
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The radioactive 103Pd and 109Pd palladium bipyridyl–bisphosphonate complexes for radionuclide therapy of bone metastatic tumor cells†
Geeva Prasanth Annamalaisamy ,
Monika Lyczko* and
Aleksander Bilewicz
,
Monika Lyczko* and
Aleksander Bilewicz
Center of Radiochemistry and Nuclear Chemistry, Institute of Nuclear Chemistry and Technology, Dorodna 16, Warsaw, 03-195, Poland
First published on 3rd June 2025
Abstract
Two radioisotopes of palladium, 103Pd and 109Pd, are considered promising candidates for therapeutic applications because they emit Auger electrons, which are known for their effectiveness in targeting and destroying cancerous cells. We synthesized complexes of 103Pd and 109Pd with two bipyridyl and one alendronate molecule. The complexes demonstrated stability and a strong affinity for the surface of hydroxyapatite grains, the main mineral component of bones. Radioactive complexes show significantly higher cytotoxicity against human prostate (DU 145) and ovarian Her2 positive (SKOV-3) cancer cell lines compared to trastuzumab labeled with the Auger electron emitter 125I and cisplatin. The biological studies showed that both 103Pd, a pure Auger electron emitter, and 109Pd, which emits both beta and Auger electrons, demonstrate high cytotoxicity. Furthermore, it was observed that in the tested complexes, 109mAg, a decay product of 109Pd, was released from the complex following the decay of 109Pd. In contrast, 103mRh, a decay product of 103Pd, remained within the structure of the complex. The release of 103mRh from the 103Pd complex is inhibited by the presence of delocalized electrons in the aromatic bipyridyl ligand. The concept of using a 109Pd/109mAg and 103Pd/103mRh generator encourages further exploration of this treatment strategy.
1. Introduction
The primary reason for treatment failure and the leading cause of death among cancer patients is the development of secondary tumours in distant organs or tissues far from the original cancer site, known as metastases. This spread is one of the most dangerous aspects of cancer, making it harder to treat and often associated with a poorer prognosis.1After the liver and lungs, bones are the most susceptible site for cancer metastasis.2 This is because the bone microenvironment provides a fertile ground for various types of cancer cells.3 Bone metastases cause accelerated bone resorption leading to serious complications such as severe pain, spinal cord compression, and pathological fracture.3 Currently, bone metastases are treated symptomatically with pain management, systemic chemotherapy, radiotherapy, surgery, or a combination thereof. According to a review,4 the clinical administration of therapies for skeletal metastases is achieved by the use of painkillers, cytostatic chemotherapy, radiotherapy and especially by administering bisphosphonates and radiopharmaceuticals based on calcium analogues and phosphonates. The main clinical goals of these therapies are to relieve pain, improve quality of life, and reduce the risk of complications such as pathological fractures, spinal cord compression, and hypercalcemia (high levels of calcium in the blood). Unfortunately, it is very rarely possible to extend the time of survival.
Radionuclide therapy has been one of the most effective treatments for bone metastases for many years. For this purpose, α and β− particle-emitting radiopharmaceuticals have been applied. Very good results are obtained in the treatment of pain as well as in the case of 223Ra (α emitter) prolongation of survival time in Castration-Resistant Prostate Cancer (CRPC) patients with bone metastases. Two classes of therapeutic bone-seeking radiopharmaceuticals are used in practice. Calcium-analogue radiopharmaceuticals such as 89SrCl2 and 223RaCl2, as well as polyphosphates of β− emitters, mainly 153Sm, 177Lu, and 188Re are available. Radiation doses to bone metastases are limited due to the radiosensitivity of bone marrow, making it unsafe to increase them without risk of destroying the bone marrow.5 The low effectiveness of radiopharmaceuticals in treating bone cancer metastases is due to the necessity of using suboptimal doses. Hence, better therapeutic outcomes are achieved by using 223Ra, which emits short-range α particles. However, there can be serious complications during 223Ra therapy because the decay of 223Ra forms α-emitting decay products 211Pb and 211Bi, which can accumulate in healthy tissues.
However, it is expected that the use of Auger and conversion electron emitters can significantly improve the efficiency of polyphosphate-based radiopharmaceuticals, allowing not only the treatment of pain but also prolonging survival. The effectiveness of radiopharmaceuticals based on Auger and conversion electron emitters in treating bone metastases is due to the fact that the emitted energy is deposited in a very short range, on the micro and nanometre scale, without causing damage to the bone marrow. For the above reasons, Auger electron therapy is a promising treatment method, especially in the case of very small tumors, such as metastatic cancer.6 In the treatment of bone metastases, Auger and conversion electron emitters allow the use of very high therapeutic doses without damaging the bone marrow.7 An attempt was made to use 117mSn for the treatment of bone cancer metastases.8 It emits monoenergetic conversion electrons, which allows for using large bone radiation doses without excessive radiation to the bone marrow. A high bone-to-marrow ratio of 11 was obtained. However, the 117mSn-DTPA complex has minimal affinity for bone and tin as a p-block metal is easily trans chelated.9 Additionally, 117mSn can only be obtained by irradiation with alpha particles in the reaction 116Cd(α,3n)117mSn or in the inefficient reaction on fast neutrons 117Sn(n,n′γ)117Sn.

In the case of Auger electron emitters, only two papers on bisphosphonate complexes of Auger electron emitters have been published,10,11 and both concern 195mPt. The authors synthesized a Pt2+ bisphosphonate (alendronate) complex in which two coordination sites of Pt2+ were occupied by the phosphonate group and two remained by ethylenediamine. Following attachment to bone cancer cells having a lower pH, the complex disintegrated and formed Pt (Et)Cl2, which diffused and penetrated the neoplastic tissue, intercalating into DNA in a similar way to cisplatin.11 Interestingly, this radiotherapeutic efficacy of 195mPt-bisphosphonate was also much higher than 223Ra, since the 32-fold increased DNA damage caused by 195mPt-bisphosphonate treatment significantly exceeded previously reported values for 223Ra treatment (only two- to threefold enhanced DNA damage).10,11 In our opinion, these studies open a new path in the treatment of cancerous bone metastases. Nevertheless, the problem in obtaining therapeutic activities of 195mPt and also 193mPt is a barrier to their widespread clinical application.12
The similar d8 electron configurations and high chemical similarity of Pd2+ and Pt2+ cations make palladium radionuclides interesting candidates for Auger electron therapy13 Additionally, numerous publications have confirmed the anti-cancer properties of Pd2+ cations and their complexes14 Therefore, the two palladium radionuclides, 103Pd (t1/2 = 16.99 d) and 109Pd (t1/2 = 13.59 h), can serve as excellent alternatives to 195mPt. 103Pd is widely used in seed form for prostate brachytherapy because it emits X-ray radiation in the 20–23 keV range. The wide use of 103Pd in brachytherapy has led to the development of a method for its large-scale production. 103Pd can be produced in a reactor by irradiating a 102Pd target with neutrons or in a cyclotron by irradiating a natural 103Rh target with protons in p, n reaction. Although 103Pd emits only a few Auger electrons, 103mRh produced by the decay of 103Pd is considered an attractive radionuclide for Auger electron therapy.15,16
Unfortunately, 103mRh has a short half-life (t½ = 56.11 min), which is challenging and makes it impractical to prepare 103mRh radiopharmaceuticals. However, it can be used in the form of an generator of 103Pd/103mRh for targeted Auger electron therapy.
