
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe impact of nanostructuring on the hemocompatibility of polysulfobetaine (PSB) coated hydrogel surfaces†
Subramanian Suriyanarayanan *a,
Nizreen Mohammad Nizama,
Linnea I. Anderssona,
Per H. Nilsson*ab,
Teodor Aastrupc,
Ulrik Palmqvistd and
Ian A. Nicholls*a
*a,
Nizreen Mohammad Nizama,
Linnea I. Anderssona,
Per H. Nilsson*ab,
Teodor Aastrupc,
Ulrik Palmqvistd and
Ian A. Nicholls*a
aLinnaeus University Centre for Biomaterials Chemistry, Linnaeus University, SE-391 82 Kalmar, Sweden. E-mail: subramanian.suriyanarayanan@lnu.se; per.h.nilsson@lnu.se; ian.nicholls@lnu.se
bDepartment of Immunology, University of Oslo and Oslo University Hospital Rikshospitalet, Sognsvannsveien 20, NO-0372 Oslo, Norway
cAttana AB, Engelbrekts väg 6, SE-19162 Sollentuna, Sweden
dLUMA Wire Tech, Amerikavägen 5, SE-39356 Kalmar, Sweden
First published on 10th June 2025
Abstract
A series of nanostructured polysulfobetaine (PSB) hydrogel-coated surfaces were fabricated and tested for hemocompatibility in contact with human blood. PSB films were grafted onto SiO2-coated silicon wafers or Au/quartz via photochemically induced polymerization of a sulfobetaine-based monomer (SBMA, [2-(methacryloylamino)propyl]dimethyl(3-sulfopropyl)ammonium hydroxide). An anodized aluminum oxide (AAO) membrane and latex beads (LB) were used as sacrificial template structures to synthesize polysulfobetaine nanowires (PSBAAO) and hyperporous (PSBLB) networks, respectively. Two soft sacrificial templates, a liquid crystalline medium (LC) and amide-based non-ionic deep eutectic solvent (ni-DESs) providing one-dimensional ordered arrays and flickering clusters, respectively, were utilized to grow nanofibrous (PSBLC) and mesoporous (PSBDES) polysulfobetaine film. Selective dissolution of the sacrificial templates affords the transposed pattern of the template with long-range periodicity from nano to micro scale (20 to 400 nm). Electron micrograph studies revealed nanostructured materials in the form of wires (198 ± 5 nm), cavities (300 nm) and fibers (20 ± 2 nm) when AAO, LB and LC-medium were used as templates, while the polymer films prepared from ni-DESs (PSBDES), water (PSBWAT) and methanol (PSBMeOH) were devoid of any noticeable topographical features. PSB-coated surfaces (except for PSBLB) inhibited non-specific adhesion of protein and biomolecules when presented with purified human proteins, i.e., albumin, fibrinogen, hemoglobin, or human plasma, down to 20–125 ng cm−2 as shown by the QCM studies. Interestingly, the hierarchical nanostructures in polymer films (PSBAAO and PSBLC) resisted the adsorption of albumin and hemoglobin (<20 ng cm−2), even at 50 mg mL−1 concentration. The hemocompatibility of the PSB nanostructures, analyzed after contact with human whole blood for one hour on the PSBAAO and PSBLC, revealed reduced complement activation, quantified as the generation of C3bc fragments and terminal complement sC5b-9 complex formation, in comparison to acrylate glass. The nanowires of PSBAAO showed significantly lower MPO release than the PSBWAT-onto surface, whereas no difference in platelet activation was seen between the surfaces. Compactly organized nanowires and fibers increase the water of hydration layers to strengthen the antifouling and hemocompatibility features, demonstrating the bio-inert nature of the PSB nanostructures. The inherent gelation (hydrophilicity) afforded by the PSB has substantial implications in designing bio-inert surfaces for hemocompatible devices.
Introduction
The design and development of bio-inert interfaces for resisting plasma protein adsorption is a key feature for developing hemocompatible devices for blood-contact based medical applications.1 Even adsorption of a small quantity of protein (<5–7 ng cm−2 of fibrinogen)2–4 is sufficient to trigger complement activation and support platelet adhesion, causing inflammation and thrombus formation5,6 on implant devices.7,8 To subdue the protein fouling effect, phospholipids,9,10 heparin,6,11 PEG,12,13 and other anti(bio)fouling coatings have been used to improve the antithrombogenicity and overall hemocompatibility of materials in blood contact applications. However, many of these coatings have limitations in terms of long-term stability and tolerance of biochemically relevant environments that can hamper their biocompatibility features.10,14 Other synthetic counterparts have been explored for constructing anti(bio)fouling layers using self-assembled monolayers,15–17 grafted copolymers,18,19 polyelectrolytes,20,21 kosmotropes,22,23 zwitterions,24,25 and carbohydrates.26,27 For this purpose, functional polymer-based nanostructures offer the potential for use in developing anti(bio)fouling coatings for hemocompatible devices, owing to their stability and ease of preparation, and their resistance to the non-specific adsorption of biomolecules, micro-organisms and living cells.28–30 Polymer hydrogels31–33 have been investigated extensively due to the organized hydration layer, in addition to their hydrophilicity and electrical neutrality.34–36 As a rule of thumb, biomolecules tend to undergo more conformational changes when adsorbed onto hydrophobic surfaces through facile displacement of the hydration layer, reducing Gibbs free energy37 than on the hydrophilic surfaces.38 Typically, the interfacial hydration layer and its arrangement play a vital role in the adsorption of biomolecules, affecting their conformational change. Essentially, a structured layer of water molecules around the polymer chains affords interfacial resistance to protein and biomolecular adhesion.39,40 Factors controlling the arrangement of this hydration layer may provide a key to improving the surface's anti(bio)fouling properties.41–43 Surface topography, for instance, can influence the hydration layer and fundamentally influence biomolecule interactions by inducing conformational changes and adhesion behavior.44–46Initial studies on improving the antifouling property of the hydrogels were directed towards the morphological features of the polymer-based materials by introducing micro- and nanostructures on the surface to reduce biomolecular adhesion.47–49 Recently, polysulfobetaines32,50–53 have been demonstrated as potential candidates for anti(bio)fouling coatings owing to their zwitterionic character, contributing a strong hydration layer.
