
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhenyltriazole-based sulfonamides: novel dual-target agents against MRSA biofilms and resistant pathogens†
Abdelrahman Hussiena,
Arafa Musa*b,
Hanzada T. Nour El-Din c,
Ahmed M. Helala,
Yosra I. Nagyc,
Hany G. Ezzata,
Ahmed S. Attiacd,
Abdelrahman S. Mayhoubae,
Khaled Shalabyf,
Della Grace Thomas Parambig and
Mohamed M. Elsebaie*a
c,
Ahmed M. Helala,
Yosra I. Nagyc,
Hany G. Ezzata,
Ahmed S. Attiacd,
Abdelrahman S. Mayhoubae,
Khaled Shalabyf,
Della Grace Thomas Parambig and
Mohamed M. Elsebaie*a
aDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy, Al-Azhar University, Cairo 11884, Egypt. E-mail: m.elsebaei@azhar.edu.eg
bDepartment of Pharmacognosy, College of Pharmacy, Jouf University, Sakaka, Aljouf 72341, Saudi Arabia. E-mail: akmusa@ju.edu.sa
cDepartment of Microbiology and Immunology, Faculty of Pharmacy, Cairo University, Cairo, 11562, Egypt
dDepartment of Microbiology and Immunology, School of Pharmacy, Newgiza University, Giza, Egypt
eBiomedical Sciences, University of Science and Technology, Zewail City of Science and Technology, Giza, Egypt
fDepartment of Pharmaceutics, College of Pharmacy, Jouf University, Sakaka, Aljouf 72341, Saudi Arabia
gDepartment of Pharmaceutical Chemistry, College of Pharmacy, Jouf University, Sakaka, Aljouf 72341, Saudi Arabia
First published on 17th June 2025
Abstract
The advent of multidrug-resistant bacteria requires the continuous development of new antimicrobial agents. A series of phenyltriazole–sulfonamide hybrid compounds (16–27) have been synthesized and evaluated for their antimicrobial properties, with a focus on combating resistant bacterial strains such as methicillin-resistant Staphylococcus aureus (MRSA) and Acinetobacter baumannii AB5075. Compounds were synthesized through a multi-step reaction, including alkylation and aminoguanidine substitution, with structural elucidation performed using NMR and elemental analysis. Antimicrobial activity was assessed through Minimum Inhibitory Concentration (MIC) measurements, which revealed that compounds with longer alkyl chains or specific functional groups had a very enhanced activity against MRSA, especially 23 and 24 analogs. The results highlighted the correlation between lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) and antimicrobial efficacy, particularly for compounds such as 23 (n-nonyl) which showed potent activity against MRSA. Further evaluation by time-killing assays demonstrated the rapid bactericidal activity of compound 23, while biofilm disruption studies showed the potential of these compounds to target biofilm-associated infections. Docking studies have shown that these compounds can interact with key bacterial targets, including PBP2a and DHPS, providing a dual-target approach for treatment of MRSA. Furthermore, in silico analysis revealed favorable pharmacokinetic and ADME properties of the synthesized compounds. The study shows promising new candidates for combating antimicrobial resistance, with the potential for further optimization and development.
P) and antimicrobial efficacy, particularly for compounds such as 23 (n-nonyl) which showed potent activity against MRSA. Further evaluation by time-killing assays demonstrated the rapid bactericidal activity of compound 23, while biofilm disruption studies showed the potential of these compounds to target biofilm-associated infections. Docking studies have shown that these compounds can interact with key bacterial targets, including PBP2a and DHPS, providing a dual-target approach for treatment of MRSA. Furthermore, in silico analysis revealed favorable pharmacokinetic and ADME properties of the synthesized compounds. The study shows promising new candidates for combating antimicrobial resistance, with the potential for further optimization and development.
1. Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) poses a significant global health threat due to its resistance to multiple antibiotics, leading to high morbidity and mortality in nosocomial and community-acquired infections.1 The emergence of vancomycin-resistant strains and biofilm-associated infections further complicates treatment, necessitating the development of novel antimicrobial agents. Sulfonamide-based compounds, historically effective against bacterial infections, offer a promising scaffold for designing new anti-MRSA agents. This study evaluates the antimicrobial activity of phenyltriazole–sulfonamides, a novel class of compounds combining triazole and sulfonamide moieties, against MRSA, with a focus on their anti-biofilm properties, time-kill kinetics, and computational ADME profiles.2,3 Studies on the different bacterial targets of existing antibiotics have revealed that antibiotics do not target all the major biochemical pathways in bacteria. Fortunately, most antibiotics work by selectively inhibiting specific bacterial processes, such as cell wall synthesis (peptidoglycan),4,5 protein synthesis (translation),6,7 DNA replication,8,9 RNA synthesis (transcription),10 and folic acid biosynthesis.11 Only a small number of antibiotics act by interfering with ion channels or promoting bacterial cell lysis.12–14According to a report by the World Health Organization (WHO), there is a global shortage of effective antibacterial agents for both Gram-negative and Gram-positive bacteria.15 Despite extensive efforts, the main reason for the persistence of bacterial infections is the development of resistance.16 Therefore, the current need to advance the development of novel antibacterial categories. The emergence of multidrug-resistant microorganisms is one of the most urgent global challenges.17 Examples of bacterial infections that have developed resistance to multiple drugs are Streptococcus pneumoniae, methicillin-resistant Staphylococcus aureus (MRSA), Pseudomonas aeruginosa, Acinetobacter baumannii, Escherichia coli, vancomycin-resistant enterococci (VRE), and Klebsiella pneumoniae.
Among these, Staphylococcus aureus is notable for its versatility. It can colonize the skin and other body parts, even without symptoms of active disease, and about 20% of the world's population are thought to be carriers of the pathogen. Normally, it remains inactive until the skin breaks or the immune system weakens, allowing the bacteria to enter the body and cause infection. This results in the appearance of diseases caused by toxins produced by the bacteria themselves, and rapid proliferation of the organism, resulting in invasion and destruction of tissues.18 The reason for our particular interest in this pathogen is the lack of comprehensive data on the distribution of MRSA clones in the Middle East, particularly in Egypt.19
Infections with S. aureus are usually self-limiting and not immediately life-threatening, but there is a growing concern that these infections are increasingly penetrating deeper into the tissues, and leading to more serious and potentially life-threatening diseases. The main problem is that S. aureus has developed resistance to the most effective antibacterial agents previously available. To address this problem, it is necessary to develop new drugs that can effectively combat this adaptable microorganism, which is very versatile.20 As S. aureus can occur naturally in human skin and in the nasal cavity, it is often associated with infections of the skin and soft tissue. However, microorganisms can spread from superficial locations to deeper and more critical areas. This dissemination may occur via the bloodstream, allowing S. aureus to establish metastatic foci of infection in distant locations.21 Diseases caused by S. aureus can be attributed to the broad spectrum of cell surface and extracellular protein toxins produced by the bacterium which actively antagonize the host defences. In addition, S. aureus may express several surface-localized proteins that bind to components of the extracellular matrix and serve as adhesins that facilitate bacterial binding and colonization. The cytolytic toxins produced by S. aureus may damage the membranes of host cells, while superantigens may cause toxic shock syndrome.22
Another example of a resistant bacterial infection is Acinetobacter baumannii, a Gram-negative bacterium. Gram-negative bacteria have an impermeable outer membrane that prevents many chemical compounds from entering. This makes porin channels a prime entry point for antibacterial agents.23 These porins are lined with highly charged residues such as arginine, aspartate, and glutamate.24 The physicochemical properties of antibacterial compounds are therefore crucial for researchers. In general, compounds that target Gram-negative bacteria must have a higher degree of polarity than compounds that have limited activity against Gram-negative strains.25
The sulfonamide moiety is present in many medications. Examples include the antibiotics sulfamethoxazole (also known as cotrimoxazole when combined with trimethoprim), chlorthalidone, a thiazide-like diuretic, zonisamide, a drug used to treat treatment of epilepsy and Parkinson's disease, and acetazolamide, carbonic anhydrase inhibitors, and dorzolamide used to treat high blood pressure in the eye, also with glaucoma, and many more,26 (Fig. 1).
 | ||
| Fig. 1 The structure of approved sulfonamide drugs. | ||
In our laboratory, our focus in recent years has been the synthesis of various antibacterial agents with potent activity against a broad spectrum of both Gram-negative and Gram-positive bacteria. This work was mainly based on the identification of our lead compound phenylthiazole, which had a lipophilic part and a polar part of the guanidine moiety. However, its activity was mainly observed against Gram-positive pathogens.27 Several structural optimization processes have been performed to improve antimicrobial activity and metabolic stability.28–42 Despite phenylthiazole's remarkable success in eliminating intracellular pathogens and reducing the bacterial biofilm burden, rapid and lethal mode of action, its activity against Gram-negative microorganisms was significantly limited and limited only to Gram-positive pathogens. However, their low activity against Gram-negative pathogens has been mainly attributed to their poor permeability to Gram-negative pathogens, as recent studies have confirmed that phenylthiazoles exert their action by inhibiting the two major proteins involved in the synthesis of undecaprenylpyrophosphate phosphatase (UppP) and undecaprenylpyrophosphate synthase (UppS) in the cell wall. Consequently, when the phenylthiazole core was replaced by phenylpyrazoles, a significant improvement in polarity and activity against Gram-negative bacteria was observed.34,43–45 As manifested in the rationalization of work (Fig. 2), introducing a polarized pharmacophore to the phenyltriazole scaffold enhances its' antimicrobial activity and pharmacokinetic properties.