109Pd also has excellent potential for radionuclide therapy. 109Pd undergoes β− decay (βmax = 1.12 MeV, 100% yield) to 109mAg (t½ = 39.6 s). The formed metastable 109mAg decays to stable isotope 109Ag, which is associated with the emission of an 88 keV photon (3.6%), which is subsequently followed by a cascade of emissions involving both conversion electrons and Auger electrons. Such properties enable its simultaneous application in both low- and high-LET internal radiation therapy. Recently published groundbreaking studies by Mueller et al.17 have demonstrated that the utilization of similar Auger and β− electron emitter – 161Tb can lead to a significantly greater therapeutic effect compared to similar studies involving 177Lu.
In this study, we propose the utilization of 103Pd and 109Pd in the form of 103Pd/103mRh and 109Pd/109mAg generators for labeling of bisphosphonate in Auger electron therapy. Due to the limited availability of 103Pd, most studies were conducted using 109Pd. However, work with 103Pd will continue as we have secured supplies of 103Pd within the PRISMAP project in 2025. 109Pd can be obtained in reactor neutron irradiation. According to Das et al.,18 irradiation of enriched 102Pd (enrichment 98%) with neutrons with a flux of 3 × 1013 n cm−2 s−1 for 72 h gives radionuclide-pure 109Pd with a specific activity of 1.85 GBq mg−1. When a high-flux reactor (>1015 n cm−2 s−1) is used, the specific activity can increase to 40 GBq mg−1.
The 103Pd and 109Pd were utilized as mixed bipyridine–bisphosphonate complexes. Two papers on palladium alendronate compounds were published recently.19,20 In these publications, mixed complexes of Pd(II) with alendronate and bipyridine or 1,10-phenanthroline were synthesized and studied. Their therapeutic effect on human prostate cancer (DU 145) and breast cancer (MDA-MB-231) cells was studied in vitro and compared with cisplatin. The results of the conducted studies indicate that the complexes exhibit greater cytotoxicity towards human prostate and breast cancer cell lines compared to normal human prostate and breast cell lines. The mechanism of the anti-cancer activity of these complexes has not been described in detail. Since Pd2+ has the same electron configuration (d8) as Pt2+, it can be assumed that the mechanism of the anti-tumor action of the Pd2+ phosphonate complexes should be similar to that of the described Pt2+ complexes21 The choice of NN co-ligands is aimed at highlighting DNA as a target. It is well known that metal complexes featuring extended planar aromatic ligands can reversibly bind to DNA by intercalating the planar aromatic portion between nucleobases.19,22 We chose the alendronate molecule as a bone-targeting ligand due to its high affinity for bone tissue. As previously mentioned, non-radioactive palladium diamine complexes with alendronate exhibited significant anti-tumor properties.19,20
In our publication, we also study the rejection of decay products 103mRh and 109mAg from 103Pd and 109Pd mixed aleodronian–bipyridine complexes. The release of Auger electron emitting decay products from the both palladium radioisotopes or their absence is crucial for the radionuclide therapy.
2. Materials and methods
2.1 Chemical reagents
Potassium tetrachloropalladium (K2PdCl4), 2,2′ bipyridyl (C10H8N2) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hydrochloric acid and sodium hydroxide were purchased from POCH (Gliwice, Poland). For cell and other biological studies phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich (St. Louis, MO, USA), Cell Titer® 96 Aqueous One solution reagent (MTS assay) from Promega (Mannheim, Germany). SKOV-3 and DU145 cell line bought from American Type Tissue culture collection (ATCC, Rockville, MD, USA) and were cultured in Mc Coy's and MEM-Eagles medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin (Beth Haemek, Isreal).2.2 Radionuclides
109Pd was produced by irradiating 3 mg of natural palladium powder or 1 mg of >99% enriched 108Pd with a thermal neutron flux of 1.5 × 1014 n cm−2 s−1 for 7 hours at the Maria nuclear reactor in Poland. After a 3 h cooling time, the targets were dissolved in 200–400 μL of aqua regia (concentrated HNO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HCl–1:3) and then heated at 130 °C until complete evaporation. To convert nitrates to chlorides, the solid residue was dissolved in 0.1 M HCl three times (100 μL) and evaporated at 130 °C. Finally, the solid residue was dissolved in 1 mL of 6 M HCl to obtain H2PdCl4 solution.
:HCl–1:3) and then heated at 130 °C until complete evaporation. To convert nitrates to chlorides, the solid residue was dissolved in 0.1 M HCl three times (100 μL) and evaporated at 130 °C. Finally, the solid residue was dissolved in 1 mL of 6 M HCl to obtain H2PdCl4 solution.
When natural palladium (Pd) is irradiated with neutrons, it can produce 111Ag as an unwanted impurity in the reaction. 110Pd(n,γ)111Pd → 111Ag. It is crucial to remove it from the solution prior to application.18 The removal of 111Ag can be achieved by precipitating it as AgCl using the addition of AgNO3, following the modified procedure described by Das et al.18 Shortly, a total of 100 μL of a 0.1 M AgNO3 solution in 0.1 M HNO3 (equivalent to 20 mg mL−1) was added to a 1 mL solution of PdCl42− in 6 M HCl. After 2 minutes, the AgCl precipitate was collected by centrifuging the mixture at 4600 rpm for 5 minutes. The resulting supernatant was separated from the AgCl precipitate, and then was evaporated until completely dry. The procedure was repeated using deionized water, and finally, palladium was suspended in 100 μL of 0.1 M HCl. The diluted solutions obtained from the radiochemical processing of the irradiated target were analyzed using a High-Purity Germanium (HPGe) detector connected to a PC-based Multichannel Analyzer (MCA, Canberra). The 88 keV gamma peak, which has an abundance of 3.67%, emitted by 109mAg, was utilized to determine the radioactivity of 109Pd. With an isotopically enriched 108Pd target, removal of silver is unnecessary due to the very low content of the 111Ag radionuclide.
103Pd was obtained as part of the European PRISMAP project from the Institute Laue–Langevin in Grenoble. The 103Pd was produced by 7 h irradiating 1.5 mg of an enriched 102Pd target (90.7%) in ILL High-Flux Reactor, with a neutron flux of >1015 n cm2 s−1. For the dissolution of the irradiated 102Pd target, the same procedure used for obtaining 109Pd was followed. After dissolution, we obtained 0.5 mL of solution with a concentration of 3.8 GBq mL−1 and with a specific activity of 1.3 GBq mg−1.
2.3 Instruments
Mass spectra were measured using the Q-TOF Premier mass spectrometer, Waters (Framingham, Massachusetts, USA) in the Institute of Biochemistry and Biophysics Polish Academy of Science.FTIR spectra were recorded on the NICOLETE iS10 Thermo Scientific spectrometer (Waltham, Massachusetts, USA) in the 4000–400 cm−1 range using KBr pellets.
Radiochemical purity was assessed by Thin Layer Chromatography (TLC) using Watman Chromatographic paper 3MM. The chromatogram was developed by water/acetonitrile mixture (1:3 v/v) dried and the radioactivity distribution on the paper strip was measured by Cyclone® Plus Storage Phosphor System.
