DOI:
10.1039/D5RA02152A
(Paper)
RSC Adv., 2025,
15, 21401-21407
Mechano-responsive color changes of a Pt(II) complex possessing triethylene glycol towards pressure sensors†
Received
27th March 2025
, Accepted 17th June 2025
First published on 24th June 2025
Abstract
Mechanochromic molecules have attracted significant attention owing to their potential in the development of pressure sensors. However, relatively few studies have investigated the detailed mechanisms of the mechano-responsive nature and the quantitative visualization of mechanical forces. Herein, we report a square-planar platinum complex possessing triethylene glycol chains that exhibits mechanocromic behavior in the amorphous phase. Its mechanochromic nature was established using a combination of spectroscopic techniques, powder X-ray diffraction analyses, and computational chemistry techniques. The continuous changes in emission intensity allowed the platinum complex to be used as a mechanical force sensor, where the output signals were readable using a luminescence spectrometer. These findings demonstrate the potential benefits of square-planar platinum complexes and triethylene glycol chains for the creation of mechanochromic material.
Introduction
A remarkable feature of mechanochromic molecules is their ability to manifest unique mechano-responsive properties that can visualize mechanical forces to be changes in absorption and emission colors, which allows for the production of pressure sensors and data storage carriers.1–6 Mechano-responsive properties have been determined to result from alterations in molecular bonds or supramolecular structures induced by mechanical forces.7–22 To date, the vast majority of studies on mechanochromic molecules have demonstrated qualitative color changes in response to mechanical forces.23 Thus, aside from a quantitative discussion of the mechanisms of color change, there remains a pivotal requirement for additional mechanochromic molecules that can quantitatively visualize mechanical forces. Recently, a few studies investigated mechanochromic molecules that can be used to quantitatively visualize the mechanical forces within polymer films.24–29 Accordingly, it is worth developing mechanochromic molecules capable of quantitatively visualizing mechanical forces to expand their scope.
Square-planar platinum(II) complexes exhibit strong luminescence emissions from a metal-to-ligand charge transfer (MLCT) excited state.30 Appropriately designed square-planar platinum(II) complexes form self-assembled constructs through intermolecular Pt–Pt and/or π–π stacking interactions, resulting in luminescence emission from a metal–metal-to-ligand charge transfer (MMLCT) excited state.31,32 The emission color of the assembled platinum(II) complexes is determined by the Pt⋯Pt distance;33,34 accordingly, the energetic inputs generating Pt⋯Pt distance changes afford emission color change of the platinum complexes.4,10,35–51 In light of these reports, we envisage that Pt complex-based mechanochromic molecules would display continuous changes in their emission color or intensity in response to mechanical forces, leading to mechanical force sensors being used as analytical tools.
Our group previously reported the self-assembly of neutral platinum(II) complexes possessing isoxazole moieties to form supramolecular assemblies in various solvents.52–57 Within these reports, we documented that the absorption and emission colors of the platinum(II) complexes were significantly altered by the solvent polarity, concentration of the platinum complexes, and solution temperature. The changes in absorption and emission colors were observed as a result of changing the Pt⋯Pt distance within the assembled structure. Therefore, the Pt⋯Pt distance of our platinum(II) complex may be altered by the application of mechanical forces. Herein, we report a square-planar platinum complex possessing a triethylene glycol (TEG) side-chain 1 that exhibits mechanochromic behavior with near-infrared emission, which is rarely reported (Fig. 1a). In addition, the investigation of the mechanochromic behavior of 1 revealed the key role of the oligo-ethylene oxide chains in the emergence of its mechano-responsive nature. Thus, the following findings help to deepen our understanding of mechano-responsive molecules.