 | ||
| Fig. 2 Rational design of phenyltriazole–sulfonamides hybrids. | ||
Taken together, phenyltriazole–sulfonamide conjugates offer a strong potential for dual interaction on MRSA targets. The chemical moieties within the novel design include phenyltriazole moiety with terminal polar guanidine to prove their ability to cross the cell wall of MRSA and cause disruption.5 Thus, the incorporation of active sulfonamide pharmacophore may potentiate the antimicrobial activity by inhibiting the folate pathway. Moreover, this design would have a higher polarity and therefore higher activity with a wider spectrum of antibacterial effectiveness. Several structural modifications have been made to improve both antibacterial activities, including anti-biofilm activity and intracellular pathogen control, and to improve the pharmacokinetic profile. However, the solubility of the resulting compounds proved to be somewhat unsatisfactory. One of the key factors in increasing the solubility of these compounds was the linker between the head and the scaffold.46–48 A new class of phenyltriazoles was therefore synthesized and evaluated for antibacterial activity. Phenyltriazoles included in this study have been designed to contain a sulfonamide moiety (Fig. 2), which is known to have antibacterial activity.49
2. Results and discussion
2.1 Chemistry
Our compounds 1–27 have been synthesised as described in (Fig. 3). The resulting structures of compounds 3–27 was elucidated by means of spectrum data and elemental analysis, as shown in the ESI.† Sulphanilamide was dissolved in 6 M HCl, and equal amount of THF and DMF, then cooled to 0 °C in an ice-bath and NaNO2 was dissolved in H2O and added dropwise to the solution and the reaction was stirred for 25 minutes. NaN3 was dissolved in H2O and then added dropwise with stirring and allowed to gradually warm to room temperature overnight. The reaction mixture is poured into H2O, then extracted three times with ethyl acetate. The combined organic layer was washed with saturated bicarbonate solution and brine solution. The organic layer was then dried over anhydrous sodium sulfate and evaporated under reduced pressure and used in the second step as a yellow solid without further purification.49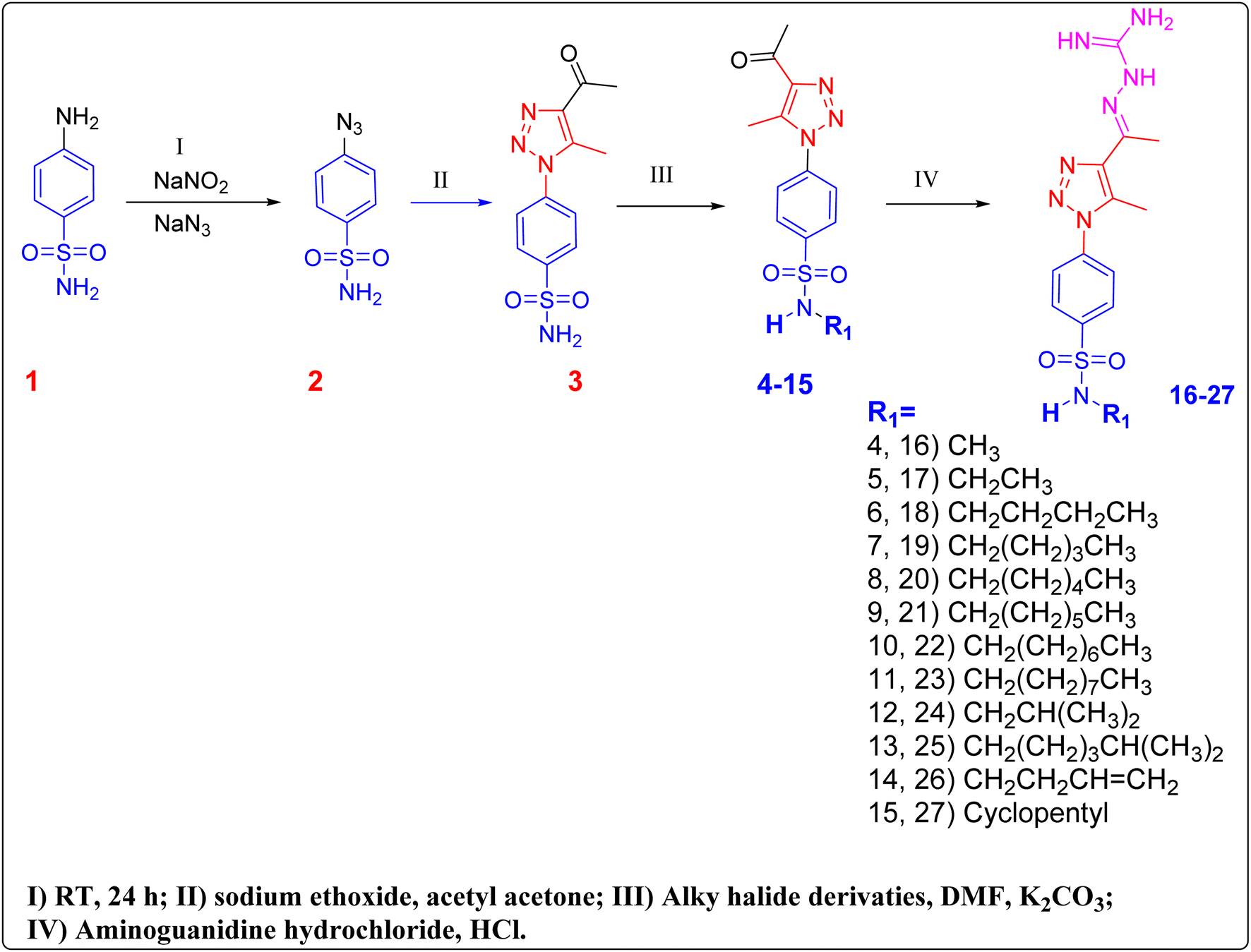 | ||
| Fig. 3 Synthesis of phenyltriazole–sulfonamides hybrids. | ||
Compound 2 was dissolved in ethanol. Acetylacetone and sodium ethoxide were added. The reaction was heated to reflux stirring for 4–6 hours. After the reaction was completed and allowed to cool to room temperature, the white solid was precipitated and then filtered to give compound 3. Compound 3 was dissolved in DMF, K2CO3 and appropriate alkyl halides were added. The reaction was then stirred at 75 °C for 1 hour. The reaction was monitored with TLC until the starting material disappeared. The reaction mixture was allowed to cool and poured into ice-cold water to afford compounds 4–15 as a white solid.
The alkylated triazole acetyl compounds 4–15 were dissolved in ethanol, 1 ml conc. hydrochloric acid and aminoguanidine bicarbonate were added. The reaction mixture was refluxed for 6 hours, after completion of the reaction, it was poured into ice-cold water and neutralized with sodium carbonate to afford compounds 16–27. The chemical structure of compounds 16–27 was confirmed based on their spectral data and elemental analysis. In addition to the signals corresponding to the phenyltriazole scaffold and its two methyl groups, which appeared in all 1H NMR and 13C NMR spectra at almost the same chemical shifts at around 2.55–2.3 ppm, the 1H NMR spectrum of compounds 11 and 23, as examples, exhibited additional multiplet signals at 1.42–0.85 ppm, in the aliphatic region due to the presence of the nonyl aliphatic moiety and the whole aliphatic chain appeared in 13C NMR spectrum as 9 signals from δ 43.04 to 15.95. Also, for the rest of the compound's NMR spectra showed obvious signals at the aliphatic region which indicates the addition of the different alkyl halides to the starting compound.
Subsequently, the reaction of 4–15 with aminoguanidine hydrochloride gave the final products 16–27 as outlined in Fig. 3. The structures of this series of novel compounds were confirmed by their spectral and elemental data. The guanidyl protons, for 24 as a representative example, displayed in the 1H NMR spectrum as broad signals at δ 5.68 ppm and these signals showed approximately the same ppm for each compound to ensure the formation of the intended aminoguanidine products.
2.2 Biological evaluation
P) values for the compounds, offering a basis for understanding the relationship between the chemical structure of the derivatives and their antimicrobial activity. A comprehensive Structure–Activity Relationship (SAR) analysis allows us to identify key structural features that influence the antimicrobial potency against these bacterial strains. The compounds tested in this study feature variations in alkyl chain length and branching, as well as the presence of specific functional groups. These variations result in substantial changes in antimicrobial efficacy, which can be explained by their effect on physicochemical properties, such as lipophilicity, and their ability to interact with bacterial targets. A clear trend emerges when examining the effect of alkyl chain length on antimicrobial activity. Shorter alkyl groups (methyl, ethyl, n-butyl, n-pentyl, and n-hexyl) generally show poor to moderate activity against both S. aureus and A. baumannii, with MIC values greater than 64 μg ml−1 for most of these compounds. Notably, compounds with shorter chains (16–20) exhibit significantly reduced activity against both bacterial strains, highlighting the importance of hydrophobicity and the optimal size of the alkyl group in penetrating bacterial membranes and affecting their target sites.
| Cp. ID | R | S. aureus USA300 | A. baumannii AB5075 | LogP value |
|---|---|---|---|---|
| a NT: not tested. | ||||
| 16 | Methyl | >64 | >64 | 0.62 |
| 17 | Ethyl | >64 | >64 | 1.15 |
| 18 | n-Butyl | >64 | >64 | 2.20 |
| 19 | n-Pentyl | >64 | >64 | 2.73 |
| 20 | n-Hexyl | 16 | 16 | 3.26 |
| 21 | n-Heptyl | 64 | >64 | 3.79 |
| 22 | n-Octyl | 64 | 64 | 4.32 |
| 23 | n-Nonyl | 1 | 64 | 4.85 |
| 24 | Isobutyl | 4 | 8 | 2.07 |
| 25 | Heptan-2-yl | >64 | >64 | 3.57 |
| 26 | But-3-en-1-yl | >64 | >64 | 1.72 |
| 27 | Cyclopentyl | 8 | 32 | 2.09 |
| Vancomycin | — | 1 | NT | −1.14 |
 | ||
| Fig. 4 Bar chart showing the antimicrobial data of phenyltriazole–sulfonamide hybrids. | ||
However, the introduction of longer alkyl chains appears to improve antimicrobial efficacy against S. aureus but not necessarily against A. baumannii. For instance, compound 23 (n-nonyl) exhibits a marked reduction in MIC against S. aureus (1 μg ml−1), but maintains a higher MIC against A. baumannii (64 μg ml−1). This suggests that while increased lipophilicity (as indicated by a higher logP value of 4.85) may enhance membrane penetration and bactericidal activity against S. aureus, it does not guarantee activity against A. baumannii, which may have different membrane properties or efflux mechanisms that limit the effectiveness of these compounds.