This study aimed to explore the anti(bio)fouling properties and hemocompatibility of polysulfobetaine (PSB) hydrogel surfaces by introducing different types of nanostructuring. The PSB nanostructures were prepared by the photochemically induced polymerization of sulfobetaine methacrylate (1, Chart 1) using a bench-top compatible template-directed polymer synthesis strategy on silicon/SiO2 surfaces. The size and dimension of the template and the experimental parameters of the polymerization process in an aqueous medium have been optimized to improve the growth and stability of the PSB nanostructures. Further, the effect of the medium of polymerization on the PSB protein resisting qualities has been studied using a non-ionic deep eutectic solvent (ni-DES) and methanol as polymerization solvents. Protein adhesion on the PSB nanostructured hydrogel surfaces was monitored using QCM and fluorescence microscopic methods. The hemocompatibility of the various PSB surfaces was investigated by incubation with human blood and the host response induced by the surface-blood interaction was examined by measuring biomarkers for complement, neutrophil and platelet activation.
 | ||
| Chart 1 Chemical structure of sulfobetaine monomer (1), cross-linker (2), initiator (3), components (4 and 5) of liquid crystalline (LC) medium and non-ionic deep eutectic solvent (ni-DES) (6 and 6′). | ||
Materials and methods
Chemicals
[2-(Methacryloylamino)propyl]dimethyl-(3-sulfopropyl)ammonium hydroxide (sulfobetaine, SBMA, 1), 1,4-bis(acryloyl)piperazine (BAP, 2), 11-mercaptoundecanoic acid (MuDA), [3-(methacryloyloxy)propyl]trimethoxysilane (silane), 2,2′-azobis(2-methylpropionamidine) dihydrochloride (ABAH, 3), cytochrome C (horse heart), human serum albumin (HSA), bovine serum albumin (BSA), hemoglobin (human), fibrinogen (from human plasma), latex beads (300 nm), sodium bis(2-ethylhexyl) sulfosuccinate (AOT, 4), p-xylene (5), N-methylurea (NMU, 6), N-methylacetamide (NMA, 6′), sodium chloride (NaCl), sodium dihydrogen phosphate (NaH2PO4), disodium hydrogen phosphate (NaH2PO4), fluorescein isothiocyanate labeled BSA (FITC-BSA), silicon wafers, sulfuric acid, sodium hydroxide, toluene, tetrahydrofuran, acetone and molecular sieves (4 Å) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Human plasma was isolated from human whole blood collected in the presence of lepirudin (50 μg mL−1), a specific thrombin inhibitor (Refludan; Celgene, Uxbridge, UK), as an anticoagulant. Plasma was isolated by centrifugation of whole blood at 3000×g for 15 minutes at 4 °C. The plasma was stored in aliquots at −80 °C until use. Hydrogen peroxide (30%, v/v), ammonia solution (25%), triethylamine, and hydrochloric acid (37%) were from Fluka (Buchs, Switzerland). Anopore® membrane (anodized aluminium oxide, AAO) disc (Catalogue No. 6809-5502) with 200-nm diameter pores was procured from Whatman (Dassel, Germany). THF was refluxed with LiAlH4 for one hour and distilled over molecular sieves to remove water, peroxides, inhibitors and other impurities. Toluene was dried over CaCl2 for 1 h and in CaH2 for 12 h, followed by distillation over molecular sieves. Acetone was distilled and dried over molecular sieves. ABAH initiator was recrystallized from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetone water mixture. The ni-DES was prepared by adding 20% (w/w) of the NMU with 80% of NMA at (35 °C) and stirring well till a homogenous solution was obtained. This mixture exists in the liquid phase till 12 ± 2 °C.
:1 acetone water mixture. The ni-DES was prepared by adding 20% (w/w) of the NMU with 80% of NMA at (35 °C) and stirring well till a homogenous solution was obtained. This mixture exists in the liquid phase till 12 ± 2 °C.
The lyotrophic liquid crystalline phase of AOT in water/p-xylene was prepared as reported previously.54 The LC phase used in this study can be described as AOT(a)/p-xylene/SBMA(b)(c), where a, b, and c denote the concentration of AOT in p-xylene, the concentration of SBMA in water and molar ratio of water to AOT, respectively. A 1.5 M AOT solution in p-xylene was used to prepare the LC phase. A 98% SBMA aqueous solution containing 1% BAP was added dropwise under vigorous stirring. The molar ratio of water to AOT was adjusted to between 12 and 15. The mixture was stirred continuously at 25 °C for a homogenous viscous liquid phase.
Surface functionalization
In this work, glass microscope slides (75 × 25 mm), polished silicon wafers (5 × 5 mm) and silica (SiO2) layer-coated Au/quartz resonator (SiO2/Au/quartz, Ø 5 mm) surfaces were functionalized with (Scheme 1A-ESI†) with [3-(methacryloyloxy)propyl]trimethoxysilane and were used as substrates for polymerization. In every step, each Si wafer and SiO2/Au/quartz substrate was activated or rinsed or washed or incubated in 1 mL of respective reagent or solvent for 10 min unless it was mentioned specifically. A glass chamber (150 mL for 10 microscope slides) was used for the activation and rinsing or washing of glass microscope slides for 10 min. Before silanization, the glass microscope slides and silicon wafers were cleaned in acid piranha solution (H2SO4 (98%) and H2O2 (30%), 7:3, v/v) at 80 °C for 30 min (caution: the piranha solution reacts violently with organic compounds and is dangerous when in contact with skin or eyes). The substrates were washed with water and dried in a stream of N2. A thin layer of silicon oxide was grown on the piranha-cleaned silicon surfaces by immersing it in an oxidizing solution containing NH4OH (25%), H2O2 (30%) and water (1:1:6, v/v) at 80 °C for 30 min. The SiO2/Au/quartz surfaces were activated by cleaning in piranha solution at room temperature for 2 min, followed by washing in deionized water and sonication in 0.1 M NaOH. Subsequently, the cleaned Si and SiO2/Au/quartz substrates were washed with water, rinsed in acetone, THF and toluene, dried in a stream of N2 (UHP-grade) and stored in a desiccator until further use.
A fresh solution containing 7.2 mL silane and 0.72 mL triethylamine in 360 mL of toluene was prepared for silanization. The activated glass microscope slides, Si wafers and SiO2/Au/quartz surfaces were immersed in this solution for 12 h at room temperature and stored in dark conditions. After that, the silanized substrates were washed successively in each solvent for three times with toluene, THF, and acetone and then dried in a stream of N2 (UHP-grade). PSBWAT-onto films were grown on the silanized substrates, as shown in Scheme 1A-ESI.† Briefly, an aliquot (200 μL for glass microscope slides and 2 μL for Si wafers and SiO2/Au/quartz surfaces) of an aqueous solution containing 98% of (1) (w/v), 1% (w/v) of (2) and 1% (w/v) of (3) initiator was dispensed on the substrate and the polymerization reaction was initiated by exposing the substrate under UV light at 354 nm for 2 h. The polymer film (PSBWAT-onto) coated surfaces were washed with water and dried in N2.
To prepare PSBAAO films, a procedure like that of PSBWAT-onto was adopted, except that the polymerization was carried out in the presence of an anopore template (Scheme 1). After the polymerization, the film-coated substrate was rinsed with water and immersed in 1.2 M HCl for 12 h at 35 °C to dissolve the nanopore membrane. The residual HCl was removed by washing in water for 30 min, followed by drying in a stream of N2 (UHP-grade).
 | ||
| Scheme 1 Graphical representation for the preparation of PSB nanowires (PSBAAO) on silicon or Au/quartz substrate. | ||
A surface pretreatment was performed when synthesizing the PSBLB films, as shown in Scheme 2-ESI.† The silanized substrate (microscope slides or Si wafer or SiO2 coated Au/quartz surfaces) was drop-coated with an aqueous polystyrene bead solution (0.25% w/v) and the solvent (water) was then allowed to evaporate by placing the setup in a fume-cupboard under constant air flow (0.56 m s−1) for 12 h. The surface was then dried with N2 gas and the PSBLB films were produced similarly using the protocol mentioned for the preparation of PSBWAT-onto films. Subsequently, the PSBLB films were immersed in toluene for 6 h on an orbital shaker to achieve selective dissolution of the polystyrene beads. The trapped toluene solvent was removed by washing in acetone and water for 30 min, and the polymer film was dried with N2 (UHP-grade). PSBLC film was, prepared (Scheme 3-ESI†) using AOT/water LC phase containing (1), (2) and (3). PSBDES and PSBMeOH films were prepared with NMA-NMU ni-DES and methanol as solvent, respectively, containing 49% of (1) (w/v), 0.5% (w/v) of (2) and 0.5% (w/v) of (3) for preparing the pre-polymerization mixture.