 |
| | Fig. 1 (a) Molecular structure of platinum complex 1 possessing TEG chains and 2 possessing decyl chains. Optical images of mechanically (b) unground (1unground) and (c) ground (1ground) powders of 1 showing the color change induced by the mechanical force. | |
Experimental
General
All solvents were commercial reagent grade and were used without further purification. Luminescence and excitation spectra were measured using a JASCO FP-6500 spectrometer. Infrared (IR) spectra were recorded on a JASCO FT/IR-4600 spectrometer. PXRD patterns were measured using Smart Lab (Rigaku Inc.) with Cu Kα radiation (λ = 1.5418 Å) at a scanning rate of 2.0° min−1, an applied voltage of 40 kV, and a current of 50 mA. NMR spectra were recorded on a Bruker APEX 400 MHz and Ascend 700 MHz spectrometers. Chemical shifts are quoted as parts per million (ppm) relative to residual chloroform (δ = 7.26 and 77.0 for 1H and 13C, respectively). Differential scanning calorimetry (DSC) analysis was conducted on a SEIKO instrument Inc., EXSTAR6000 (DSC6200 TG/DTA6200). The DSC curves were recorded at a heating rate of 15 °C min−1. Melting points were determined using a Yanagimoto micro melting point apparatus, which were uncorrected. ESI-Mass spectra were reported with a Thermo Fisher Scientific LTQ Orbitrap XL. Elemental analyis was performed on a Perkin-Elmer 2400II elemental analyzer. Recycling preparative GPC-HPLC separations were carried out on a JAI LC-5060 system using preparative JAIGEL-2.5HH and 2HH columns in series. Previously synthesized 1 was used in this study; purity data and detailed experimental procedures are provided in our previous paper.55
Synthesis of complex 2
To a solution of 3 (120 mg, 0.17 mmol) and (6-phenyl-2,2′-bipyridine)platinum (4) chloride (80 mg, 0.17 mmol) in dry CH2Cl2 (13 mL) and dry DMF (8 mL) was added dry NEt3 (190 μL, 0.9 mmol). The resulting solution was deoxygenated by bubbling nitrogen for 30 min, and then CuI (7 mg, 3 μmol) was added. After being stirred at room temperature for 21 h under an argon atmosphere in the dark, the reaction mixture was quenched with a portion of water. The resulting mixture was extracted with CH2Cl2. The organic layer was washed with saturated aqueous NaCl, dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was separated by column chromatography on silica gel (5% methanol in CH2Cl2, eluent), followed by the purification using gel permeation chromatography to give the desired product 2 (101 mg, 53%) as an orange solid (Scheme 1): m.p. 265–268 °C; 1H NMR (700 MHz, CDCl3): δ 9.24 (d, 1H, J = 4.7 Hz), 8.08 (s, 1H), 8.06 (s, 2H), 8.05 (d, 1H, J = 7.0 Hz), 7.98 (d, 1H, J = 7.4 Hz), 7.90 (d, 1H, J = 8.0 Hz), 7.84–7.80 (m, 5H), 7.62–7.60 (m, 2H), 7.56 (d, 1H, J = 8.0 Hz), 7.37 (d, 1H, J = 7.4 Hz), 7.23 (t, 1H, J = 6.8 Hz), 7.09 (t, 1H, J = 6.8 Hz), 7.00 (d, 4H, J = 8.4 Hz), 6.89 (s, 2H), 4.02 (t, 4H, J = 6.6 Hz), 1.82 (m, 4H), 1.48 (m, 4H), 1.37 (m, 4H), 1.34–1.26 (m, 20H), 0.89 (t, 6H, J = 7.1 Hz) ppm; 13C NMR (176 MHz, CDCl3) δ 169.4, 165.8, 162.7, 160.7, 158.0, 154.5, 151.8, 146.7, 141.8, 138.9, 138.8, 138.5, 131.7, 130.5, 130.4, 128.2, 128.0, 127.7, 124.6, 124.0, 122.5, 121.3, 119.4, 118.5, 117.5, 114.9, 109.5, 104.9, 98.0, 77.2, 77.0, 76.8, 68.1, 31.9, 29.6, 29.6, 29.4, 29.3, 29.2, 26.0, 22.7, 14.1 ppm; HRMS (ESI+) calcd for [C62H66N4O4Pt + H]+ m/z 1126.48048, found m/z 1126.47930; anal. calcd for C62H66N4O4Pt: C 66.0, H 5.91, N 4.97, found C 65.84, H 5.83, N 4.87%.
 |
| | Scheme 1 Synthesis of platinum complex 2. | |
Results and discussion
Synthesis and mechanochromic behaviors
The platinum complex 1 was synthesized according to the synthetic pathway described previously.55 1 was dissolved in dichloromethane (DCM), and hexane was added to the resulting solution, yielding a yellow precipitate (1unground, Fig. 1b). 1unground exhibited a discernible color change from yellow to red upon grinding the powder of 1unground using a mortar and pestle (1ground, Fig. 1c). During the grinding process, 1ground became stickier compared to 1unground. The dissolve-in-DCM then reprecipitate-by-hexane process allowed a color change of 1ground from red-to-yellow, indicating that 1ground reverted back to the original 1unground. Notably, no discernible changes in the 1H NMR spectra were observed between the spectra of yellow-colored powder 1unground and red-colored powder 1ground in CDCl3 (Fig. S1†). These results indicate that the color change originated from neither a chemical reaction nor molecular decomposition; therefore, the molecular arrangement of 1 in the solid state was primarily responsible for the color change.