Additionally, cyclopentyl analog 27 and branched alkyl groups (24, Isobutyl) also demonstrate notable activity. Cyclopentyl (MIC = 8 μg ml−1 for S. aureus and 32 μg ml−1 for A. baumannii) and isobutyl (MIC = 4 μg ml−1 for S. aureus and 8 μg ml−1 for A. baumannii) derivatives show moderate to good antimicrobial activity, suggesting that such substitutions might provide steric or electronic benefits that enhance bacterial cell interaction, potentially via changes in the binding or affinity to bacterial targets. These branched and cyclic groups may also hinder bacterial efflux mechanisms, contributing to their improved activity. A noteworthy observation is the correlation between lipophilicity (logP) and antimicrobial activity. As the alkyl chain length increases, the logP value also increases, suggesting an enhanced ability of these compounds to partition into the lipid bilayer of bacterial membranes. For example, compound 16 (methyl group, logP = 0.62) exhibits no antimicrobial activity, whereas compound 23 (n-nonyl, logP = 4.85) demonstrates significant activity against S. aureus (MIC = 1 μg ml−1). However, the correlation between logP and MIC is not straightforward, as shown by compound 21 (n-heptyl, logP = 3.79) which has high MIC values (64 μg ml−1) against both bacterial strains. This suggests that while logP is a contributing factor to antimicrobial activity, other structural features, such as the nature of the alkyl group, might play a more dominant role in determining activity. It also indicates that the compounds in this study, while effective against S. aureus, may not be as effective against A. baumannii unless further structural modifications are made to address specific resistance mechanisms in A. baumannii.
 | ||
| Fig. 5 Time-kill analysis of compounds against MRSA. Compounds 31, 32 and vancomycin (4× MIC) against MRSA USA300 over a 24-hour incubation period at 37 °C. | ||
To address this issue, the development of antibacterial agents with potent anti-biofilm activity is essential. Compounds 23 and 24 were investigated for their ability to eradicate preformed, mature biofilms of methicillin-resistant Staphylococcus aureus (MRSA), Fig. 6. The green bars show that compound 23 achieves a high percentage of biofilm disruption, with around 70–80% indicating it is effective in disrupting MRSA biofilms, which are known to protect bacteria from antibiotic effects. While compound 24, the blue bars show that compound 24 also disrupts biofilm, although to a slightly lesser extent than compound 23, with disruption percentages typically lower than compound 23. These results suggest that these compounds are promising alternatives or complements. These compounds may offer advantages over current MRSA treatments especially in treating infections associated with biofilms where traditional antibiotics are less effective.
 | ||
| Fig. 6 Disruption of mature MRSA USA300 biofilms by phenyltriazole compounds and ceftriaxone (at 4× MIC). Data are shown as the percentage of biofilm disruption relative to untreated controls. | ||
2.3 In silico studies
Therefore, the binding affinity of potent analogs based on the inhibitory effect on MRSA and other microbial strains to both targets was therefore examined using inhibition constant data; Ki is an effective parameter for cell wall destruction and inhibition of folate biosynthesis. The docking score of potent analogs 23 and 24 are estimated and the inhibitor constant, Ki, is calculated using the equation (Table 2). The equation used for Ki calculation is as follows:
| Ki = exp(ΔG/(R × T)), (T = 298 K) |
| Cp. | PBP2a | DHPS | Hemolysis LC [mg mL−1] | PSA [Å2] | HEK293 cytotoxicity IC50 [μM] | ||
|---|---|---|---|---|---|---|---|
| ΔG (kcal mol−1) | Ki (nM) | ΔG (kcal mol−1) | Ki (nM) | ||||
| a Two targets from methicillin-resistant Staphylococcus aureus; G acyl-penicillin binding protein 2a (PDB ID: 1MWT) and dihydropteroate synthase (PDB ID: 6CLV). MTT assay for cytotoxicity against human embryonic kidney cell line. LC lytic concentration 30%. | |||||||
| 23 | −8.16 | 33.25 | −8.54 | 26.91 | >10 | 149 | 52.4 |
| 24 | −7.99 | 34.67 | −8.12 | 30.55 | >10 | 148 | 18.9 |
Data revealed potent inhibitory data activity of both compounds on both target cell walls and DNA synthesis pathways; 34–26 nM consistent with antimicrobial activity against target strains which enforce the targets probability. The presence of such structural pharmacophores as triazole, sulfonamid, and guanidine base predispose it to be the same mechanism. The Polar Surface Area (PSA), an important descriptor of molecular polarity, is often associated with key pharmacokinetic properties such as bioavailability, solubility, and membrane permeability. Both compounds showed uniform PSA that assist in the enzyme interactions and antimicrobial activity reflecting molecular size and polarity. The results show that both compounds have hemolytic concentrations (LC) of more than 10 mg ml−1, indicating minimal impact on red blood cell integrity and low toxicity to erythrocytes. The cytotoxicity assays revealed IC50 values greater than 18 μM for all compounds, suggesting low cytotoxicity and indicating that these compounds can potentially exhibit antimicrobial activity without causing significant damage to host cells, Table 2.
First, the interaction analysis between the phenyltriazole 23 analog and dihydropteroate synthase (DHPS) reveals several similarities and differences compared to the reference bound Pterin–sulfa conjugate. Key insights into these interactions are derived from the analysis of hydrogen bonding, ionic interactions, and π-interactions with the receptor residues. First of all, both the phenyltriazole analog 23 and Pterin–sulfa conjugate engages in strong hydrogen bonding with conserved residues in DHPS, specifically ASN 103 and ASP 167. The analog shows hydrogen bond interactions with ASN 103 at 2.83 Å and E = −4.3 kcal mol−1 and ASP 167 at 2.73 Å, E = −5.9 kcal mol−1, which are similar to the interactions seen in the reference substrate with ASN 103 at 3.02 Å, E = −4.3 kcal mol−1 and ASP 167 at 2.87 Å, E = −6.7 kcal mol−1. This suggests that these residues play a central role in stabilizing the ligand in both cases, with particularly strong interactions between the analog and ASP 167, reflected in a highly favorable binding energy. In addition, it also forms hydrogen bonds with SER 16 at 2.95 Å, E = −3.4 kcal mol−1 and LYS 203 at 3.17 Å, E = −8.9 kcal mol−1, whereas the bound inhibitor forms hydrogen bonds with LYS 203 at 3.71 Å, E = −1.7 kcal mol−1 which suggests a more favorable fit for the analog at this binding site. Both the 23 and reference bound compound formed an ionic interaction with ASP 84 and ASP 167; (2.83 Å, E = −3.3 kcal mol−1) and (2.84 Å, E = −4.9 kcal mol−1) and (3.78 Å, E = −1.0 kcal mol−1) (3.88 Å, E = −0.7 kcal mol−1) respectively. Moreover, both the analog 23 and Pterin–sulfa conjugate show π-interactions with LYS 203 and PHE 172 through the aromatic fragments, which in turn is less energetically significant than hydrogen bonding or ionic interactions but still contributes to ligand stability. In conclusion, the phenyltriazole analog forms similar interactions with DHPS as the reference substrate, with notable improvements in the binding affinity for LYS 203 and ASP 167 (Fig. 7).

| Code | Solubility (mg L−1) | Drug likeness model score | Lipinski's rule violation | Bioavailability score |
|---|---|---|---|---|
| 23 | 1.3 | 0.33 | 0 | 0.55 |
| 24 | 4.83 | 0.98 | 0 | 0.55 |
The molecular weights of the selected compounds are all below 500, their logP values (octanol/water partition coefficient) are under 5, and they possess fewer than 5 hydrogen bond donors and acceptors. Based on these characteristics, compounds 23 and 24 are expected to exhibit good absorption. Additionally, the number of rotatable bonds as an indicator of molecular flexibility and a factor influencing oral bioavailability was analyzed. This value is determined by counting non-ring, non-terminal, non-hydrogen-bonded single bonds, and is categorized into three groups: ≤7, 8–10, and >10. Compound 24 has 8 rotatable bonds, indicating generally favorable bioavailability, while compound 23, with 14 rotatable bonds, suggests moderate bioavailability. All three candidates have fewer than 5 hydrogen bond donors, and compounds 23 and 24 each have 7 hydrogen bond acceptors. These findings support the potential for good absorption in all three compounds.57
All three compounds have identical values for tPSA (159.52), hydrogen bond donors,4 and hydrogen bond acceptors,7 indicating similar polarity and hydrogen bond formation potential. However, they differ in the number of rotatable bonds (NORTB), molecular weight (MW), and logP values. Compound 23 has the highest NORTB,14 MW (462.61), and logP (3.08), suggesting greater flexibility, higher molecular mass, and higher lipophilicity compared to the others. While compound 24 has lower NORTB,8 lower MW, and lower logP values (1.23), suggesting that they are less flexible and less lipophilic than compound 23. This suggests that despite having similar hydrogen bonding capabilities, the structural differences between the compounds (in terms of flexibility and lipophilicity) could influence their pharmacokinetic behavior. On the other hand, Table 4 shows the predicted drug-like properties of three compounds (23 and 24) using Molsoft and SwissADME. The data shows different solubility levels, with compound 23 displaying the lowest solubility (1.3 mg L−1), while compound 24 has higher solubility values of 4.83 mg L−1. Notably, compound 24 exhibits the highest drug-like value in the model (0.98), followed by compound 23 (0.33), indicating that compound 24 has the most favorable drug-like properties. The two compounds meet Lipinski's rule, without violations, suggesting good potential for oral bioavailability. The bioavailability value is consistent for all compounds and is, with a value of 0.55 for all. Overall, compound 24 stands out as the most promising candidate due to its high solubility and superior drug likeness, making it a favorable candidate for further development.