For comparison, a graft from approach was adopted to prepare PSBWAT-from films as shown in Scheme 4-ESI.† Here, the films were grown on the piranha-cleaned gold surface. The gold surface was functionalized with MuDA by incubating in a 9:1 ethanol acetic acid mixture containing 1 mM MuDA for 12 h. After rinsing the gold surface with ethanol, acetic acid (10%) and water, the functionalized surface was activated by immersing it in an aqueous solution containing 15 mM NHS and 60 mM EDC for 5 min. The activated surface was immediately incubated in an aqueous solution of ABAH initiator (5 mM) for 30 min to immobilize the initiator covalently. The initiator-modified surface was used immediately thereafter to grow PSBWAT-from films. An aliquot (∼2 μL) of an aqueous solution containing 98% of (1) (w/v) and 1% (w/v) of (2) was dispensed on the substrate, and the polymerization reaction was initiated under UV light at 354 nm for 2 h. The prepared film (PSBWAT-from) was washed with water and dried in N2 (UHP-grade).
Surface characterization
Reflection absorption infrared spectroscopy (RAIRS), fluorescence microscopy and scanning electron microscopy measurements were employed for the structural characterization of the polymer films. Images of the water droplet on modified and unmodified silicon surfaces were captured with a Canon (PowerShot A710 IS) camera. They were utilized to evaluate the contact angle values using Image J 1.46r software.55Infrared spectroscopy
Briefly, the IR spectroscopic measurements were recorded on a Bruker Hyperion 3000 IR microscope attached to a Tensor 27 IR spectrometer and furnished with a computerized sample stage. A grazing angle objective enabling double surface reflections with incident angles of 52° and 83° relative to the surface normal was used. The mercury–cadmium–telluride (MCT) detector collected the spectra at 4 cm−1 resolution, and 1000 interferograms were recorded to obtain a spectrum. The sample chamber was saturated with UHP-grade nitrogen gas throughout the measurement. A three-term Blackmann–Harris apodization function was applied to the interferograms before the Fourier transformation. An unmodified gold surface was used as the reference to record the background spectrum.Scanning electron microscope (SEM)
Scanning electron microscopy (SEM) analyses were carried out using a Leo 1550 Gemini instrument (Zeiss, Oberkochen, Germany) equipped with a field emission electron gun. Polymer particles were applied on black carbon tape attached to alumina stubs and coated with a thin layer of platinum using a platinum sputtering unit (LEICA EM SCD 500) before being inserted in the SEM instrument. A 3 kV potential was applied to the electron gun to generate the electron beam to scan the polymer particles. The pressure inside the measurement chamber was maintained at 2 × 10−5 mbar.Fluorescence microscopy
Non-specific protein adsorption was studied with a fluorescence microscope equipped with a CCD camera. The PSB hydrogels-coated surfaces were immersed in a 1 mL solution of FITC-BSA (25 mg mL−1 in PBS) at 25 °C for 1 h and rinsed extensively with water to remove the loosely bound protein.Quartz crystal microbalance (protein adsorption studies)
A continuous flow quartz crystal microbalance system (Attana Cell 200, Attana AB, Stockholm, Sweden) equipped with a degasser for flow injection analysis (FIA) was used for piezoelectric microgravimetric analysis. Attester software provided by the same manufacturer was used to control the system. AT-cut, 8 mm diameter quartz resonators, operating at a fundamental frequency of 10 MHz and sputtered on both sides with a 100 nm thick gold film (underlined with 10 nm Ti or Cr as an adhesive layer), were used as substrates. A thin silica (SiO2) layer of gold-coated quartz resonators was coated on the active side (the side exposed to the protein solution). These resonators were procured from Attana AB, Stockholm, Sweden. For surface cleaning, pretreatment, activation and modification of the resonator surfaces, please refer to section “surface functionalization”.The polymer film-modified gold surface was mounted in the flow cell holder to monitor the adsorption and desorption of proteins under FIA conditions. Phosphate buffer solution (10 mM, pH 7.4) containing 10 mM NaH2PO4, 10 mM Na2HPO4 and 150 mM NaCl was prepared with ultrapure water. An in-built hydraulic pump with dual-piston control afforded the linear flow of the carrier buffer solution at 25 mL min−1 over the PSB-modified SiO2/Au/quartz substrate. The carrier PBS solution was pumped through the flow cell holder at this flow rate. The system was allowed to equilibrate at this condition until the frequency change was less than 0.5 Hz over 400 s, as specified by the manufacturer. Solid protein samples were dissolved and diluted in PBS, the same as the carrier solution composition, and frozen at −20 °C. For each experiment, an aliquot of 180 μL was introduced in the flow cell using two 6-port injection valves (dual channel) provided in the system. Protein adsorption measurements were obtained by averaging the results from two surfaces prepared under identical conditions.
Stability studies
The PSBAAO modified surfaces, which were prepared as shown in Scheme 1, were stored in PBS (pH 7.4) at 25 °C for a definite time interval. Subsequently, the sample was washed in water, dried under a N2 stream and protein adsorption (50 mg per mL hemoglobin and 100% plasma) studies were carried out by QCM under the FIA conditions as described previously (section “Quartz crystal microbalance (protein adsorption studies)”).Blood chamber experiments (hemocompatibility studies)
The blood chambers were incubated at 37 °C in an incubator for 1 h with manual agitation every 15 min. Following incubation, the blood in each chamber was pooled into a 1.5 mL Eppendorf tube, and activation was interrupted by using 10 mM EDTA at the final concentration. A 20 μL-blood sample was used for complete blood cell count in a Swelab hematology instrument (Swelab, Stockholm, Sweden) according to the manufacturer's instructions. The remaining whole blood samples were centrifuged at 4 °C for 15 min at 3000×g. Plasma was isolated from each sample and frozen at −80 °C until further analysis.
