
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereocontrolled synthesis of 3-hydroxy-2-piperidinone carboxamides by catalytic ring-opening aminolysis of bridged δ-lactam-γ-lactones†
Timothy K. Beng *,
Katharyn Curry,
Alan Gitonga,
Keegan Yu and
Samuel Edwards
*,
Katharyn Curry,
Alan Gitonga,
Keegan Yu and
Samuel Edwards
Department of Chemistry, Central Washington University, Ellensburg, WA 98926, USA. E-mail: Timothy.beng@cwu.edu
First published on 14th May 2025
Abstract
The α-hydroxy-δ-valerolactam, 3-hydroxypiperidine, and piperidine-3-carboxamide topologies are resident in several natural products and pharmaceuticals, including anticonvulsant and antithrombotic agents. A modular and stereocontrolled strategy that merges these privileged scaffolds into one motif could facilitate the discovery of more small molecules with medicinal value. Here, we demonstrate that bridged valerolactam-butyrolactones can be skeletally remodelled to highly decorated 3-hydroxy-2-piperidinone carboxamides by catalytic and site-selective deconstructive aminolysis with primary and secondary amines. The products are obtained in a sterocontrolled manner following oxidative addition and concomitant trapping with the amine. The scaffold hopping proceeds with exclusive acyl C–O bond cleavage under palladium catalysis and represents the first catalytic method for activating the acyl C–O bonds of γ-lactones.
Introduction
The 3-hydroxy-2-piperidinone topology is resident in natural products and pharmaceuticals, including anticonvulsant A, natural alkaloids B and C, as well as (+)-awajanomycin (Fig. 1).1 Additionally, polysubstituted saturated piperidines bearing the β-hydroxypiperidine substructure constitute the core of several natural products and pharmaceuticals, including deoxocassine, spectaline, azimic acid, and afegostat.2,3 These β-hydroxypiperidines also serve as cyclic choline analogs.4 Meanwhile, the piperidine-3-carboxamide scaffold is prevalent in antithrombotic agents5 and is known to induce senescence-like phenotypic changes in human melanoma A375 cells6 (see D and E, Fig. 1).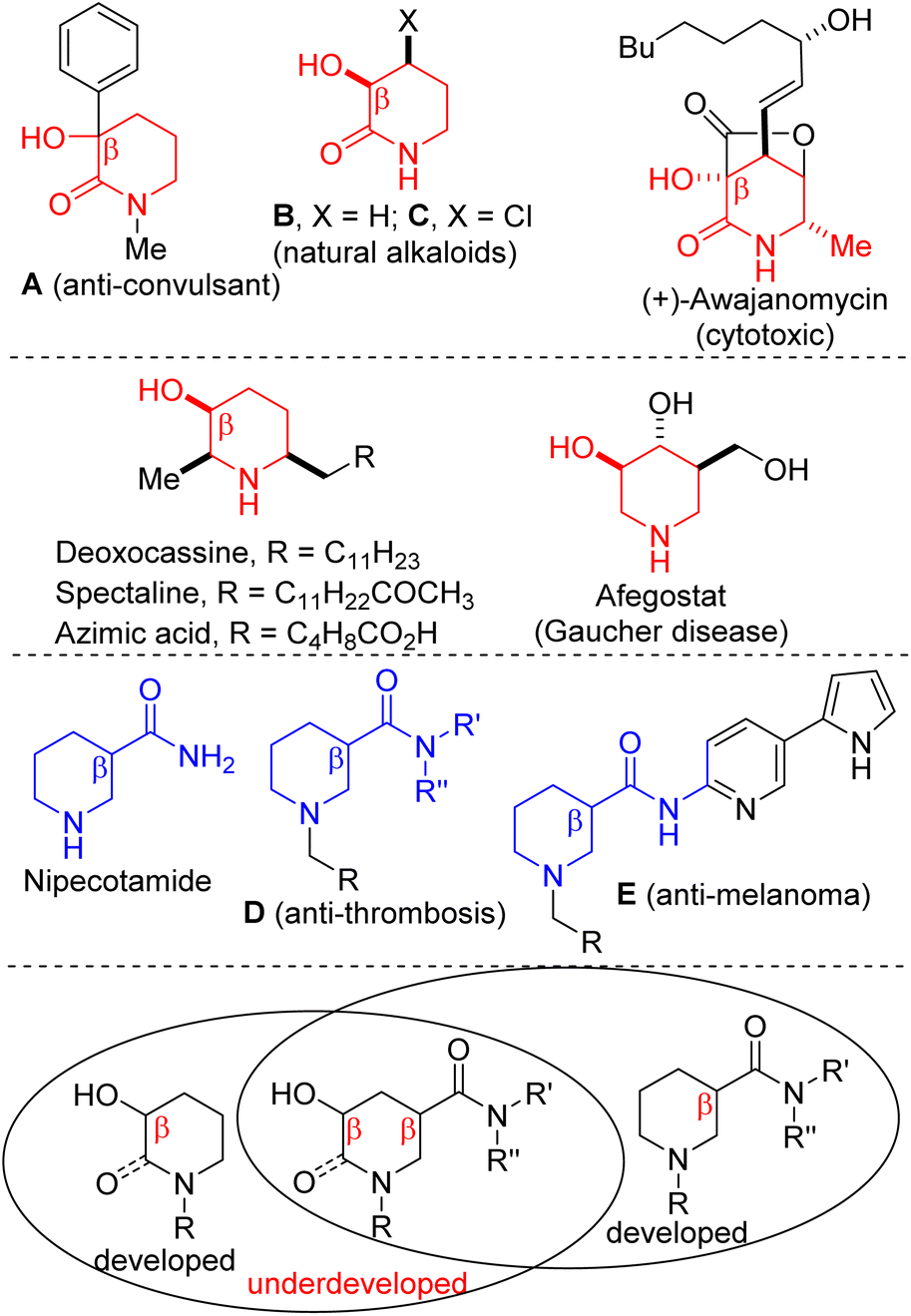 | ||
| Fig. 1 Examples of bioactive 3-hydroxy-2-piperidinones, β-hydroxypiperidines, and piperidine-3-carboxamides. | ||
We surmised that a strategy that merges these pharmaceutically pertinent topologies into one motif (Fig. 1, bottom) would likely expand the 3D-chemical space for the discovery of more small molecules with medicinal value. Although the literature is replete with several approaches to 3-hydroxy-2-piperidinones,7 β-hydroxypiperidines,8 and piperidine-3-carboxamides,5,6 we are not aware of any report on the stereocontrolled and the catalytic construction of the 3-hydroxy-2-piperidinone carboxamide scaffold.