Table 5 summarizes the predicted pharmacokinetic properties of compounds 23, 24, and the reference drug vancomycin using pre-ADME analysis. Notably, compound 24 and vancomycin have much lower BBB penetration (0.06 and 0.03, respectively), which indicates limited access to central nervous system. Regarding Caco-2 permeability, vancomycin exhibits a much higher rate (20.64 × 106 cm s−1) compared to all three test compounds, indicating better intestinal absorption. However, compound 23 shows moderate permeability (1.07 × 106 cm s−1), surpassing compound 24. All compounds show good human intestinal absorption (HIA), with compound 23 being the highest at 70.1% HIA achieved, and also plasma protein binding (PPB) is highest (84.25%), which may influence its bioavailability and distribution, while compound 24 shows the lowest PPB at 52.1%.
| Code | Pharmacokinetics | |||||
|---|---|---|---|---|---|---|
| BBBa | CACO-2b (×106 cm s−1) | HIAc (%) | MDCKd (nm s−1) | PPBe (%) | CYP2D6f | |
| a BBB: blood–brain barrier penetration.b CACO-2: permeability through cells derived from human colon adenocarcinoma.c HIA: percentage human intestinal absorption.d MDCK: permeability through Madin–Darby canine kidney cells.e PPB: plasma protein binding.f CYP2D6: cytochrome P450 2D6. | ||||||
| 23 | 0.21 | 1.07 | 70.10 | 0.08 | 84.25 | Substrate |
| 24 | 0.06 | 0.38 | 60.01 | 12.33 | 52.10 | Substrate |
| Vancomycin | 0.0.26 | 20.59 | 1.19 | 0.039 | 42.18 | No |
All test compounds function as CYP2D6 substrates, unlike vancomycin, which does not interact with this enzyme.
2.4 Prediction of toxicity
Predicting the toxicity of a compound is a critical step in the development of new drug candidates, making in silico toxicity studies a faster and cheaper procedure than in vivo animal toxicity testing or in vitro testing in cell lines. It also helps significantly reduce the number of animals used in experimental assays. Several online programs access toxicities that use in silico models to predict mean lethal dose, carcinogenicity, mutagenicity, and more.The Pro-Tox II web server58 predicts the mean lethal dose (LD50) in rodents. According to this program, all compounds can be classified into six GHS (Globally Harmonized System of Classification and Labeling of Chemicals) Categories59 according to their toxicity and LD50 value.
Toxicity classes are defined according to the globally harmonized system of classification of labeling of chemicals (GHS). LD50 was noted in mg kg−1 units.
■ Class I: fatal if swallowed (LD50 ≤ 5)
■ Class II: fatal if swallowed (5 < LD50 ≤ 50)
■ Class III: toxic if swallowed (50 < LD50 ≤ 300)
■ Class IV: harmful if swallowed (300 < LD50 ≤ 2000)
■ Class V: may be harmful if swallowed (2000 < LD50 ≤ 5000)
■ Class VI: non-toxic (LD50 > 5000)
Toxicity assessment is an important part of the development of therapeutic agents as it directly impacts their safety profile and potential clinical applications (Table 6) reveals critical insights into the predicted toxicity of the listed compounds. All three compounds (23 and 24) have the same predicted LD50 value of 500 mg kg−1, and are therefore classified in toxicity class IV, which indicates low toxicity. However, there are clear differences in their specific toxicity profiles. Compound 24 was predicted to have active carcinogenicity with values of 0.59, while compound 23 shows a slightly lower activity (0.55). In contrast, two compounds show inactivity regarding hepatotoxicity, mutagenicity, and cytotoxicity, although the values indicate slight differences in their relative safety. While the compounds exhibit low toxicity overall, the Table 6 highlights the importance of evaluating toxicity when developing new compounds. Compound 23 appears to be the most promising candidate in terms of safety, while compound 24 may require further investigation to address their active toxicity profiles. Understanding these factors is crucial for advancing these compounds toward therapeutic applications.
| No | Predicted LD50 (mg kg−1) | Predicted toxicity class | Hepatotoxicity | Carcinogenicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|---|
| 23 | 500 | IV | Inactive (0.63) | Active (0.55) | Inactive (0.54) | Inactive (0.55) |
| 24 | 500 | IV | Inactive (0.68) | Active (0.59) | Inactive (0.59) | Inactive (0.59) |
3. Conclusion
Bacterial infections remain a significant challenge in the medical field due to the rapid development of bacterial resistance to existing antibacterial agents. Therefore, there is an urgent need to continuously discover new scaffolds of antimicrobial agents to combat these infections. While sulfonamides have limited activity against methicillin-resistant Staphylococcus aureus (MRSA), our laboratory has already developed antibacterial agents from phenyltriazoles that are specifically effective against MRSA. The synthesis and biological evaluation of phenyltriazole–sulfonamide hybrids have led to the identification of several promising compounds with significant antimicrobial activity, particularly against MRSA. The structure–activity relationship (SAR) analysis revealed that modifications in alkyl chain length and branching, along with functional group variations, significantly influenced the antimicrobial potency of these compounds. In this study, we found that analog 23 showed even better results with an MIC of 1 μg ml−1 against Staphylococcus aureus USA300, indicating that the addition of a nonyl group gave the best results. However, compound 24 showed good activity against Acinetobacter baumannii AB5075, with a minimum inhibitory concentration (MIC) of 8 μg ml−1. Compound 23, featuring an n-nonyl group, demonstrated the best antimicrobial performance, exhibiting rapid bactericidal activity and effective disruption of MRSA biofilms that outperformed reference vancomycin drug. In silico studies further supported the potential of these compounds to target key enzymes involved in bacterial survival, including PBP2a and DHPS, with favorable binding affinities. These findings suggest that phenyltriazole–sulfonamide hybrids could serve as viable alternatives or complements to existing antibiotics, particularly in addressing drug-resistant infections. Further research and optimization of these compounds could lead to the development of novel therapies to combat resistant bacterial pathogens and biofilm-related infections.4. Experimental section
4.1 Chemistry
1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded in CDCl3 or DMSO-d6 using a Varian Mercury VX-400 spectrometer. Chemical shifts (δ, ppm) were referenced to solvent peaks. Flash chromatography was carried out on 230–400 mesh silica, and reaction progress was monitored using Merck silica gel IB2-F plates (0.25 mm). Mass spectra were obtained at 70 eV, and high-resolution mass spectra were recorded on a Finnigan MAT XL95. MIC values for the compounds and linezolid (control) were determined at the Department of Microbiology and Immunology, Faculty of Pharmacy, Cairo University. Melting points were measured with a Stuart SMP30 apparatus using capillary tubes and are uncorrected. Reported yields correspond to isolated products.4-Azidobenzenesulfonamide 2 was prepared as reported procedures.23
:1) and then washed. Yields, physical properties, and spectral data of the purified compounds are provided below.
4.1.2.1 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-methylbenzenesulfonamide 4. White solid (286 mg, 84%). mp = 115–116 °C; 1H NMR-DMSO-d6, δ: 8.01 (d, J = 8.1 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H), 7.86 (brs, 1H), 2.71 (s, 3H), 2.56 (s, 3H), 2.37 (s, 3H); 13C NMR (DMSO-d6) δ: 199.70, 145.25, 139.67, 139.19, 136.01, 131.53, 129.51, 126.33, 29.17, 14.89, 11.54; anal. calc. for: C12H14N4O3S (294): C, 48.97; H, 4.79; N, 19.04%; found: C, 49.08; H, 4.86; N, 18.94%.
4.1.2.2 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-ethylbenzenesulfonamide 5. Buff solid (203 mg, 82%). mp = 117–119 °C; 1H NMR-DMSO-d6, δ: 8.05 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.80 (brs, 1H), 2.9 (q, J = 8.0 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.05 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 194.40, 154.88, 145.19, 141.76, 139.10, 135.22, 131.48, 128.44, 126.31, 120.46, 38.00, 14.90, 11.50, 9.99; anal. calc. for: C13H16N4O3S (308): C, 50.64; H, 5.23; N, 18.17%; found: C, 50.71; H, 5.31; N, 18.05%.
4.1.2.3 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-butylbenzenesulfonamide 6. White solid (322 mg, 81%). mp = 116–118 °C; 1H NMR-DMSO-d6, δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.76 (brs, 1H), 2.84 (t, J = 8.1 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.42–1.21 (m, 4H), 0.83 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 199.34, 145.24, 144.43, 141.82, 139.07, 131.42, 128.41, 126.30, 42.73, 31.57, 19.64, 14.88, 13.92, 11.49; anal. calc. for: C15H20N4O3S (336): C, 53.56; H, 5.99; N, 16.65%; found: C, 53.59; H, 6.07; N, 16.54%.
4.1.2.4 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-pentylbenzenesulfonamide 7. White solid (261 mg, 85%). mp = 119–120 °C; 1H NMR-DMSO-d6, δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.74 (brs, 1H), 2.84 (t, J = 8.1 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.43–1.19 (m, 6H), 0.83 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 200.85, 145.17, 144.57, 141.88, 139.06, 131.48, 128.42, 126.29, 43.01, 29.13, 28.67, 22.08, 14.90, 14.26, 11.48; anal. calc. for: C16H22N4O3S (350): C, 54.84; H, 6.33; N, 15.99%; found: C, 54.94; H, 6.39; N, 15.89%.
4.1.2.5 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-hexylbenzenesulfonamide 8. White solid (392 mg, 88%). mp = 120–121 °C; 1H NMR-DMSO-d6, δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.76 (brs, 1H), 2.84 (t, J = 8.0 Hz, 2H), 2.51 (s, 3H), 2.37 (s, 3H), 1.49–1.14 (m, 8H), 0.86 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 192.48, 145.20, 141.88, 139.06, 131.45, 128.42, 128.23, 126.28, 120.43, 43.05, 31.19, 29.40, 29.30, 26.10, 22.39, 14.89, 14.30, 11.48; anal. calc. for: C17H24N4O3S (364): C, 56.02; H, 6.64; N, 15.37%; found: C, 56.22; H, 6.76; N, 15.17%.
4.1.2.6 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-heptylbenzenesulfonamide 9. White solid (324 mg, 83%). mp = 119–120 °C; 1H NMR-DMSO-d6 δ: 8.04 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.27 (brs, 1H), 3.15 (t, J = 8.1 Hz, 2H), 2.55 (s, 3H), 2.41 (s, 3H), 1.51–1.21 (m, 10H), 0.86 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 196.81, 145.33, 144.62, 141.96, 138.95, 131.99, 128.45, 128.23, 126.31, 43.04, 31.56, 29.44, 28.65, 26.39, 22.44, 15.07, 14.35, 11.43; anal. calc. for: C18H26N4O3S (378): C, 57.12; H, 6.92; N, 14.80%; found: C, 57.22; H, 6.99; N, 14.70%.
