
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiverse rare ginsenosides derived from ginsenoside Re in aqueous methanol solution via heterogeneous catalysis and identified by HPLC-MS†
Yanyan Chang,
Bing Li,
Yusheng Xiao,
Mengya Zhao,
Yujiang Zhou,
Huanxi Zhao* and
Yang Xiu *
*
Jilin Ginseng Academy, Changchun University of Chinese Medicine, Changchun 130117, P. R. China. E-mail: phoenix8713@sina.com; xiuyang@ccucm.edu.cn
First published on 16th May 2025
Abstract
Rare ginsenosides, known for their significant pharmacological effects, are found in only trace amounts in natural ginseng, making it necessary to produce them through transformation processes. In this study, ginsenoside Re was chemically transformed into 30 rare ginsenosides using a novel heterogeneous catalyst HSiW@MeSi in aqueous methanol solution. The HSiW@MeSi catalyst was synthesized by the incorporation of silicotungstic acid (H4SiW12O40, HSiW) into a mesoporous silica (MeSi) carrier. The resulting rare ginsenosides, which included six pairs of isomers, three sets of four isomers and one set of six isomers, were separated and identified using high-performance liquid chromatography coupled with multistage tandem mass spectrometry through characteristic neutral loss, product ions, and chromatographic retention times. The transformation pathways involved deglycosylation, epimerization, elimination, addition, and cyclization reactions. Water and methanol molecules competitively participated in the reaction, forming 8 hydroxylated and 14 methoxylated products at the C-20(21) or C-24(25) position, respectively. Notably, the HSiW@MeSi catalyst could be recycled and maintained an 83.3 ± 0.3% transformation rate after three cycles. This study represents the successful chemical transformation to produce protopanaxatriol-type rare ginsenosides featuring methoxyl groups at either the C-20(21) or C-24(25) positions. It highlights the potential of heteropolyacid-based heterogeneous transformation strategies in the generation of structurally diverse rare ginsenosides and demonstrates the expanded utility of HPLC-MS in the structural identification of these compounds.
Introduction
Ginseng, a traditional Chinese medicinal herb with a history of thousands of years, is renowned for its ability to maintain human vitality and enhance overall health.1 Ginsenosides are the principal active constituents of ginseng and exhibit a wide range of pharmacological effects, including memory enhancement, immunoregulation, anti-aging, and antioxidant activity.2–5 Ginsenosides are composed of an aglycone and glycosyl substituents. They are categorized into the dammarane type, the oleanolic acid type, and the ocotillol type based on the aglycone structures. The dammarane type can be further subdivided into protopanaxatriol (PPT) and protopanaxadiol (PPD) types, both of which are characterized by the tetracyclic skeleton.6,7 To date, more than 150 ginsenosides have been identified from ginseng roots, stems, leaves, and flowers, as well as processed products.8 Rare ginsenosides, which normally have superior biological activities compared to the major ginsenosides, are usually present in minimal amounts or are absent in natural sources. Consequently, they are mainly obtained through the transformation of major ginsenosides such as PPD-type Rb1, and PPT-type Re and Rg1.9,10Given the limited natural occurrence of rare ginsenosides, the development of convenient, economical, and efficient transformation methods is essential for large-scale preparation and subsequent activity studies.11,12 Chemical transformation represents one of the most common methods used to transform major ginsenosides into rare ones. Traditional chemical transformation approaches primarily use homogeneous catalysts such as hydrochloric acid, sulfuric acid, citric acid, etc. due to their high efficiency and low cost. However, these homogeneous catalysts are difficult to recover from the aqueous reaction mixture, thus leading to challenges in product separation and catalyst reuse.13–15
To address these limitations, heteropolyacids (HPAs) offer a promising alternative. HPAs are oxygen-containing polyacids composed of heteroatoms (such as P, Si, Fe, Co) and polyatomic elements (such as Mo, W, V, Nb, Ta) connected by oxygen bridges in a specific structure.16–19 Their oxygen-bridged frameworks, such as the Keggin type, provide high thermal and chemical stability, which ensures their recyclability and sustained catalytic performance.20 The potent Brønsted acidity of these compounds exceeds that of conventional acids, thus facilitating highly efficient catalysis in both homogeneous and heterogeneous reaction systems.21 Furthermore, their reversible redox behaviour supports oxidation reactions with easy regeneration.20,21 Additionally, they can function as environmentally friendly phase transfer catalysts. Although many HPAs dissolve readily in polar solvents, similar to traditional homogeneous catalysts, they can be immobilized on porous solid carriers to reduce solubility and facilitate a heterogeneous reaction environment while retaining catalytic activity. Mesoporous silica (MeSi) is considered an ideal carrier candidate for this purpose due to its chemical inertness, large specific surface area, high pore volume, and tunable pore size.22,23 It has proven to be a highly versatile support material for organometallics, inorganic acids, metals, inorganic salts, and organic ligands, and it has been widely used for dehydration,24 oxidation,25 drug delivery,26 adsorption,27 and degradation.28 For instance, Hou et al. synthesized Fe3O4@SiO2@HPW nanoparticles by embedding Fe3O4 and phosphotungstic acid in a silica carrier using a simple solvothermal and impregnation process. The obtained nanoparticles exhibited excellent photocatalytic efficiency for the degradation of Rhodamine B under UV light irradiation and good recyclability.29
Silica-supported HPAs thus have great potential to overcome the challenges associated with the separation and recovery of catalysts from the chemical transformation products of ginsenosides by forming a heterogeneous catalytic system. The heterogeneous catalytic system provides an ideal platform for precise regulation of ginsenoside molecular modification, particularly through the selective incorporation of small organic molecules as structural modifiers.12,30,31 Specifically, these supported catalysts can be easily separated from the reaction mixture through filtration or centrifugation, significantly simplifying the processing steps and enabling catalyst reuse.32,33 Moreover, structural modifications using small organic molecules in solvents are predicted to generate more kinds of rare ginsenosides with novel structures. This strategy is particularly effective for targeting the double bond at the C-24(25) position of the ginsenoside aglycone, which is highly susceptible to electrophilic addition reactions in an acidic environment. For instance, Zhao et al. employed methanol molecules to modify the aglycone moiety, leading to the synthesis of rare ginsenosides novel structural configurations at the C-20 and C-24(25) positions.30 By selectively introducing functional groups at this site, it is possible to generate new ginsenoside derivatives with more promising structures.
Advanced analytical techniques are therefore essential for the structural identification of complicated unknown ginsenosides. Over the past decade, high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS) has become the primary analytical method for identifying the structural features of ginsenosides.34–36 High-resolution mass spectrometry (HRMS) provides precise quasi-molecular and fragmentation ion information, enabling accurate determination of molecular formulas and relative molecular weights.37 Multistage tandem mass spectrometry (MSn) combined with collision-induced dissociation (CID) consistently isolates and dissociates precursor ions, allowing for accurate assignment of fragment ions.38,39 Compared to other analytical methods such as thin-layer chromatography and HPLC, HPLC-MS offers more detailed and accurate structural information of ginsenosides.
In this study, 12-silicotungstic acid (H4SiW12O40, HSiW), composed of one silicon atom and twelve tungsten atoms connected by oxygen bridges, was loaded into MeSi to form the heterogeneous catalyst HSiW@MeSi. This catalyst was used to chemically transform PPT type ginsenoside Re in methanol aqueous solution. The transformation products were rapidly differentiated and identified using HPLC-MSn and HRMS. A total of 30 rare ginsenosides were obtained through deglycosylation, addition, elimination, cyclization, and epimerization reactions. The synthesized HSiW@MeSi catalyst exhibits favorable stability and insolubility in polar solvents, which facilitates its separation from the reaction solution and recycling.
Experimental
Chemicals and materials
Analytical grade tetraethyl orthosilicate (TEOS), hexadecyl trimethyl ammonium bromide (CTAB) and ammonium hydroxide (NH3·H2O) were obtained from Shanghai Macklin Biochemical Co., Ltd. Analytical grade 12-silicotungstic acid (H4SiW12O40) and authentic ginsenoside standards, including Re, 20(S)-Rg2, 20(R)-Rg2, 20(S)-Rh1, 20(R)-Rh1, Rh4, Rk3, Rg6, and F4, all with over 98% purity, were purchased from Shanghai Yuanye Biological Technology Co., Ltd (Shanghai, China). HPLC-grade acetonitrile and methanol were acquired from Tedia (Fairfield, USA), while HPLC-grade formic acid was obtained from Thermo Fisher (Waltham, USA). Distilled water was prepared using a Milli-Q system from Millipore (Bedford, MA).Instruments and conditions
The powder X-ray diffraction (XRD) patterns were collected using a TDM-10 X-ray diffractometer (Dandong Tongda Technology Co., Ltd, Liaoning, China) equipped with Cu Kα radiation (λ = 1.5418 Å). Measurements were performed over a 2θ range of 1° to 5° for small-angle XRD and 10° to 70° for wide-angle XRD, at an operating voltage of 40 kV and a current of 30 mA. Fourier transform infrared spectroscopy (FTIR) spectra were obtained using a Thermo Scientific Nicolet iS10 FTIR spectrometer with KBr pellets. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-2100F transmission electron microscope operated at an accelerating voltage of 200 kV.The HPLC analysis was conducted on an Ultimate 3000 system from Thermo Scientific (San Jose, CA, USA), using a Thermo Syncronis C18 column (100 × 2.1 mm, 1.7 μm) for chromatographic separation at a temperature of 35 °C. The mobile phases consisted of 0.1% formic acid in water (v/v) as phase A and acetonitrile as phase B, delivered at a flow rate of 0.2 mL min−1. The elution gradient was programmed as follows: 0–5 min (30% B), 5–8 min (30–36% B), 8–15 min (36–48% B), 15–20 min (48–70% B), 20–23 min (70–90% B), 23–24 min (90–90% B), and 24–25 min (25% B). The injection volume was 2 μL.