:300 in PBS containing 10 mM EDTA. A rabbit anti-C3c was used for detection, followed by a peroxidase-labeled anti-rabbit antibody (GE Healthcare). The absorbance of each sample was related to a fully activated serum standard set to contain 1000 complement arbitrary units per mL.56 The sC5b-9 was quantified as for C3bc, but the monoclonal antibody aE11 was used as a capturing antibody, and a biotinylated monoclonal antibody 9C4-1 followed by streptavidin horseradish peroxidase (GE Healthcare, UK Limited) for detection. The samples were diluted 1:10 in PBS containing 10 mM EDTA.:10000, and samples for MPO were diluted 1:1000, all in PBS containing 1% BSA.Results and discussion
In this study, a series of PSB nanostructured hydrogel-coated surfaces (Table 1) were prepared by photochemically induced polymerization of sulfobetaine methacrylate monomer (1) on SiO2 coated Si or Au/quartz surfaces (Scheme 1A-ESI†). Hard and/or soft templates, such as AAO (Scheme 1), LB (Scheme 2-ESI†) and LC medium (Scheme 3-ESI) of p-xylene/AOT/water reverse micellar phase were used as sacrificial scaffolds during the polymerization process. Prior to polymer synthesis, the nature of organized structures in the sacrificial scaffolds was determined with SEM and optical microscopic measurement. The AAO membrane comprised vertically aligned, 200 nm-sized, uniformly distributed cylindrical nanopores (Fig. 1A-ESI†). Template latex beads, when self-assembled on Si wafer, displayed lattice-like compactly arranged packing with long-range uniformity (Fig. 1B-ESI†). The liquid crystal template revealed birefringence patterns of the broken mosaic textures of the hexagonal phase (Fig. 1D-ESI†) when visualized under an optical polarizing microscope (OPM). The phase diagram of the sub-room temperature ni-DES system, derived from NMA and NMU (Fig. 1E-ESI†), was constructed to identify the eutectic point as 12 ± 2 °C and has been used to prepare PSBDES films. To improve the stability of the PSB films against wear and tear, we functionalized the substrates with a silane layer bearing the polymerizable moiety of the functional monomer (acrylate group) on the substrate. The apparent increase in the water contact angle (Table 1-ESI†) on the Si/SiO2 surfaces from 22 ± 4° to 70 ± 9° (Fig. 2-ESI†) following silane modification revealed the long-range uniformity of the highly oriented acrylate terminal group of the silane adsorbent. Polymerization using these terminal acrylate groups ensures the covalent attachment of the polymer to the underlying substrate.54 For uniformly coated continuous polymer films, an optimized concentration of the sulfobetaine (99%) and BAP (2) (1%) monomers in aqueous or specified medium was used. Schemes 1-ESI, 2-ESI and 3-ESI† represent the preparation of PSBAAO, PSBLB and PSBLC nanostructured films. In each case, the polymer films were grown via a graft-onto approach using azo initiators with UV irradiation (254 nm) for 2 h. Subsequent dissolution of the sacrificial templates leaves behind its transposed form implanted in the PSB film.| System | Medium | Sacrificial template |
|---|---|---|
| PSBAAO | Water | AAO |
| PSBLB | Water | Latex beads |
| PSBLC | Water | AOT micelle |
| PSBDES | NMA-NMU ni-DES | — |
| PSBMeOH | Methanol | — |
| PSBWAT-onto | Water | — |
| PSBWAT-from | Water | — |
Characterization of the PSB films
SEM images displayed the distinct topographical features of the grafted PSB films (Fig. 1A). Particularly, the PSBAAO film, with its homogeneously grown nanowires in a long-range order (Fig. 3A-ESI†), stood out for its unique properties. Nanowires were distinctly separated from each other evidencing the stability of the anopore template during the polymerization (Fig. 2B-ESI†). The porous features on the nanowire surface are plausibly due to the absorbent qualities to withhold large amounts of water molecules for a hydration layer. A subtle decrease in the water contact angle on PSBAAO (14 ± 3°), when compared to the underlying silane layer, substantiates the formation of a hydration layer (Table 1-ESI†). | ||
| Fig. 1 Electron micrograph of (A) PSBAAO, (B) PSBLC, (C) PSBLB and (D) PSBWAT-onto films observed with scanning electron microscope. Inset to (A) is the histogram showing the size distribution of nanowires in PSBAAO. | ||
The liquid-crystalline templated PSBLC film (Fig. 1B) contained a loosely packed, thin fibrous structure of 100 nm diameter, arranged continuously over a 10-micron scale (Fig. 4-ESI†). Most importantly, analogous birefringence patterns of the p-xylene/AOT/water LC medium with and without the sulfobetaine monomer (Fig. 1C- and 1D-ESI†) evidence for the stability of liquid-crystalline template during polymerization and in achieving the nanofibrous-like texture in PSBLC. The rough surface of the fibers and the underlying corrugated structure elevates the surface area of the polymer film and the hydration layer that was complemented by the contact angle for water on this surface (16 ± 3°).
The latex bead templated PSBLB film (Fig. 1C) also revealed uniformly coated macroporous structures (Fig. 5-ESI†) reflecting the dimension and packing of the interstitial voids created by the template latex beads. The water contact angle for this surface was 48 ± 3°, revealing a moderately hydrophobic surface, probably due to some entrapped latex bead residues and toluene solvent from the extraction process.
The polymer films prepared using the ni-DES (PSBDES) stand out with their unique porous features (Fig. 6A-ESI†), a result of the flickering cluster-like57 structures formed by the components (NMA and NMU). In contrast, PSB prepared in methanol and aqueous medium (PSBMeOH and PSBWAT-onto) were observed to be continuously coated surfaces (Fig. 7A-ESI†) with high hydrophilic character, i.e. low contact angle values for water (Table 1-ESI†). The PSBWAT-from film, synthesized via the graft-from approach, affords a thin and discontinuously grown polymer texture (Fig. 7B-ESI†), substantiated by the higher contact angle for water than for PSBWAT-onto (Table 1-ESI†).
The survey spectra for all the PSB films confirmed the proleptic elements, such as O, C, N and S (Fig. 8-ESI†). The band around 400 eV (Fig. 2A), representing N 1s electrons of nitrogen moieties, upon deconvolution revealed three different nitrogen species at 399.2 (green), 399.8 (dark yellow) and 401.4 eV (blue) corresponding to the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH, OC–NR and C–N+ moieties. A band at 168 eV (Fig. 2B) corresponds to the S 2p electrons confirmed the presence of sulfonic acid groups. Similar binding energy profiles for all the PSB films revealed uniform composition throughout the polymer matrix. The ratio of N+/N peaks is higher especially in PSB nanostructures (PSBAAO, PSBLC, PSBLB, and PSBDES) than the compact thin films (PSBMeOH and PSBwater-onto) which may be due to the presence of initiator-residues from the polymerization process. These residues are more efficiently removed in the case of nanostructured PSB matrices, due to the enhance mass transfer provided by the nanostructures.