Characterization of the luminescence
To determine the emission colors of 1, the solid-state emission spectra of 1unground and 1ground were monitored. Based on our previous report,55 1 showed characteristic absorption band at 444 nm in solution; thus, the solution-phase emission spectra were recorded at an excitation wavelength of 444 nm within the work. According to the absorption band of 1, the solid-state emission spectrum 1unground was recorded at an excitation wavelength of 444 nm. 1unground showed an emission band at approximately 570 nm with a quantum emission yield (Φ) of 3.4% (Fig. 2, black line). In contrast, emission bands at approximately 700 nm (Φ = 2.7%) and 570 nm were observed in the emission spectrum of 1ground (Fig. 2, red line). In light of our previous study, the emission band at approximately 700 nm was ascribed to the MMLCT emission band derived from Pt–Pt interactions. This observation suggests that the mechanical grinding induced the reorientation of the packing structures of 1 in the solid state, resulting in the molecular arrangement with an appropriate Pt⋯Pt distance to promote intermolecular Pt–Pt interactions. Further support for the MMLCT emission was provided by the excitation spectra of 1unground and 1ground. The excitation spectrum of 1unground (λem = 568 nm) corroborated that the emission band at approximately 570 nm was derived primarily from the absorption band at approximately 444 nm, which is characteristic of π–π* and MLCT transitions52 (Fig. 2, dotted black line). In contrast, the excitation spectrum of 1ground (λem = 710 nm) clearly showed a characteristic MMLCT-absorption band (approximately 535 nm52), which supports the Pt–Pt interactions between molecules (Fig. 2, dotted red line). To further research the mechanochromic behavior of 1, the solid-state emission spectra of platinum complexes possessing decyl side chains 2 (Fig. S2†) were monitored. No apparent changes in the luminescence band were observed, indicating that the TEG chains played a central role in the mechanochromic behavior.
 |
| | Fig. 2 Solid-state excitation (dashed lines) and emission (solid lines) spectra58 of 1unground (black lines, λex = 444 nm, λem = 568 nm) and 1ground (red lines, λex = 444 nm, λem = 710 nm) at room temperature. | |
Mechanisms of the mechanochromic behavior
To detail the mechano-responsive color change, the shear stress was applied to 1unground. The powder of 1unground was placed on the lattice-grooved plate and subjected to the shear stress by the lattice-grooved plate jig of 12 mm in diameter. Under the normal force FN of 49.2 ± 0.1 N, by applying the strain of 100% for the sample thickness of 0.136 mm, the resultant shear stress of 91.7 ± 0.7 kPa was given to the sample at a rotational frequency of 1 Hz for 10 min (MCR-302 Anton Paar Inc.). The resulting powder displayed a relatively weak MMLCT band, leading to an inference that the pressing forces within the grinding process primarily contributed to the emission color change of 1 (Fig. S3†).