As part of a program aimed at leveraging the synthetic versatility of lactam acids of type 1 (Fig. 2A),9 our group has identified catalytic and stereocontrolled sp3 C–H lactonization as a means to access conformationally restricted bicyclic lactam-lactones (see 2).9k Organic synthesis strategies that seek to achieve flexible peripheral editing of heterocycles as well as skeletal editing are becoming increasingly popular in medicinal chemistry programs. Within this context and with 2 in hand, we next sought to explore its amenability to skeletal diversification by means of catalytic deconstructive functionalization, in view of assembling highly decorated 3-hydroxy-2-piperidinone carboxamides. We hypothesized that 2 could undergo site-selective and stereocontrolled ring-opening with amine nucleophiles under transition metal catalysis.
 | ||
| Fig. 2 (A) Our prior efforts to prepare lactam-lactone 2, (B and C) our designed plan for accessing 3-hydroxy-2-piperidinone carboxamides from 2, (D) existing report on Pd-catalyzed ring-opening of methylene-β-lactones. | ||
As depicted in Fig. 2B, a major challenge associated with the transition metal (TM)-catalyzed ring-opening of conformationally restricted lactones such as 2 is the possibility of formation of two constitutionally isomeric products due to competing acyl C–O bond cleavage (pathway A) and alkyl C–O bond cleavage (pathway B).10 Some reports exist on the alkyl C–O bond activation of 4-membered ring lactones.11 However, TM-catalyzed ring opening of esters,12 especially lactones, at the acyl C–O bond, is still at the incipient stages. Another challenge that needs to be addressed is that of diastereoselectivity (Fig. 2C). One of the most important aspects for the reaction to proceed in high diastereoselectivity is for the OH-bearing stereocenter in the opened chain to retain the same configuration as the original stereocenter in the lactone (stereoretentive ring-opening aminolysis, see 7) or for the OH-bearing stereocenter to have the opposite configuration (stereoinvertive ring-opening aminolysis, see 8). Inspired by the only existing report on the transition metal-catalyzed deconstructive functionalization of a strained β-lactone with amines (Fig. 2D),13 we herein describe efforts toward the elicitation of our ideals. We find that the ring-opening with amines proceeds with exclusive site-selective acyl C–O bond activation and complete stereoretention at the OH-bearing stereocenter.