The identity studies of the radioactive 109Pd2(bpy)2ale complex (trace level) with the non-radioactive complex, synthesized from a macro amount of Pd and previously identified by mass spectroscopy, were performed using high performance liquid chromatography Merck Hitachi HPLC system (Tokyo, Japan) equipped with a reversed-phase C-18 column (Aeris Peptide 3.6μ XB-C18 150 × 4.60 mm) with water (A) and water/acetonitrile mixture (50/50) with 0.001 M EDTA as mobile phase. The following methods were used for complex separation (0–20 min 95% A to 5% B, 20–25 min 5% A to 95% B, 25–30 min 95% A to 5% B) with a flow rate of 1 mL min−1.
2.4 Synthesis of Pd2(bpy)2ale complex
The Pd2(bpy)2ale complex was prepared using a modified method based on the procedure reported by Cipriani et al.20 Briefly, aqueous solutions of 0.15 mmol of K2PdCl4 and 0.015 mmol of bipyridine ligand were added dropwise in equal molar proportions to the reaction mixture (total volume 5 mL) and stirred at room temperature for 1 h. In the meantime, molar excess of alendronate (0.30 mmol) and 0.30 mmol of triethylamine were prepared in 10 mL of aqueous solution. The mixture was added dropwise until the solution become clear brown and next was heated at 100 °C under reflux. The synthesis was maintained at the pH = 7. Once the solution was clear, it was concentrated to half its initial volume. Acetone was added to precipitate the product and the obtained precipitate was filtered and washed with diethyl ether and ethanol to remove unreacted bipyridine.2.5 Synthesis of radioactive 103Pd2(bpy)2ale and 109Pd2(bpy)2ale complexes
To synthesize the radioactive complex, 420 MBq of 109Pd was added to the bipyridine (equal molar ratio) in 100 μL of water. After adjusting the pH to 7 using 0.1 M NaOH and HCl, the solution was sealed in an Eppendorf tube and left at room temperature for 1 hour. A 200 μL aqueous solution containing 100 times the excess of alendronate and triethylamine was added to the mixture dropwise while maintaining a pH of 7. The solution was then sealed again and heated to 60 °C for 1 h.2.6 Stability of the complex
The stability of the complex was analyzed at 37 °C in human serum (HS) and PBS over 24 hours (∼2 half-lives of 109Pd). 50 μL of a radioactive complex was added to freshly prepared HS and PBS, and incubated at 37 °C for 24 hours. The stability was analyzed at specific time intervals using radio ITLC.2.7 Release of 109mAg from 109Pd2(bpy)2ale and 103mRh from 103Pd2(bpy)2ale complexes
To study the release of 109mAg from the 109Pd2(bpy)2ale complex, we employed an original procedure involving forming an insoluble AgCl precipitate, in which 109mAgCl is co-precipitated. We added 10 μL of the 109Pd2(bpy)2ale complex to a centrifuge tube containing 1 mL of NaCl solution, resulting in a radioactivity level of approximately 7.5 × 104 cpm. After, to this solution, 100 μL of 0.1 M AgNO3 in 0.1 M HNO3 was added. The sample was mixed thoroughly, and the AgCl precipitate was separated from the solution using a centrifuge for 40 seconds. The supernatant was carefully pipetted away from the precipitate, and the AgCl was then dispersed in 1 mL of H2O. Activity measurements began 124 seconds after the precipitation of AgCl, which corresponds to three half-lives of 109mAg, and were taken at 15 second intervals.To investigate the release of 103mRh from the 103Pd2(bpy)2ale complex, the ITLC technique was employed (refer to the stability section). The Cyclone® Plus Storage Phosphor System was utilized to measure the X-ray and low-energy γ radiation emitted by 103Pd and 103mRh with high sensitivity.
2.8 Sorption on hydroxyapatite
Sorption on hydroxyapatite was performed according to the procedure described by Majkowska et al.23 Samples of hydroxyapatite weighing 50 mg, 75 mg, and 100 mg were added to 2 mL of saline and allowed to equilibrate for 1 hour. Following this, 50 μL of the radioactive 109Pd2(bpy)2ale complex was added, and the mixture was shaken at room temperature for 12 hours. The dispersion was centrifuged, and two aliquots of supernatant (2 × 500 μL) were taken to measure the activity using a scintillation gamma counter. The percentage sorption of 109Pd2(bpy)2ale complex was calculated using the formulae sorption (%) = (1 − A/B) × 100, where A is the average activity of the sample after equilibration and B is the initial activity of the solution.2.9 Cytotoxicity studies
The MTS assay was employed to evaluate the ability of cells to proliferate. Tests were conducted on SKOV-3 (ovarian adenocarcinoma) and DU 145 (prostate cancer) cells. SKOV-3 and DU145 cells were maintained in McCoy's and MEM Eagle's media, respectively. Both cell types were seeded in 96-well plates at a density of 3 × 103 cells per well, suspended in 100 μL of culture medium. After 24 hours of seeding, the 109/103Pd2(bpy)2ale complex with the desired activities (6 MBq mL−1, 12 MBq mL−1, 25 MBq mL−1, 50 MBq mL−1) was suspended in the medium and incubated for a period of 24 to 72 hours. Before adding 20 μL of MTS cells were washed with PBS and fresh medium was added (100 μL). Absorbance was measured at 490 nm.2.10 DNA break (γH2AX)
γH2AX assay was performed to evaluate the double-strand break of DNA caused by the influence of 109Pd2(bpy)2ale complex. DU145 cells with density of 0.25 million cells per well were seeded in six well plates. Five sterile coverslips were placed in each well. After 24 h of incubation the medium was removed and treated with 109Pd2(bpy)2ale complex with two concentrations (25 MBq mL−1 and 6 MBq mL−1) and control cells. The cells were incubated for 4 h and 24 h. At proper time intervals the coverslips were transferred to 24 well plates (1 slip per well for each concentration). The slips were washed with PBS several times and 350 μL per well of primary antibody (anti-phospho-histone H2A.X (ser139)), clone JBW301 with dilution of 1:100 in blocking buffer and left at 4 °C overnight. Next day, wells were washed and treated with 250 μL per well of anti-mouse IgG(H + L) conjugated with CFTM633 and incubated for 2 h with time to time shaking at room temperature. Then the cells were washed with water several times and Hoechst 33258 was used for nuclei staining. The stained nuclei and breaks were analysed by FV-1000 confocal microscopy (Olympus corporation, Tokyo, Japan) with ex/em maxima: 630/650 nm for CF633 and ex/em maxima: 325/454 nm for Hoechst 33258. The images were then analysed by Fiji ImageJ.