A Q-Exactive quadrupole orbitrap mass spectrometer and an LTQ XL mass spectrometer were used for HRMS and MSn analysis, respectively. Both mass spectrometers were purchased from Thermo Scientific (San Jose, CA, USA) and equipped with an electrospray ionization (ESI) source operated in negative ion mode. The ESI source settings were configured as follows: sheath gas flow at 35 arbitrary units, auxiliary gas flow at 10 arbitrary units, sweep gas flow at 1 arbitrary unit, and a capillary voltage of −3200 V. The scan range was set from m/z 100 to 2000 in full scan mode and from m/z 200 to 1000 in tandem MS mode.
Prior to analyzing the transformation products of ginsenoside Re, the developed HPLC-MS method was validated for the determination of authentic ginsenoside standards. The calibration curve, R2 value, linear range, limit of detection (LOD), limit of quantification (LOQ), precision, reproducibility, stability and accuracy were evaluated using previously reported methodological validation method.40 The validation results, as shown in Tables S1 and S2,† demonstrate that the developed HPLC-MS method is suitable for the qualitative and quantitative analysis of ginsenosides.
Sample preparation
Results and discussion
Characterization of HSiW@MeSi
As shown in Fig. 1a, several distinct diffraction peaks appear in the XRD patterns of the synthesized HSiW@MeSi, matching well with the standard patterns of HSiW (PDF#01-0559). In particular, the main diffraction peaks at 25.2°, 34.7°, 53.2°, and 61.4° correspond to the (222), (332), (550), and (651) crystal planes of HSiW, respectively. This indicates that HSiW has been successfully immobilized in MeSi while retaining its intrinsic structural characteristics.41 In the small-angle XRD patterns shown in Fig. 1b, a single diffraction peak is observed at approximately 2.1° for MeSi, indicating the presence of mesoporous channels with non-uniform pore sizes. After loading HSiW, this peak shifts to 1.9°, suggesting that the loading of HSiW not only retains the mesoporous structure of MeSi but also slightly increases its pore size.42 This enhancement facilitates better contact between the loaded HSiW and ginsenoside molecules, thereby reducing steric hindrance effects.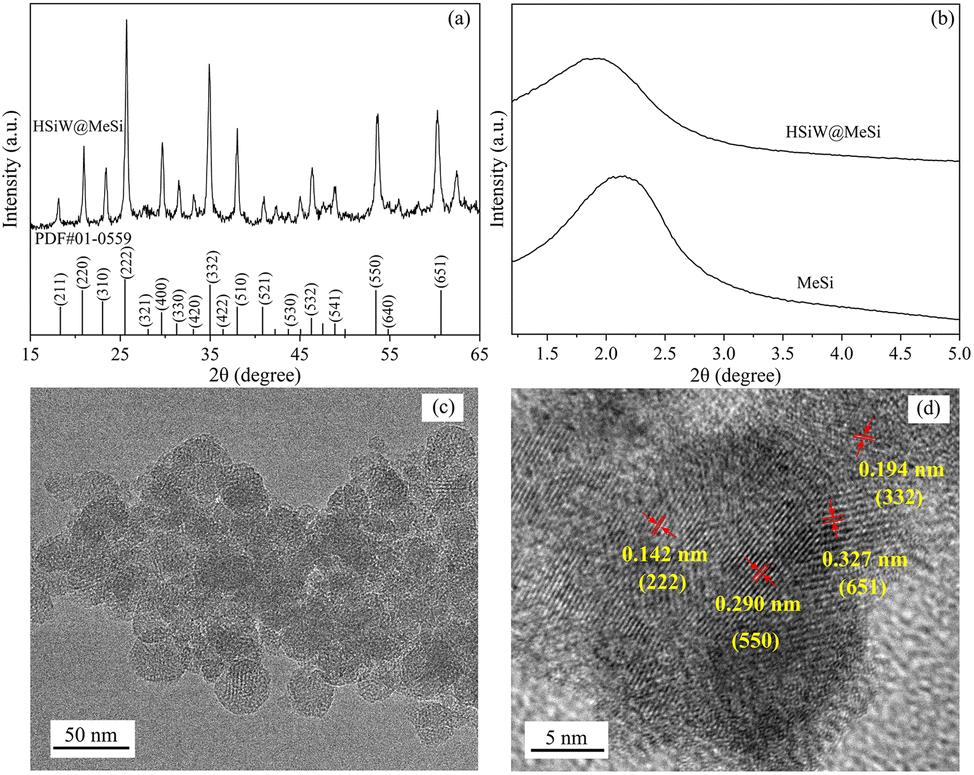 | ||
| Fig. 1 XRD patterns of MeSi and HSiW@MeSi over the 2θ range of 15–65° (a) and 1.2–5° (b). TEM (c) and HRTEM images (d) of HSiW@MeSi. | ||
As shown in the TEM image of HSiW@MeSi in Fig. 1c, the synthesized MeSi framework exhibits a spherical morphology with a diameter of approximately 50 nm. The ordered mesoporous channels are clearly visible, indicating that the loading of HSiW does not significantly affect the mesoporous structure of the MeSi framework. Well-aligned lattices are also clearly observed in the high-resolution TEM image presented in Fig. 1d. The measured lattice spacings correspond mainly to the (222), (232), (550), and (651) lattice planes of HSiW, further confirming the successful incorporation of HSiW into the MeSi framework.43
Fig. S1† shows the FTIR spectra of HSiW, MeSi, and HSiW@MeSi. The synthesized MeSi exhibits a broad band in the range of 1300 to 1000 cm−1, which corresponds to the asymmetric stretching of Si–O–Si bridges in its framework. Additionally, the bands observed at 803 cm−1 and 465 cm−1 are attributed to the symmetric stretching and bending vibrations of Si–O–Si, respectively.44–46 The absorption bands of HSiW are observed at 980 cm−1, 930 cm−1, 883 cm−1, and 800 cm−1. The first two bands correspond to the characteristic vibrations of W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Si–O bonds in HSiW, respectively, while the latter two correspond to the characteristic vibrations of W–O–W bonds involving corner-sharing oxygen atoms connecting the W3O13 units and edge-sharing oxygen atoms connecting the tungsten atoms, respectively.47,48 The characteristic bands of both MeSi and HSiW can be clearly observed simultaneously in the spectrum of HSiW@MeSi, further proving that the synthesized HSiW@MeSi retains the characteristic structures of both MeSi and HSiW.
O and Si–O bonds in HSiW, respectively, while the latter two correspond to the characteristic vibrations of W–O–W bonds involving corner-sharing oxygen atoms connecting the W3O13 units and edge-sharing oxygen atoms connecting the tungsten atoms, respectively.47,48 The characteristic bands of both MeSi and HSiW can be clearly observed simultaneously in the spectrum of HSiW@MeSi, further proving that the synthesized HSiW@MeSi retains the characteristic structures of both MeSi and HSiW.
The N2 adsorption was carried out for MeSi and HSiW@MeSi in order to evaluate their permanent porosity. As shown in Fig. S2a,† both the synthesized MeSi and HSiW@MeSi exhibit reversible type IV isotherms and H1 hysteresis loops, which are the main characteristics of mesoporous materials.49 Furthermore, the surface areas of MeSi and HSiW@MeSi are calculated to be 701.5 m2 g−1 and 588.8 m2 g−1, respectively, using the Brunauer–Emmett–Teller (BET) model. These features indicate that both MeSi and HSiW@MeSi have typical mesoporous structures, in accordance with the results of XRD. The loading of HSiW resulted in a reduction in the surface area of MeSi, which is attributed to the deposition of HSiW inside the pores and its fine dispersion on the surface, partially blocking the adsorption sites for N2 molecules on the MeSi framework. The pore size distributions of MeSi and HSiW@MeSi are shown in Fig. S2b.† It can be seen that they both have narrow pore size distributions with the maximum value at about 2.8 nm, which is consistent with the characteristics of MeSi loaded with HPAs.50,51
All the above characterization results demonstrate that the synthesized HSiW@MeSi retains both the crystalline structure of HSiW and the mesoporous channels of MeSi. Additionally, its stability and insolubility in aqueous methanol solution suggest its potential for use in the heterogeneous transformation of ginsenosides.