Results and discussion
As articulated previously, the development of practical and stereocontrolled synthesis strategies for the construction of the carboxamide motif is a responsibility entrusted on the organic synthesis community given that it is commonly found in peptides, natural products, pharmaceuticals, and active ingredients in crop protection.14 The sustainable construction of the amide scaffold has been highlighted by the American Chemical Society (ACS) Green Chemistry Institute as one of the top impactful areas.15 Within this context, the catalytic ring-opening aminolysis of a cyclic ester is a highly desirable transformation given that it obviates the need for stoichiometric activation and side-steps the undesirable ester hydrolysis step. We commenced these studies by benchmarking our optimization efforts toward the synthesis of α-hydroxy-δ-valerolactam-carboxamides with the reaction conditions described in Table 1. In the event, using acid 2a as a model substrate, 2-methyltetrahydrofuran (2-MeTHF) emerged as the solvent of choice (entries 1–9). Of note, ethyl acetate, itself an ester, performs satisfactorily as a solvent in this reaction (entry 9). Pd(TFA)2 and PPh3 emerged as the most effective catalyst system (entries 10–21). No background ring-opening aminolysis is observed in the absence of Pd(TFA)2 (entry 10) or in the absence of the phosphine ligand (entry 11). Therefore, these results support the notion that palladium catalysis is indeed operative in the lactone acyl C–O bond-cleavage process. Increasing the Pd(TFA)2 loading to 5 mol% only leads to a marginal increase in the yield of 7a (entry 18). However, lowering the loading to 1 mol% has a deleterious effect on the efficiency of the transformation (entry 19). Other catalytic aminolysis conditions that were developed primarily for methyl-bearing noncyclic esters have been surveyed and none of them promoted ring-opening aminolysis of 2a to 7a (entries 22–24). Base-mediated aminolysis of 2a with isopropylamine using conventional bases such as Cs2CO3 (entry 25), K2CO3, and CsF led to complete epimerization at the OH-bearing stereocenter. These results further highlight the merit of our newly developed protocol from both the standpoints of regioselectivity and diastereoselectivity. The atom efficiency of this transformation is commendable given that all atoms originating from the bicyclic lactam-lactone and the amine are incorporated into the product structure.
