potential anticancer agents: synthesis, biological evaluation and molecular docking study, Int. J. Mol. Sci., 2021, 22(8), 3825, DOI:10.3390/ijms22083825.
- R. M. Keshk, Z. A. Salama and S. K. Elsaedany, et al., Synthesis, antimicrobial, anti-inflammatory, antioxidant and cytotoxicity of new pyrimidine and pyrimidopyrimidine derivatives, Sci. Rep., 2025, 15, 9328, DOI:10.1038/s41598-025-92066-w.
- M. A. Aziz, I. M. Salem and M. A. Al-Awadh, et al., Exploration of anti-inflammatory activity of pyrazolo[3,4-d]pyrimidine/1,2,4-oxadiazole hybrids as COX-2, 5-LOX and NO release inhibitors: design, synthesis, in silico and in vivo studies, Bioorg. Chem., 2025, 156, 108181, DOI:10.1016/j.bioorg.2025.108181.
- Y. J. Jian, Q. Lv, L. Du, C. C. Lei, L. P. Zhi and X. H. Liu, Discovery of a novel pyrimidine derivative for treatment of acute lung injury through reducing oxidative stress and inflammatory response, RSC Med. Chem., 2025, 16(3), 1441–1458, 10.1039/d4md00858h.
- B. Nammalwar and R. A. Bunce, Recent advances in pyrimidine-based drugs, Pharmaceuticals, 2024, 17, 104, DOI:10.3390/ph17010104.
- A. Jadhav, S. G. Shingade, P. G. Dessai, B. S. Biradar and S. MamleDesai, Design, docking, characterisation, and synthesis of pyrimidine derivatives for antidepressant activity, Curr. Drug Discovery Technol., 2023, 21(3) DOI:10.2174/0115701638243835230925161546.
- R. Fioravanti, E. Proia and I. N. Tyurenkov, et al., Pyrimidine thioethers: a novel class of antidepressant agents, endowed with anxiolytic, performance enhancing and nootropic activity, Eur. J. Med. Chem., 2022, 245, 114902, DOI:10.1016/j.ejmech.2022.114902.
- M. Ban, H. Taguchi, T. Katsushima, S. Aoki and A. Watanabe, Novel antiallergic agents. Part I: synthesis and pharmacology of pyrimidine amide derivatives, Bioorg. Med. Chem., 1998, 6(7), 1057–1067, DOI:10.1016/s0968-0896(98)00064-9.
- E. A. Georgiou, K. Paraskevas and C. Koutra, et al., Exploring 4,7-disubstituted pyrimido[4,5-d]pyrimidines as antiviral and anticancer agents, Molecules, 2024, 29, 5549, DOI:10.3390/molecules29235549.
- S. F. Mohamed, D. H. Elnaggar and H. S. Abd-Elghaffar, et al., Synthesis, antiviral screening, and in silico investigations of new pyrimidine and pyridopyrimidine derivatives: finding new therapeutic prospects to combat the Herpes simplex virus (HSV-1), J. Mol. Struct., 2024, 140246, DOI:10.1016/j.molstruc.2024.140246.
- E. C. S. Costa, Z. S. Monte and E. P. S. Falcão, et al., Pharmacological potential of pyrimidine derivatives: a review with emphasis on antiviral effects and virtual screening against SARS-CoV-2 molecular targets, ChemistrySelect, 2023, 8(23) DOI:10.1002/slct.202300132.
- A. D. Bagul, M. Kumar and A. M. Alanazi, et al., Experimental and computational evaluation of anti-malarial and antioxidant potential of transition metal(II) complexes with tridentate Schiff base derived from pyrrolopyrimidine, BioMetals, 2024, 37, 1713–1737, DOI:10.1007/s10534-024-00636-8.
- O. R. Vanukuri, M. Gundluru, P. Bellala, R. S. R. Dachuru and S. R. Cirandur, Facile one-pot green synthesis, antioxidant and antimicrobial activities of 7-(substituted phenyl)-7,11-dihydro-6H, 8H-chromeno[3′,4′:5,6]pyrano[2,3-d]pyrimidine-6,8,10(9H)-trione derivatives, Synth. Commun., 2025, 1–13, DOI:10.1080/00397911.2025.2455521.