The role of the TEG chains in the mechanochromic behavior was established by a combination of infrared (IR) spectroscopy and density functional theory (DFT) calculations. The IR spectra of 1unground and 1ground were recorded using a JASCO J-1500 spectrometer with an attenuated total reflection (ATR) accessory. 1unground showed characteristic IR bands at 764 and 1102 cm−1 (Fig. 3a, black line). Mechanical grinding resulted in the low-wavenumber shift of the two characteristic peaks to 761 and 1097 cm−1, respectively (Fig. 3a, red line). To characterize these bands and their shifts, DFT calculations of monomeric M1 and dimeric M1·M1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59 were performed at the B3LYP/Lanl2DZ computational level using the 6-31G(d) basis set60 (Fig. 3b–d, S6, S7, Tables S1 and S2†). The raw calculation data were processed using a scale factor of 0.9614 for B3LYP/6-31G(d).61 Considering that 1ground was densely packed in the solid state to generate intermolecular Pt–Pt interactions, dimeric M1·M1 should be suitable for estimating the electric structure of 1ground. In contrast, monomeric 1 is a good model for estimating the electric structure of 1unground because luminescence measurements of 1unground provided no evidence of Pt–Pt interactions. The calculated IR spectrum of monomeric M1 indicated that the CH out-of-plane bending vibration of the phenylbipyridine ligand moiety displayed a band at 763 cm−1, which is in good agreement with the experimentally observed band at 764 cm−1 (1unground). Calculations of dimeric M1·M1 revealed that the CH out-of-plane bending vibration band emerged in the low-wavenumber region compared to that of monomeric M1. A low-wavenumber shift was experimentally observed upon grinding 1; thus, this low-wavenumber shift supports the inference that the packing structure of 1ground is more condensed than that of 1unground, which permits intermolecular Pt–Pt interactions. Based on the calculations, the bands at 1102 cm−1 (1unground) and 1097 cm−1 (1ground) correspond to C–O–C asymmetric vibrations.62 The band (≈1100 cm−1) was not observed in the IR spectrum of 2, leading to an inference that the bands were derived from C–O–C asymmetric vibrations of the TEG chains (Fig. S4†). Thus, the low-wavenumber shift upon grinding suggests that mechanical grinding induced a conformational change in the TEG chains from a chain-folded conformation to a fully or partially extended conformation.62
59 were performed at the B3LYP/Lanl2DZ computational level using the 6-31G(d) basis set60 (Fig. 3b–d, S6, S7, Tables S1 and S2†). The raw calculation data were processed using a scale factor of 0.9614 for B3LYP/6-31G(d).61 Considering that 1ground was densely packed in the solid state to generate intermolecular Pt–Pt interactions, dimeric M1·M1 should be suitable for estimating the electric structure of 1ground. In contrast, monomeric 1 is a good model for estimating the electric structure of 1unground because luminescence measurements of 1unground provided no evidence of Pt–Pt interactions. The calculated IR spectrum of monomeric M1 indicated that the CH out-of-plane bending vibration of the phenylbipyridine ligand moiety displayed a band at 763 cm−1, which is in good agreement with the experimentally observed band at 764 cm−1 (1unground). Calculations of dimeric M1·M1 revealed that the CH out-of-plane bending vibration band emerged in the low-wavenumber region compared to that of monomeric M1. A low-wavenumber shift was experimentally observed upon grinding 1; thus, this low-wavenumber shift supports the inference that the packing structure of 1ground is more condensed than that of 1unground, which permits intermolecular Pt–Pt interactions. Based on the calculations, the bands at 1102 cm−1 (1unground) and 1097 cm−1 (1ground) correspond to C–O–C asymmetric vibrations.62 The band (≈1100 cm−1) was not observed in the IR spectrum of 2, leading to an inference that the bands were derived from C–O–C asymmetric vibrations of the TEG chains (Fig. S4†). Thus, the low-wavenumber shift upon grinding suggests that mechanical grinding induced a conformational change in the TEG chains from a chain-folded conformation to a fully or partially extended conformation.62
 |
| | Fig. 3 (a) Observed IR spectra of 1unground (black line) and 1ground (red line) and DFT-calculated IR intensities of monomeric M1 (black bar) and dimeric M1·M1 (red bar). The calculated frequencies were scaled by 0.9614. The intensities are listed in Tables S1 and S2.† Calculated structures of (b) monomeric M1 and (c) dimeric M1·M1 optimized at the B3LYP/Lanl2DZ computational level with the 6-31G(d) basis set for the estimation of the vibrational mode shown in panel a and Tables S1 and S2.† (d) Comparison of the experimental and calculated (scaled) vibrational wavenumbers (cm−1) of 1 and M1 (CH out-of-plane bending mode). | |
The phase information of 1 was obtained using solid-phase powder X-ray diffraction (PXRD). 1unground exhibited scattering in the small-angle region (2θ < 10°), suggesting the presence of large-scale heterogeneous structures. The broad scattering at approximately 2θ = 23° indicates the amorphous nature of 1unground (Fig. 4, black line). Grinding resulted in minimal changes63 in the PXRD pattern (1ground) (Fig. 4, red line), which clearly indicated that mechanical grinding did not induce a phase transition of 1 in the solid state. These results were obtained for a three-dimensionally aggregated bulk solid of 1; thus, it can be concluded that the mechanochromic behavior of 1 was driven by a change in the molecular arrangement within the amorphous phase.