|
||
|---|---|---|
| Entry | Deviation from conditions A | %Yield 7a |
| 1 | Tetrahydrofuran (THF) as solvent | 76 |
| 2 | 2,2,5,5-Tetramethyloxolane (TMO) as solvent | 70 |
| 3 | Acetonitrile (MeCN) as solvent | 52 |
| 4 | N,N-Dimethylformamide (DMF) as solvent | 39 |
| 5 | N,N-Dimethylacetamide (DMA) as solvent | 22 |
| 6 | N,N-Dimethylsulfoxide (DMSO) as solvent | 13 |
| 7 | Dichloromethane (DCM) as solvent | 65 |
| 8 | Dichloroethane (DCE) as solvent | 60 |
| 9 | Ethyl acetate (EtOAc) as solvent | 71 |
| 10 | No Pd(TFA)2 | 0 |
| 11 | No PPh3 | 0 |
| 12 | Pd(OAc)2 in place of Pd(TFA)2 | 82 |
| 13 | Pd2(dba), in place of Pd(TFA)2 | 58 |
| 14 | Pd(PPh), in place of Pd(TFA)2 | 41 |
| 15 | Pd[(allyl)Ci]2 in place of Pd(TFA)2 | 67 |
| 16 | Pd(MeCN)2Cl2 in place of Pd(TFA)2 | 0 |
| 17 | Pd(PhCN)2Cl2 in place of Pd(TFA)2 | 0 |
| 18 | Using 5 mol% Pd(TFA)2 | 90 |
| 19 | Using 1 mol% Pd(TFA)2 | 70 |
| 20 | P(OEt), in place of PPh3 | 0 |
| 21 | PC3, in place of PPh3 | 45 |
| 22 | Conditions B in place of conditions A | 0 |
| 23 | Conditions C in place of conditions A | 0 |
| 24 | Conditions D in place of conditions A | 0 |
| 25 | Conditions E in place of conditions A | 34(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) :1 dr) |
 |
||

The exclusive formation of 7a and the failure to observe side products of type 6 are a testament to the fact that alkyl C–O bond activation (see pathway B, Fig. 2) is not operative. Support for this assertion comes from spectroscopic data (see Fig. 3). Representative COSY, HSQC, HMBC, NOESY, and IR spectra for 7a and several other compounds depicted in Scheme 1 are provided in the (ESI†). Decarbonylation was also not observed in this palladium-catalyzed conversion of conformationally restricted bridged lactam-lactones into 3-hydroxy-2-piperidinone carboxamides.
 | ||
| Fig. 3 Spectroscopic evidence for the exclusive formation of 7a by regioselective acyl C–O bond cleavage. | ||
 | ||
| Scheme 1 Construction of α-hydroxy-δ-valerolactam carboxamides by catalytic deconstructive functionalization of [3-2-1] bicyclic lactam-lactones with primary and secondary amines. | ||
Using the optimized conditions, the scope of the catalytic and site-selective ring-opening aminolysis protocol was explored. We began by surveying various diversely substituted bridged lactam-lactones using isopropylamine as the nucleophile. In the event, the differentially substituted hydroxypiperidinone carboxamides depicted in Scheme 1 were obtained (see 7a–s). The functional group tolerance exhibited by the transformation is impressive given that alkenes, allyl ethers, tertiary anilines, anisoles, thioethers, and thiophenes are well tolerated in this transformation. As a testament to the mildness of these reaction conditions, ring-opening of methylbenzoate-bearing substrates occurs exclusively on the lactone moiety (see 7j and 7p). Such chemoselectivity is noteworthy and speaks to the enhanced reactivity of the inherently strained bridged lactam-lactone scaffold. We envision that this would pave the way for orthogonal aminolysis given that amide formation at the methyl ester terminus should be achievable using the well established Ni-catalyzed conditions.12a–f Piperidinone carboxamides bearing four contiguous stereocenters have been prepared in a stereocontrolled manner (see 7l/m). Our studies have revealed that other primary alkyl amines undergo excellent deconstructive aminolysis without complications arising from epimerization at the OH-bearing stereocenter (see 7t–7z2). In the field of organic synthesis, alkynes have become a staple given that they are often employed as synthons and as catalysts in enantioselective studies.16 We were therefore pleased to find that the ring-opening aminolysis with propargyl amine proceeds uneventfully leading to products such as 7x/y. An inherently less nucleophilic primary aryl amine afforded the product in high yield and diastereoselectivity (see 7z3). When both enantiomers of α-methyl benzylamine are employed in this transformation, the optically active 3-hydroxy-2-piperidinone carboxamides are obtained in high stereochemical fidelity. These results indicate that epimerization at the methyl-bearing stereocenter did not occur in either case. Ring-opening aminolysis with sterically challenged secondary amines proceeds satisfactorily (see 7z6–7z8). When 4-substituted piperidines are employed as the amine nucleophile, the lactam carboxamides are obtained in variable diastereoselectivities (see 7z9 and 7z10) as judged