- M. Abbas and N. Arshad, Synthesis, highly potent α-glucosidase inhibition, antioxidant and molecular docking of various novel dihydropyrimidine derivatives to treat diabetes mellitus, Bioorg. Med. Chem. Lett., 2024, 115, 130016, DOI:10.1016/j.bmcl.2024.130016.
- A. I. Sayed, Y. E. Mansour and M. A. Ali, et al., Novel pyrrolopyrimidine derivatives: design, synthesis, molecular docking, molecular simulations and biological evaluations as antioxidant and anti-inflammatory agents, J. Enzyme Inhib. Med. Chem., 2022, 37(1), 1821–1837, DOI:10.1080/14756366.2022.2090546.
- A. E. Rashad, T. E. Malah and A. H. Shamroukh, Developments of pyrrolo[2,3-d]pyrimidines with pharmaceutical potential, Curr. Org. Chem., 2024, 28(16), 1244–1264, DOI:10.2174/0113852728306820240515054401.
- D. Chaturvedi and M. Kamboj, Role of Schiff base in drug discovery research, Chem. Sci. J., 2016, 7(2), e114 Search PubMed.
- D. Tatlidil, M. A. Raza and N. Dege, et al., Therapeutical potential of imines: synthesis, single crystal structure, computational, molecular modeling, and ADMET evaluation, ACS Omega, 2022, 7(12), 10568–10579, DOI:10.1021/acsomega.2c00102.
- K. Rajimon, D. S. Nair, D. Srinivasaragavan and R. Thomas, Integrated experimental and computational study of a fluorescent Schiff base: synthesis, characterization, electronic structure properties, and biological potentials of (1E,1′E)-1,1′-(1,4-phenylene)bis(N-(2-chlorophenyl)methanimine) with a focus on molecular docking and dynamics simulation, Chem. Phys. Impact., 2023, 8, 100435, DOI:10.1016/j.chphi.2023.100435.
- P. X. Kolagkis, E. M. Galathri and C. G. Kokotos, Green and sustainable approaches for the Friedel–Crafts reaction between aldehydes and indoles, Beilstein J. Org. Chem., 2024, 20, 379–426, DOI:10.3762/bjoc.20.36.
- J. L. Tyler, F. Katzenburg and F. Glorius, A focus on sustainable method development for greener synthesis, Chem. Sci., 2023, 14(27), 7408–7410, 10.1039/d3sc90120c.
- I. T. Horváth and P. T. Anastas, Innovations and green chemistry, Chem. Rev., 2007, 107(6), 2169–2173, DOI:10.1021/cr078380v.
- P. Gupta and A. Mahajan, Green chemistry approaches as sustainable alternatives to conventional strategies in the pharmaceutical industry, RSC Adv., 2015, 5(34), 26686–26705, 10.1039/c5ra00358j.
- J. Becker, C. Manske and S. Randl, Green chemistry and sustainability metrics in the pharmaceutical manufacturing sector, Curr. Opin. Green Sustain. Chem., 2021, 33, 100562, DOI:10.1016/j.cogsc.2021.100562.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Green chemistry in the synthesis of pharmaceuticals, Chem. Rev., 2022, 122(3), 3637–3710, DOI:10.1021/acs.chemrev.1c00631.
- A. De La Hoz, A. Díaz-Ortiz and P. Prieto, Microwave-assisted Green Organic Synthesis, in Royal Society of Chemistry eBooks, 2016, pp. 1–33, 10.1039/9781782623632-00001.
- G. Murbach, S. A. Biyani and D. H. Thompson, High throughput experimentation as a tool to guide the microwave assisted catalytic amidation of aryl amines with aryl acids, Eur. J. Org Chem., 2025, 28(13) DOI:10.1002/ejoc.202400764.
- S. C. Ameta, P. B. Punjabi, R. Ameta and C. Ameta, in Microwave-assisted Organic Synthesis, Apple Academic Press eBooks, 2014, DOI:10.1201/b17953.
- S. Mallakpour and M. Dinari, in Ionic Liquids as Green Solvents: Progress and Prospects, Springer eBooks, 2012, pp. 1–32, DOI:10.1007/978-94-007-2891-2_1.