 |
| | Fig. 4 Powder X-ray diffraction (PXRD) patterns of 1unground (black line) and 1ground (red line). | |
The thermal behaviors of 1unground and 1ground were studied using differential scanning calorimetry (DSC), providing insights into the mechanism of color change upon grinding. 1unground displayed endothermic peaks at 69.5, 79.3, and 159.4 °C in the heating run (Fig. 5, black line), whereas the DSC trace of 1ground showed endothermic peaks at 75.8, 94.0, 156.8, and 165.4 °C in the heating run (Fig. 5, red line). In addition to the endothermic peaks, exothermic peaks were observed at 118.7 °C and 121.2 °C in the bulk solid specimens of 1unground and 1ground, respectively. The equilibrium melting point (Tm) of polyethylene glycol (PEG) is known to be at approximately 60 °C.64 Accordingly, the broad endothermic peaks of 1unground and 1ground observed between 65 and 95 °C can be attributed to the changes in the molecular arrangement of 1 in the bulk solid derived from melting of the TEG chains. Moreover, the broad peak between 65 and 95 °C of 1ground was more intense compared with that of 1unground, which indicates that the TEG chains of 1ground were packed with higher affinity, forming conformationally rigid packing structures in the solid state. The exothermic peak at approximately 120 °C can be rationalized by the reorientation of the TEG chains. The endothermic peak at 159.4 °C in 1unground corresponds to the Tm of 1. Additionally, two separate endothermic peaks were observed at 156.9 °C and 165.2 °C. A split DSC peak such as this corresponding to Tm is caused by the reorientation of the molecular arrangement in bulk. In the case of 1gournd, one corresponds to the Tm of 1 and the other is derived from the dissociation of intermolecular Pt–Pt interactions within the bulk solid of 1ground. This consideration is supported by the experimental results shown in Fig. 2. No Pt–Pt interactions were evidenced in 1unground, whereas 1ground was densely packed through intermolecular Pt–Pt interactions. Moreover, 2ground did not show the endothermic peak at approximately 160 °C characteristic of the cleavage of Pt–Pt interactions, indicating that grounding did not induce the formation of Pt–Pt interactions within the powder 2 in bulk (Fig. S5†). These results indicates that the color change of 1 was driven by the conformational changes of the TEG chains and square-planar platinum moiety, as well as the reorientation of the molecular arrangement in bulk by the application of mechanical forces.
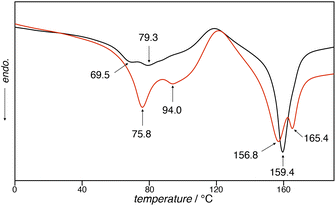 |
| | Fig. 5 DSC curves of 1unground (black line) and 1ground (red line) showing the 1st heating run at a heating rate of 15 °C min−1. | |
Visualization of the mechanical forces
The mechano-responsive nature of 1 allows the quantification of mechanical forces using a luminescence spectrometer. The MMLCT emission band grew upon the application of pressure (Fig. 6). The plot of the intensity of the MMLCT emission (720 nm) against pressure showed a continuous increase in the emission intensity, which can be read using a luminescence spectrometer. The steep increase in the emission intensity in the pressure range 0–20 MPa was gradually saturated when the total pressure reached approximately 80 MPa. This observation highlights the potential of 1 as a pressure sensor in the pressure range 0–20 MPa.
 |
| | Fig. 6 (a) Changes in the emission spectra58 of 1 (λex = 444 nm) in the solid state. The pressures applied are: (i)–(viii) 0, 5.5, 11, 16, 27, 55, 110, and 165 MPa, respectively. The blue and red lines represent the first point and the last point of the measurement, respectively. (b) Plot of the normalized emission intensity of 1 at 720 nm against the pressure applied, showing the continuous changes in the emission intensity upon pressing. | |
Conclusions
In summary, we demonstrated that a platinum(II) complex possessing TEG chains 1 showed a significant change in absorption and emission colors upon the application of mechanical forces. Color changes were found to be driven by a combination of conformational changes within the molecule and changes in the molecular arrangement in bulk. PXRD measurements confirmed that a mechano-responsive color change occurred in the amorphous phase. Moreover, luminescence spectroscopy, IR spectroscopy, and DSC measurements revealed that the conformational flexibility of the TEG chains imparted mechanochromic features to the Pt complex. Many mechanochromic molecules contain oligo-ethylene oxide chains within the molecular skeleton;65–68 thus, our detailed studies elucidated the mechano-responsive color transformation with the participation of TEG chains in bulk. This work underscores the potential benefits of both TEG chains and square-planar platinum complexes for the creation of mechanochromic materials capable of quantitatively visualizing mechanical forces. These findings are expected to serve as a design guide for luminophore/dye-based mechanical force sensors.