C–NH, OC–NR and C–N+ moieties. A band at 168 eV (Fig. 2B) corresponds to the S 2p electrons confirmed the presence of sulfonic acid groups. Similar binding energy profiles for all the PSB films revealed uniform composition throughout the polymer matrix. The ratio of N+/N peaks is higher especially in PSB nanostructures (PSBAAO, PSBLC, PSBLB, and PSBDES) than the compact thin films (PSBMeOH and PSBwater-onto) which may be due to the presence of initiator-residues from the polymerization process. These residues are more efficiently removed in the case of nanostructured PSB matrices, due to the enhance mass transfer provided by the nanostructures.
 | ||
| Fig. 2 Binding energy profiles in electron volts (eV) of (A) N 1s and (B) S 2p electrons, measured with XPS, for different PSB nanostructured films prepared in this study (Table 1). | ||
FT-IR measurements observed in ATR mode also complement this trend regarding the PSB-coated surfaces. IR spectra of all the PSB systems (Fig. 3) revealed comparable spectral patterns denoting the similarity in chemical structure for all the PSB films and nanostructures irrespective of the presence or absence of sacrificial templates and polymerization medium. The presence of the sulfonic acid group can be inferred from the sharp bands at 1038 and 1169 cm−1 for the symmetric and asymmetric stretching modes of the ν(SO3−) group.58 Bands at 1479, 1528 and 1630 cm−1 correspond to the ν(C–N+), ν(N–H) and ν(CO) modes of vibrations. The broad bands around ∼2900 and ∼3400 cm−1 denote the presence of alkyl and hydroxyl groups. The comparable spectral features of the template-extracted films demonstrate the efficient extraction of the sacrificial template or solvent from the PSB matrices. The peaks around 1350 and 1700 cm−1 may be due to the residual artifacts mostly present in the thin films (PSBMeOH and PSBwater-onto) than the PSB nanostructures (PSBAAO, PSBLC, PSBLB, and PSBDES) concurrent with the XPS measurements discussed previously.
 | ||
| Fig. 3 Infrared spectra of the all the PSB films prepared in this study (Table 1), measured in the attenuated total reflection mode. | ||
Anti(bio)fouling studies on the PSB films
The anti(bio)fouling features of the PSB surfaces in relation to protein adsorption were examined using a series of proteins possessing different molecular weights and pIs (Table 2-ESI†). Piezoelectric microgravimetric analysis using QCM under FIA conditions was used to investigate the protein adsorption trend on the series of PSB nanostructures coated SiO2 coated Au/quartz surfaces pH 7.4. The effect of PSB films on protein adhesion phenomena has been reported elsewhere, emphasizing the impact of molecular weight and hydrogelation properties.59 The zwitterionic PSB coated nanostructures synthesized in this study should be neutral at pH 7 and associated with an efficiently bound hydration layer. The frequency versus time curve for the FIA injection of different proteins on the PSBAAO grafted gold-coated quartz resonator is shown in Fig. 4. The mass of the PSB-coated resonator increased upon interaction of the proteins, showing a rapid decrease in the resonant frequency (Δf) (Fig. 4). The carrier buffer solution, subsequently, eluted the loosely bound proteins to restore the frequency (signal) of the PSB-coated resonator to its equilibrium value. Complete recovery in resonance frequency demonstrates the protein-free polymer surface and resistance to irreversible or non-specific adhesion of injected protein samples offered by the nanowires of the PSB surface. Noticeably, the adsorption of the smaller (RNase A and cyt C) and larger proteins (HSA) was more efficiently suppressed on the PSB surface than in the case of both hemoglobin and fibrinogen (Fig. 8-ESI†), and whole plasma. The effect of PSB coating on resisting protein adsorption was evident when compared to underlying silanized and unmodified SiO2 substrates (Fig. 8-ESI†) displaying significant protein binding. This eliminates the possibility of the underlying substrate being exposed or contributing to the observed non-specific protein adsorption.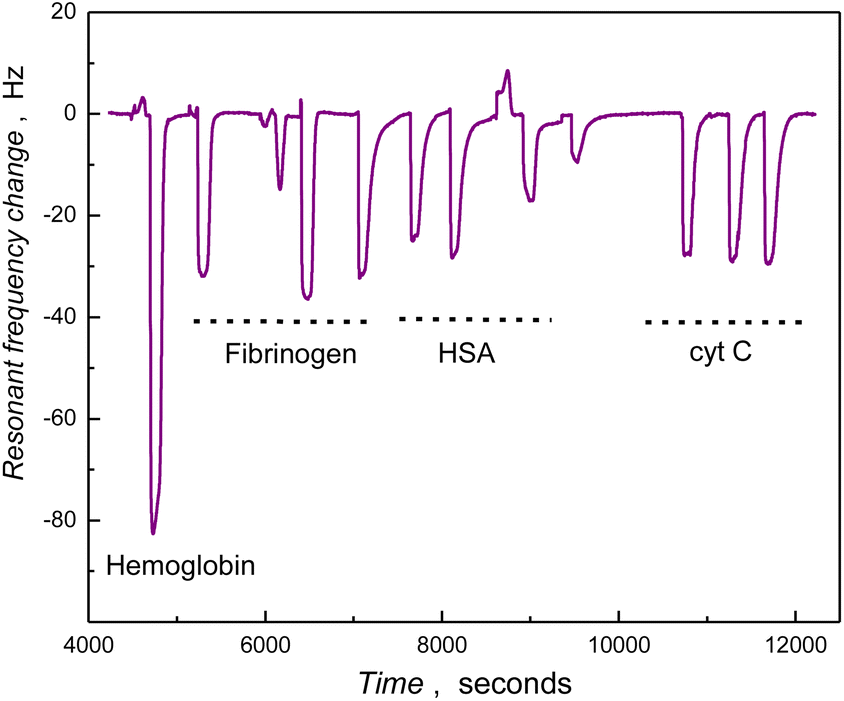 | ||
| Fig. 4 QCM trace (frequency vs. time) of the PSBAAO deposited SiO2 coated Au/quartz resonator, for the injected purified protein samples (2 mg mL−1) indicated in the curve (one injection of hemoglobin and three injections each for fibrinogen, HSA and cytochrome C). | ||
Contiguous polymer films with long-range uniformity are essential for resisting the protein adsorption, as revealed by low protein binding on the polymer film grafted from 90% sulfobetaine monomer solution, as compared to the one grown from 10% monomer concentration (Fig. 9-ESI†). The graft-to method appears more reliable for synthesizing homogeneous polymer surfaces (PSBWAT-onto) with a moderately low level of protein adsorption than for surfaces fabricated by the graft-from approach (PSBWAT-from). In comparison, the PSBAAO film efficiently resisted the adhesion of human blood/plasma proteins; albumin, hemoglobin and fibrinogen, even at higher concentrations (50 mg mL−1) (Fig. 10-ESI†). Compactly arranged, well-oriented PSB nanowires enhance the hydration layer and virtually inhibit the adsorption of fibrinogen, hemoglobin and plasma proteins, even at higher concentrations. The PSB film, deprived of any distinct topographical features (PSBWAT-onto), though suppresses the protein adhesion at lower concentrations (10 mg mL−1) but failed to resist at higher concentrations of the human blood/plasma proteins (Fig. 9-ESI†).