by GC-MS and 1H NMR of the crude reaction mixture. In both cases, the diastereomeric mixture was inseparable in our hands. Unsurprisingly, when inherently less nucleophilic tertiary amines such as N-methylpiperidine and triethylamine were employed, no ring-opening aminolysis occurred even at elevated temperatures (80 °C, for 18 h). In these cases, the mass balance was mostly accounted for by the recovered bridged lactam-lactone precursor. The addition of fluorine-containing scaffold to standard organic frameworks tends to enhance properties such as metabolic stability and dissolution, which justifies why ∼25% of current preclinical medicines harbor one or more fluorine atoms.17 For instance, the well-heralded trifluoromethyl (CF3) group facilitates drug metabolism.18 We were therefore thrilled to see that compounds 7z and 7z3 were produced in high efficiencies and selectivities. The synthesis of compounds capable of serving as drug candidates in a scalable manner is an important objective as it makes it relatively easier for clinical tests to be performed. Fittingly, we have synthesized compounds such as 7a and 7z4 in gram-scale quantities (starting with as much as 5 mmol of 2), with little to no compromise in efficiency and diastereoselectivity. When morpholinone-bridged lactone 2z11 was engaged in this ring-opening aminolysis protocol, complete epimerization at the OH-bearing stereocenter was observed as cyclic hemiacetal 7z11 was obtained in 1:1 dr. The diastereomeric mixture was inseparable in our hands. These results further exemplify why efforts to extend reactivity trends from one class of a nitrogen heterocycle to another can sometimes seem like a wild goose chase.
Conclusions
In summary, the stereocontrolled deconstructive amination of bridged δ-lactam-γ-lactones has been accomplished under palladium catalysis, leading to the synthesis of 3-hydroxy-2-piperidinone carboxamides. The approach enables a formal α-hydroxylation of inherently inert lactams in a way that is modular and amenable to rapid diversity incorporation. The target compounds are obtained in high yields and diastereoselectivities. Primary and secondary amines are suitable reactive partners. This skeletal diversification of strained lactones bridged to lactams proceeds with exclusive acyl C–O bond scission and stereoretentive ring-opening aminolysis is implicated. We anticipate that the epimerization-free nature of the reaction conditions would endear this approach to the synthesis and medicinal chemistry communities. Detailed mechanistic studies as well as the development of an enantioselective variant of the transformation are underway and the results will be disclosed in due course.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. K. B. – conceptualized, administered, and supervised the project. Also carried our exploration, data analysis, and optimization studies. Wrote the original draft manuscript and acquired internal funds. K. C., A. G., K. Y., and S. E. carried our investigation, data curation, and exploration of the scope of the reaction. K. C., A. G., K. Y., and S. E. contributed equally.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Central Washington University (CWU) for supporting this project through startup funds awarded to T. K .B. We are grateful to CWU's School of Graduate Studies and Research for partially funding this work through a Faculty Research Award to T. K. B. We greatly acknowledge the Office of the Provost at CWU for Faculty-student research fellowships awarded to T. K. B., K. Y., and S. E. Both K. C. and A. G. express their gratitude to the Office of University Research (OUR) at CWU for financial support.Notes and references
- (a) I. Choudhury-Mukherjee, H. A. Schenck, S. Cechova, T. N. Pajewski, J. Kapur, J. Ellena, D. S. Cafiso and M. L. Brown, J. Med. Chem., 2003, 46, 2494–2501 CrossRef CAS PubMed; (b) I. Krasteva, V. Bratkov, F. Bucar, O. Kunert, M. Kollroser, M. KondevaBurdina and I. Ionkova, J. Nat. Prod., 2015, 78, 2565–2571 CrossRef CAS; (c) J. Masschelein, M. Jenner and G. L. Challis, Nat. Prod. Rep., 2017, 34, 712–783 RSC; (d) G. J. Patrick, L. Fang, J. Schaefer, S. Singh, G. R. Bowman and T. A. Wencewicz, Biochemistry, 2018, 57, 117–135 CrossRef CAS PubMed; (e) K. E. Harding and M. W. Jones, Heterocycles, 1989, 663–668 CrossRef CAS.
- (a) A. Grauer and B. König, Eur. J. Org Chem., 2009, 30, 5099–5111 CrossRef; (b) S. Hanessian, G. McNaughton-Smith, H.-G. Lombart and W. D. Lubell, Tetrahedron, 1997, 53, 12789–12854 CrossRef CAS.