3. Results and discussion
3.1 Synthesis of nonradioactive Pd2(bpy)2ale and radioactive 103Pd2(bpy)2ale and 109Pd2(bpy)2ale complexes
The synthesis procedure for the Pd alendronate bipyridyl mixed complexes is illustrated in Fig. 1. The initial step results in the formation of Pd(bpy)Cl2 complex, which is insoluble in water. The subsequent addition of alendronate leads to the formation of dinuclear species that contain one bisphosphonate for every two Pd(bpy) units. The complex obtained was yellow and soluble in water. Under similar synthetic conditions, Cipriani et al.20 also obtained a soluble in water yellow complex to which, based on elemental analysis, IR, and 31P NMR, assigned a structure in which two alendronate molecules bind to the Pd2+ cation via the terminal amine nitrogen. However, the authors did not perform identification by mass spectrometry. Since N-coordination is strongly favored under basic conditions, the synthesis was conducted at high pH. However, unlike Ciprani, we carried out the synthesis at pH = 7, where O coordination is preferred.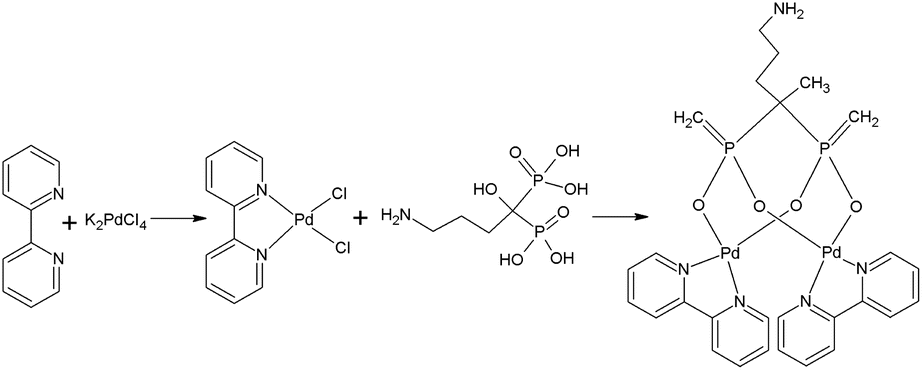 | ||
| Fig. 1 Synthesis of palladium complex with bipyridyl and alendronate ligands 109Pd2(bpy)2ale. | ||
The recorded MS spectra unambiguously indicate the presence of the dinuclear complex with M+H+ 771.9360 m/z (calc. 771.9388) (Fig. S1†). In a similar synthetic procedure, such a complex was obtained and characterized in the work of Fathy et al.19 The IR spectrum recorded by us (Fig. 2) closely resembled the spectrum obtained by Fathy et al., displaying characteristic band patterns that confirm the presence of the corresponding bipyridyl and bisphosphonate ligands in the synthesized palladium complex. The characteristic band between 1200 and 1600 cm−1 confirms the presence of bipyridyl ligand related to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN vibrations. Phosphonate band region was observed in the region from 900–1300 cm−1. The most important band from υ(Pd–O) occurring at a wavenumber of 550 cm−1 confirms that the palladium in the complex is bonded to bisphosphonates through an oxygen atom. This band was not present in the studies of Ciprani et al.,20 indicating a difference in the structure of the obtained products.
C and CN vibrations. Phosphonate band region was observed in the region from 900–1300 cm−1. The most important band from υ(Pd–O) occurring at a wavenumber of 550 cm−1 confirms that the palladium in the complex is bonded to bisphosphonates through an oxygen atom. This band was not present in the studies of Ciprani et al.,20 indicating a difference in the structure of the obtained products.
 | ||
| Fig. 2 IR spectrum for non-radioactive Pd2(bpy)2ale complex, bipyridine, and sodium alendronate. | ||
The same procedure was used to synthesize the radioactive 103Pd2(bpy)2ale and 109Pd2(bpy)2ale complexes. The progress of the reaction was monitored by ITLC using water: acetonitrile (1:3 v/v), in which Pd2(bpy)2ale moved towards the solvent front (Rf = 0.7), while free PdCl2 remained at the origin (Rf = 0.1). The complexation efficiencies were over 80% for 109Pd and exceeded 85% for 103Pd. The highest yield was obtained at a temperature of 60 °C and a pH of 7.
HPLC chromatography was employed to compare the nonradioactive complexes with the synthesized ones at trace level radioactive ones. The chromatograms are presented in Fig. S2.† The presence of the same peak at 3.5 minutes in both the UV-Vis detector and the radio detector indicates that in both cases, the dinuclear Pd2(bpy)2ale complex presented in Fig. 1 is formed. The synthesized radioactive complexes maintain stability at 37 °C in human serum and PBS buffer for 24 hours, approximately two half-lives of 109Pd.
3.2 Liberation of 109mAg from 109Pd2(bpy)2ale and 103mRh from 103Pd2(bpy)2ale complexes
In nuclear medicine, when using in vivo generators, it is crucial to consider the behavior of the daughter radionuclide following radioactive decay. In our previously published papers, we studied liberation of 109mAg from 5 nm 109Pd-PEG nanoparticles and the 109Pd-cyclam complex.24 Using the original procedure, we showed that while 109mAg is completely released from the 109Pd-cyclam complex, in the case of 5 nm Pd nanoparticles 109mAg remains in the nanoparticle.The same procedure was used to study the release of 109mAg from the 109Pd2(bpy)2ale complex. Fig. 5 illustrates the decay curve of 109mAgCl precipitated from the complex solution. Based on the decay curve it is found that the calculated half-life is similar to that of t1/2 39 s of 109mAg. Extrapolating the activity of the AgCl precipitate to time zero, we observed that the calculated activity of 109mAg is close to the initial activity of 109Pd in the complex, evidencing the complete release of 109mAg from the complex and coprecipitation as 109mAgCl. This aligns with the interpretation presented by Wang et al.,25 who hypothesized, based on the behaviour of 166Ho in the 166Dy/166Ho generator, that the loss of daughter radionuclides was due to their de-excitation through internal conversion. This process results in the emission of Auger electrons rather than the recoil energy associated with the emission of β− particles and neutrinos. As a result the de-excited daughter radionuclides become highly charged, leading to electron uptake from surrounding chelator donor atoms.
When electron transfer occurs to highly charged atoms, the donor atoms in chelators become positively charged. This positive charge creates a repulsive force between the atoms, leading to breaking metal–ligand bonds. As a result, daughter radionuclides are released as free cations. In the case of the 109Pd–109mAg system, this release also happens because formed Ag+ forms significantly weaker complexes than Pd2+.The longer half-life of 103mRh (t½ = 56.11 min) compared to 109mAg (t½ = 39 s) makes the release of Auger electron-emitting 103mRh from 103Pd conjugates, or the absence of this release, significantly more important than that of 109mAg.