Further, the effect of nanostructures on the PSB toward the non-specific adhesion of proteins was investigated with QCM and fluorescence microscopy. The amount of blood/plasma proteins adsorbed on PSB-modified and unmodified SiO2 surfaces were evaluated with QCM under FIA conditions (Fig. 5 and 6). In the case of the thin film formats of PSB (PSBMeOH and PSBWAT-onto), without the presence of nanostructures (surfaces prepared in different media), though significantly reducing the adhesion of smaller proteins they failed to resist the adhesion of blood/plasma proteins (Fig. 5). The nanostructured entities on the PSB coatings overall reduced protein adsorption compared to the SiO2 surface. Notably, the polymer film PSBAAO and PSBLC constituting nanowires and nanofibers demonstrated significant resistance to protein adhesion, while the macroporous texture of PSBDES moderately suppressed the protein adhesion when compared with PSBWAT-onto (Fig. 6). In the case of PSBLB significant protein adhesion was observed, we propose that this arises due to the higher hydrophobicity of these surfaces (contact angle 48 ± 3°, Table 1-ESI†). This phenomenon may arise due to the hydrophobic-latex beads driving the accumulation of monomer hydrophobic domains on their surface, which after bead removal renders the inside of the resulting cavities with a more hydrophobic character.
 | ||
| Fig. 5 Plot showing the amount of protein adsorbed on the PSB modified SiO2/Au/quartz surfaces due to the repetitive injection of protein samples under flow injection analysis conditions. The blood plasma was diluted with PBS (pH 7.4). | ||
 | ||
| Fig. 6 Histogram of the resonant frequency change for the (A) PSBWAT-onto, (B) PSBDES, (C) PSBLC and (D) PSBAAO modified SiO2/Au/quartz surfaces observed for the repeated injections of different protein samples under flow injection analysis conditions. The blood plasma was diluted with PBS (pH 7.4) (dilution factor was 50, 10, 5, 2.5 or undiluted in the order mentioned in the figure). | ||
The results from fluorescence microscope studies of the adhesion of fluorescein isothiocyanate-tagged bovine serum albumin (FITC-BSA) on the PSB-coated silicon surfaces support the conclusions from the QCM-studies (Fig. 7). Initial studies with FITC-BSA revealed a clustered and discontinuous adsorption, while the fluorescence intensity was drastically diminished on the PSB-modified surfaces (except for PSBLB), prepared with the graft-to approach, demonstrating the resistance displayed by the PSB hydrogels for protein adhesion when compared to the underlying Si or SiO2 surfaces. In the case of the PSB surfaces containing nano-edifices (PSBAAO and PSBLC), these exhibited very low fluorescent intensities, reflecting the superior resistance for the protein adhesion of FITC-BSA.
 | ||
| Fig. 7 Fluorescence microscope images showing the adsorption of fluorescein isothiocyanate tagged bovine serum albumin (FITC-BSA) onto (A) Si wafer, (B) hydroxylated silicon (SiO2/Si), (C) silanized silicon, as well as (D) PSBWAT-onto, (E) PSBAAO, (F) PSBLB, (G) PSBLC, (H) PSBDES, (I) PSBWAT-from and (J) PSBMeOH coated silicon wafers. | ||
Essentially, protein adhesion on unmodified and silanized silicon wafers was significantly reduced with the PSB coating. Presence of an organized hydration layer on the PSB surfaces shields the charged sites of the protein from the polymer.34,60 Apparently, any adsorption of protein in the vicinity of zwitterionic PSB hydrogels due to charge inversion phenomenon, as postulated earlier, can be excluded.61,62 In addition, the contribution of the hydration layer in reducing hydrophobic interactions between the proteins and the polymer coating is essential.63,64 In the case of the PSB nanostructures, the effect of the hydration layer is more pronounced due to the increase in the surface area. The presence of densely packed nanowires increased the surface area of the polymer hydrogel, which managed to form a highly organized hydration layer. Evidently, the surface roughness of the polymer abruptly decreases due to the hydrogelating nature of PSB. Overall, the remarkable protein-resistant character of these PSB nanostructures can be attributed to a combination of the zwitterionic character and the hydrogelation property of PSB nanogels associated with the well-constructed hydration layer.
The stability of the PSB films, during storage in aqueous media (pH 7.4 25 °C, PBS) over ten months, was intermittently tested by examining protein binding characteristics using QCM under FIA conditions (Fig. 11-ESI†). PSBAAO-coated substrates showed minimal variation in the adsorption of hemoglobin and plasma proteins over this time frame. Essentially, the amount of the adsorbed protein was less than 10 ng cm−2 from 50 mg mL−1 concentrations and comparable with previously reported values on antifouling surfaces.65,66 The covalent attachment of the PSB nanowires PSBAAO using the graft-to approach affords enough stability to maintain the antifouling property.
Surface-hemocompatibility was monitored by incubating human whole blood on PSB-coated glass slides (Scheme 1B-ESI†). Adhesion of plasma proteins determines the activation of the plasma cascade systems with subsequent cellular activation.67 Hemocompatibility was analyzed in relation to complement, platelet, and neutrophil activation by incubating the PSB nanostructured coated glass slides with human lepirudin-anticoagulated whole blood for one hour at 37 °C. C3-activation, analyzed as the generation of C3bc, was significantly lower on all modified surfaces compared to acrylate glass (p < 0.05–p < 0.001) (Fig. 8A), which indicates that the PSB film itself reduced C3-activation while the influence of PSB nanostructures on the degree of activation was negligible. There was a trend towards a lower level of sC5b-9-formation on the modified surfaces, however, the differences were not statistically significant (Fig. 8B). Platelet activation, analyzed as the release of PF4 and NAP-2 from platelet alpha-granules, was comparable for the different PSB modified surfaces and acrylate glass. Nevertheless, the PF4-levels on PSBWAT-onto and PSBAAO were reduced of PF4 (Fig. 8C and D) in relation to acrylate glass and nearly significant (p = 0.067 and p = 0.053 respectively). MPO, as a marker of primarily neutrophil activation and oxidative burst, increased significantly on the PSBWAT-onto-surface (p < 0.05), whereas the PSBAAO-surface showed significantly reduced levels of MPO after incubation with human whole blood (p < 0.01) (Fig. 8E). Incubation on PSBLC did not influence the degree of MPO-release. Collectively, the results presented here provide the first study of the impact of PSB surfaces on complement activation and the influence of the nanostructuring of PSB surfaces. This study complements previous reports examining the impact of PSB-based materials on hemocompatibility. By employing strategies such as nanostructuring,68 coatings,69 shape modifications70 to limit or shape the degree of protein adhesion, plasma protein cascade- and cell activation, a deleterious host thromboinflammatory response to a blood-contacting biomaterial can be avoided.67
 | ||
| Fig. 8 Generation of (A) C3bc, (B) soluble terminal complement C5b-9 complex, (C) platelet factor 4 (PF4), (D) neutrophil activating peptide-2 (NAP-2) and (E) myeloperoxidase (MPO) on nanostructured PSB modified glass surfaces and acrylate glass as control after incubation with human whole blood at 37 °C for one hour. Data is shown as mean ± SD from n = 6. *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Conclusions
Nanostructured PSB hydrogels effectually inhibit non-specific adsorption of proteins and have been investigated for activation of plasma complement and cellular activation through human blood incubation studies. Sacrificial template-directed synthesis using soft and hard templates affords uniform, highly organized and stable nanostructures. Nanowires and nanofibrous features in the PSB hydrogels strengthen the interfacial hydration layer to resist protein adsorption to less than 10 ng cm−2 for 10 mg mL−1 concentrations of the plasma and blood proteins. The activation of C3 was drastically reduced to less than 50% with the PSB nanostructured entities (PSBAAO and PSBLC), while neutrophil activation has been suppressed when compared with thin film hydrogels (PSBWAT-onto) devoid of any nanostructuring. Nanowire and fibrous features in PSB are verified to enhance the biocompatibility of the contact surface by minimizing the inflammatory responses. The use of nanostructuring of PSB surfaces can provide access to more hemocompatible surfaces, potentially being of use in the development of medical devices for use in contact with blood.Ethical statement
All experiments were conducted in accordance with the guidelines of the Declaration of Helsinki and were approved by the Ethical Review Board in Linköping, Sweden (Dnr 03-520). Informed consent was obtained from all human participants involved in the study.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
SS – conceptualization, experimental work, analysis and manuscript preparation, NN – hemocompatibility studies, analysis, manuscript preparation, LA – hemocompatibility studies, manuscript preparation, PN – conceptualization and manuscript preparation, TA – characterization and analysis, UP – characterization and analysis, IAN – conceptualization and manuscript preparation.Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
The authors thank Horizon (MindGAP 829040), Swedish Research Council (Vetenskapsrådet grant number 2023-03406), Swedish Knowledge Foundation (ULTRA, Grant Number 20190114 and Synergi22, Grant Number 20230019) and Linnaeus University for financial support assistance.Notes and references
- J. Feng, J. H. Wang, H. H. Wang, X. Y. Cao, X. L. Ma, Y. Rao, H. M. Pang, S. L. Zhang, Y. H. Zhang, L. Wang, X. L. Liu and H. Chen, ACS Appl. Mater. Interfaces, 2023, 15, 35860–35871 CrossRef CAS PubMed.