Data availability
The data that supports the findings of this study are available in the ESI† of this article.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors are grateful to Ms Naomi Kawata and Ms Tomoko Amimoto of the Natural Science Center for Basic Research Development (N-BARD) at Hiroshima University for facilitating the PXRD experiments and HRMS measurements, respectively. The authors thank Mr Motonari Kobayashi of the Department of Instrumental Analysis & Cryogenics Division of Instrumental Analysis at Okayama University for facilitating the elemental analysis. This work was supported by JSPS KAKENHI Grants-in-Aid for Transformative Research Areas, “Condensed Conjugation” Grant Number JP21H05491 and “Materials Science of Meso-Hierarchy” Grant Number JP23H04873, Grants-in-Aid for Scientific Research (A) Grant Number JP21H04685, a Grant-in-Aid for Young Scientists Grant Number JP22K14727, and a Grant-in-Aid for JSPS Fellow JP23KJ1640. We acknowledge the support of KEIRIN JKA (Grant Number 2023M-419). Funding from the Hosokawa Powder Technology Foundation, Kumagai Foundation for Science and Technology, Mukai Science Technology Foundation, Shorai Foundation for Science and Technology, Amano Institute of Technology, and Old River Foundation is gratefully acknowledged.
Notes and references
- Y. Sagara and T. Kato, Nat. Chem., 2009, 1, 605–610 CrossRef CAS PubMed.
- F. Ciardelli, G. Ruggeri and A. Pucci, Chem. Soc. Rev., 2013, 42, 857–870 RSC.
- Y. Sagara, S. Yamane, M. Mitani, C. Weder and T. Kato, Adv. Mater., 2016, 28, 1073–1095 CrossRef CAS PubMed.
- P. Xue, J. Ding, P. Wang and R. Lu, J. Phys. Chem. C, 2016, 4, 6688–6706 CAS.
- G. Chen and W. Hong, Adv. Opt. Mater., 2020, 8, 2000984 CrossRef CAS.
- Y. Sun, Z. Lei and H. Ma, J. Phys. Chem. C, 2022, 10, 14834–14867 CAS.
- Y.-A. Lee and R. Eisenberg, J. Am. Chem. Soc., 2003, 125, 7778–7779 CrossRef CAS PubMed.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
- H. Ito, T. Saito, N. Oshima, N. Kitamura, S. Ishizaka, Y. Hinatsu, M. Wakeshima, M. Kato, K. Tsuge and M. Sawamura, J. Am. Chem. Soc., 2008, 130, 10044–10045 CrossRef CAS PubMed.
- T. Abe, T. Itakura, N. Ikeda and K. Shinozaki, Dalton Trans., 2009, 711–715 RSC.
- S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed.
- G. Zhang, J. Lu, M. Sabat and C. L. Fraser, J. Am. Chem. Soc., 2010, 132, 2160–2162 CrossRef CAS PubMed.
- H. Ito, M. Muromoto, S. Kurenuma, S. Ishizaka, N. Kitamura, H. Sato and T. Seki, Nat. Commun., 2013, 4, 2009 CrossRef PubMed.
- Q. Benito, X. F. Le Goff, S. Maron, A. Fargues, A. Garcia, C. Martineau, F. Taulelle, S. Kahlal, T. Gacoin, J.-P. Boilot and S. Perruchas, J. Am. Chem. Soc., 2014, 136, 11311–11320 CrossRef CAS PubMed.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952–2955 CrossRef CAS PubMed.
- Y. Lv, Y. Liu, X. Ye, G. Liu and X. Tao, CrystEngComm, 2015, 17, 526–531 RSC.
- X.-P. Zhang, J.-F. Mei, J.-C. Lai, C.-H. Li and X.-Z. You, J. Phys. Chem. C, 2015, 3, 2350–2357 CAS.