In our studies, we used the ITLC technique to investigate the release of 103mRh from the 103Pd2(bpy)2ale complex. The ITLC recorded radioactivity on the strip using the Cyclone® Plus Storage Phosphor System, which measures X-ray radiation with high sensitivity. This allowed us to record the X-ray emitting positions on the strip emitted of both 103Pd and 103mRh radionuclides. The results of stability studies conducted using ITLC in a water-acetonitrile system (Fig. 3). The liberation of 103mRh from 103Pd complexes was previously examined during the development of the 103smRh radionuclide generator. In the studies of Jensen et al.,26 it was shown that after the decay of 103Pd, approximately 7% of 103mRh is liberated from the macrocyclic [103Pd]16aneS4-O-octyl complexes. This phenomenon can be explained by Zeevaart and coworkers,27,28 who conducted detailed calculations of recoil energy associated with the emission of Auger electrons, photons, and neutrinos. They reported that the total energy generated is lower than the chemical bond energy in palladium–macrocycle complexes and is insufficient to allow the escape of particles from the complex. However, the highly positive charge of the Rhn+ cation after the emission of Auger electrons caused partial release of Rh3+ cations. The complete retention of Rh3+ in the 103Pd(bpy)ale complex observed in Fig. 4 is related to the presence of a bipyridyl ligand containing delocalized electrons. According to Nath et al.29 these ligands, such as for example heme, show the ability to quench the observed phenomenon known as the “Coulomb explosion” by transporting delocalized electrons to the metal core within time scales equivalent to the Auger cascade. It is also possible that after the decay of 103Pd, the resulting 103mRh will be complexed only by bipyridyl ligand, forming the complex 103mRh(bpy)3+. However, this cannot be confirmed by ITLC analysis.
 | ||
| Fig. 3 Stability of 109Pd2(bpy)2ale in human serum and PBS buffer. | ||
 | ||
| Fig. 4 Retention study of Rh3+ in 103Pd2(bpy)2ale complex. | ||
 | ||
| Fig. 5 Radioactivity of AgCl precipitated from the 109Pd2(bpy)2ale complex solution. The measurement started 133 s after the addition of AgNO3. The radioactivity of the AgCl was measured successively at 15 second intervals. Activity in cpm. | ||
3.3 Sorption of 109Pd2(bpy)2ale and 103Pd2(bpy)2ale on hydroxyapatite (HA)
The study of how effectively bone-seeking radiopharmaceuticals bind to bone matrix is a key indicator of the efficacy of any bone therapeutic agent or pain reliever. Hydroxyapatite (HA) particles, the fundamental chemical component of bone matrix, typically serve as the primary material for binding studies.30 HA is an ionic compound containing Ca2+, PO43−, and OH− ions on its surface. In bone cancer, the metabolism of bone is altered, leading to an increased uptake of calcium and phosphorus species, such as bisphosphonates. Since bisphosphonates have a strong affinity for Ca2+ ions, the primary mechanism for the adsorption of bisphosphonates is coordination to Ca2+ ions on the surface, as well as the potential substitution of surface phosphate anions.31 The adsorption is then described as bidentate coordination via two phosphonate oxygen atoms. In bisphosphonates complexes, the metal cation is coordinated by oxygen atoms from phosphonate. The same atoms are also responsible for the adsorption on the surface of HA. The complexes are kinetically labile and after adsorption on the HA surface, their transformation may occur. Thus, the adsorption of metal bisphosphonate complexes involves the dissociation and formation of the complexes, as well as the adsorption of both the complex species and its components (i.e., the ligand and metal ion) separately.32 For example, the adsorption of Sn(II) diphosphonate complex on hydroxyapatite occurs as the separate adsorption of Sn2+ and diphosphonate after dissociation of the complex.33The hydroxyapatite binding ability, represented as the percentage adsorption of 109Pd2(bpy)2ale zoledronates in relation to the amount of hydroxyapatite (HA), is illustrated in Fig. 6. The adsorption curve exhibits a profile similar to that observed for 153Sm-labelled alendronate; however, the percentage of binding is higher. Fig. 7 shown kinetic of adsorption of trace level 109Pd2(bpy)2ale complex on HAP samples. Pd2(bpy)2ale uptake onto HAP reached maximum after 72 h with 96% adsorption. The course of the curve is analogous to that observed for the non-radioactive (1 μM) complex.19
 | ||
| Fig. 6 Adsorption of 109Pd2(bpy)2ale on HA grains. | ||
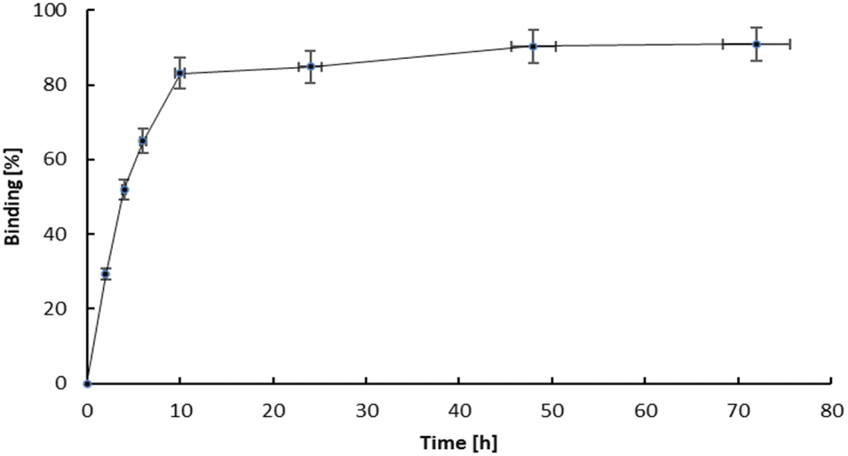 | ||
| Fig. 7 Adsorption kinetics of 109Pd2(bpy)2ale on HA grains. | ||
However, in contrast to the results of these studies, we did not observe the desorption of 109Pd species from early adsorbed 109Pd2(bpy)2ale on HAP. This could be probably due to the shorter duration of the experiment, due to the half-life of 109Pd of 13.59 h.
3.4 In vitro toxicity
The cytotoxicity of nonradioactive Pd2(bpy)2ale and radioactive 103Pd2(bpy)2ale, 109Pd2(bpy)2ale was tested against cancer cells exhibiting the ability to form bone metastases. The viability of SKOV-3 cells overexpressing Her2 receptors, incubated with non-radioactive Pd2(bpy)2ale, was evaluated using the MTS assay. The Pd2(bpy)2ale was tested at concentrations ranging from 30 μg mL−1 (39 nmol mL−1) to 70 μg mL−1 (90 nmol mL−1). This study aimed to determine the cytotoxicity of the obtained bioconjugate toward the studied cells. The impact of non-radioactive Pd2(bpy)2ale on the viability of SKOV-3 cells is shown in Fig. 8. As the concentration of the Pd2(bpy)2ale increases, the viability of the cells gradually decreases. However, cell viability remains stable between concentrations of 50 and 70 μg mL−1. Additionally, in all concentrations examined, toxicity significantly increases with longer incubation times. This toxicity profile is similar to that reported by Fathy et al.19 for Pd2(bpy)2ale on MDA-MB-231 cells. The results also show that the cytotoxicity of the Pd2(bpy)2ale toward SKOV-3 cells is similar to that of cisplatin. Specifically, at a concentration of 20 μg mL−1, cisplatin achieves a cell survival rate of 25%.34 In comparison, the same level of cell survival with Pd2(bpy)2ale is reached at a concentration of 30 μg mL−1. | ||
| Fig. 8 Metabolic viability of SKOV-3 cells after treatment with different concentrations of Pd2(bpy)2ale non-radioactive conjugates. | ||
The effects of different concentrations of radioactivity in 103Pd2(bpy)2ale and 109Pd2(bpy)2ale conjugates on the metabolic viability of SKOV-3 cells over 24, 48, and 72 hours of incubation are shown in Fig. 9. It is evident that the use of 103Pd2(bpy)2ale and 109Pd2(bpy)2ale radio conjugates significantly reduced the metabolic activity of SKOV-3 cells, with the reduction depending on the activity concentration and time interval after administration. An increase in the radioactivity of the radiobioconjugate progressively decreased mitochondrial activity, ultimately resulting in near-total cell death for an activity of 25 MBq mL−1 103Pd2(bpy)2ale after 48 h. Even at the lowest radioactivity, metabolic activity was below 30% after 72 hours. In the case of Pd2(bpy)2ale complex labelled with 109Pd, the cytotoxic effect is slightly smaller. This is primarily due to the shorter half-life of 109Pd, which is significant for longer incubation times. Also, the type of emitted radiation must be considered. The in vivo generator 109Pd/109mAg emits β− radiation as well as conversion and Auger electrons. In contrast, 103Pd/103mRh emits only low-energy conversion and Auger electrons, although the number of electrons emitted by 103Pd/103mRh is much higher.