- I. H. Jaffer, J. C. Fredenburgh, J. Hirsh and J. I. Weitz, J. Thromb. Haemostasis, 2015, 13, S72–S81 CrossRef PubMed.
- D. Kwak, Y. Wu and T. A. Horbett, J. Biomed. Mater. Res., Part A, 2005, 74, 69–83 Search PubMed.
- W.-B. Tsai, J. M. Grunkemeier and T. A. Horbett, J. Biomed. Mater. Res., Part A, 1999, 44, 130–139 CrossRef CAS.
- M. Douglass, M. Garren, R. Devine, A. Mondal and H. Handa, Prog. Mater. Sci., 2022, 130, 100997 CrossRef CAS PubMed.
- Y. N. Zhang, L. J. Zhang, S. Duan, Y. Hu, X. K. Ding, Y. C. Zhang, Y. Li, Y. Z. Wu, X. J. Ding and F. J. Xu, J. Mater. Chem. B, 2022, 10, 1795–1804 RSC.
- A. Abdelrasoul and A. Shoker, Chem. Eng. Res. Des., 2022, 177, 615–624 CrossRef CAS.
- F. Altaf, S. R. Wu and V. Kasim, Front. Mol. Biosci., 2021, 8, 680397 CrossRef CAS PubMed.
- P. C. Li, X. J. Li, W. H. Cai, H. Q. Chen, H. Chen, R. Wang, Y. C. Zhao, J. Wang and N. Huang, Mater. Sci. Eng., C, 2020, 116, 111237 CrossRef CAS PubMed.
- K. Ishihara, Sci. Technol. Adv. Mater., 2022, 23, 498–524 CrossRef CAS.
- J. C. Lei, H. Y. Wang, Q. Dang, X. Wang, C. G. Li, J. Huang, J. Z. Hou, K. J. Fang and S. X. Zhao, Coatings, 2023, 13, 1248 CrossRef CAS.
- M. C. Sin, W. L. Lin, J. C. H. Chen, A. Higuchi, J. Zheng, A. Chinnathambi, S. A. Alharbi and Y. Chang, Int. J. Polym. Mater. Polym. Biomater., 2016, 65, 409–420 Search PubMed.
- T. P. Ribeiro, F. J. Monteiro and M. S. Laranjeira, Eur. Polym. J., 2020, 137, 109934 CrossRef CAS.
- A. Willers, J. Arens, S. Mariani, H. Pels, J. G. Maessen, T. M. Hackeng, R. Lorusso and J. Swol, Membranes, 2021, 11, 617 CrossRef CAS PubMed.
- C. Sperling, R. B. Schweiss, U. Streller and C. Werner, Biomaterials, 2005, 26, 6547–6557 CrossRef CAS PubMed.
- X. Hu, T. Wang, F. Q. Li and X. Mao, RSC Adv., 2023, 13, 20495–20511 RSC.
- H. Y. Zhang, F. Y. Wang and Z. G. Guo, Adv. Colloid Interface Sci., 2024, 325, 103097 CrossRef CAS PubMed.
- M. C. Sin, S. H. Chen and Y. Chang, Polym. J., 2014, 46, 436–443 CrossRef CAS.
- H. Y. Qiu, K. Feng, A. Gapeeva, K. Meurisch, S. Kaps, X. Li, L. M. Yu, Y. K. Mishra, R. Adelung and M. Baum, Prog. Polym. Sci., 2022, 127, 101516 CrossRef CAS.
- M. K. Peng, X. D. Zhang, X. Xiao, M. J. Dong, G. W. Zhao, P. Liu, Y. S. Chen and C. H. Wang, J. Coat. Technol. Res., 2019, 16, 857–868 CrossRef CAS.
- J. Akintola, Y. H. Chen, Z. A. Digby and J. B. Schlenoff, ACS Appl. Mater. Interfaces, 2023, 15, 50058–50068 CrossRef CAS PubMed.
- R. S. Kane, P. Deschatelets and G. M. Whitesides, Langmuir, 2003, 19, 2388–2391 CrossRef CAS.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- J. Baggerman, M. M. J. Smulders and H. Zuilhof, Langmuir, 2019, 35, 1072–1084 CrossRef CAS PubMed.
- A. B. Asha, Y. J. Chen and R. Narain, Chem. Soc. Rev., 2021, 50, 11668–11683 RSC.
- D. H. Cai, L. R. Shi, R. M. Long, G. Ren, S. B. Wang and Y. G. Liu, Carbohydr. Polym., 2021, 261, 117847 CrossRef CAS.
- Y. Duan, J. Wu, W. Qi and R. Su, Carbohydr. Polym., 2023, 304, 120504 CrossRef CAS.
- D. Campoccia, L. Montanaro and C. R. Arciola, Biomaterials, 2013, 34, 8533–8554 CrossRef CAS PubMed.
- J. E. Gittens, T. J. Smith, R. Suleiman and R. Akid, Biotechnol. Adv., 2013, 31, 1738–1753 CrossRef CAS PubMed.
- L. Tauhardt, K. Kempe, M. Gottschaldt and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 7998–8011 RSC.
- S. Suriyanarayanan, H.-H. Lee, B. Liedberg, T. Aastrup and I. A. Nicholls, J. Colloid Interface Sci., 2013, 396, 307–315 CrossRef CAS PubMed.