- K. Ohno, S. Yamaguchi, A. Nagasawa and T. Fujihara, Dalton Trans., 2016, 45, 5492–5503 RSC.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- W. Yang, C. Liu, S. Lu, J. Du, Q. Gao, R. Zhang, Y. Liu and C. Yang, J. Phys. Chem. C, 2018, 6, 290–298 CAS.
- F. Khan, M. Mahmoudi, P. K. Gupta, D. Volyniuk, J. V. Grazulevicius and R. Misra, J. Phys. Chem. C, 2023, 127, 1643–1654 CrossRef CAS.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
- Y. Xiong, J. Huang, Y. Liu, B. Xiao, B. Xu, Z. Zhao and B. Z. Tang, J. Phys. Chem. C, 2020, 8, 2460–2466 CAS.
- K. Ogumi, K. Nagata, Y. Takimoto, K. Mishiba and Y. Matsuo, J. Phys. Chem. C, 2022, 10, 11181–11186 CAS.
- S. Thazhathethil, T. Muramatsu, N. Tamaoki, C. Weder and Y. Sagara, Angew. Chem., Int. Ed., 2022, 61, e202209225 CrossRef CAS PubMed.
- V. C. Ritter, S. M. McDonald, A. V. Dobrynin, S. L. Craig and M. L. Becker, Adv. Mater., 2023, 35, 2302163 CrossRef CAS PubMed.
- X. Yang, N. Li, B. Wang, P. Chen, S. Ma, Y. Deng, S. Lü and Y. Tang, Angew. Chem., Int. Ed., 2025, 64, e202419114 CrossRef CAS PubMed.
- Y. Zhu, M. Pan, L. Ma and Y. Wang, Chem. Eng. J., 2025, 505, 159245 CrossRef CAS.
- J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055–3066 CrossRef CAS PubMed.
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- M. Nakagaki, S. Aono, M. Kato and S. Sakaki, J. Phys. Chem. C, 2020, 124, 10453–10461 CrossRef CAS.
- M. Yoshida and M. Kato, Coord. Chem. Rev., 2018, 355, 101–115 CrossRef CAS.
- D. Saito, T. Ogawa, M. Yoshida, J. Takayama, S. Hiura, A. Murayama, A. Kobayashi and M. Kato, Angew. Chem., Int. Ed., 2020, 59, 18723–18730 CrossRef CAS PubMed.
- J. Ni, Y.-H. Wu, X. Zhang, B. Li, L.-Y. Zhang and Z.-N. Chen, Inorg. Chem., 2009, 48, 10202–10210 CrossRef CAS PubMed.
- J. Ni, X. Zhang, N. Qiu, Y.-H. Wu, L.-Y. Zhang, J. Zhang and Z.-N. Chen, Inorg. Chem., 2011, 50, 9090–9096 CrossRef CAS PubMed.
- S. J. Choi, J. Kuwabara, Y. Nishimura, T. Arai and T. Kanbara, Chem. Lett., 2012, 41, 65–67 CrossRef CAS.
- X. Zhang, J.-Y. Wang, J. Ni, L.-Y. Zhang and Z.-N. Chen, Inorg. Chem., 2012, 51, 5569–5579 CrossRef CAS PubMed.
- X. Zhang, Z. Chi, Y. Zhang, S. Liu and J. Xu, J. Phys. Chem. C, 2013, 1, 3376–3390 CAS.
- A. Han, P. Du, Z. Sun, H. Wu, H. Jia, R. Zhang, Z. Liang, R. Cao and R. Eisenberg, Inorg. Chem., 2014, 53, 3338–3344 CrossRef CAS PubMed.
- C.-J. Lin, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2017, 56, 4978–4989 CrossRef CAS PubMed.
- L. Liu, X. Wang, N. Wang, T. Peng and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 9160–9164 CrossRef CAS PubMed.
- C.-Y. Lien, Y.-F. Hsu, Y.-H. Liu, S.-M. Peng, T. Shinmyozu and J.-S. Yang, Inorg. Chem., 2020, 59, 11584–11594 CrossRef CAS PubMed.
- J. Ni, G. Liu, M. Su, W. Zheng and J. Zhang, Dyes Pigm., 2020, 180, 108451 CrossRef CAS.