 | ||
| Fig. 9 Metabolic viability of SKOV-3 cells after treatment with different concentrations of 103Pd2(bpy)2ale (up) and 109Pd2(bpy)2ale complexes (down). | ||
When analysing the results presented in Fig. 9, it is important to note that the individual solutions of 103Pd2(bpy)2ale and 109Pd2(bpy)2ale used in the cytotoxicity tests were prepared by diluting initial solutions of 50 MBq mL−1 Pd2(bpy)2ale, which were labelled with carrier-added 103Pd and 109Pd. In the solution of the 50 MBq mL−1 Pd2(bpy)2ale complex labelled with carrier-added 109Pd, the concentration of the conjugate was 36.6 μg mL−1 and in the same activity of 103Pd2(bpy)2ale complex, the concentration of the conjugate was 63.9 μg mL−1. Consequently, the observed total cytotoxic effect consists of a chemo toxic effect caused by the non-radioactive conjugate (Fig. 8) and a radiotoxic effect from both 103Pd and 109Pd. Comparing the Fig. 8 and 9 we can see that the radiotoxic impact of the radio conjugates is much greater than the cytotoxic one. For SKOV-3, obtained IC50 values 103Pd2(bpy)2ale 18.86 MBq mL−1 at 24 h, 3.77 MBq mL−1 at 48 h and 2.233 MBq mL−1 at 72 h. In case of 109Pd2(bpy)2ale 10.62 MBq mL−1 at 24 h, 6.8 MBq mL−1 at 48 h and 0.1 MBq mL−1 at 72 h.
Similar results were obtained in the cytotoxicity studies against DU-145 prostate cancer cells (Fig. 10). Like breast cancer, advanced-stage prostate cancer can also lead to numerous bone metastases. Increasing the radioactivity of the 103Pd2(bpy)2ale and 109Pd2(bpy)2ale caused a gradual reduction in mitochondrial activity, leading to almost complete cell death for 50 MBq mL−1 radioactivity for both radio conjugates. The impact of 103Pd and 109Pd labelled conjugates on DU-145 cells are nearly identical. As can be observed, the sensitivity for chemo and radiotherapeutics of SKOV-3 and DU-145 cells is similar. However, the toxicity of both radio conjugates is much higher than that of cisplatin administered in the same doses. It is even higher than in the case of the Auger electron emitter 111In-trastuzumab tested on Her2 positive SK-BR-3 cells.35
 | ||
| Fig. 10 Metabolic viability of DU-145 cells after treatment with different concentrations of 103Pd2(bpy)2ale (up) and 109Pd2(bpy)2ale complexes (down). | ||
The strong anticancer properties of the non-radioactive Pd2(bpy)2ale complexes were explained by Fathy et al.19 due to their ability to bind to DNA. As an alternative to classical coordinate binding to DNA, metal complexes with planar aromatic ligands can also bind in a noncovalent π-stacking interaction, arising from the intercalation of a planar aromatic ring from the ligand between the base pairs of the DNA double helix.36 Such intercalating ligands are pyridine, 2,2′-bipyridine, 1,10-phenanthroline derivatives, and other similar ligands. Additionally, the increasing lipophilicity of the complexes makes them suitable for entering cells.36 This feature is further supported by the reported cytotoxicity of Pd2+ complexes with 2,2′-bipyridyl and 1,10-phenanthroline ligands, which show lower IC50 values than cisplatin in MTS assays of leukaemia cell line.37 For DU145, obtained IC50 values 103Pd2(bpy)2ale 20.31 MBq mL−1 at 24 h, 10.57 MBq mL−1 at 48 h and 4.48 MBq mL−1 at 72 h. In case of 109Pd2(bpy)2ale 24.68 MBq mL−1 at 24 h, 8.19 MBq mL−1 at 48 h and 3.45 MBq mL−1 at 72 h.
The possibility of intercalation of the Auger electron emitter complex into the double strand of DNA is crucial for the efficacy of the Auger electron therapy. Due to the range of Auger electrons being approximately in the range of the nanometre scale, the intercalation of 103Pd2(bpy)2ale and 109Pd2(bpy)2ale facilitates direct interaction of Auger electrons with DNA, as well as indirect interaction through reactive oxygen species formed by water radiolysis.38 As has been repeatedly stated, the short range of the emitted Auger electron cascade results in high levels of linear energy transfer (LET) 4–26 keV μm−1 allowing for double-stranded and irreparable DNA breakage.39 In the case of 109Pd, additionally, emitted β− particles can interact with neighbouring cells through the so-called crossfire effect. Of course, the therapeutic effectiveness will be lower than Auger electrons due to the much smaller LET of β− radiation.
3.5 DNA break
Lethal and irreparable damage to genetic material is considered one of the most sought-after outcomes of the anticancer activity of radiopharmaceuticals.40 This damage may result either from the direct ionization of DNA by ionizing radiation or indirectly through interactions between DNA and reactive oxygen species (ROS) produced from water radiolysis. In our study, we examined the induction of DNA double-strand breaks (DSBs) following exposure to 109Pd2(bpy)2ale complex. Double-strand break experiment was performed in prostate cancer cell line DU145 treated with 109Pd2(bpy)2ale complex whose concentration was 25 MBq mL−1 and 6 MBq mL−1 (18.2 μg mL−1 and 4 μg mL−1). DNA double-strand breaks in DU145 cells are visualized with γH2AX foci after treatment with 6 MBq mL−1 109Pd2(bpy)2ale (Fig. 11). | ||
| Fig. 11 Microscopic images of nuclei (blue), γH2AX foci (red) and merging of both nuclei and γH2AX for the treated DU145 cells with 109Pd2(bpy)2ale after 4 h and 24 h of incubation. | ||
Quantification of these foci, represented as the integrated density of γH2AX foci per nucleus area is shown in Fig. 12. Cells with untreated doses showed average of about 1 double strand break up to 24 h. For 6 MBq mL−1 only 1.5 γH2AX foci was recorded after 4 h of treatment only slight difference from that of untreated ones. When comparing it to the longer incubation time say 24 h the number of γH2AX is in increasing pattern. Fig. 12 shows that signals of DNA damage are dose and time-dependent manners, which indicates the interaction between the high LET radiation exposure and DNA break formation. The presence of DSBs in the DU145 cells treated with 109Pd2(bpy)2ale is consistent with the cytotoxicity results shown in Fig. 9. In addition to DNA breaks, at higher concentration (25 MBq mL−1) even at 4 h of treatment there were decrease in cell count, potentially due to increased DNA damage weakens the cellular repair.