4.1.2.7 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-octylbenzenesulfonamide 10. Pale white solid (357 mg, 84%). mp = 120–122 °C; 1H NMR-DMSO-d6 δ: 8.05 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.27 (brs, 1H), 2.84 (t, J = 8.1 Hz, 2H), 2.56 (s, 3H), 2.49 (s, 3H), 1.40–1.35 (m, 2H), 1.26–1.19 (m, 10H), 0.85 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 193.80, 147.36, 143.17, 142.19, 138.69, 133.36, 128.5, 126.4, 43.04, 31.62, 29.47, 28.95, 26.45, 22.51, 15.52, 14.38, 11.31; anal. calc. for: C19H28N4O3S (392): C, 58.14; H, 7.19; N, 14.27%; found: C, 58.24; H, 7.29; N, 14.17%.
4.1.2.8 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-nonylbenzenesulfonamide 11. Buff solid (271 mg, 72%). mp = 122–123 °C; 1H NMR-DMSO-d6 δ: 8.06 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.1 Hz, 2H), 7.76 (brs, 1H), 2.84 (t, J = 8.1 Hz, 2H), 2.56 (s, 3H), 2.54 (s, 3H), 1.42–1.36 (m, 2H), 1.23–1.19 (m, 12H), 0.85 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 186.20, 148.33, 142.40, 142.31, 138.55, 134.09, 128.55, 126.47, 43.04, 31.70, 29.45, 29.30, 28.99, 26.43, 22.52, 15.95, 14.39, 11.24; anal. calc. for: C20H30N4O3S (406): C, 59.09; H, 7.44; N, 13.78%; found: C, 59.29; H, 7.54; N, 13.68%.
4.1.2.9 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-isobutylbenzenesulfonamide 12. White solid (204 mg, 85%). mp = 121–122 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.0 Hz, 2H), 7.87 (d, J = 8.0 Hz, 2H), 7.84 (brs, 1H), 2.65 (d, J = 8.0 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.71 (m, 1H), 0.85 (d, J = 8.0 Hz, 6H); 13C NMR (DMSO-d6) δ: 194.37, 144.48, 141.94, 139.04, 131.44, 128.38, 126.29, 50.5, 28.59, 20.32, 14.89, 11.51; anal. calc. for: C15H20N4O3S (336): C, 53.56; H, 5.99; N, 16.65%; found: C, 53.60; H, 6.05; N, 16.60%.
4.1.2.10 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-(5-methylhexyl)benzenesulfonamide 13. Buff solid (284 mg, 83%). mp = 122–123 °C; 1H NMR-DMSO-d6, δ: 8.03 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.27 (brs, 1H), 2.95–2.67 (m, 1H), 2.54 (s, 3H), 2.38 (s, 3H), 1.67–1.09 (m, 8H), 0.86 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 193.03, 144.78, 142.04, 138.98, 131.63, 128.39, 126.30, 49.21, 37.77, 30.71, 30.54, 26.37, 25.72, 14.94, 11.46; anal. calc. for: C18H26N4O3S (378): C, 57.12; H, 6.92; N, 14.80%; found: C, 57.22; H, 6.99; N, 14.69%.
4.1.2.11 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-(but-3-en-1-yl)benzenesulfonamide 14. White solid (254 mg, 86%). mp = 111–112 °C; 1H NMR-DMSO-d6 δ: 8.04 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.85 (brs, 1H), 5.79–5.68 (m, 2H), 5.01 (dd, J = 12.0 Hz, 2H), 2.92 (t, J = 8.0 Hz, 2H), 2.52 (s, 3H), 2.38 (s, 3H), 2.19–2.14 (m, 2H); 13C NMR (DMSO-d6) δ: 195.49, 144.77, 141.76, 139.09, 135.68, 131.63, 128.46, 126.32, 117.27, 42.62, 33.92, 14.93, 11.49; anal. calc. for C15H18N4O3S (334): C, 53.88; H, 5.43; N, 16.76%; found: C, 53.94; H, 5.48; N, 16.70%.
4.1.2.12 4-(4-Acetyl-5-methyl-1H-1,2,3-triazol-1-yl)-N-cyclopentylbenzenesulfonamide 15. Buff solid (273 mg, 83%). mp = 129–130 °C; 1H NMR-DMSO-d6 δ: 8.05 (d, J = 8.1 Hz, 2H), 8.03 (brs, 1H), 7.86 (d, J = 8.1 Hz, 2H), 3.52–3.48 (m, 1H), 2.54 (s, 3H), 2.37 (s, 3H), 1.68–1.34 (m, 8H); 13C NMR (DMSO-d6) δ: 195.13, 145.22, 144.46, 142.77, 139.01, 131.42, 128.43, 126.26, 55.02, 32.95, 23.28, 14.89, 11.53; anal. calc. for: C16H20N4O3S (348): C, 55.16; H, 5.79; N, 16.08%; found: C, 55.21; H, 5.85; N, 16.01%.
4.1.3.1 2-(1-(5-Methyl-1-(4-(N-methylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 16. White solid (322 mg, 88%). mp = 162–163 °C; 1H NMR-DMSO-d6 δ: 8.01 (d, J = 8.1 Hz, 2H), 7.92 (d, J = 8.1 Hz, 2H), 7.86 (brs. 1H), 5.67 (brs, 4H), 2.71 (s, 3H), 2.56 (s, 3H), 2.37 (s, 3H); 13C NMR (DMSO-d6) δ: 145.25, 139.67, 139.19, 136.01, 131.43, 129.51, 126.33, 29.17, 14.89, 11.54; anal. calc. for: C13H18N8O2S (350.40): C, 44.56; H, 5.18; N, 31.98%; found: C, 44.66; H, 5.28; N, 31.88%.
4.1.3.2 2-(1-(1-(4-(N-Ethylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carboximidamide 17. Buff solid (276 mg, 85%). mp = 160–161 °C; 1H NMR-DMSO-d6 δ: 8.05 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.77 (brs, 1H), 5.83 (brs, 4H), 2.90 (q, J = 8.0 Hz, 2H), 2.56 (s, 3H), 2.37 (s, 3H), 1.07 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 162.85, 160.77, 159.89, 154.88, 145.19, 144.53, 141.76, 139.10, 135.22, 131.66, 128.44, 126.31, 120.46, 38.11, 17.74, 15.27, 11.50, 9.99; anal. calc. for: C14H20N8O2S (364.43): C, 46.14; H, 5.53; N, 30.75%; found: C, 46.34; H, 5.75; N, 30.55%.
4.1.3.3 2-(1-(1-(4-(N-Butylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carboximidamide 18. White solid (393 mg, 89%). mp = 169–171 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 5.69 (brs, 5H), 2.84 (t, J = 8.1 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.42–1.21 (m, 4H), 0.83 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 159.98, 145.24, 144.43, 141.82, 139.07, 131.42, 128.41, 126.30, 42.73, 31.57, 19.64, 14.88, 13.92, 11.49; anal. calc. for: C16H24N8O2S (392.48): C, 48.96; H, 6.16; N, 28.55%; found: C, 49.06; H, 6.26; N, 28.35%.
4.1.3.4 2-(1-(5-Methyl-1-(4-(N-pentylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 19. White solid (326 mg, 90%). mp = 162–163 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 5.69 (brs, 5H), 2.84 (t, J = 8.1 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.43–1.19 (m, 6H), 0.83 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 159.87, 145.17, 144.57, 141.88, 139.06, 131.48, 128.42, 126.29, 43.01, 29.13, 28.67, 28.62, 22.08, 14.90, 14.26, 11.48; anal. calc. for: C17H26N8O2S (406.51): C, 50.23; H, 6.45; N, 27.57%; found: C, 50.43; H, 6.55; N, 27.37%.
4.1.3.5 2-(1-(1-(4-(N-Hexylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carboximidamide 20. White solid (441 mg, 90%). mp = 163–165 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 7.76 (brs, 1H), 5.81 (brs, 4H), 2.84 (t, J = 8.0 Hz, 2H), 2.51 (s, 3H), 2.37 (s, 3H), 1.49–1.14 (m, 8H), 0.86 (t, J = 8.0 Hz, 3H); anal. calc. for: C18H28N8O2S (420.54): C, 51.41; H, 6.71; N, 26.65%; found: C, 51.61; H, 6.81; N, 26.55%.
4.1.3.6 2-(1-(1-(4-(N-Heptylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene hydrazine-1-carboximidamide 21. White solid (449 mg, 87%). mp = 160–162 °C; 1H NMR-DMSO-d6 δ: 8.04 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.74 (brs, 1H), 6.14 (brs, 4H), 2.83 (t, J = 8.1 Hz, 2H), 2.57 (s, 3H), 2.41 (s, 3H), 1.42–1.19 (m, 10H), 0.85 (t, J = 8.0 Hz, 3H); anal. calc. for: C19H30N8O2S (434.56): C, 52.51; H, 6.96; N, 25.79%; found: C, 52.55; H, 7.05; N, 25.59%.
4.1.3.7 2-(1-(5-Methyl-1-(4-(N-octylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carboximidamide 22. Pale-white solid (465 mg, 88%). mp = 174–176 °C; 1H NMR-DMSO-d6 δ: 8.05 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 7.27 (brs, 5H), 2.84 (t, J = 8.1 Hz, 2H), 2.56 (s, 3H), 2.49 (s, 3H), 1.40–1.35 (m, 2H), 1.26–1.19 (m, 10H), 0.85 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 157.38, 147.36, 143.17, 142.19, 138.69, 133.36, 128.5, 126.4, 43.04, 31.62, 29.47, 29.01, 28.95, 26.45, 22.51, 15.52, 14.38, 11.31; anal. calc. for: C20H32N8O2S (448.59): C, 53.55; H, 7.19; N, 24.98%; found: C, 53.75; H, 7.29; N, 24.88%.