Structural characterization and identification of ginsenoside Re transformation products in aqueous methanol by HPLC-MS
The synthesized HSiW@MeSi was used for the heterogeneous transformation of ginsenoside Re in a 70% methanol aqueous solution. The reaction was conducted for 8 h at 80 °C, followed by detection using an HPLC-MS method. The total ion chromatogram (TIC) shown in Fig. 2a displays 30 peaks with different retention times, labeled compounds 1 to 30, indicating the ability of HSiW@MeSi to transform ginsenosides. The structures of these transformation products were further determined using MSn and HRMS techniques.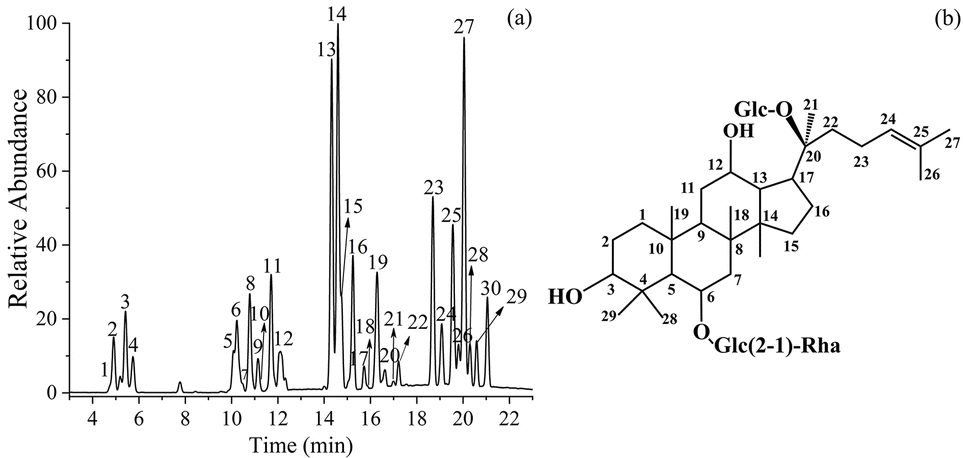 | ||
| Fig. 2 TIC of the transformation products of ginsenoside Re with methanol (a) and the structural formula of ginsenoside of Re (b). | ||
Compounds 5, 6, 8, and 10 have an identical [M − H]− ion at m/z 815.7 and [M + HCOO]− ion at m/z 861.7, indicating that they are one set of four isomers with a relative molecular mass of 816.7 and the molecular formula C43H74O14. Among these four compounds, the fragment ions observed in the tandem mass spectra of compounds 5 and 6 are identical to each other, as are those of compounds 8 and 10. This suggests that compounds 5 and 6 form one pair of epimers, while compounds 8 and 10 form another pair, both resulting from the chirality at the C-20 position (Fig. 2b). The MS2 spectra of compound 5 are employed as an example to identify the structures of the first pair of isomers, as shown in Fig. 3a. The two neutral losses of 146.1 Da and 162.1 Da between the precursor ion at m/z 815.7 and the product ions at m/z 669.6 and m/z 507.5 correspond to the rhamnose and glucose substituents, respectively, suggesting that the two product ions are formed by the successive removal of one rhamnose and one glucose substituent from the precursor ion. This also indicates that the disaccharide substituent of ginsenoside Re at the C-6 position was retained during the transformation, while the glucose substituent at the C-20 position was lost. Therefore, the product ion at m/z 507.5 is considered as the aglycone ion of compound 5.
 | ||
| Fig. 3 MS2 spectrum of the [M − H]− ion at m/z 815.7 (a), fragmentation pathways, and MS3 spectrum of the product ion at m/z 507.5 (b) from ginsenoside 20(S)-25-OCH3-Rg2. | ||
Further MS3 analysis was conducted on the aglycone ion at m/z 507.5 to investigate its detailed structure. As shown in Fig. 3b, the product ion at m/z 475.5 is the typical deprotonated aglycone ion of PPT-type ginsenoside.52 It differs from the precursor ion by a mass difference of 32.0 Da, corresponding to one methanol molecule. This suggests that compound 5 is derived from the methanol addition reaction of the reactant ginsenoside Re, with the possible reaction site being the double bond at the C-24(25) position or the double bond formed by the elimination of the hydroxyl group at the C-20 position. The neutral loss of 84.1 Da between the ion at m/z 475.5 and m/z 391.4 corresponds to the molecular formula C6H12, formed by the loss of the entire olefin chain through single bond cleavage at the C-20(22) position. The ion at m/z 391.4 is the characteristic product ion of the PPT-type ginsenoside. Its presence indicates that the tetracyclic skeleton of ginsenoside Re was preserved during the transformation and that the hydroxyl group at the C-20 position was not dehydrated to form a new double bond. Therefore, the extra methanol molecule is identified as being added at the C-24(25) double bond. According to the rules of addition reactions of olefins in acidic environments, the methoxyl group is preferentially attached to the C-25 position to form a 2-methoxypropyl group. The ion at m/z 457.5 is derived from the removal of one H2O molecule of from the ion at m/z 475.5. The lost H2O molecule is supposed to originate from the dissociation of the retained hydroxyl group at the C-20 position and the proton transfer from the precursor ion. Therefore, compounds 5 and 6 are the transformation products of ginsenoside Re via the hydrolysis of the glucose substituent at the C-20 position and the addition of one methanol molecule to the double bond at the C-24(25) position. Since the 20(S)-ginsenoside epimer is eluted before its 20(R)-counterpart in the C18 column,52–54 compounds 5 and 6 can be identified as 20(S)-25-OCH3-Rg2 and 20(R)-25-OCH3-Rg2, respectively, with their structures illustrated in Fig. 3 and S3.†
Compound 8 was used as an example to analyze the structures of the other pair of epimers, compounds 8 and 10. As shown in Fig. 4, compound 8 exhibits four identical product ions at m/z 669.6, m/z 651.6, m/z 507.5, and m/z 475.5 to those of compound 5, suggesting that it has the same glycosyl substituents and methanol-added aglycone as compound 5. Therefore, the only structural difference between compounds 8 and 5 should be the positions of the methoxyl group generated by the addition reaction of one methanol molecule. The distinctive product ion at m/z 417.5 provides decisive information.53 It is 58.0 Da less than the ion at m/z 475.5, which is formed by the dissociation of one methanol molecule from the aglycone ion. The 58.0 Da corresponds to the molecular formula C3H6O, indicating the presence of a 2-hydroxypropyl group at the C-25 position of dammarane-type ginsenosides. This suggests a definitive hydration reaction on the double bond at the C-24(25) position. Consequently, the methanol molecule can only add to the double bond formed by the elimination of the hydroxyl group at the C-20 position, generating a methoxyl group attached to the C-20, which dissociates more easily during CID. Moreover, the absence of the dehydrated ion at m/z 457.5 also confirms that the hydroxyl group at the C-20 position is not retained. It can therefore be concluded that both compounds 8 and 10 are composed of one methoxyl group at the C-20 position, one hydroxyl group at the C-25 position, and one rutinose disaccharide substituent at the C-6 position. They are identified as 20(S)-OCH3-25-OH-Rg2 and 20(R)-OCH3-25-OH-Rg2, respectively, the structures of which are shown in Fig. 4 and S4.†
 | ||
| Fig. 4 MS2 spectrum of the [M − H]− ion at m/z 815.7 from ginsenoside 20(S)-OCH3-25-OH-Rg2. | ||
Compounds 18, 19, 23, and 24 are found to be another set of four isomers with a relative molecular mass of 798.7 and the molecular formula C43H72O13. Based on their relatively close retention times, it can be inferred that compounds 18 and 19, as well as compounds 23 and 24, form two pairs of structurally similar isomers. As shown in Fig. 5, S5–S7,† these four compounds exhibit identical tandem MS spectra. Their [M − H]− ions at m/z 797.7 sequentially dissociate one rhamnose and one glucose substituent to generate the aglycone ions at m/z 489.5. This indicates that they retain the rutinose disaccharide substituent from the reactant ginsenoside Re at the C-6 position.