 | ||
| Fig. 12 Analysis of γH2AX foci per cell with different time and concentration. | ||
4. Conclusion
In summary, nonradioactive binuclear Pd2(bpy)2ale complexes was synthesized and their structures were evaluated on the basis of mass spectroscopy and infrared analysis. The same procedure was used to synthesize the radioactive 103Pd2(bpy)2ale and 109Pd2(bpy)2ale complexes. Comparing HPLC chromatographs of macro amount non-radioactive complex with those of synthesized in trace amounts radioactive complexes shows that the same complexes are formed in both cases. Using ITLC we found in the 103Pd2(bpy)2ale complex complete retention of the daughter isotope 103mRh. In the case of the 109Pd complex, the situation is different. Due to the higher recoil energy and the chemical differences between Pd2+ and Ag+, we observe the complete release of 109mAg. However, its t½ = 39 s is short, so there should be no significant displacement of radioactivity outside the targeting area. The tested Pd2(bpy)2ale complexes show high affinity to the surface of hydroxyapatite grains, which is the main mineral component of bone. Nonradioactive and radioactive complexes exhibit high cytotoxicity against the human prostate (DU 145) and ovarian Her2+ (SKOV-3) cancer cell lines, higher than trastuzumab labelled with Auger electron emitter 125I and also cisplatin. Both the radioactive 103Pd2(bpy)2ale and 109Pd2(bpy)2ale conjugates exhibit multimodal toxicity, emitting Auger electrons, and demonstrate chemotoxicity comparable to the commonly used chemotherapeutic cisplatin. Additionally, 109Pd, like the popular 161Tb, emits β− radiation. However, the radiotoxic effects of Auger electrons become dominant due to the relatively high internalization of the radio bioconjugate into the cell nucleus, and expected intercalation to DNA. The results presented in this publication are preliminary and need to be confirmed through studies on induced bone metastases. However, in our opinion, these findings are interesting and promising, which is why we chose to publish them. We plan to conduct further studies in the near future in collaboration with a team specializing in in vivo preclinical studies.Data availability
The data supporting this article have been included in the ESI.†Author contributions
Conceptualization: A. B. and M. L. formal analysis: G. P. A., M. L. and A. B; funding acquisition: A. B.; investigation: G. P. A., M. L.; methodology: G. P. A., M. L. and A. B.; supervision: A. B. and M. L.; visualization: G. P. A., M. L.; writing—original draft: A. B., G. P. A., M. L.; writing—review and editing: M. L., G. P. A. and A. B. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This project was funded by the National Science Center Poland, grant no. 2022/45/B/ST4/00825. The radionuclide 103Pd was produced as part of the European Medical Isotope Program: PRISMAP.References
- A. S. Maheswaran and D. A. Haber, Circulating tumor cells: a window into cancer biology and metastases, Curr. Opin. Genet. Dev., 2010, 20, 96–99 CrossRef PubMed.
- G. Selvaggi and G. V. Scagliotti, Management of bone metastases in cancer: a review, Crit. Rev. Oncol. Hematol., 2005, 56, 365–378 CrossRef PubMed.
- M. van Driel and J. P. van Leeuwen, Cancer and bone: a complex complex, Arch. Biochem. Biophys., 2014, 561, 159–166 CrossRef CAS PubMed.
- G. Selvaggi and G. V. Scagliotti, Management of bone metastases in cancer: a review, Crit. Rev. Oncol. Hematol., 2005, 56, 365–378 CrossRef PubMed.
- J. R. Zeevaart, D. R. Jansen, M. F. Botelho, A. Abrunhosa, C. Gomes, L. Metello, Z. I. Kolar, G. C. Krijger, W. K. A. Louw and I. C. Dormehl, Comparison of the predicted in vivo behaviour of the Sn (II)–APDDMP complex and the results as studied in a rodent model, J. Inorg. Biochem., 2004, 98, 1521–1530 CrossRef CAS PubMed.
- A. Ku, V. J. Facca, Z. Cai and R. M. Reilly, Auger electrons for cancer therapy – a review, EJNMMI Radiopharm. Chem., 2019, 4, 27 CrossRef PubMed.
- S. N. Sioshansi, K. E. Huber and D. E. Wazer, The implications of breast cancer molecular phenotype for radiation oncology, Front. Oncol., 2021, 1, 28 Search PubMed.
- S. C. Srivastava, H. L. Atkins, G. T. Krishnamurthy, I. Zanzi, E. B. Silberstein, G. Meinken, L. Mausner, F. Swailem, T. D'Alessandro, J. J. Cabahug, Y. Lau, T. Park and S. Madajewicz, Treatment of metastatic bone pain with tin-117m Stannic diethylenetriaminepentaacetic acid: a phase I/II clinical study, Clin. Cancer Res., 1998, 4, 61–68 CAS.
- W. A. Volkert and T. J. Hoffman, Therapeutic radiopharmaceuticals, Chem. Rev., 1999, 99, 2269–2292 CrossRef CAS PubMed.
- R. A. Nadar, G. M. Franssen, N. W. M. Van Dijk, K. Codee-van der Schilden, M. de Weijert, E. Oosterwijk, M. Iafisco, N. Margiotta, S. Heskamp, J. J. J. P. van den Beucken and S. C. G. Leeuwenburgh, Bone tumor–targeted delivery of theranostic 195mPt-bisphosphonate complexes promotes killing of metastatic tumor cells, Mater. Today Bio, 2021, 9, 100088 CrossRef CAS PubMed.
- R. A. Nadar, K. Farbod, K. Codee-van der Schilden, K. Karlijn, L. Schlatt and B. Crone, Targeting of radioactive platinum-bisphosphonate anticancer drugs to bone of high metabolic activity, Sci. Rep., 2020, 10, 5889 CrossRef CAS PubMed.
- F. F. (Russ) Knapp Jr, S. Mirzadeh, A. L. Beets and M. Du, Production of therapeutic radioisotopes in the ORNL High Flux Isotope Reactor (HFIR) for applications in nuclear medicine, oncology and interventional cardiology, J. Radioanal. Nucl. Chem., 2005, 263, 503–509 CrossRef.
- E. Hindié, A. Larouze, M. Alcocer-Ávila, C. Morgat and C. Champion, Palladium-103 (103Pd/103mRh), a promising Auger- electron emitter for targeted radionuclide therapy of disseminated tumor cells – absorbed doses in single cells and clusters, with comparison to 177Lu and 161Tb, Theranostics, 2024, 14, 4318–4330 CrossRef PubMed.
- B. B. Zmejkovski, N. D. Pantelic and G. N. Kaluđerovic, Palladium(II) complexes: Structure, development and cytotoxicity from cisplatin analogues to chelating ligands with N stereocenters, Inorg. Chim. Acta, 2022, 534, 120797 CrossRef CAS.
- D. Filosofov, E. Kurakina and V. Radchenko, Potent candidates for Targeted Auger Therapy: Production and radiochemical considerations, Nucl. Med. Biol., 2021, 94–95, 1–19 CrossRef CAS PubMed.
- P. Bernhardt, E. Forssell-Aronsson, L. Jacobsson and G. Skarnemark, Low-energy electron emitters for targeted radiotherapy of small tumours, Acta Oncol., 2001, 40(5), 602–608 CrossRef CAS PubMed.
