
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in focal adhesion kinase (FAK)-targeting antitumor agents
Si-Kai Zhua,
Qi Wub,
Guang-Xin Liub,
An-Qi Geng*b and
Ping-An Wang *a
*a
aDepartment of Medicinal Chemistry and Pharmaceutic Analysis, School of Pharmacy, The Fourth Military Medical University, Changle West Road 169, Xi'an, 710032, P. R. China. E-mail: ping_an1718@outlook.com
bInstitute of Medical Research, Northwestern Polytechnical University, Youyi West Road 127, Xi'an, 710072, P. R. China. E-mail: anqi@nwpu.edu.cn
First published on 20th June 2025
Abstract
Focal adhesion kinase (FAK) is a non-receptor intracellular tyrosine kinase that plays an important role in cell adhesion, survival, proliferation, and other processes. Research studies have shown that overexpression of FAK can promote the proliferation and migration of tumor cells. Therefore, FAK has become a promising target for small molecular anticancer drugs with great clinical potential. In recent years, research studies on small molecular FAK inhibitors have been increasing. With the rise of proteolysis-targeting chimera (PROTAC) technology, FAK PROTACs have also been developed to induce FAK degradation. This article reviews the design and biological activity of FAK-targeting antitumor agents, with a focus on their chemical structures, antitumor effects, and clinical progress.
Focal adhesion kinase (FAK) is a protein encoded and synthesized by the PTK2 gene. It plays an important role in cell adhesion, migration, survival, proliferation, and embryonic development.1 FAK is a class of cytoplasmic non-receptor protein tyrosine kinases (PTKs) consisting of three domains, including an N-terminal FERM domain, a central kinase domain, and a C-terminal domain. There are multiple tyrosine phosphorylation sites (Y194, Y397, Y407, Y576, Y577, Y861, Y925, Y1007) in FAK, which play a key regulatory role in the molecular function of FAK. Their phosphorylation can provide binding sites for signal proteins and regulate the catalytic activity of FAK. FAK is widely expressed in human tissues and can regulate various cellular processes. Overexpression of FAK can be detected in many different sources of human cancer cells. Biological studies have shown that the activity of FAK is crucial for the occurrence and progression of human cancer. Therefore, targeting FAK is considered one of the effective means for development of novel antitumor drugs.2,3 At present, many small molecular FAK inhibitors have been developed. Although there are no small molecular FAK inhibitors on market yet, some chemical entities such as VS-6063 (Defictinib) and CT-707 (Contertinib) have entered the phase III clinical research stage. In addition, with the rise of Proteolysis-Targeting Chimera (PROTAC) technology,4 PROTAC targeting FAK has also been developed to induce FAK degradation. Based on a brief introduction to the structure and function of FAK protein, this review covers the design, and biological activity of targeted FAK agents, with a focus on their chemical structures, antitumor effects, and clinical research progress.
1. Structure and function of FAK
The FAK protein consists of 1052 amino acids, with a molecular weight of approximately 125–130 kDa, and contains three domains: N-terminal FERM domain, central kinase domain, and C-terminal domain (Fig. 1).5,6 The N-terminal FERM domain of FAK consists of three subdomains F1, F2, and F3, resembling a clover-like structure, with tyrosine residue Y397 located on it as a phosphorylation site. The FERM domain can interact with intracellular parts of many upstream membrane proteins such as growth factor receptors and c-Met, as well as with many downstream cytoplasmic proteins such as P53, MDM2, SRC kinase, etc., mediating protein–lipid and protein–protein interactions through scaffold function. The FAK central kinase domain has high homology with other tyrosine kinases, and the tyrosine residues Y576 and Y577 located on it are phosphorylation sites that can mediate the activation of the PI3K/AKT/mTOR pathway. The C-terminal domain of FAK consists of a FAT (focal adhesion targeting) domain and two proline rich regions (PRRs) subdomains, on which tyrosine residues Y861 and Y925 are phosphorylation sites. Proline rich motifs can interact with proteins containing SH3 domains, while FAT domains can interact with adhesion complex proteins such as paxillin, talin, vinculin, and GRb2. In addition, the FAT domain can be combined with the F2 subdomain of the FERM domain, leading to the dimerization of FAK. | ||
| Fig. 1 FAK structure and FAK-related signaling pathways in cancer development. | ||
FAK is the intersection of multiple signalling pathways within cells, promoting the survival and growth of tumor cells through multiple signalling pathways, including kinase dependent and non-kinase dependent pathways, involving physiological processes such as apoptosis, drug resistance, and cell cycle of tumor cells. After entering the cell, FAK receives signals from integrins, growth factors, receptor tyrosine kinases (RTKs), G-protein coupled receptors, and other stimuli, and undergoes phosphorylation activation, thereby mediating downstream changes in P53, MDM2, LATS1/2, PI13K, Ras, etc., which have a significant impact on tumor cell growth, migration, proliferation, etc. (Fig. 1).