 | ||
| Fig. 5 MS2 spectrum of the [M − H]− ion at m/z 797.7 (a), fragmentation pathways, and MS3 spectrum of the product ion at m/z 651.6 (b) from ginsenoside 25-OCH3-Rg6. | ||
The difference of 32.0 Da between the product ions at m/z 457.4 and the aglycone ions at m/z 489.5 suggests that the aglycone has been modified by the addition of one methanol molecule, forming a methoxyl group. The methoxyl group can be located either at the C-20 or C-25 position, as in the set of four isomers discussed above. Therefore, this set of four isomers should consist of two pairs: one pair with methoxylation at the C-20 position and another pair with methoxylation at the C-25 position. Moreover, the aglycone ions of compounds 18, 19, 23, and 24 at m/z 489.5 have a mass difference of −18.0 Da compared to those of compounds 5, 6, 8, and 10 at m/z 507.5, indicating that they are the dehydration products of compounds 5, 6, 8, and 10 with an additional double bond attached to the aglycone. When the methoxyl group is attached at the C-20 position, as in compounds 8 and 10, the double bond is located at the C-24(25) position, retaining the olefin chain structure characteristic of dammarane-type ginsenosides. In contrast, when the methoxyl group is attached at the C-25 position, as in compounds 5 and 6, the double bond is formed through the elimination of the hydroxyl group at the C-20 position, resulting in double bonds at the C-20(21) or C-20(22) positions. By comparing the retention times of 20(S)-OCH3-25-OH-Rg2 and 20(S)-25-OCH3-Rg2, it is observed that C-25 methoxylated products elute earlier than C-20 methoxylated products on a C18 column. Additionally, Δ20(21) ginsenosides elute earlier than their Δ20(22) isomers. Therefore, compounds 18, 19, 23, and 24 are identified as 25-OCH3-Rg6, 25-OCH3-F4, 20(S)-OCH3-Rg2 and 20(R)-OCH3-Rg2, respectively, with their structures illustrated in Fig. 5b, S5b, S6b, and S7b.†
Compounds 7 and 9 are epimers with a relative molecular mass of 670.6 and the chemical formula C37H65O10. The tandem MS spectra of their [M − H]− ions at m/z 669.6, as shown in Fig. S8a and S9a,† exhibit only one neutral loss of 162.1 Da, corresponding to a single glucose substituent. The MS3 fragment ions generated from the aglycones at m/z 507.5 are identical to those of compounds 5 and 6, as shown in Fig. S8b and S9b,† suggesting that compounds 7 and 9 share the same aglycone structure as compounds 5 and 6. This evidence indicates that compounds 7 and 9 are derived from further removal of the external rhamnose substituent at the C-6 position of compounds 5 and 6. Therefore, they can be identified as 20(S)-25-OCH3-Rh1 and 20(R)-25-OCH3-Rh1, respectively, the structures of which are shown in Fig. S8 and S9.†
Similarly, compounds 21, 22, 26, and 28 are another set of four isomers with a relative molecular mass of 652.6 and the molecular formula C37H63O9. As shown in Fig. S10,† their tandem MS spectra exhibit identical product ions, similar to those of compounds 18, 19, 23, and 24, except for the presence of only one product ion resulting from the dissociation of a glycosyl group. This indicates that they share the same aglycone structure as compounds 18, 19, 23, and 24. The only difference is that they consist of only one glucose substituent. Therefore, it can be inferred that compounds 21, 22, 26, and 28 are derived from compounds 18, 19, 23, and 24 by the further removal of the rhamnose substituent at the C-6 position, resulting in the formation of ginsenosides 25-OCH3-Rk3, 25-OCH3-Rh4, 20(S)-OCH3-Rh1 and 20(R)-OCH3-Rh1, respectively, with their structures shown in Fig. S10.† The 14 compounds identified above are all methoxylated at the C-20 or C-25 position, as deduced from the neutral loss of 32.0 Da relative to the aglycone ion.
For the remaining 16 transformation products, the authentic standards of 20(S)-Rg2, 20(R)-Rg2, 20(S)-Rh1, 20(R)-Rh1, Rg6, F4, Rk3, and Rh4 are used for their identification by comparison of retention times and tandem MS spectra. The compounds 13, 14, 15, 16, 25, 27, 29, and 30 have the identical retention times and tandem MS spectra with the respective authentic standards. Therefore, they are identified as 20(S)-Rg2, 20(R)-Rg2, 20(S)-Rh1, 20(R)-Rh1, Rg6, F4, Rk3, and Rh4, respectively. Their tandem mass spectra and structural formulas are shown in Fig. S11–S14.†
In particular, 20(S)-Rg2 and 20(R)-Rg2 are epimers with a relative molecular mass of 784.7, produced by the removal of the glucose substituent at the C-20 position from the reactant ginsenoside Re. Further removal of the external rhamnose substituent at the C-6 position generates the epimers 20(S)-Rh1 and 20(R)-Rh1, with a relative molecular mass of 638.6. These two pairs of epimers retain the olefin chain characteristic of dammarane-type ginsenosides. Rg6 and F4 are the Δ20(21) and Δ20(22) ginsenosides formed by the removal of the hydroxyl group at the C-20 position from Rg2. As shown in Fig. S13a and b,† their aglycone ions at m/z 457.5 are exactly 18.0 Da less than those of Rg2. Subsequent removal of the external rhamnose substituent at the C-6 position results in the formation of Rk3 and Rh4. For these two pairs of positional isomers, no product ions resulting from the dissociation of the aglycone ions at m/z 457.5 are observed in their tandem MS spectra. This is due to their rigid framework formed by the two double bonds, which remains stable during CID. Additionally, their relative retention times also confirm the identification of the other two sets of Δ20(21) and Δ20(22) ginsenoside isomers of 25-OCH3-Rg6 and 25-OCH3-F4, as well as 25-OCH3-Rk3 and 25-OCH3-Rh4.
Compounds 2 and 3 have a relative molecular mass of 802.7 and the chemical formula C42H73O14. The tandem MS spectra shown in Fig. S15 and S16† indicate that they possess a rutinose disaccharide substituent. Their aglycone ions at m/z 493.5 are 18.0 Da higher than those of the PPT-type ginsenosides, suggesting that they are hydrated products of ginsenoside Re. The neutral loss of 58.0 Da between the ions at m/z 475.5 and m/z 417.5 confirms the presence of a 2-hydroxypropyl group at the C-25 position. Therefore, compounds 2 and 3 can be identified as 20(S)-Rf2 and 20(R)-Rf2, respectively. Compounds 1 and 4 are isomers with a relative molecular mass of 656.6, which is 146.1 Da less than that of compounds 2 and 3, indicating the absence of one rhamnosyl substituent. The product ions observed in their tandem MS spectra (Fig. S17 and S18†) are all included in those of compounds 2 and 3, suggesting that they share the same aglycone structure with ginsenoside Rf2. Therefore, compounds 1 and 4 are identified as 20(S)-25-OH-Rh1 and 20(R)-25-OH-Rh1, respectively.
Compounds 11 and 12 are the isomers of ginsenoside Rg2. Their tandem MS spectra in Fig. S19† differ from those of Rg2 only by the presence of the ion at m/z 417.5 instead of the characteristic ion at m/z 391.4 observed for Rg2. The ion at m/z 417.5 exhibits a neutral loss of 58.0 Da compared to the aglycone ion at m/z 475.5, indicating a hydration reaction to the C-24(25) double bond. To counterbalance the additional degree of unsaturation introduced by this hydration, a dehydration reaction to the hydroxyl group at the C-20 position is expected to occur, resulting in the formation of Δ20(21) and Δ20(22) isomers. This structural rearrangement is supported by the absence of the m/z 391.4 ion. Therefore, compounds 11 and 12 can be identified as 25-OH-Rg6 and 25-OH-F4, respectively, with their structures shown in Fig. S19.†
Compounds 16 and 19 represent another pair of isomers of ginsenoside Rg2. Their tandem MS spectra in Fig. 6b and S20b† exhibit both the characteristic product ions at m/z 417.5 and m/z 391.4, as well as the same aglycone ion at m/z 475.5 observed for Rg2. This suggests that after the additional degree of unsaturation introduced by the hydration reaction is counterbalanced, it remains possible to dissociate the olefin chain, indicating that no double bond is formed at the C-20 position. The simultaneous appearance of the two characteristic product ions can only be achieved through a cycloaddition reaction initiated by the electrophilic attack of the C-20 hydroxyl group to the C-24(25) double bond. As shown in Fig. 6b, the ion at m/z 391.4 is generated through the simultaneous cleavage of the single bonds at C-20(22) and C-25(O), while the ion at m/z 417.5 is generated through that at C-20(O) and C-24(25). Therefore, compounds 16 and 19 are identified as 20(S, 25)-epoxy-Rg2 and 20(R, 25)-epoxy-Rg2, respectively. It is seen that 20(S)-Rg2, 20(R)-Rg2, 25-OH-Rg6, 25-OH-F4, 20(S, 25)-epoxy-Rg2, and 20(R, 25)-epoxy-Rg2 are six isomers with a relative molecular mass of 784.7. Furthermore, the presence or absence of the two characteristic product ions at m/z 417.5 and m/z 391.4 could be used to identify and differentiate these six isomers.