- C. Müller, C. A. Umbricht, N. Gracheva, V. J. Tschan, G. Pellegrini, P. Bernhardt, J. R. Zeevaart, U. Köster, R. Schibli and N. P. van der Meulen, Terbium-161 for PSMA-targeted radionuclide therapy of prostate cancer, Eur. J. Nucl. Med. Mol. Imaging, 2019, 46, 1919–1930 CrossRef PubMed.
- T. Das, S. Chakraborty, H. D. Sarma and S. Banerjee, A novel [109Pd] palladium labeled porphyrin for possible use in targeted radiotherapy, Radiochim. Acta, 2008, 96(7), 427–433 CrossRef CAS.
- A. A. Fathy, I. S. Butler, M. A. Elrahman, B. J. Jean-Claude and S. I. Mostafa, Anticancer evaluation and drug delivery of new palladium (II) complexes based on the chelate of alendronate onto hydroxyapatite nanoparticles, Inorg. Chim. Acta, 2018, 473, 44–50 CrossRef CAS.
- M. Cipriani, S. Rostán, I. León, Z.-H. Li, J. S. Gancheff, U. Kemmerling, C. Olea Azar, S. Etcheverry, R. Docampo, D. Gambino and L. Otero, Multi-target heteroleptic palladium bisphosphonate complexes, J. Biol. Inorg. Chem., 2020, 25, 509–519 CrossRef CAS PubMed.
- L. Todorov and I. Kostova, Recent Trends in the Development of Novel Metal-Based Antineoplastic Drugs, Molecules, 2023, 28, 1959 CrossRef CAS PubMed.
- M. Fanelli, M. Formica, V. Fusi, L. Giorgi, M. Micheloni and P. Paoli, New trends in platinum and palladium complexes as antineoplastic agents, Coord. Chem. Rev., 2016, 310, 41–79 CrossRef CAS.
- A. Majkowska, M. Neves, I. Antunes and A. Bilewicz, Complexes of low energy beta emitters 47Sc and 177Lu with zoledronic acid for bone pain therapy, Appl. Radiat. Isot., 2009, 67, 11–13 CrossRef CAS PubMed.
- N. Abbasi Gharibkandi, K. Wawrowicz, R. Walczak, A. Majkowska-Pilip, M. Wierzbicki and A. Bilewicz, 109Pd/109mAg in-vivo generator in the form of nanoparticles for combined β- - Auger electron therapy of hepatocellular carcinoma, EJNMMI Radiopharm. Chem., 2024, 9, 59 CrossRef PubMed.
- R. Wang, B. Ponsard, H. Wolterbeek and A. Denkova, Core–shell structured gold nanoparticles as carrier for 166Dy/166Ho in vivo generator, EJNMMI Radiopharm. Chem., 2022, 7, 16 CrossRef PubMed.
- A. I. Jensen, F. Zhuravlev, G. Severin, C. B. Magnus, J. Fonslet, U. Koester and M. Jensen, A solid support generator of the Auger electron emitter rhodium-103m from [103Pd] palladium, Appl. Radiat. Isot., 2020, 156, 108985 CrossRef PubMed.
- J. Rooyen, Z. Szucs and J. R. Zeevaart, A possible in vivo generator 103Pd/103mRh-recoil considerations, Appl. Radiat. Isot., 2008, 66, 1346–1349 CrossRef PubMed.
- Z. Szucs, J. van Rooyen and J. R. Zeevaart, Recoil effect on β- decaying in vivo generators, interpreted for 103Pd/103mRh, Appl. Radiat. Isot., 2009, 67, 1401–1404 CrossRef CAS PubMed.
- A. Nath, M. Prushan and J. Gilbert, Can super-excited molecules survive fragmentation?, J. Radioanal. Nucl. Chem., 2001, 247, 589–591 CrossRef CAS.
- International Atomic Energy Agency, Pain Palliation of Bone Metastases: Production, Quality Control and Dosimetry of Radiopharmaceuticals, IAEA Radioisotopes and Radiopharmaceuticals Series No. 9, Vienna, 2023 Search PubMed.
- R. L. Hilderbrand, The Role of Phosphonates in Living Systems, CRC Press, Boca Raton, FL, 1983 Search PubMed.
- T. Vitha, V. Kubíček, P. Hermann, Z. I. Kolar, H. T. Wolterbeek, J. A. Peters and I. Lukeš, Complexes of DOTA−Bisphosphonate Conjugates: Probes for Determination of Adsorption Capacity and Affinity Constants of Hydroxyapatite, Langmuir, 2008, 24, 1952–1958 CrossRef CAS PubMed.
- R. A. M. J. Claessens and Z. I. Kolar, „Affinity of Tin (II) and Tin (II) Diphosphonates for Hydroxyapatite: An Experimental and Model Study, Langmuir, 2000, 16, 1360–1367 CrossRef CAS.
- L. Zhao, S. Liu, D. Liang, T. Jiang, X. Yan, S. Zhao, Y. Liu, W. Zhao and H. Yu, Resensitization of cisplatin resistance ovarian cancer cells to cisplatin through pretreatment with low-dose fraction radiation, Cancer Med., 2019, 8(5), 2442–2448 CrossRef CAS PubMed.
- D. L. Costantini, C. Chan, Z. Cai, K. A. Vallis and R. M. Reilly, 111In-Labeled Trastuzumab (Herceptin) Modified with Nuclear Localization Sequences (NLS): An Auger Electron-Emitting Radiotherapeutic Agent for HER2/neu-Amplified Breast Cancer, J. Nucl. Med., 2007, 48, 1357–1368 CrossRef CAS PubMed.
- M. Fanelli, M. Formica, V. Fusi, L. Giorgi, M. Micheloni and P. Paoli, New trends in platinum and palladium complexes as antineoplastic agents, Coord. Chem. Rev., 2016, 310, 41–79 CrossRef CAS.
- H. Mansouri-Torshizi, M. Saeidifar, A. Divsalar, A. Saboury and S. Shahraki, DNA-Binding and Thermodynamic Parameters, Structure and Cytotoxicity of Newly Designed Platinum (II) and Palladium(II) Anti-Tumor Complexes, Bull. Korean Chem. Soc., 2010, 31, 435 CrossRef CAS.
- A. I. Kassis, R. S. Harapanhalli and S. J. Adelstein, Strand breaks in plasmid DNA after positional changes of Auger electron-emitting iodine-125: direct compared to indirect effects, Radiat. Res., 1999, 152, 530–538 CrossRef CAS PubMed.
- J. Bolcaen, M. A. Gizawy, S. Y. A. Terry, A. Paulo, B. Cornelissen, A. Korde, J. Engle, V. Radchenko and R. W. Howell, Marshalling the Potential of Auger Electron Radiopharmaceutical Therapy, J. Nucl. Med., 2023, 64, 1344–1351 CrossRef CAS PubMed.
- F. Buchegger, F. Perillo-Adamer, Y. M. Dupertuis and A. Bischof Delaloye, Auger radiation targeted into DNA: a therapy perspective, Eur. J. Nucl. Med. Mol. Imaging, 2006, 33, 1352–1363 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra02172c |
| This journal is © The Royal Society of Chemistry 2025 |