- Q.-Y. Yang, H.-H. Zhang, X.-W. Qi, S.-S. Sun, D.-S. Zhang, L.-Z. Han, X.-P. Zhang and Z.-F. Shi, Dalton Trans., 2021, 50, 8938–8946 RSC.
- H.-H. Zhang, Q.-Y. Yang, X.-W. Qi, S.-S. Sun, B.-S. Li, D.-S. Zhang, X.-P. Zhang and Z.-F. Shi, Inorg. Chim. Acta, 2021, 523, 120411 CrossRef CAS.
- D. Gómez de Segura, E. Lalinde and M. T. Moreno, Inorg. Chem., 2022, 61, 20043–20056 CrossRef PubMed.
- Q.-Z. Yuan, F.-S. Wan, T.-T. Shen and D.-K. Cao, RSC Adv., 2022, 12, 148–153 RSC.
- H.-H. Zhang, S.-X. Wu, Y.-Q. Wang, T.-G. Xie, S.-S. Sun, Y.-L. Liu, L.-Z. Han, X.-P. Zhang and Z.-F. Shi, Dyes Pigm., 2022, 197, 109857 CrossRef CAS.
- H. Sogawa, M. Abe, R. Shintani, T. Sotani, K. Tabaru, T. Watanabe, Y. Obora and F. Sanda, Polym. J., 2023, 55, 1119–1128 CrossRef CAS.
- B.-C. Tzeng, C.-C. Liao, P.-Y. Jung, S.-Y. Chen, B.-J. Sun, W.-C. Cheng, A. H. H. Chang and G.-H. Lee, Inorg. Chem., 2023, 62, 916–929 CrossRef CAS PubMed.
- T. Ikeda, M. Takayama, J. Kumar, T. Kawai and T. Haino, Dalton Trans., 2015, 44, 13156–13162 RSC.
- T. Ikeda and T. Haino, Polymer, 2017, 128, 243–256 CrossRef CAS.
- T. Haino and T. Hirao, Chem. Lett., 2020, 49, 574–584 CrossRef CAS.
- T. Hirao, H. Tsukamoto, T. Ikeda and T. Haino, Chem. Commun., 2020, 56, 1137–1140 RSC.
- M. Yoshida, T. Hirao and T. Haino, Org. Biomol. Chem., 2021, 19, 5303–5311 RSC.
- M. Yoshida, T. Hirao and T. Haino, Chem. Commun., 2022, 58, 8356–8359 RSC.
- The spike peaks found in the emission and excited spectra arise from excitation light.
- Dimeric M1·M1 in a head-to-tail stacking arrangement prevents the formation of intermolecular Pt–Pt interactions; thus, we studied with dimeric M1·M1 in a head-to-head stacking arrangement shown in Fig. 3c.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef CAS.
- J. J. Shephard, P. J. Bremer and A. J. McQuillan, J. Phys. Chem. B, 2009, 113, 14229–14238 CrossRef CAS PubMed.
- The grinding induced a slight wide-angle shift of the peak top of 2θ = 23° and a decrease in intensity of diffraction around at 2θ = 14°. The slight changes in the diffraction pattern appear to be reorientation of molecular arrangement of 1 to form densely packed structures in bulk.
- T. Jayaramudu, G. M. Raghavendra, K. Varaprasad, G. V. S. Reddy, A. B. Reddy, K. Sudhakar and E. R. Sadiku, J. Appl. Polym. Sci., 2016, 133, 43027 CrossRef.
- S. Yagai, T. Seki, H. Aonuma, K. Kawaguchi, T. Karatsu, T. Okura, A. Sakon, H. Uekusa and H. Ito, Chem. Mater., 2016, 28, 234–241 CrossRef CAS.
- J. Zessin, M. Schnepf, U. Oertel, T. Beryozkina, T. A. F. König, A. Fery, M. Mertig and A. Kiriy, Adv. Opt. Mater., 2020, 8, 1901410 CrossRef CAS.
- H. Sakai, K. Nonaka, R. Hayasaka, S. Thazhathethil, Y. Sagara and T. Hasobe, Chem. Commun., 2024, 60, 4084–4087 RSC.
- S. Thazhathethil, F. S. Thuluvanchery, S. Shimizu, I. Scarlat, J. M. Clough, C. Weder and Y. Sagara, J. Phys. Chem. C, 2024, 12, 6170–6176 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab