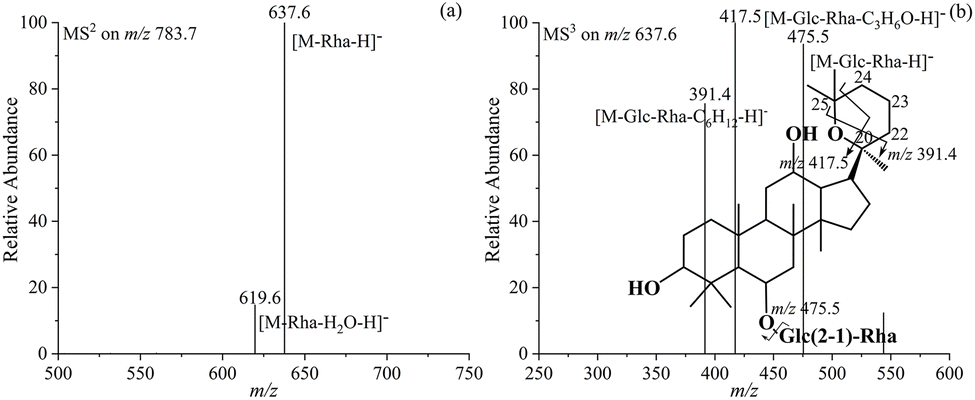 | ||
| Fig. 6 MS2 spectrum of the [M − H]− ion at m/z 783.7 (a), fragmentation pathways, and MS3 spectrum of the product ion at m/z 637.6 (b) from ginsenoside 20(S,25)-epoxy-Rg2. | ||
HRMS analysis was further performed in both AIF and dd-MS2 mode using normalized collision energy to verify the accuracy of the product ions obtained from MSn analysis for all identified transformation products. As listed in Table 1, the product ions of all 30 transformation products are consistent between HRMS and MSn analyses. These results confirm the reliability and accuracy of the HPLC-MSn structural identification method used in this study. Moreover, the yields of the 30 transformation products are also estimated using the peak areas from the extracted ion chromatograms and are shown in Table 1.
| Peak | Identification | Relative molecular mass | Molecular formula | Measured [M − H]− (m/z) | Fragment ions | Yield (%) |
|---|---|---|---|---|---|---|
| 1 | 20(S)-25-OH-Rh1 | 656.6572 | C36H63O10 | 655.6572 | 493.5664[M-Glc-H]−, 475.5215[M-Glc-H2O-H]−, 417.5839[M-Glc-C3H8O2-H]−, 391.4672[M-Glc-C6H14O-H]− | 2.51 |
| 2 | 20(S)-Rf2 | 802.7563 | C42H73O14 | 801.7563 | 655.6321[M-Rha-H]−, 637.6381[M-Rha-H2O-H]−, 493.5871[M-Rha-Glc-H]−, 475.5267[M-Glc-Rha-H2O-H]−, 417.5835[M-Rha-Glc-C3H8O2-H]−, 391.4662[M-Glc-Rha-C6H14O]− | 4.11 |
| 3 | 20(R)-Rf2 | 802.7561 | C42H73O14 | 801.7561 | 655.6329[M-Rha-H]−, 637.6385[M-Rha-H2O-H]−, 493.5873[M-Rha-Glc-H]−, 475.5361[M-Glc-Rha-H2O-H]−, 417.5830[M-Rha-Glc-C3H8O2-H]−, 391.4669[M-Glc-Rha-C6H14O]− | 6.44 |
| 4 | 20(R)-25-OH-Rh1 | 656.6577 | C36H63O10 | 655.6577 | 493.5668[M-Glc-H]−, 475.5210[M-Glc-H2O-H]−, 417.5830[M-Glc-C3H8O2-H]−, 391.4678[M-Glc-C6H14O-H]− | 3.72 |
| 5 | 20(S)-25-OCH3-Rg2 | 816.7441 | C43H74O14 | 815.7441 | 669.6326[M-Rha]−, 651.6182[M-Rha-H2O-H]−, 507.5326[M-Glc-Rha-H]−, 475.5263[M-Glc-Rha-H]−, 457.5[M-Glc-Rha-CH3OH-H2O-H]−, 391.4663[M-Glc-Rha-C7H16O-H]− | 2.47 |
| 6 | 20(R)-25-OCH3-Rg2 | 816.7440 | C43H74O14 | 815.7440 | 669.6325[M-Rha-H]−, 651.6180[M-Rha-H2O-H]−, 507.329[M-Glc-Rha-H]−, 475.5261[M-Glc-Rha-CH3OH-H]−, 457.5[M-Glc-Rha-CH3OH-H2O-H]−, 391.4662[M-Glc-Rha-C7H16O-H]− | 4.55 |
| 7 | 20(S)-25-OCH3-Rh1 | 670.6310 | C37H65O10 | 669.6310 | 507.5321[M-Glc-H]−, 475.5263[M-Glc-CH3OH-H]−, 457.5132[M-Glc-CH3OH-H]−, 391.4661[M-Glc-C7H16O-H]− | 2.25 |
| 8 | 20(S)-OCH3-25-OH-Rg2 | 816.7442 | C43H74O14 | 815.7442 | 669.6321[M-Rha-H]−, 651.6183[M-Rha-H2O-H]−, 507.5326[M-Glc-Rha-H]−, 475.5262[M-Glc-Rha-CH3OH-H]−, 417.5[M-Glc-Rha-C4H10O2-H]− | 7.91 |
| 9 | 20(R)-25-OCH3-Rh1 | 670.6311 | C37H65O10 | 669.6311 | 507.5325[M-Glc-H]−, 475.5264[M-Glc-CH3OH-H]−, 457.5133[M-Glc-CH3OH-H]−, 391.4662[M-Glc-C7H16O-H]− | 3.25 |
| 10 | 20(R)-OCH3-25-OH-Rg2 | 816.7441 | C43H74O14 | 815.7441 | 669.6324[M-Rha-H]−, 651.6185[M-Rha-H2O-H]−, 507.5327[M-Glc-Rha-H]−, 475.5263[M-Glc-Rha-CH3OH-H]−, 417.5[M-Glc-Rha-C4H10O2-H]− | 0.74 |
| 11 | 25-OH-Rg6 | 784.7760 | C42H71O13 | 783.7760 | 637.6122[M-Rha-H]−, 619.6321[M-Rha-H2O-H]−, 475.5131[M-Glc-Rha-H]−, 417.5831[M-Glc-Rha-C3H6O]− | 6.33 |
| 12 | 25-OH-F4 | 784.7761 | C42H71O13 | 783.7761 | 637.6121[M-Rha-H]−, 619.6322[M-Rha-H2O-H]−, 475.5132[M-Glc-Rha-H]−, 417.5832[M-Glc-Rha-C3H6O]− | 4.11 |
| 13 | 20(S)-Rg2 | 784.7763 | C42H71O13 | 783.7763 | 637.6124[M-Rha-H]−, 619.6324[M-Rha-H2O-H]−, 475.5133[M-Glc-Rha-H]−, 391.4661[M-Glc-Rha-C6H12-H]− | 17.93 |
| 14 | 20(R)-Rg2 | 784.7761 | C42H71O13 | 783.7761 | 637.6121[M-Rha-H]−, 619.6320[M-Rha-H2O-H]−, 475.5134[M-Glc-Rha-H]−, 391.4663[M-Glc-Rha-C6H12-H]− | 22.53 |
| 15 | 20(S)-Rh1 | 638.6552 | C36H62O9 | 637.6552 | 475.5260[M-Glc-H]− | 7.07 |
| 16 | 20(R)-Rh1 | 638.6551 | C36H62O9 | 637.6551 | 475.5261[M-Glc-H]− | 10.42 |
| 17 | 20(S,25)-epoxy-Rg2 | 784.7760 | C42H71O13 | 783.7760 | 637.6125[M-Rha-H]−, 619.6320[M-Rha-H2O-H]−, 475.5132[M-Glc-Rha-H]−, 417.5830[M-Glc-Rha-C3H6O-H]−, 391.4[M-Glc-Rha-C6H12-H]− | 1.68 |
| 18 | 25-OCH3-Rg6 | 798.7566 | C43H72O13 | 797.7566 | 651.6443[M-Rha-H]−, 633.6321[M-Rha-H2O-H]−, 489.5431[M-Glc-Rha-H]−, 457.5131[M-Glc-Rha-CH3OH-H]− | 7.25 |
| 19 | 25-OCH3-F4 | 798.7567 | C43H72O13 | 797.7567 | 651.6441[M-Rha-H]−, 633.6320[M-Rha-H2O-H]−, 489.5432[M-Glc-Rha-H]−, 457.5132[M-Glc-Rha-CH3OH-H]− | 1.67 |
| 20 | 20(R,25)-epoxy-Rg2 | 784.7761 | C42H71O13 | 783.7761 | 637.6121[M-Rha-H]−, 619.6324[M-Rha-H2O-H]−, 475.5131[M-Glc-Rha-H]−, 417.5831[M-Glc-Rha-C3H6O-H]−, 391.4[M-Glc-Rha-C6H12-H]− | 1.72 |
| 21 | 25-OCH3-Rk3 | 652.6441 | C37H63O9 | 651.6441 | 489.5432[M-Glc-H]−, 457.5134[M-Glc-CH3OH-H]− | 1.09 |
| 22 | 25-OCH3-Rh4 | 652.6442 | C37H63O9 | 651.6442 | 489.5430[M-Glc-H]−, 457.5132[M-Glc-CH3OH-H]− | 3.02 |
| 23 | 20(S)-OCH3-Rg2 | 798.7563 | C43H72O13 | 797.7563 | 651.6441[M-Rha-H]−, 633.6320[M-Rha-H2O-H]−, 489.5430[M-Glc-Rha-H]−, 457.5134[M-Glc-Rha-CH3OH-H]− | 12.31 |
| 24 | 20(R)-OCH3-Rg2 | 798.7562 | C43H72O13 | 797.7562 | 651.6442[M-Rha-H]−, 633.6321[M-Rha-H2O-H]−, 489.5433[M-Glc-Rha-H]−, 457.5131[M-Glc-Rha-CH3OH-H]− | 4.89 |
| 25 | Rg6 | 766.7134 | C42H69O12 | 765.7134 | 619.6321[M-Rha-H]−, 601.6550[M-Rha-H2O-H]−, 457.5131[M-Glc-Rha-H]− | 17.54 |
| 26 | 20(S)-OCH3-Rh1 | 652.6443 | C37H63O9 | 651.6443 | 489.5430[M-Glc-H]−, 457.5131[M-Glc-CH3OH-H]− | 3.54 |
| 27 | F4 | 766.7132 | C42H69O12 | 765.7132 | 619.6324[M-Rha-H]−, 601.6551[M-Rha-H2O-H]−, 457.5133[M-Glc-Rha-H]− | 16.63 |
| 28 | 20(R)-OCH3-Rh1 | 652.6445 | C37H63O9 | 651.6445 | 489.5431[M-Glc-H]−, 457.5135[M-Glc-CH3OH-H]− | 0.76 |
| 29 | Rk3 | 620.6442 | C36H60O8 | 619.6442 | 457.5138[M-Glc-H]− | 6.97 |
| 30 | Rh4 | 620.6441 | C36H60O8 | 619.6441 | 457.5139[M-Glc-H]− | 5.38 |
Transformation pathways and mechanisms of ginsenoside Re in aqueous methanol
The use of HSiW@MeSi facilitated the transformation of ginsenoside Re into 30 rare ginsenosides, including ten pairs of epimers, five pairs of positional isomers, and notably one set of six isomers as well as three sets of four isomers. Based on the characterized structures of these 30 products, the transformation pathways of ginsenoside Re in aqueous methanol solution are summarized in Fig. 7, involving deglycosylation, epimerization, addition, elimination, and cyclization reactions. Both methanol and water molecules played key roles in the transformation. Methanol molecules participated in the formation of 14 rare ginsenosides by played attaching methoxyl groups at the C-20 or C-25 positions, while water molecules facilitated the generation of 8 rare ginsenosides through hydroxylation at the C-25 position. | ||