- M. Zhu, Ionic-liquid/metal–organic-framework composites: synthesis and emerging sustainable applications, Inorg. Chem. Front., 2025, 12, 39–84, 10.1039/d4qi02383h.
- A. Tzani, M. Karadendrou, S. Kalafateli, V. Kakokefalou and A. Detsi, Current trends in green solvents: biocompatible ionic liquids, Crystals, 2022, 12(12), 1776, DOI:10.3390/cryst12121776.
- T. Welton, Ionic liquids in green chemistry, Green Chem., 2011, 13(2), 225, 10.1039/c0gc90047h.
- K. Liu, Z. Wang, L. Shi, S. Jungsuttiwong and S. Yuan, Ionic liquids for high performance lithium metal batteries, J. Energy Chem., 2020, 59, 320–333, DOI:10.1016/j.jechem.2020.11.017.
- A. Berthod, M. Ruiz-Ángel and S. Carda-Broch, Ionic liquids in separation techniques, J. Chromatogr. A, 2008, 1184(1–2), 6–18, DOI:10.1016/j.chroma.2007.11.109.
- J. Zhou, H. Sui, Z. Jia, Z. Yang, L. He and X. Li, Recovery and purification of ionic liquids from solutions: a review, RSC Adv., 2018, 8(57), 32832–32864, 10.1039/c8ra06384b.
- A. Maia, Room temperature ionic liquids: a “green” alternative to conventional organic solvents?, Mini-Rev. Org. Chem., 2011, 8(2), 178–185, DOI:10.2174/157019311795177826.
- R. L. Vekariya, A review of ionic liquids: applications towards catalytic organic transformations, J. Mol. Liq., 2016, 227, 44–60, DOI:10.1016/j.molliq.2016.11.123.
- N. Poonam, N. Geetanjali and R. Singh, Applications of ionic liquids in organic synthesis, in Nanotechnology in the Life Sciences, 2020, pp. 41–62, DOI:10.1007/978-3-030-44176-0_3.
- N. Jain, A. Kumar, S. Chauhan and S. Chauhan, Chemical and biochemical transformations in ionic liquids, Tetrahedron, 2004, 61(5), 1015–1060, DOI:10.1016/j.tet.2004.10.070.
- S. K. Singh and A. W. Savoy, Ionic liquids synthesis and applications: an overview, J. Mol. Liq., 2019, 297, 112038, DOI:10.1016/j.molliq.2019.112038.
- A. S. Amarasekara, Acidic ionic liquids, Chem. Rev., 2016, 116(10), 6133–6183, DOI:10.1021/acs.chemrev.5b00763.
- S. Magaldi, S. Mata-Essayag and C. H. De Capriles, et al., Well diffusion for antifungal susceptibility testing, Int. J. Infect. Dis., 2003, 8(1), 39–45, DOI:10.1016/j.ijid.2003.03.002.
- I. Gajic, J. Kabic and D. Kekic, et al., Antimicrobial susceptibility testing: a comprehensive review of currently used methods, Antibiotics, 2022, 11(4), 427, DOI:10.3390/antibiotics11040427.
- J. M. Andrews, Determination of minimum inhibitory concentrations, J. Antimicrob. Chemother., 2002, 49(6), 1049, DOI:10.1093/jac/dkf083.
- S. Kumar, J. Devi and A. Dubey, et al., Co(II), Ni(II), Cu(II) and Zn(II) complexes of Schiff base ligands: synthesis, characterization, DFT, in vitro antimicrobial activity and molecular docking studies, Res. Chem. Intermed., 2022, 49(3), 939–965, DOI:10.1007/s11164-022-04941-0.
- C. N. Banti and S. K. Hadjikakou, Evaluation of toxicity with brine shrimp assay, Bio-Protoc., 2021, 11(2) DOI:10.21769/bioprotoc.3895.
- W. S. Abbott, A method of computing the effectiveness of an insecticide, J. Econ. Entomol., 1925, 18(2), 265–267, DOI:10.1093/jee/18.2.265a.
- M. J. Frisch, G. W. Trucks and H. B. Schlegel, et al., Gaussian 09, Revision E.01, Gaussian Inc., Wallingford, CT, USA, 2016 Search PubMed.