The N-terminal FERM domain of FAK protein is connected to P53 in the F1 and F2 lobes, and to the ubiquitin ligase MDM2 in the F3 lobe. It promotes the ubiquitination and inactivation of P53 through a non-kinase dependent signalling pathway. This path can slow down the rate of tumor cell apoptosis, and promote tumor cell growth and proliferation.7 The key phosphorylation site Y397 is in the FERM domain of the N-terminus of FAK protein. This is also a key site for small molecular FAK inhibitors design.8 The extracellular matrix (ECM) connecting to cells through the integrin receptor group results to FAK dimerization. This interaction causes autophosphorylation at the Y397 site.9 It connects to the SRC family kinase to mediate the activation of the LATS1/2-YAP signaling pathway and prevent tumor cell apoptosis.10 In various tumor cells, the increase of FAK expression and activity lead to the promotion of cell antiapoptotic effects. The tyrosine residues Y576 and Y577 located on the FAK central domain are also key phosphorylation sites. Phosphorylation activates the PI3K/AKT/mTOR signaling pathway,11,12 promoting tumor growth and migration. Therefore, the FAK central kinase domain has become a highly useful area for the design of FAK inhibitors. The C-terminal FAT domain of FAK protein is responsible for interacting with proteins such as talin, paxilin, and GRb2 to form focal adhesion complexes,13,14 which play an important role in the localization and regulation of FAK protein. After phosphorylation of tyrosine residues Y861 and Y925 located in the FAT domain, the Ras/RAF/MEK-ERK pathway is activated through the complex, leading to tumor cell proliferation and migration. After FAK is phosphorylated and activated, it mediates multiple intracellular signalling pathways, promoting the formation and development of tumor cells. Therefore, the development of inhibitors targeting FAK can achieve the goal of treating tumors.
2. Development of small molecular FAK inhibitors
Nitrogen-containing heterocycles are usually used as main motif of small molecular FAK inhibitors, such as pyrimidines, triazines, and others, as well as some dual and multi-target inhibitors and inhibitors targeting FAK through protein–protein interactions (PPIs). A description of the chemical structure characteristics of small molecular FAK inhibitors is described below.2.1 FAK inhibitors based on pyrimidines
FAK inhibitors containing pyrimidine structures, with 2,4-diaminopyrimidine as the most common backbone, can be divided into two categories: monocycle and fused ring. There are several reports on FAK inhibitors with 2-aminopyrimidine structure.
In 2010, A. Schultze et al.17 reported ATP competitive FAK inhibitor 1 (PF-573228). Research has found that PF-573228 exhibits a strong inhibitory effect on FAK (IC50 = 4.0 nM), effectively reducing Y397 phosphorylation in REF52 and PC3 and inhibiting cell migration. It also dose-dependently inhibits the phosphorylation of the downstream substrate paxillin in Tyr31. PF-573228 can affect the survival rate of OVCAR-3 cells, and its combination with metformin can enhance its cytotoxicity. Therefore, the combination of PF-573228, metformin and carboplatin can improve the therapeutic effect of platinum resistant ovarian cancer. By optimizing the structure of PF-573228, Pfizer researchers also obtained multiple FAK inhibitors, including two lead compounds VS-6062 and VS-6063. Compound 2 is an analog of TAE226,18 which differs from TAE226 in that a carbonyl group is inserted between the morpholine ring and the benzene ring. Compared with TAE226, 2 exhibited higher activity in inhibiting FAK (IC50 = 5.17 nM). Compound 2 also exhibits excellent antiproliferative activity against AsPC-1, BxPC-3, and MCF-7/ADR cells, but with very low cytotoxicity against normal human cells. In addition, the expression of MEK and ERK proteins is rarely affected by compound 2. In the AsPC-1 cancer xenograft mouse model, compound 2 at a continuous oral dose of 60 mg kg−1 for 7 days resulted in tumor regression, indicating that compound 2 has strong antitumor activity in vivo. Docking studies have shown that the carbonyl group introduced in compound 2 forms hydrogen bonds with the amino group of Gln438, which forms a new hydrogen bond with FAK central kinase domain, explaining that compound 2 is more potent than TAE226.