| Fig. 7 Transformation pathways of ginsenoside Re in aqueous methanol. | ||
It is evident that all the 30 products are free of glycosyl substituent at the C-20 position, indicating that deglycosylation at this site is a necessary step in the transformation of ginsenoside Re by HSiW@MeSi. Of these products, 18 retain the disaccharide substituent at the C-6 position inherited from the reactant ginsenoside Re, while the remaining 12 lose the external rhamnosyl substituent. This observation suggests that the quaternary carbon at the C-20 position is more reactive than the tertiary carbon at the C-6 position. Furthermore, under the acidic conditions generated by HSiW@MeSi, the internal glycosidic bond of the disaccharide substituent at the C-6 position is more stable than the external one. Deglycosylation at the C-20 position introduces chirality to this carbon atom, enabling to the possibility of epimerization. The almost equal peak areas observed for 20(S)-Rg2 and 20(R)-Rg2 indicate that the epimerization reaction lacks stereoselective. Moreover, the Rg2 epimers require the fewest reaction steps and emerge as the predominant products in the initial stages of the reaction. They also act as intermediates for the formation of other products.
The hydroxyl group at the C-20 position of the Rg2 epimers can undergo elimination reactions with adjacent hydrogen atoms at the C-21 and C-22 positions, resulting in the formation of Δ20(21) and Δ20(22) ginsenoside isomers, including Rg6, F4, Rk3, and Rh4. In acidic conditions, the elimination of hydroxyl groups may proceed via either an E1 or E2 mechanism. However, due to the high stability of the tertiary carbenium ion formed from the tertiary alcohol at the C-20 position of Rg2, the transformation is more likely to follow the E1 unimolecular elimination mechanism.12,55 The formation of the carbenium ion represents the rate-determining step, meaning the reaction rate depends solely on the concentration of the reactant and not on the concentration of H+ ions released by HSiW@MeSi. As a result, Rg6 and F4 are rapidly generated in significant amounts during the initial stages of the reaction. The elimination reactions introduce double bonds at the C-20(21), C-20(22), and C-24(25) positions, creating potential sites for subsequent addition reactions involving water and methanol molecules. These addition reactions further diversify the transformation pathways, leading to the formation of additional rare ginsenosides.
In an acidic environment, the addition of water and methanol molecules to double bonds typically follows Markovnikov's rule, where the electrophilic group attaches to the carbon atom with more substituents, forming a more stable carbenium ion intermediate.32,56 Consequently, among the 20 products involving methoxylation and hydroxylation, both methoxyl and hydroxyl groups are attached at the C-20 and C-25 positions, such as the 25-OCH3-Rg2 epimers and the Rf2 epimers. The addition reactions of water and methanol molecules are competitive, with water molecules being more prone to undergo addition due to their stronger nucleophilicity. However, the concentration of methanol in the aqueous solution significantly affects the outcome of these competitive reactions. Moreover, the hydroxyl group at the C-20 position can also act as a nucleophilic reagent and undergo addition to the double bond at the C-24(25) position, leading to the formation of cycloaddition products of the 20(25)-epoxy-Rg2 epimers. Notably, the electrophilic addition to double bonds is reversible under acidic conditions, with the elimination of tertiary alcohols restoring the original double bonds. This establishes a dynamic equilibrium between Rg2, Rh1, Rg6, F4, and their C-25 hydroxylated products. In contrast, the methoxyl group is an unfavorable leaving group under acidic conditions, making its direct elimination difficult. As a result, the addition reactions of methanol molecules to the double bonds at the C-20(21), C-20(22), and C-24(25) positions are irreversible.
Effects of reaction conditions on ginsenoside Re transformation in aqueous methanol
Time-course experiments were performed to elucidate the formation process of the 30 transformation products. As shown in Fig. 8A, after 1 h of reaction in a 70% aqueous methanol solution, most of the reactant has been transformed, leaving a small amount of unreacted ginsenoside Re remaining detectable. Meanwhile, the dominant products observed are the Rg2 epimers, along with the main products of Rg6, F4, and the 20-OCH3-Rg2 epimers. This observation further confirms that the Rg2 epimers serve as the primary intermediates in the transformation. The elimination reaction of the C-20 hydroxyl group takes precedence over the addition reaction of the C-24(25) double bond, resulting in the formation of products Rg6 and F4 containing both the C-20 and C-24(25) double bonds. Methanol molecules exhibit a preference for adding to the double bond at the C-20 position rather than the C-24(25) position in Rg6 and F4, leading to the formation of the 20-OCH3-Rg2 isomers. In addition, the deglycosylated products of the Rh1 epimers and their dehydrated products of Rk3 and Rh4 were observed in trace amounts, further confirming the preference of the hydroxyl elimination reaction at the C-20 position. | ||
| Fig. 8 TIC of ginsenoside Re transformation products in aqueous methanol after 1 h (A), 4 h (B), and 8 h (C). Peak areas of the transformation products at varied temperature (D), catalyst amount (E), and methanol concentration (F). | ||
As shown in Fig. 8B, the reactant ginsenoside Re is completely transformed after 4 h of reaction. The amounts of hydroxylated products at the C-25 position gradually increased during this time. The Rf2 epimers and 25-OCH3-Rg2 epimers, the 25-OH-Rh1 epimers and 25-OCH3-Rh1 epimers, the 25-OH-Rg6/F4 and 25-OCH3-Rg6/F4 are the products of competitive addition reactions of water and methanol molecules to the double bond at the C-24(25) position, respectively. The peak areas of the corresponding products do not show significant differences over time, indicating that the probability of the two competitive addition reactions is similar in 70% aqueous methanol solution.