- S. Bharadwaj, A. Dubey and U. Yadava, et al., Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro, Briefings Bioinf., 2021, 22, 1361–1377 CrossRef CAS PubMed.
- A. D. Bagul, M. Kumar and A. M. Alanazi, et al., Experimental and computational evaluation of anti-malarial and antioxidant potential of transition metal(II) complexes with tridentate Schiff base derived from pyrrolopyrimidine, BioMetals, 2024, 1–25 Search PubMed.
- M. Dalal, A. Dubey and A. Tufail, et al., Organyltellurium (IV) complexes incorporating Schiff base ligand derived from 2-hydroxy-1-naphthaldehyde: preparation, spectroscopic investigations, antimicrobial, antioxidant activities, DFT, MESP, NBO, molecular docking and ADMET evaluation, J. Mol. Struct., 2023, 1287, 135590, DOI:10.1016/j.molstruc.2023.135590.
- D. Majumdar, A. Dubey and A. Tufail, et al., Synthesis, spectroscopic investigation, molecular docking, ADME/T toxicity predictions, and DFT study of two trendy ortho vanillin-based scaffolds, Heliyon, 2023, 9(6), e16057 CrossRef CAS PubMed.
- D. Majumdar, J. E. Philip and A. Dubey, et al., Synthesis, spectroscopic findings, SEM/EDX, DFT, and single-crystal structure of Hg/Pb/Cu–SCN complexes: in silico ADME/T profiling and promising antibacterial activities, Heliyon, 2023, 9(5), e16103 CrossRef CAS PubMed.
- G. M. Morris, R. Huey and W. Lindstrom, et al., AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791 CrossRef CAS PubMed.
- A. Dubey, A. Marabotti, P. W. Ramteke and A. Facchiano, In silico approach to find chymase inhibitors among biogenic compounds, Future Med. Chem., 2016, 8, 841–851 CrossRef CAS PubMed.
- A. Dubey, A. Marabotti, P. W. Ramteke and A. Facchiano, Interaction of human chymase with ginkgolides, terpene trilactones of Ginkgo biloba investigated by molecular docking simulations, Biochem. Biophys. Res. Commun., 2016, 473, 449–454 CrossRef CAS PubMed.
- A. Dubey, M. M. Alawi and T. A. Alandijany, et al., Viruses, 2023, 15(1), 251 CrossRef CAS PubMed.
- S. Bharadwaj, A. Dubey and N. K. Kamboj, et al., Drug repurposing for ligand-induced rearrangement of Sirt2 active site-based inhibitors via molecular modeling and quantum mechanics calculations, Sci. Rep., 2021, 11(1), 10169, DOI:10.1038/s41598-021-89627-0.
- A. Dubey, S. Dotolo and P. W. Ramteke, et al., Searching for chymase inhibitors among chamomile compounds using a computational-based approach, Biomolecules, 2018, 9(1), 5, DOI:10.3390/biom9010005.
- D. A. Case, H. M. Aktulga and K. Belfon, et al., Amber 2018, University of California, San Francisco, 2018 Search PubMed.
- J. Wang, R. M. Wolf and J. W. Caldwell, et al., Development and testing of general AMBER force field, J. Comput. Chem., 2004, 25(9), 1157–1174 CrossRef CAS PubMed.
- V. K. Mishra and S. Mishra, Origin of regio- and stereospecific catalysis by 8-lipoxygenase, J. Phys. Chem. B, 2019, 123(50), 10605–10621 CrossRef CAS PubMed.
- V. K. Mishra and S. Mishra, Flipped regiospecificity in L434F mutant of 8-lipoxygenase, Phys. Chem. Chem. Phys., 2020, 22(28), 16013–16022 RSC.
- R. Singh, V. K. Mishra and A. K. Das, Crystal structure of a thiolase from Archaeal Pyrococcus furiosus and its in silico functional annotation, Biochem. Biophys. Res. Commun., 2024, 693, 149377 CrossRef CAS PubMed.