4.1.3.8 2-(1-(5-Methyl-1-(4-(N-nonylsulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)ethylidene)hydrazine-1-carboximidamide 23. Buff solid (305 mg, 74%). mp = 165–166 °C; 1H NMR-DMSO-d6 δ: 11.46 (brs, 1H), 8.06 (d, J = 8.1 Hz, 2H), 7.89 (d, J = 8.1 Hz, 2H), 7.76 (brs, 4H), 2.84 (t, J = 8.1 Hz, 2H), 2.56 (s, 3H), 2.54 (s, 3H), 1.42–1.36 (m, 2H), 1.23–1.19 (m, 12H), 0.85 (t, J = 8.0 Hz, 3H); 13C NMR (DMSO-d6) δ: 156.61, 148.33, 142.40, 142.31, 138.55, 134.09, 128.55, 126.47, 43.04, 31.70, 29.45, 29.30, 29.05, 28.99, 26.43, 22.52, 15.95, 14.39, 11.24; anal. calc. for: C21H34N8O2S (462.62): C, 54.52; H, 7.41; N, 24.22%; found: C, 54.71; H, 7.51; N, 24.02%.
4.1.3.9 2-(1-(1-(4-(N-Isobutylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 24. White solid (270 mg, 90%). mp = 161–163 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.0 Hz, 2H), 7.87 (d, J = 8.0 Hz, 2H), 5.68 (brs, 5H), 2.65 (d, J = 8.0 Hz, 2H), 2.54 (s, 3H), 2.37 (s, 3H), 1.71 (m, 1H), 0.85 (d, J = 8.0 Hz, 6H); 13C NMR (DMSO-d6) δ: 159.91, 145.2, 144.48, 141.94, 139.04, 131.44, 128.38, 126.29, 50.5, 28.59, 20.32, 14.89, 11.51; anal. calc. for: C16H24N8O2S (392.48): C, 48.96; H, 6.16; N, 28.55%; found: C, 49.06; H, 6.28; N, 28.35%.
4.1.3.10 2-(1-(5-Methyl-1-(4-(N-(5-methylhexyl)sulfamoyl)phenyl)-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 25. Buff solid (329 mg, 85%). mp = 174–176 °C; 1H NMR-DMSO-d6 δ: 8.03 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 5.85 (brs, 5H), 2.67–2.66 (m, 1H), 2.51 (s, 3H), 2.38 (s, 3H), 1.67–1.09 (m, 8H), 0.86 (t, J = 8.0 Hz, 3H); anal. calc. for: C19H30N8O2S (434.56): C, 52.51; H, 6.96; N, 25.79%; found: C, 52.71; H, 7.03; N, 25.59%.
4.1.3.11 2-(1-(1-(4-(N-(But-3-en-1-yl)sulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 26. White solid (310 mg, 89%). mp = 154–156 °C; 1H NMR-DMSO-d6 δ: 8.04 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.1 Hz, 2H), 5.79–5.68 (m, 6H), 5.01 (dd, J = 12.0 Hz, 2H), 2.92 (t, J = 8.0 Hz, 2H), 2.52 (s, 3H), 2.38 (s, 3H), 2.19–2.14 (m, 2H); 13C NMR (DMSO-d6) δ: 159.68, 145.03, 144.77, 141.76, 139.09, 135.68, 131.63, 128.46, 126.32, 124.67, 117.27, 42.62, 33.92, 14.93, 11.49; anal. calc. for C16H22N8O2S (390): C, 49.22; H, 5.68; N, 28.70%; found: C, 49.42; H, 5.78; N, 28.59%.
4.1.3.12 2-(1-(1-(4-(N-Cyclopentylsulfamoyl)phenyl)-5-methyl-1H-1,2,3-triazol-4-yl)ethylidene) hydrazine-1-carboximidamide 27. Buff solid (321 mg, 89%). mp = 189–190 °C; 1H NMR-DMSO-d6 δ: 8.05 (d, J = 8.1 Hz, 2H), 7.86 (d, J = 8.1 Hz, 2H), 5.68 (brs, 3H), 5.56 (brs, 2H), 3.52–3.48 (m, 1H), 2.54 (s, 3H), 2.37 (s, 3H), 1.68–1.34 (m, 8H); 13C NMR (DMSO-d6) δ: 159.96, 145.22, 144.46, 142.77, 139.01, 131.42, 128.43, 126.26, 55.02, 32.95, 23.28, 14.89, 11.53; anal. calc. for: C17H24N8O2S (404): C, 50.48; H, 5.98; N, 27.70%; found: C, 50.58; H, 6.07; N, 27.50%.
4.2 Biology screening
:100 and inoculated into sterile, polypropylene 96-well microtiter plates. Plates were sealed and incubated statically at 37 °C for 24 hours to allow biofilm formation. Each assay included negative control (media only) and a positive control (media with 1% inoculum). All experiments were performed in triplicate unless otherwise stated.62Biofilm inhibition was quantified using the crystal violet staining method. After incubation, non-adherent cells and culture media were removed, and wells were washed three times with distilled water to eliminate planktonic cells. Adherent biofilms were stained with 125 μL of 0.1% crystal violet for 30 minutes at room temperature. Excess stain was rinsed off with distilled water, and the bound dye was solubilized in 30% acetic acid. Absorbance was measured at 550 nm.52,63,64 The percentage of biofilm inhibition was calculated using the formula:65
| Biofilm inhibition (%) = [1 − (OD550 of treated wells/OD550 of untreated control wells)] × 100 |
Ligands were constructed using AutoDock's ligand builder, with geometry optimization performed using the CHARMm force field.72 Docking calculations were prepared using Python scripts from the AutoDock suite. Each ligand underwent 50 docking runs, and the resulting poses were clustered using a 1.8 Å RMSD cutoff. The conformational search utilized the Lamarckian Genetic Algorithm,51,73–76 with a starting population of 150 individuals and up to 25000000 energy evaluations. The most populated low-energy clusters were selected for further analysis.
Molecular dynamics (MD) simulations were subsequently carried out to assess the stability of compound 23 in complex with the target proteins, compared to reference drugs. Desmond version 3.8, utilizing the OPLS2005 force field77 and developed by D. E. Shaw Research, was used for the simulations. Production MD was performed under isothermal-isobaric (NPT) conditions at 300 K and 1 bar using Langevin dynamics, over three independent 100 ns simulation runs.
Data availability
The data supporting this article have been included in the main manuscript and ESI.†Author contributions
I confirm that the contribution of each author is outlined in the manuscript below, with some authors contributing to multiple roles. I am responsible for ensuring the accuracy of these descriptions, which have been agreed upon by all authors involved. A. H. and M. M. E.: synthesis and refinement of methodology, spectroscopic analysis, and drafting the original manuscript. A. N. H., H. G. E., and A. S. M.: conceptualization, supervision, manuscript writing and review, and project administration. All authors have read and approved the final version of the manuscript. H. T. N. E. and Y. N.: antimicrobial screening, methodology development, and drafting the original manuscript. M. A.: molecular modeling, biochemical data evaluation and discussion, manuscript writing, review and editing, and funding acquisition. A. M., K. S., and D. G. T. P.: biological evaluation of target compounds, molecular modeling, data analysis, and partial funding acquisition. M. M. E. and A. S. M.: proposal writing, supervision, and synthesis.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was funded by the Deanship of Graduate Studies and Scientific Research at Jouf University under grant no. (DGSSR-2024-01-01038) and supported by grant number C21-113 from the Science, Technology & Innovation Funding Authority (STDF).References
- M. Dulon, F. Haamann, C. Peters, A. Schablon and A. Nienhaus, MRSA prevalence in European healthcare settings: a review, BMC Infect. Dis., 2011, 11, 1–13 CrossRef PubMed.
- J. M. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. Piddock, Molecular mechanisms of antibiotic resistance, Nat. Rev. Microbiol., 2015, 13(1), 42–51 CrossRef CAS PubMed.
- M. Terreni, M. Taccani and M. Pregnolato, New antibiotics for multidrug-resistant bacterial strains: latest research developments and future perspectives, Molecules, 2021, 26(9), 2671 CrossRef CAS PubMed.
- S. Boyle-Vavra, S. Yin, M. Challapalli and R. S. Daum, Transcriptional induction of the penicillin-binding protein 2 gene in Staphylococcus aureus by cell wall-active antibiotics oxacillin and vancomycin, Antimicrob. Agents Chemother., 2003, 47(3), 1028–1036 CrossRef CAS PubMed.
- M. M. Elsebaei, H. G. Ezzat, A. M. Helal, M. H. El-Shershaby, M. S. Abdulrahman and M. Alsedawy, et al., Rational design and synthesis of novel phenyltriazole derivatives targeting MRSA cell wall biosynthesis, RSC Adv., 2024, 14(54), 39977–39994 RSC.
- W. S. Champney and R. Burdine, Macrolide antibiotics inhibit 50S ribosomal subunit assembly in Bacillus subtilis and Staphylococcus aureus, Antimicrob. Agents Chemother., 1995, 39(9), 2141–2144 CrossRef CAS PubMed.
- M. Almaghrabi, A. Musa, A. K. B. Aljohani, H. E. A. Ahmed, M. Alsulaimany and S. F. Miski, et al., Introducing of novel class of pyrano[2,3-c]pyrazole-5-carbonitrile analogs with potent antimicrobial activity, DNA gyrase inhibition, and prominent pharmacokinetic and CNS toxicity profiles supported by molecular dynamic simulation, J. Biomol. Struct. Dyn., 2024, 42(18), 9529–9546 CrossRef CAS PubMed.
- A. M. Fouda, A. H. Hassan, E. M. Eliwa, H. E. A. Ahmed, A.-A. M. Al-Dies and A. M. Omar, et al., Targeted potent antimicrobial benzochromene-based analogues: Synthesis, computational studies, and inhibitory effect against 14α-Demethylase and DNA Gyrase, Bioorg. Chem., 2020, 105, 104387 CrossRef CAS PubMed.
- M. M. Elsebaei, H. Mohammad, M. Abouf, N. S. Abutaleb, Y. A. Hegazy and A. Ghiaty, et al., Alkynyl-containing phenylthiazoles: Systemically active antibacterial agents effective against methicillin-resistant Staphylococcus aureus (MRSA), Eur. J. Med. Chem., 2018, 148, 195–209 CrossRef CAS PubMed.
- S. M. Swaney, H. Aoki, M. C. Ganoza and D. L. Shinabarger, The oxazolidinone linezolid inhibits initiation of protein synthesis in bacteria, Antimicrob. Agents Chemother., 1998, 42(12), 3251–3255 CrossRef CAS PubMed.