When the reaction proceeded for 8 h, as shown in Fig. 8C, the content of all 24 products, except for the six main products, increased significantly. Additionally, trace amounts of the cyclization reaction product, 20(25)-epoxy-Rg2, were observed. This indicates that the hydroxyl group at the C-20 position requires more stringent reaction conditions for electrophilic addition to the C-24(25) double bond compared to free solvent molecules such as water and methanol.12,30 The results of the time-course experiments demonstrate that the transformation of ginsenoside Re by HSiW@MeSi in aqueous methanol solution begins with deglycosylation and epimerization reactions, followed by elimination reactions. Subsequently, methanol and water molecules add to the double bonds formed, accompanied by further deglycosylation at the C-6 position.
The effects of reaction temperature, catalyst amount, and methanol concentration on the transformation of ginsenoside Re by HSiW@MeSi were further investigated. As shown in Fig. 8D, the main products observed are the Rg2 epimers, 20-OCH3-Rg2 epimers, Rf2 epimers, Rg6, and F4 after 8 h of reaction at 40 °C, indicating that HSiW@MeSi can effectively catalyze the deglycosylation, elimination, and addition reactions of ginsenoside Re at lower temperatures. When the reaction temperature is gradually increased from 40 °C to 80 °C, the transformation rate is significantly accelerated, resulting in the formation of all 30 transformation products. The amounts of products derived from the addition reactions of water and methanol molecules as well as those from deglycosylation at the C-6 position increase substantially. While the amounts of the Rg2 epimers, Rg6, and F4 slightly decrease due to their involvement as reactants in the subsequent transformation reactions.
Increasing the amount of HSiW@MeSi enhances the concentration of H+ ion in the reaction environment, which is conducive to the forward transformation reaction, similar to increasing the reaction temperature. As shown in Fig. 8E, increasing the amount of HSiW@MeSi from 15.7 mg to 37.8 mg effectively promotes the addition reactions of water and methanol molecules to the double bonds at the C-20 and C-24(25) positions. However, further increasing the catalyst amount to 78.7 mg results in little change in the transformation products, indicating that an excess concentration of H+ ion no longer significantly accelerates the transformation reaction.30
As shown in Fig. 8F, when the methanol concentration is increased from 30% to 70%, the amounts of hydroxylated products at the C-25 position, such as the Rf2 epimers, 25-OH-Rg6, and 25-OH-F4, decrease significantly. In contrast, the amounts of the corresponding methoxylated products, such as the 20-OCH3-Rg2 epimers, 25-OCH3-Rg2 epimers, 25-OCH3-Rg6, and 25-OCH3-F4, markedly increased. These observations further confirm that the addition reactions of water and methanol molecules to the double bonds at the C-20 and C-24(25) positions are competitive, and that the methanol concentration directly affects the distribution of these addition products.
Cyclic transformation of ginsenoside Re using HSiW@MeSi in aqueous methanol
The synthesized HSiW@MeSi is insoluble in the aqueous methanol solution of ginsenoside Re, enabling it to be efficiently separated from the reaction system via centrifugation. The recovered HSiW@MeSi can be reused for the transformation of ginsenoside Re after vacuum drying at 50 °C. To evaluate its recyclability, the transformation of ginsenoside Re was carried out in a 70% methanol aqueous solution for 4 h over multiple cycles, each cycle being carried out in triplicates. As shown in Fig. 9, the transformation rate of ginsenoside Re remains above 80% after three cycles but significantly decreases to 51.4 ± 0.5% after five cycles. The decrease in the transformation rate is due to the deactivation of HSiW@MeSi, which may result from the release of the loaded HSiW from the MeSi framework or a reduction in its crystallinity with increasing cycle numbers. This result demonstrates that while the synthesized HSiW@MeSi effectively facilitates the heterogeneous cyclic transformation of ginsenoside Re, its recyclability still needs further improvement.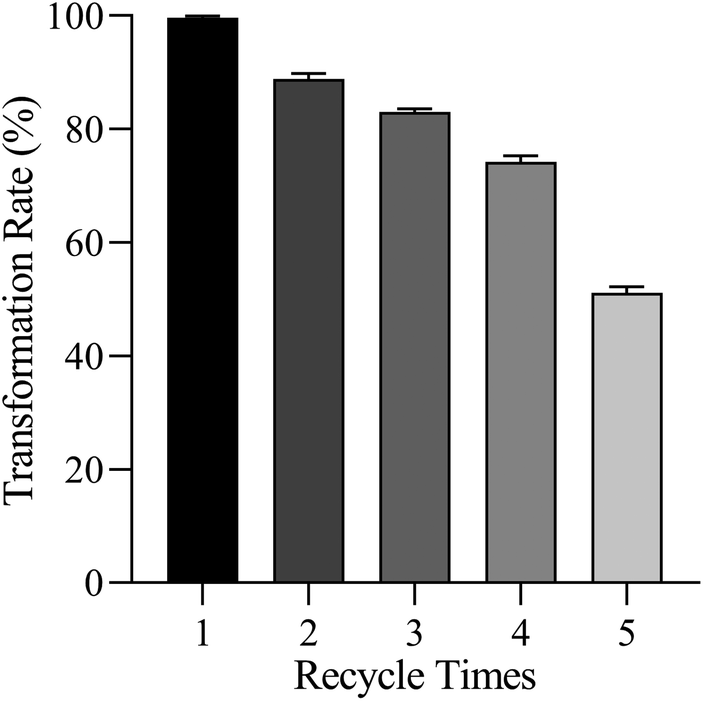 | ||
| Fig. 9 Transformation rate of ginsenoside Re catalyzed by HSiW@MeSi at different cycle times. | ||
Conclusions
In this study, HSiW was successfully loaded into MeSi to form the heterogeneous catalyst, HSiW@MeSi, which was characterized and used for the transformation of ginsenoside Re in an aqueous methanol solution. A total of 30 transformation products were obtained and identified by HPLC-MSn and HRMS. Ginsenoside Re undergoes deglycosylation, epimerization, elimination, addition, and cyclization reactions to produce diverse rare ginsenosides, including six pairs of isomers, three sets of four isomers, and one set of six isomers. The competitive addition reactions between water and methanol molecules produced 14 methoxylated rare ginsenosides. HSiW@MeSi exhibited moderate recyclability, maintaining a transformation rate of over 80% after three cycles. These findings indicate that the use of composite heterogeneous catalysts in the transformation of ginsenosides with organic molecules provides new pathways for generating rare ginsenosides with novel structures.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yanyan Chang: writing – original draft, data curation and formal analysis; Bing Li: methodology; Yusheng Xiao: software; Mengya Zhao: investigation; Yujiang Zhou: investigation; Huanxi Zhao: writing – reviewing & editing, supervision; Yang Xiu: writing – review & editing, funding acquisition and conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Science and Technology Development Plan Project of Jilin Province, China (YDZJ202201ZYTS261) and the Scientific Research Project of Jilin Provincial Education Department, China (JJKH20241090KJ).Notes and references
- H. Ito and M. Ito, J. Nat. Med., 2024, 78, 455–466 CrossRef.
- G. Zhou, C. Wang, S. Mohammadi, W. R. Sawadogo, Q. Ma and C. Yuan, Am. J. Chin. Med., 2023, 51, 1085–1104 CrossRef CAS PubMed.
- J. Li, J. Zhao, X. Wang, Z. Lin, H. Lin and Z. Lin, Food Funct., 2024, 15, 1825–1839 RSC.
- Y. Y. Chen, Q. P. Liu, P. An, M. Jia, X. Luan, J. Y. Tang and H. Zhang, Phytomedicine, 2022, 95, 153883 CrossRef CAS PubMed.
- X. Xu, J. Bi, L. Ping, P. Li and F. Li, Drug Des., Dev. Ther., 2018, 12, 967–979 CrossRef CAS PubMed.
- P. Liang, J. Zhang, J. Hou, R. Feng and J. Yin, Heliyon, 2024, 10, e9906 Search PubMed.
- D. Han, Z. Zhao, T. Mao, M. Gao, X. Yang and Y. Gao, CNS Neurosci. Ther., 2024, 30, e70150 CrossRef CAS PubMed.
- S. Qiu, W. Yang, X. Shi, C. Yao, M. Yang, X. Liu, B. Jiang, W. Wu and D. Guo, Anal. Chim. Acta, 2015, 893, 65–76 CrossRef CAS.
- M. Zheng, F. Xu, Y. Li, X. Xi, X. Cui, C. Han and X. Zhang, BioMed Res. Int., 2017, 2017, 1–10 Search PubMed.
- S. Luo, D. Tian, R. Gao, C. Cui, B. Yang and Z. Wu, Int. J. Biol. Macromol., 2025, 291, 139230 CrossRef.