- (a) M. C. Maillard, F. A. Brookfield, S. M. Courtney, F. M. Eustache, M. J. Gemkow, R. K. Handel, L. C. Johnson, P. D. Johnson, M. A. Kerry, F. Krieger, M. Meniconi, I. Munoz-Sanjuán, J. J. Palfrey, H. Park, S. Schaertl, M. G. Taylor, D. Weddell and C. Dominguez, Bioorg. Med. Chem., 2011, 19, 5833–5851 CrossRef CAS PubMed; (b) M. Quibell, A. Benn, N. Flinn, T. Monk, M. Ramjee, Y. Wang and J. Watts, Bioorg. Med. Chem., 2004, 12, 5689–5710 CrossRef CAS.
- M. Bollenbach, M. Ortega, M. Orman, C. L. Drennan and E. P. Balskus, ACS Med. Chem. Lett., 2020, 11, 1980–1985 CrossRef CAS PubMed.
- (a) A. Lasslo, E. O. Dillingham, J. C. McCastlain, G. Carter- Burks, R. P. Quintana, R. W. Johnson and J. N. Naylor, Med. Prog. Technol., 1986, 11, 109 CAS; (b) R. Gollamudi, E. O. Dillingham and S. E. Bond, Thromb. Haemost., 1993, 69, 1322 Search PubMed; (c) A. Lasslo, R. P. Quintana, D. Crisan, R. E. Baier, A. E. Meyer and M. S. Fornalik, Med. Prog. Technol., 1984, 10, 71–78 CAS; (d) W. H. Lawrence, R. D. Howell and R. Gollamudi, J. Pharm. Sci., 1994, 83, 222 CrossRef CAS; (e) Z. Guo, X. Zheng, W. Thompson, M. Dugdale and R. Gollamudi, Bioorg. Med. Chem., 2000, 8, 1041–1058 CrossRef CAS; (f) K. S. Anil Kumar, A. Misra, T. I. Siddiqi, S. Srivastava, M. Jain, R. S. Bhatta, M. Barthwal, M. Dikshit and D. K. Dikshit, Eur. J. Med. Chem., 2014, 81, 456–472 CrossRef CAS PubMed.
- S. Oh, D. Y. Kwon, I. Choi, Y. M. Kim, J. Y. Lee, J. Ryu, H. Jeong, M. J. Kim and R. Song, ACS Med. Chem. Lett., 2021, 12, 563–571 CrossRef CAS PubMed.
- (a) P. R. Krishna, P. V. A. Kumar, V. S. Mallula and K. V. S. Ramakrishna, Tetrahedron, 2013, 69, 2319 CrossRef CAS; (b) A. Kamal, K. V. Ramana, A. V. Ramana and A. H. Babu, Tetrahedron Asymmetry, 2003, 14, 2587 CrossRef CAS; (c) J. Romero-Ibanez, S. Cruz-Gregorio, L. Quintero and F. Sartillo-Piscil, Synthesis, 2018, 50, 2878–2886 CrossRef CAS.
- J.-D. Ha, D. Lee and J.-K. Cha, J. Org. Chem., 1997, 62, 455024551 Search PubMed.
- (a) H. Braunstein, S. Langevin, M. Khim, J. Adamson, K. Hovenkotter, L. Kotlarz, B. Mansker and T. K. Beng, Org. Biomol. Chem., 2016, 14, 8864–8872 RSC; (b) T. K. Beng and A. Moreno, New J. Chem., 2020, 44, 4257–4261 RSC; (c) T. K. Beng, M. Bauder, M. J. Rodriguez and A. Moreno, New J. Chem., 2018, 42, 16451–16455 RSC; (d) K. Hovenkotter, H. Braunstein, S. Langevin and T. K. Beng, Org. Biomol. Chem., 2017, 15, 1217–1221 RSC; (e) T. K. Beng and A. Moreno, RSC Adv., 2020, 10, 8805–8809 RSC; (f) J. Garcia, J. Eichwald, J. Zesiger and T. K. Beng, RSC Adv., 2022, 12, 309–318 RSC; (g) T. K. Beng, J. Garcia, J. Eichwald and C. Borg, RSC Adv., 2023, 13, 14355–14360 RSC; (h) T. K. Beng, J. Eichwald, J. Fessenden, K. Quigley, S. Sharaf, N. Jeon and M. Do, RSC Adv., 2023, 13, 21250–21258 RSC; (i) T. K. Beng, M. Rodriguez and C. Borg, RSC Adv., 2022, 12, 17617–17620 RSC; (j) T. K. Beng, C. Borg and M. Rodriguez, RSC Adv., 2022, 12, 28685–28691 RSC; (k) T. K. Beng, V. Shearer and A. O. Farah, J. Am. Chem. Soc., 2025 Search PubMed , under review.