- S. A. Grim, R. P. Rapp, C. A. Martin and M. E. Evans, Trimethoprim-sulfamethoxazole as a viable treatment option for infections caused by methicillin-resistant Staphylococcus aureus, Pharmacotherapy, 2005, 25(2), 253–264 CrossRef CAS PubMed.
- C. Rayner and W. J. Munckhof, Antibiotics currently used in the treatment of infections caused by Staphylococcus aureus, Intern. Med. J., 2005, 35(Suppl 2), S3–S16 CAS.
- H. E. A. Ahmed, S. K. Ihmaid, A. M. Omar, A. M. Shehata, H. S. Rateb and M. F. Zayed, et al., Design, synthesis, molecular docking of new lipophilic acetamide derivatives affording potential anticancer and antimicrobial agents, Bioorg. Chem., 2018, 76, 332–342 CrossRef CAS PubMed.
- A. A. Abuelkhir, Y. I. Nagy, T. Gamal, A. M. Abdelhalim, A. S. Attia and A. S. Mayhoub, et al., Small Molecule Alkynyl-Phenylaminoguanidines: A New Weapon Against Multi-Drug Resistant Bacteria, ChemistrySelect, 2025, 10(4), e202404320 CrossRef CAS.
- Organization WH, 2019 Antibacterial Agents in Clinical Development: an Analysis of the Antibacterial Clinical Development Pipeline, 2019 Search PubMed.
- R. A. Fisher, B. Gollan and S. Helaine, Persistent bacterial infections and persister cells, Nat. Rev. Microbiol., 2017, 15(8), 453–464 CrossRef CAS PubMed.
- E. Pontali, M. C. Raviglione and G. B. Migliori, Regimens to treat multidrug-resistant tuberculosis: past, present and future perspectives, Eur. Respir. Rev., 2019, 28(152), 190035 CrossRef PubMed.
- K. Iwamoto, M. Moriwaki, R. Miyake and M. Hide, Staphylococcus aureus in atopic dermatitis: Strain-specific cell wall proteins and skin immunity, Allergol. Int., 2019, 68(3), 309–315 CrossRef CAS PubMed.
- R. M. Hassan, M. G. Elanany, M. M. Mostafa, R. H. A. Yousef and S. T. Salem, Whole genome characterization of methicillin resistant Staphylococcus aureus in an Egyptian Tertiary Care Hospital, J. Microbiol. Immunol. Infect., 2023, 802–814 CrossRef CAS PubMed.
- R. Pavillard, K. Harvey, D. Douglas, A. Hewstone, J. Andrew and B. Collopy, et al., Epidemie of hospital-acquired infection due to methicillin-resistant Staphylococcus aureus in major Victorian hospitals, Med. J. Aust., 1982, 1(11), 451–454 CrossRef CAS PubMed.
- J. M. Kwiecinski and A. R. Horswill, Staphylococcus aureus bloodstream infections: pathogenesis and regulatory mechanisms, Curr. Opin. Microbiol., 2020, 53, 51–60 CrossRef CAS PubMed.
- T. Foster, 39 - Staphylococcus aureus, Mol. Med. Microbiol., 2002, 2, 839–888 CAS.
- H. Nikaido, Prevention of drug access to bacterial targets: permeability barriers and active efflux, Science, 1994, 264(5157), 382–388 CrossRef CAS PubMed.
- E. Yamashita, M. V. Zhalnina, S. D. Zakharov, O. Sharma and W. A. Cramer, Crystal structures of the OmpF porin: function in a colicin translocon, EMBO J., 2008, 27(15), 2171–2180 CrossRef CAS PubMed.
- R. O'Shea and H. E. Moser, Physicochemical properties of antibacterial compounds: implications for drug discovery, J. Med. Chem., 2008, 51(10), 2871–2878 CrossRef PubMed.
- C. Zhao, K. Rakesh, L. Ravidar, W.-Y. Fang and H.-L. Qin, Pharmaceutical and medicinal significance of sulfur (SVI)-Containing motifs for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 162, 679–734 CrossRef CAS PubMed.
- S. S. Atwa, M. Hagras, A. S. Mayhoub and M. Elsebaei, Synthesis Of Some New Azole Derivatives As Antibacterial Agents, Al-Azhar J. Pharm. Sci., 2024, 69(1), 108–129 CrossRef.
- I. Eid, M. M. Elsebaei, H. Mohammad, M. Hagras, C. E. Peters and Y. A. Hegazy, et al., Arylthiazole antibiotics targeting intracellular methicillin-resistant Staphylococcus aureus (MRSA) that interfere with bacterial cell wall synthesis, Eur. J. Med. Chem., 2017, 139, 665–673 CrossRef CAS PubMed.
- M. M. Elsebaei, H. Mohammad, M. Abouf, N. S. Abutaleb, Y. A. Hegazy and A. Ghiaty, et al., Alkynyl-containing phenylthiazoles: Systemically active antibacterial agents effective against methicillin-resistant Staphylococcus aureus (MRSA), Eur. J. Med. Chem., 2018, 148, 195–209 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, M. Cushman and M. N. Seleem, Anti-biofilm activity and synergism of novel thiazole compounds with glycopeptide antibiotics against multidrug-resistant staphylococci, J. Antibiotics, 2015, 68(4), 259–266 CrossRef CAS PubMed.
- H. Mohammad, P. V. Reddy, D. Monteleone, A. S. Mayhoub, M. Cushman and M. N. Seleem, Synthesis and antibacterial evaluation of a novel series of synthetic phenylthiazole compounds against methicillin-resistant Staphylococcus aureus (MRSA), Eur. J. Med. Chem., 2015, 94, 306–316 CrossRef CAS PubMed.
- H. Mohammad, W. Younis, L. Chen, C. E. Peters, J. Pogliano and K. Pogliano, et al., Phenylthiazole Antibacterial Agents Targeting Cell Wall Synthesis Exhibit Potent Activity in Vitro and in Vivo against Vancomycin-Resistant Enterococci, J. Med. Chem., 2017, 60(6), 2425–2438 CrossRef CAS PubMed.
- M. Hagras, N. S. Abutaleb, A. O. Ali, J. A. Abdel-Aleem, M. M. Elsebaei and M. N. Seleem, et al., Naphthylthiazoles: Targeting Multidrug-Resistant and Intracellular Staphylococcus aureus with Biofilm Disruption Activity, ACS Infect. Dis., 2018, 4(12), 1679–1691 CrossRef CAS PubMed.
- A. M. Sayed, N. S. Abutaleb, A. Kotb, H. G. Ezzat, M. N. Seleem and A. S. Mayhoub, et al., Arylpyrazole as selective anti-enterococci; synthesis and biological evaluation of novel derivatives for their antimicrobial efficacy, J. Heterocycl. Chem., 2023, 60(1), 134–144 CrossRef CAS.
- I. G. Shahin, K. O. Mohamed, A. T. Taher, M. M. Elsebaei, A. S. Mayhoub and A. E. Kassab, et al., New Phenylthiazoles: Design, Synthesis, and Biological Evaluation as Antibacterial, Antifungal, and Anti-COVID-19 Candidates, Chem. Biodiversity, 2023, e202301143 CrossRef CAS PubMed.
- M. Omara, M. Hagras, M. M. Elsebaie, N. S. Abutaleb, H. T. N. El-Din and M. O. Mekhail, et al., Exploring novel aryl/heteroaryl-isosteres of phenylthiazole against multidrug-resistant bacteria, RSC Adv., 2023, 13(29), 19695–19709 RSC.
- A. Mancy, N. S. Abutaleb, M. M. Elsebaei, A. Y. Saad, A. Kotb and A. O. Ali, et al., Balancing physicochemical properties of phenylthiazole compounds with antibacterial potency by modifying the lipophilic side chain, ACS Infect. Dis., 2019, 6(1), 80–90 CrossRef PubMed.
- M. M. Elsebaei, H. Mohammad, A. Samir, N. S. Abutaleb, A. B. Norvil and A. R. Michie, et al., Lipophilic efficient phenylthiazoles with potent undecaprenyl pyrophosphatase inhibitory activity, Eur. J. Med. Chem., 2019, 175, 49–62 CrossRef CAS PubMed.
- H. T. N. El-Din, M. M. Elsebaie, N. S. Abutaleb, A. M. Kotb, A. S. Attia and M. N. Seleem, et al., Expanding the structure–activity relationships of alkynyl diphenylurea scaffold as promising antibacterial agents, RSC Med. Chem., 2023, 14(2), 367–377 RSC.
- Y. Hosny, N. S. Abutaleb, M. Omara, M. Alhashimi, M. M. Elsebaei and H. S. Elzahabi, et al., Modifying the lipophilic part of phenylthiazole antibiotics to control their drug-likeness, Eur. J. Med. Chem., 2020, 185, 111830 CrossRef CAS PubMed.
- I. G. Shahin, K. O. Mohamed, A. T. Taher, M. M. Elsebaei, A. S. Mayhoub and A. E. Kassab, et al., New Phenylthiazoles: Design, Synthesis, and Biological Evaluation as Antibacterial, Antifungal, and Anti-COVID-19 Candidates, Chem. Biodiversity, 2023, 20(11), e202301143 CrossRef CAS PubMed.
- A. Mancy, Synthesis And Evaluation Of Antimicrobial Activity Of Arylazole Derivatives, Al-Azhar J. Pharm. Sci., 2019, 60(2), 43–50 CrossRef.
- A. Hammad, N. S. Abutaleb, M. M. Elsebaei, A. B. Norvil, M. Alswah and A. O. Ali, et al., From phenylthiazoles to phenylpyrazoles: broadening the antibacterial spectrum toward carbapenem-resistant bacteria, J. Med. Chem., 2019, 62(17), 7998–8010 CrossRef CAS PubMed.
- M. M. Elsebaei, N. S. Abutaleb, A. A. Mahgoub, D. Li, M. Hagras and H. Mohammad, et al., Phenylthiazoles with nitrogenous side chain: An approach to overcome molecular obesity, Eur. J. Med. Chem., 2019, 182, 111593 CrossRef PubMed.