- H. Wang, J. Chen, Z. Pei, J. Huang, J. Wang, S. Yang and H. Li, Biofuel Res. J., 2025, 12, 2306–2318 CrossRef CAS.
- M. Zhao, L. Tian, Y. Xiao, Y. Chang, Y. Zhou, S. Liu, H. Zhao and Y. Xiu, ACS Omega, 2023, 8, 43285–43294 CrossRef CAS PubMed.
- R. Wang, J. Li, H. Hu, J. Li, Y. Yang, L. Yang and Z. Wang, J. Ginseng Res., 2018, 42, 270–276 CrossRef.
- J. Cao, C. Liu, Q. Wang, Y. Li and Q. Yu, Sci. China: Chem., 2017, 60, 748–753 CrossRef CAS.
- J. Huang, Y. Ding, J. Li, Z. Hu, S. Saravanamurugan, J. Wang, Y. Su, S. Yang and H. Li, Carbon Energy, 2025, 7, e675 CrossRef CAS.
- M. José da Silva, J. A. Vergara Torres and C. B. Vilanculo, RSC Adv., 2022, 12, 11796–11806 RSC.
- Z. Shen, F. Long, T. Ma, H. Li, A. Li, Q. Feng, J. Liu and Y. Sun, ChemSusChem, 2020, 13, 6016–6027 CrossRef CAS PubMed.
- C. N. Dias, I. C. M. S. Santos Vieira, C. R. Gomes, F. Mirante and S. S. Balula, Nanomaterials, 2024, 14, 733 CrossRef CAS PubMed.
- C. B. Hong, T. Wang and H. Liu, Inorg. Chem., 2023, 62, 4054–4065 CrossRef CAS.
- M. B. Colombo Migliorero, V. Palermo, A. Ponzinibbio and G. P. Romanelli, Rev. Chem. Eng., 2025 DOI:10.1515/revce-2024-0085.
- E. G. Zhizhina, L. L. Gogin, Y. A. Rodikova and V. I. Bukhtiyarov, Kinet. Catal., 2021, 62, 197–232 CrossRef CAS.
- Y. Feng, Z. Liao, M. Li, H. Zhang, T. Li, X. Qin, S. Li, C. Wu, F. You, X. Liao, L. Cai, H. Yang and Y. Liu, Adv. Healthcare Mater., 2023, 12, 2201884 CrossRef CAS PubMed.
- A. Barkat, S. Beg, S. K. Panda, K. S Alharbi, M. Rahman and F. J. Ahmed, Semin. Cancer Biol., 2021, 69, 365–375 CrossRef CAS PubMed.
- I. W. Zapelini, L. L. Silva and D. Cardoso, Braz. J. Chem. Eng., 2023, 41, 409–416 CrossRef.
- X. Chen, J. Wang, Y. Yu, H. Chen, X. Wu and S. Gao, Catal. Lett., 2022, 152, 3807–3813 CrossRef CAS.
- S. Saroj and S. Rajput, Indian J. Pharm. Educ. Res., 2020, 54, 590–601 CrossRef.
- W. M. Saod, I. W. Oliver, D. F. Thompson, A. Contini and V. Zholobenko, Int. J. Environ. Anal. Chem., 2023, 104, 9772–9784 CrossRef.
- B. Huang, Y. Chang, H. Wang and Z. Qu, Chem. Eng. J., 2024, 484, 149658 CrossRef CAS.
- Y. Hou, J. Ma, T. Wang and Q. Fu, Mater. Sci. Semicond. Process., 2015, 39, 229–234 CrossRef CAS.
- M. Zhao, Y. Xiao, Y. Chang, L. Tian, Y. Zhou, S. Liu, H. Zhao and Y. Xiu, J. Ginseng Res., 2024, 48, 366–372 CrossRef PubMed.
- R. Ren, H. Li, Q. Jiang, X. Wang and D. D. Y. Chen, Anal. Bioanal. Chem., 2022, 415, 887–897 CrossRef.
- A. Mollar Cuni, S. Martín, G. Guisado Barrios and J. A. Mata, Carbon, 2023, 206, 314–324 CrossRef CAS.
- L. Rahimi, Y. Mansoori, A. Nuri, B. Koohi Zargar and D. Esquivel, Appl. Organomet. Chem., 2020, 35, e6078 CrossRef.
- Y. Yang, S. Xu, K. Yang, Y. Sun, R. Yang, Y. Hu, G. Chen and H. Cai, Foods, 2023, 12, 4316 CrossRef CAS.
- Y. Qiu, N. Wang, Z. Yu, X. Guo and M. Yang, Front. Pharmacol, 2024, 15, 1425794 CrossRef CAS.
- M. Li, Z. Fan, Q. Gao, Y. He, A. Xu, Z. Gu, S. Wang, H. Bai, Y. Liao and R. Zhang, Adv. Sci., 2025, 2500430 CrossRef CAS PubMed.
- S. Shi, K. Li, J. Peng, J. Li, L. Luo, M. Liu, Y. Chen, Z. Xiang, P. Xiong, L. Liu and W. Cai, Biomed. Pharmacother., 2022, 149, 112828 CrossRef CAS.
- M. Y. Li, Y. Li, L. L. Wang, F. Xu, X. Y. Guo, J. Zhang, Y. Lv, P. P. Wang, S. Q. Wang, J. Min, X. Zou and S. Q. Cai, Front. Chem., 2023, 11, 1179956 CrossRef CAS.
- J. Song, R. Dai, Y. Deng and F. Lv, Phytochemistry, 2018, 147, 147–157 CrossRef CAS.
- Y. Xiu, X. Li, X. Sun, D. Xiao, R. Miao, H. Zhao and S. Liu, J. Ginseng Res., 2019, 43, 508–516 CrossRef.
- G. Chen, J. Li, X. Yang and Y. Wu, Appl. Catal., A, 2006, 310, 16–23 CrossRef CAS.
- Y. Chen, X. Zhang, M. Dong, Y. Wu, G. Zheng, J. Huang, X. Guan and X. Zheng, J. Taiwan Inst. Chem. Eng., 2016, 61, 147–155 CrossRef CAS.
- M. H. Firouzjaee and M. Taghizadeh, Ind. Eng. Chem. Res., 2023, 62, 1322–1337 CrossRef.
- W. Cheng, J. Nie, L. Xu, C. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Huang and X. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 CrossRef CAS.
- A. A. Ansari, A. Khan, J. P. Labis, M. Alam, M. Aslam Manthrammel, M. Ahamed, M. J. Akhtar, A. Aldalbahi and H. Ghaithan, Mater. Sci. Eng., C, 2019, 96, 365–373 CrossRef CAS.
- P. Hongmanorom, J. Ashok, G. Zhang, Z. Bian, M. H. Wai, Y. Zeng, S. Xi, A. Borgna and S. Kawi, Appl. Catal., B, 2021, 282, 119564 CrossRef CAS.
- M. Zhang, S. Huang, W. Liu, J. Yang, M. Zhu and S. Ho, Chemosphere, 2023, 344, 140322 CrossRef CAS PubMed.
- W. Hu, H. Xu, Z. Zhang, Y. Duan, X. Lu, L. Lu, C. Si, Y. Peng and X. Li, Biomass Bioenergy, 2024, 186, 107275 CrossRef CAS.
- Y. C. Chen, B. W. Lin, W. C. Hsu and Y. S. Wu, Mater. Lett., 2014, 118, 72–75 CrossRef CAS.
- A. E. R. S. Khder, H. M. A. Hassan and M. S. El-Shall, Appl. Catal., A, 2012, 411–412, 77–86 CrossRef CAS.
- H. Ding, H. Sun and Y. Shan, J. Photochem. Photobiol., A, 2005, 169, 101–107 CrossRef CAS.
- Y. Xiu, H. Zhao, Y. Gao, W. Liu and S. Liu, New J. Chem., 2016, 40, 9073–9080 RSC.
- Y. Gao, Y. Feng, Y. Chang, Z. Zhu, H. Zhao, W. Xu, M. Zhao, Y. Xiao, L. Tian and Y. Xiu, ACS Omega, 2024, 9, 22744–22753 CrossRef CAS PubMed.
- Y. Z. Liang, M. Guo, Y. F. Li, L. J. Shao, X. M. Cui and X. Y. Yang, ACS Omega, 2022, 7, 14910–14919 CrossRef CAS PubMed.
- C. Bockisch, E. D. Lorance, H. E. Hartnett, E. L. Shock and I. R. Gould, J. Org. Chem., 2022, 87, 14299–14307 CrossRef CAS PubMed.
- S. M. Jeong, J. H. Lee, J. H. Kim, B. H. Lee, I. S. Yoon, J. H. Lee, D. H. Kim, H. Rhim, Y. Kim and S. Nah, Mol. Cells, 2004, 18, 383–389 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02261d |
| This journal is © The Royal Society of Chemistry 2025 |