- (a) S. G. Nelson, R. D. Dura and T. J. Peelen, Org. React., 2013, 82, 471 CAS; (b) E. H. Wiedemann, F. A. Mandl, I. D. Blank, C. Ochsenfeld, A. R. Ofial and S. A. Sieber, ChemPlusChem, 2015, 80, 1673 CrossRef CAS; (c) A. Noel, B. Delpech and D. Crich, Org. Biomol. Chem., 2012, 10, 6480 RSC; (d) M. A. Calter and X. Guo, J. Org. Chem., 1998, 63, 5308 CrossRef CAS; (e) E. S. Ratemi and J. C. Vederas, Tetrahedron Lett., 1994, 35, 7605 CrossRef CAS; (f) A. Griesbeck and D. Seebach, Helv. Chim. Acta, 1987, 70, 1326 CrossRef CAS; (g) L. D. Arnold, T. H. Kalantar and J. C. Vederas, J. Am. Chem. Soc., 1985, 107, 7105 CrossRef CAS.
- (a) K. T. Aye, D. Colpitts, G. Ferguson and R. J. Puddephatt, Organometallics, 1988, 7, 1454 CrossRef CAS; (b) A. F. Noels, J. J. Herman and P. Teyssie, J. Org. Chem., 1976, 41, 2527 CrossRef CAS; (c) T. Hattori, Y. Suzuki, O. Uesugi, S. Oi and S. Miyano, Chem. Commun., 2000, 1, 73–74 RSC; (d) T. Hattori, Y. Suzuki, Y. Ito, D. Hotta and S. Miyano, Tetrahedron, 2002, 58, 5215 CrossRef CAS.
- (a) L. Hie, N. F. Fine Nathel, X. Hong, Y.-F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., Int. Ed., 2016, 55, 2810–2814 CrossRef CAS; (b) L. Hie, N. F. Fine Nathel, X. Hong, Y.-F. Yang, K. N. Houk and N. K. Garg, Angew. Chem., 2016, 128, 2860–2864 CrossRef; (c) T. Ben Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., Int. Ed., 2018, 57, 12915–12929 CrossRef; (d) T. Ben Halima, J. Masson-Makdissi and S. G. Newman, Angew. Chem., 2018, 130, 13107–13111 CrossRef; (e) Y.-L. Zheng and S. G. Newman, ACS Catal., 2019, 9, 4426–4433 CrossRef CAS; (f) T. Ben Halima, J. K. Vandavasi, M. Shkoor and S. G. Newman, ACS Catal., 2017, 7, 2176–2180 CrossRef CAS; (g) W. Zhang, J. Smillovich and V. Albert, Tetrahedron, 2022, 114, 154242 CrossRef; (h) Z. Fu, X. Wang, S. Tao, Q. Bu, D. Wei and N. Liu, J. Org. Chem., 2021, 86, 2339–2358 CrossRef CAS PubMed; (i) A. Mondal, M. Subaramanian, A. Nandakumar and E. Balaraman, Org. Lett., 2018, 20, 3381–3384 CrossRef CAS; (j) S. Sultan, M. Kumar, S. Devari, D. Mukherjee and B. A. Shah, ChemCatChem, 2016, 8, 703–707 CrossRef CAS.
- C. A. Malapit, D. R. Caldwell, N. Sassu, S. Milbin and A. R. Howell, Org. Lett., 2018, 19, 1966–1969 CrossRef.