- M. M. Elsebaie, H. T. N. El-Din, N. S. Abutaleb, A. A. Abuelkhir, H.-W. Liang and A. S. Attia, et al., Exploring the structure-activity relationships of diphenylurea as an antibacterial scaffold active against methicillin-and vancomycin-resistant Staphylococcus aureus, Eur. J. Med. Chem., 2022, 234, 114204 CrossRef CAS PubMed.
- M. Alswah, Synthesis And Biological Evaluation Of Novel Pyrazolo [3, 4-D] Pyrimidine Derivatives Of Expected Anticancer Activity, Al-Azhar J. Pharm. Sci., 2021, 64(2), 80–92 CrossRef.
- A. M. Helal, A. M. Sayed, M. Omara, M. M. Elsebaei and A. S. Mayhoub, Peptidoglycan pathways: there are still more, RSC Adv., 2019, 9(48), 28171–28185 RSC.
- K. El-Gamal, F. Sherbiny, A. El-Morsi, H. Abu-El-khair, I. Eissa and M. El-Sebaei, Design, synthesis and antimicrobial evaluation of some novel quinoline derivatives, Pharm. Pharmacol. Int. J., 2015, 2(5), 165–177 Search PubMed.
- S. A. Grim, R. P. Rapp, C. A. Martin and M. E. Evans, Trimethoprim-sulfamethoxazole as a viable treatment option for infections caused by methicillin-resistant Staphylococcus aureus, Pharmacotherapy, 2005, 25(2), 253–264 CrossRef CAS PubMed.
- T. A. Łeski and A. Tomasz, Role of penicillin-binding protein 2 (PBP2) in the antibiotic susceptibility and cell wall cross-linking of Staphylococcus aureus: evidence for the cooperative functioning of PBP2, PBP4, and PBP2A, J. Bacteriol., 2005, 187(5), 1815–1824 CrossRef PubMed.
- M. Almaghrabi, A. Musa, A. K. B. Aljohani, H. E. A. Ahmed, M. Alsulaimany and S. F. Miski, et al., Introducing of novel class of pyrano[2,3-c]pyrazole-5-carbonitrile analogs with potent antimicrobial activity, DNA gyrase inhibition, and prominent pharmacokinetic and CNS toxicity profiles supported by molecular dynamic simulation, J. Biomol. Struct. Dyn., 2023, 1–18 Search PubMed.
- A. M. Omar, S. Ihmaid, E. E. Habib, S. S. Althagfan, S. Ahmed and H. S. Abulkhair, et al., The rational design, synthesis, and antimicrobial investigation of 2-Amino-4-Methylthiazole analogues inhibitors of GlcN-6-P synthase, Bioorg. Chem., 2020, 99, 103781 CrossRef CAS PubMed.
- N. Lv, Q. Kong, H. Zhang and J. Li, Discovery of novel Staphylococcus aureus penicillin binding protein 2a inhibitors by multistep virtual screening and biological evaluation, Bioorg. Med. Chem. Lett, 2021, 41, 128001 CrossRef CAS PubMed.
- H. Lade and J.-S. Kim, Bacterial Targets of Antibiotics in Methicillin-Resistant Staphylococcus aureus, Antibiotics, 2021, 10(4), 398 CrossRef CAS PubMed.
- N. H. Amin, M. T. El-Saadi, A. A. Ibrahim and H. M. Abdel-Rahman, Design, synthesis and mechanistic study of new 1,2,4-triazole derivatives as antimicrobial agents, Bioorg. Chem., 2021, 111, 104841 CrossRef CAS PubMed.
- C. Y. Jia, J. Y. Li, G. F. Hao and G. F. Yang, A drug-likeness toolbox facilitates ADMET study in drug discovery, Drug Discov. Today, 2020, 25(1), 248–258 CrossRef CAS PubMed.
- D. H. O'Donovan, C. De Fusco, L. Kuhnke and A. Reichel, Trends in molecular properties, bioavailability, and permeability across the bayer compound collection: miniperspective, J. Med. Chem., 2023, 66(4), 2347–2360 CrossRef PubMed.
- P. Banerjee, A. O. Eckert, A. K. Schrey and R. Preissner, ProTox-II: a webserver for the prediction of toxicity of chemicals, Nucleic Acids Res., 2018, 46(W1), W257–W63 CrossRef CAS PubMed.
- A. Angeli, A. Petrou, V. Kartsev, B. Lichitsky, A. Komogortsev and C. Capasso, et al., Synthesis, Biological and In Silico Studies of Griseofulvin and Usnic Acid Sulfonamide Derivatives as Fungal, Bacterial and Human Carbonic Anhydrase Inhibitors, Int. J. Mol. Sci., 2023, 24(3), 2802 CrossRef CAS PubMed.
- J. An, G. Y. Zuo, X. Y. Hao, G. C. Wang and Z. S. Li, Antibacterial and synergy of a flavanonol rhamnoside with antibiotics against clinical isolates of methicillin-resistant Staphylococcus aureus (MRSA), Phytomedicine, 2011, 18(11), 990–993 CrossRef CAS PubMed.
- N. Rezki, S. A. Al-Sodies, H. E. A. Ahmed, S. Ihmaid, M. Messali and S. Ahmed, et al., A novel dicationic ionic liquids encompassing pyridinium hydrazone-phenoxy conjugates as antimicrobial agents targeting diverse high resistant microbial strains, J. Mol. Liq., 2019, 284, 431–444 CrossRef CAS.
- G. A. O’Toole, Microtiter dish biofilm formation assay, J. Vis. Exp., 2011,(47), 2437 Search PubMed.
- K. Alam, D. A. A. Farraj, E. F. S. Mah, M. A. Yameen, M. S. Elshikh and R. M. Alkufeidy, et al., Anti-biofilm activity of plant derived extracts against infectious pathogen-Pseudomonas aeruginosa PAO1, J. Infect. Public Health., 2020, 13(11), 1734–1741 CrossRef PubMed.
- H. E. A. Ahmed, H. A. Abdel-Salam and M. A. Shaker, Synthesis, characterization, molecular modeling, and potential antimicrobial and anticancer activities of novel 2-aminoisoindoline-1, 3-dione derivatives, Bioorg. Chem., 2016, 66, 1–11 CrossRef CAS PubMed.
- A. Hend and Y. A. F. El-sayed, Correlation between biofilm formation and multidrug resistance in clinical isolates of Pseudomonas aeruginosa, Microbes Infect. Dis., 2021, 2(3), 541–549 Search PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS PubMed.
- F. Demirci and K. H. C. Başer, Bioassay Techniques for Drug Development, ed. Atta-ur-Rahman, M. Iqbal Choudhary and William J. Thomsen, Harwood Academic Publishers, Amsterdam, The Netherlands, 2001, xii + 223 pp. 15.5 × 23.5 cm. $79.00, ISBN 90-5823-051-1.J. Nat. Prod., 2002, 65(7):1086– Search PubMed.
- S. Howard, V. Berdini, J. A. Boulstridge, M. G. Carr, D. M. Cross and J. Curry, et al., Fragment-Based Discovery of the Pyrazol-4-yl Urea (AT9283), a Multitargeted Kinase Inhibitor with Potent Aurora Kinase Activity, J. Med. Chem., 2009, 52(2), 379–388 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart and R. K. Belew, et al., Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function, J. Comput. Chem., 1998, 19(14), 1639–1662 CrossRef CAS.
- S. Dallakyan and A. J. Olson, Small-molecule library screening by docking with PyRx, Methods Mol. Biol., 2015, 1263, 243–250 CrossRef CAS PubMed.
- M. A. Soliman, H. E. Ahmed, E. H. Eltamany, A. T. Boraei, A. Aljuhani, S. A. Salama, R. Alghamdi, A. K. Aljohani, M. Almaghrabi and M. R. Aouad, Novel bis-benzimidazole-triazole hybrids: anticancer study, in silico approaches, and mechanistic investigation, Future Med. Chem., 2025, 17(1), 93–107 CrossRef PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong and J. Shim, et al., CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields, J. Comput. Chem., 2010, 31(4), 671–690 CrossRef CAS PubMed.
- A. A. Awaji, W. A. Z. E. Zaloa, M. A. Seleem, M. Alswah, M. M. Elsebaei and A. H. Bayoumi, et al., N- and s-substituted Pyrazolopyrimidines: A promising new class of potent c-Src kinase inhibitors with prominent antitumor activity, Bioorg. Chem., 2024, 145, 107228 CrossRef CAS PubMed.
- A. M. Malebari, H. E. A. Ahmed, S. K. Ihmaid, A. M. Omar, Y. A. Muhammad and S. S. Althagfan, et al., Exploring the dual effect of novel 1,4-diarylpyranopyrazoles as antiviral and anti-inflammatory for the management of SARS-CoV-2 and associated inflammatory symptoms, Bioorg. Chem., 2023, 130, 106255 CrossRef CAS PubMed.
- M. T. Khayat, H. E. A. Ahmed, A. M. Omar, Y. A. Muhammad, K. A. Mohammad and A. M. Malebari, et al., A novel class of phenylpyrazolone-sulphonamides rigid synthetic anticancer molecules selectively inhibit the isoform IX of carbonic anhydrases guided by molecular docking and orbital analyses, J. Biomol. Struct. Dyn., 2023, 1–19 Search PubMed.
- M. Alsehli, A. Aljuhani, S. K. Ihmaid, S. M. El-Messery, D. I. A. Othman and A.-A. A. A. El-Sayed, et al., Design and Synthesis of Benzene Homologues Tethered with 1,2,4-Triazole and 1,3,4-Thiadiazole Motifs Revealing Dual MCF-7/HepG2 Cytotoxic Activity with Prominent Selectivity via Histone Demethylase LSD1 Inhibitory Effect, Int. J. Mol. Sci., 2022, 23(15), 8796 CrossRef CAS PubMed.
- D. E. Shaw, P. Maragakis, K. Lindorff-Larsen, S. Piana, R. O. Dror and M. P. Eastwood, et al., Atomic-level characterization of the structural dynamics of proteins, Science, 2010, 330(6002), 341–346 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02412a |
| This journal is © The Royal Society of Chemistry 2025 |