- D. R. Roe and T. E. Cheatham III, PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- P. Kadyan, M. Kumar and A. Tufail, et al., Microwave-assisted green synthesis of fluorescent graphene quantum dots (GQDs) using Azadirachta indica leaves: enhanced synergistic action of antioxidant and antimicrobial effects and unveiling computational insights, Mater Adv., 2025, 6, 805–826 RSC.
- B. Kumar, J. Devi, A. Dubey, A. Tufail and N. Antil, Biological and computational investigation of transition metal(II) complexes of 2-phenoxyaniline-based ligands, Future Med. Chem., 2023, 15(21), 1919–1942, DOI:10.4155/fmc-2023-0046.
- B. Kumar, J. Devi, A. Dubey, A. Tufail and B. Taxak, Investigation of antituberculosis, antimicrobial, anti-inflammatory efficacies of newly synthesized transition metal(II) complexes of hydrazone ligands: structural elucidation and theoretical studies, Sci. Rep., 2023, 13(1), 15906, DOI:10.1038/s41598-023-42180-4.
- S. S. Malunavar, A. Anchi and P. Prabhala, et al., A copper-catalysed facile synthesis of highly functionalized aryl sulphones in guanidinium IL (GIL) aided with ultrasound, ChemistrySelect, 2022, 7(37) DOI:10.1002/slct.202202033.
- Y. Tao, R. Dong, I. V. Pavlidis, B. Chen and T. Tan, Using imidazolium-based ionic liquids as dual solvent-catalysts for sustainable synthesis of vitamin esters: inspiration from bio- and organo-catalysis, Green Chem., 2015, 18(5), 1240–1248, 10.1039/c5gc02557e.
- A. Anchi, R. G. Kalkhambkar and S. M. Sutar, et al., Tropylium-BF4 as organocatalyst for microwave-assisted Beckmann rearrangement in [TMG][BF4]: one-pot conversion of ketones to amides, ChemistrySelect, 2023, 8(23) DOI:10.1002/slct.202301431.
- S. K. Singh, A. Bagul and A. Tufail, et al., Synthesis, characterization, and bioactivity of Cu(II), Fe(II), Co(II), Ni(II), and Mn(II) complexes with benzilmonoximethiocarbohydrazide-O-methoxybenzaldehyde: experimental and computational insights, BioMetals, 2025, 38, 245–274, DOI:10.1007/s10534-024-00652-8.
- B. Kumar, J. Devi, A. Dubey, A. Tufail and N. Antil, Biological and computational investigation of transition metal(II) complexes of 2-phenoxyaniline-based ligands, Future Med. Chem., 2023, 15(21), 1919–1942, DOI:10.4155/fmc-2023-0046.
- M. S. Alam and D. U. Lee, Molecular structure, spectral (FT-IR, FT-Raman, UV-vis, and fluorescent) properties and quantum chemical analyses of azomethine derivative of 4-aminoantipyrine, J. Mol. Struct., 2021, 1227, 129512 CrossRef CAS.
- A. R. Moosavi-Zare, H. Goudarziafshar and K. Saki, Synthesis of pyranopyrazoles using nano-Fe-[phenylsalicylaldiminemethylpyranopyrazole]Cl2 as a new Schiff base complex and catalyst, Appl. Organomet. Chem., 2017, 32(1) DOI:10.1002/aoc.3968.
- M. Singh, N. Vaishali, A. K. Paul and V. Singh, Isatin as a 2-aminobenzaldehyde surrogate: transition metal-free efficient synthesis of 2-(2′-aminophenyl)benzothiazole derivatives, Org. Biomol. Chem., 2020, 18(23), 4459–4469, 10.1039/d0ob00888e.
- S. Abu-Melha, Design, synthesis and DFT/DNP modeling study of new 2-amino-5-arylazothiazole derivatives as potential antibacterial agents, Molecules, 2018, 23(2), 434, DOI:10.3390/molecules23020434.
- A. Bagul, D. Gaikwad, Y. Patil and N. Bhusari, Synthesis, characterization, antimicrobial and cytotoxic investigations of some transition metal(II) complexes with tridentate Schiff base derived from pyrrolopyrimidine, Asian J. Chem., 2023, 35(11), 2815–2821, DOI:10.14233/ajchem.2023.30256.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, John Wiley & Sons, 7th edn, 2005 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02622a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2025 |