- (a) H. Charville, D. Jackson, G. Hodges and A. Whiting, Chem. Commun., 2010, 46, 1813–1823 RSC; (b) L. J. Goossen, D. M. Ohlmann and P. P. Lange, Synthesis, 2009, 209, 160–164 CrossRef; (c) B. S. Jursic and Z. Zdravkovski, Synth. Commun., 1993, 23, 2761–2770 CrossRef CAS; (d) L. Zhang, X. J. Wang, J. Wang, N. Grinberg, D. Krishnamurthy and C. H. Senanayake, Tetrahedron Lett., 2009, 50, 2964–2966 CrossRef CAS; (e) N. Goyal, Synlett, 2010, 20, 335–336 Search PubMed; (f) J. W. Bode and L. J. Goossen, Top. Organomet. Chem., 2012, 44, 13–33 CrossRef; (g) H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 714–2742 RSC; (h) F. Ye, Y. Ge, A. Spannenberg, H. Neumann and M. Beller, Nat. Commun., 2020, 11, 5383 CrossRef CAS; (i) D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443–4458 CrossRef CAS PubMed; (j) R. W. Dugger, J. A. Ragan and D. H. B. Ripin, Org. Process Res. Dev., 2005, 9, 253–258 CrossRef CAS; (k) J. Magano, Org. Process Res. Dev., 2022, 26, 1562–1589 CrossRef CAS; (l) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC; (m) R. M. De Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (n) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org Chem., 2020, 30, 4641–4651 CrossRef; (o) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS; (p) W. Muramatsu, T. Hattori and H. Yamamoto, J. Am. Chem. Soc., 2019, 141, 12288–12295 CrossRef CAS PubMed; (q) D. T. Nguyen, D. C. Lenstra and J. Mecinović, RSC Adv., 2015, 5, 77658–77661 RSC; (r) M. A. Ali, S. M. A. H. Siddiki, K. Kon and K.-I. Shimizu, ChemCatChem, 2015, 7, 2705–2710 CrossRef CAS; (s) H. Morimoto, R. Fujiwara, Y. Shimizu, K. Morisaki and T. Ohshima, Org. Lett., 2014, 16, 2018–2021 CrossRef CAS; (t) D. C. Lenstra, D. T. Nguyen and J. Mecinović, Tetrahedron, 2015, 71, 5547–5553 CrossRef CAS; (u) M. N. Rashed, K. Masuda, T. Ichitsuka, N. Koumura, K. Sato and S. Kobayashi, Adv. Synth. Catal., 2021, 363, 2529–2535 CrossRef CAS; (v) B. D. Mkhonazi, M. Shandu, R. Tshinavhe, S. B. Simelane and P. T. Moshapo, Molecules, 2020, 25, 1040–1048 CrossRef CAS PubMed; (w) H. Tsuji and H. Yamamoto, J. Am. Chem. Soc., 2016, 138, 14218–14221 CrossRef CAS PubMed.
- (a) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC; (b) M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- (a) H. Ehrhorn and M. Tamm, Chem.–Eur. J., 2019, 25, 3190 CrossRef CAS; (b) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed; (c) U. H. F. Bunz, K. Seehafer, M. Bender and M. Porz, Chem. Soc. Rev., 2015, 44, 4322 RSC; (d) R. Chinchilla and C. Najera, Chem. Rev., 2014, 114, 1783 CrossRef CAS; (e) X. Ren, G. Li, S. Wei and H. Du, Org. Lett., 2015, 17, 990 CrossRef CAS PubMed; (f) A. Ahlers, T. de Haro, B. Gabor and A. Furstner, Angew. Chem., Int. Ed., 2016, 55, 1406 CrossRef CAS; (g) P. M. Cromm, S. Schaubach, J. Spiegel, A. Furstner, T. N. Grossmann and H. Waldmann, Nat. Commun., 2016, 7, 11300 CrossRef CAS PubMed.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- K. Muller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. Experimental procedures and spectroscopic data. See DOI: https://doi.org/10.1039/d5ra02161h |
| This journal is © The Royal Society of Chemistry 2025 |