- M. Yao, Z. Wei, J. Li, Z. Guo, Z. Yan, X. Sun, Q. Yu, X. Wu, C. Yu, F. Yao, S. Feng, H. Zhang and J. Li, Nat. Commun., 2022, 13, 5339 CrossRef CAS PubMed.
- A. Venault, M. W. Lai, J. F. Jhong, C. C. Yeh, L. C. Yeh and Y. Chang, ACS Appl. Mater. Interfaces, 2018, 10, 17771–17783 CrossRef CAS PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
- Q. L. Cheng, A. B. Asha, Y. Liu, Y. Y. Peng, D. Diaz-Dussan, Z. S. Shi, Z. C. Cui and R. Narain, ACS Appl. Mater. Interfaces, 2021, 13, 9006–9014 CrossRef CAS.
- L. Y. Deng, S. L. Li, Y. W. Qin, L. J. Zhang, H. N. Chen, Z. G. Chang and Y. X. Hu, J. Membr. Sci., 2021, 619, 118564 CrossRef CAS.
- W. Norde, Macromol. Symp., 1996, 103, 5–18 CrossRef CAS.
- Y. F. Yano, J. Condens Matter. Phys., 2012, 24, 503101 CrossRef PubMed.
- Y. He, Y. Chang, J. C. Hower, J. Zheng, S. Chen and S. Jiang, Phys. Chem. Chem. Phys., 2008, 10, 5539–5544 RSC.
- P. J. Molino, D. Yang, M. Penna, K. Miyazawa, B. R. Knowles, S. MacLaughlin, T. Fukuma, I. Yarovsky and M. J. Higgins, ACS Nano, 2018, 12, 11610–11624 CrossRef CAS PubMed.
- Y. He, J. Hower, S. Chen, M. T. Bernards, Y. Chang and S. Jiang, Langmuir, 2008, 24, 10358–10364 CrossRef CAS PubMed.
- H. Kitano, M. Imai, T. Mori, M. Gemmei-Ide, Y. Yokoyama and K. Ishihara, Langmuir, 2003, 19, 10260–10266 CrossRef CAS.
- A. M. C. Maan, A. H. Hofman, W. M. de Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- E. Luong-Van, I. Rodriguez, H. Y. Low, N. Elmouelhi, B. Lowenhaupt, S. Natarajan, C. T. Lim, R. Prajapati, M. Vyakarnam and K. Cooper, J. Mater. Res., 2013, 28, 165–174 CrossRef CAS.
- L. Rizzello, R. Cingolani and P. P. Pompa, Nanomedicine, 2013, 8, 807–821 CrossRef CAS PubMed.
- X. Yu, W. F. Yang, Y. Yang, X. G. Wang, X. Liu, F. Zhou and Y. Zhao, J. Mater. Sci., 2020, 55, 14544–14557 CrossRef CAS.
- F. W. Y. Myan, J. Walker and O. Paramor, Biointerphases, 2013, 8, 30 CrossRef PubMed.
- C. M. Kirschner and A. B. Brennan, Annu. Rev. Mater. Res., 2012, 42, 211–229 CrossRef CAS.
- A. J. Scardino and R. de Nys, Biofouling, 2011, 27, 73–86 CrossRef CAS PubMed.
- Y. Chang, W.-J. Chang, Y.-J. Shih, T.-C. Wei and G.-H. Hsiue, ACS Appl. Mater. Interfaces, 2011, 3, 1228–1237 CrossRef CAS PubMed.
- R. Quintana, M. Gosa, D. Janczewski, E. Kutnyanszky and G. J. Vancso, Langmuir, 2013, 29, 10859–10867 CrossRef CAS PubMed.
- W.-H. Kuo, M.-J. Wang, H.-W. Chien, T.-C. Wei, C. Lee and W.-B. Tsai, Biomacromolecules, 2011, 12, 4348–4356 Search PubMed.
- S. Jiang and Z. Cao, Adv. Mater., 2010, 22, 920–932 CrossRef CAS PubMed.
- N. Ndizeye, S. Suriyanarayanan and I. A. Nicholls, Eur. Polym. J., 2018, 106, 223–231 CrossRef CAS.
- W. S. Rasband, ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, 2018 Search PubMed.
- G. Bergseth, J. K. Ludviksen, M. Kirschfink, P. C. Giclas, B. Nilsson and T. E. Mollnes, Mol. Immunol., 2013, 56, 232–239 CrossRef CAS PubMed.
- S. Suriyanarayanan, G. D. Olsson, S. Kathiravan, N. Ndizeye and I. A. Nicholls, Int. J. Mol. Sci., 2019, 20, 2857 CrossRef CAS PubMed.
- T. Xiang, R. Wang, W.-F. Zhao, S.-D. Sun and C.-S. Zhao, Langmuir, 2014, 30, 5115–5125 CrossRef CAS PubMed.
- Y.-J. Shih and Y. Chang, Langmuir, 2010, 26, 17286–17294 CrossRef CAS PubMed.
- J. Wu, W. Lin, Z. Wang, S. Chen and Y. Chang, Langmuir, 2012, 28, 7436–7441 CrossRef CAS PubMed.
- P. M. Biesheuvel and A. Wittemann, J. Phys. Chem. B, 2005, 109, 4209–4214 Search PubMed.
- J. J. Ramsden, R. Kurrat, D. J. Roush, D. S. Gill and R. C. Willson, J. Am. Chem. Soc., 1995, 117, 8511–8516 Search PubMed.
- J. M. Park, B. B. Muhoberac, P. L. Dubin and J. Xia, Macromolecules, 1992, 25, 290–295 Search PubMed.
- V. Lesins and E. Ruckenstein, Colloid Polym. Sci., 1988, 266, 1187–1190 CrossRef CAS.
- W. Feng, S. P. Zhu, K. Ishihara and J. L. Brash, Langmuir, 2005, 21, 5980–5987 CrossRef CAS PubMed.
- Z. Zhang, S. Chen, Y. Chang and S. Jiang, J. Phys. Chem. B, 2006, 110, 10799–10804 CrossRef CAS PubMed.
- K. N. Ekdahl, J. D. Lambris, H. Elwing, D. Ricklin, P. H. Nilsson, Y. Teramura, I. A. Nicholls and B. Nilsson, Adv. Drug Delivery Rev., 2011, 63, 1042–1050 CrossRef CAS PubMed.
- N. Ferraz, M. K. Ott and J. Hong, Microsc. Res. Tech., 2010, 73, 1101–1109 CrossRef CAS PubMed.
- F. Weber, H. Q. Quach, M. Reiersen, S. Y. Sarraj, D. N. Bakir, V. A. Jankowski, P. H. Nilsson and H. Tiainen, J. Biomed. Mater. Res., Part A, 2022, 110, 1341–1355 CrossRef CAS PubMed.
- P. P. Wibroe, A. C. Anselmo, P. H. Nilsson, A. Sarode, V. Gupta, R. Urbanics, J. Szebeni, A. C. Hunter, S. Mitragotri, T. E. Mollnes and S. M. Moghimi, Nat. Nanotechnol., 2017, 12, 589–594 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02435h |
| This journal is © The Royal Society of Chemistry 2025 |