Molecular hybridization is one of the commonly used methods in drug design.19 Thiocarbamate esters are widely used in the design of novel anticancer drugs.20–22 Yin et al.23 combined dithiocarbamate with the classical 2,4-diaminopyrimidine structure to design and synthesize a novel FAK inhibitor. Compound 3 exhibited FAK inhibition with IC50 value of 0.07 nM and demonstrated good anti-cancer activity in multiple cancer cell lines. Further research has shown that compound 3 can induce apoptosis in MCF-7 and HCT116 cells in a dose-dependent manner, and lead to cell cycle retard in the G2/M phase. In addition, compound 3 can effectively inhibit the migration of MCF-7 and suppress microtubule formation in HUVEC cells at low concentrations, demonstrating its antiangiogenic effect. Due to their advantages such as enhanced water solubility and improved bioavailability, phosphate and sulfonamide groups have been widely used in drug design.24,25 Ma et al.26 used fragment-based drug design (FBDD) strategy to design and synthesize a series of 2,4-diaminopyrimidine derivatives containing phosphorus amides as ATP competitive FAK inhibitors. Among them, compound 4 showed stronger FAK inhibitory activity than TAE226, effectively inhibiting the proliferation of AsPC cells, inducing apoptosis of AsPC cells in a dose-dependent and time-dependent manner, and exhibiting low cytotoxicity in HPDE6-C7 cells. Their research group also designed and synthesized a series of compounds containing sulfonamide substituted pyrimidine structures, among them, compound 5 (ref. 27) had an IC50 value of 86.7 nM for FAK, which could inhibit the cell growth of AsPC-1 and Panc-1 cell lines and induce apoptosis of AsPC cells in a dose-dependent manner. Farand et al.28 designed and synthesized a macrocyclic compound 6 containing a sulfonamide structure, which showed high selectivity towards PYK2. The IC50 values for FAK and PYK2 were 4.34 nM and 0.84 nM, respectively. The macrocyclic structure also endowed compound 6 with good metabolic stability. In 2022, Ma et al. replaced the formamide group and morpholine ring in the TAE226 structure with isopropyl sulfonate group and long chain phosphoryl amine group, respectively, and designed and synthesized compound 7.29 Compound 7 with 0.12 nM IC50 value to FAK demonstrated higher FAK inhibitory activity than TAE226, and showed significant antiproliferation effect on six different cancer cell lines. In vivo model studies showed that compound 7 at a dose of 10 mg per kg per day could significantly inhibit tumor growth with an inhibition rate of up to 85%, which expected to be a candidate drug for targeted treatment of pancreatic cancer (Fig. 3).
 | ||
| Fig. 3 FAK inhibitors based on 2,4-diaminopyrimidine core. | ||
Covalent inhibition has become one of the important strategies in drug development.30 Since the discovery of the oldest covalent drug aspirin in 1895, the FDA has approved nearly 1700 small molecule drugs, of which over 120 are covalent drugs, accounting for 7%. In recent years, the development of covalent inhibitors targeting the undruggable G12C mutant KRAS protein has brought about a revival of covalent drugs.31 In 2018, Chen et al.32,33 designed and synthesized the first FAK covalent inhibitor 8 (IC50 = 0.6 nM) using TAE226 as the parent structure and introducing acrylamide covalent warheads. The inhibitor exhibited strong inhibitory activity against human glioblastoma cells U87 MG, U251, and A172 (Fig. 4). Research has shown that compound 8 can block the G2/M phase of U87 cells, thereby delaying the cell cycle progression. It has a significant inhibitory effect on the autophosphorylation of FAK and its downstream signal molecules AKT and Erk, confirming that FAK covalent inhibitors have the potential application for the treatment of malignant gliomas. The cocrystal structure reveals a strong binding affinity between 8 and FAK protein (PDB ID: 6GCX), with its acrylamide head forming a stable covalent interaction with the thiol group (–SH) of Cys427 residue in the FAK central kinase domain (Fig. 5). In 2021, Chen et al.34 retained the pyrimidine ring and ortho-aminobenzamide structure in TAE226 and replaced the morpholine ring with a piperazine ring, developing a novel covalent FAK inhibitor 9 with IC50 value of 35 nM. The research team replaced the covalent warhead in compound 9 with sulfonamide group35 to prepare compounds 10, which has similar inhibitory activity against FAK with IC50 values of 48 nM. They have also prepared compound 11 with one CF3 group to replace Cl at pyrimidine ring and connecting 2,3-dihydro-1H-inden-1-one group on the left side. Compound 11 displayed good inhibition of FAK (IC50 = 45 nM) with at least 22 fold of selectivity over insulin receptor (IR, IC50 > 1000 nM). It also exhibited potent anticancer activity against HeLa, HCT116 and MDA-MB-231 cell lines with IC50 values of 0.27, 0.19 and 0.26 μM respectively. Compound 11 can stimulate cell cycle arrest in the G2/M phase through the inhibition of FAK-Src-ERK signaling pathway in a dose-dependent manner. Moreover, compound 11 showed adequate oral bioavailability and good pharmacokinetic property. In 2021, Zhou et al.36 designed and synthesized FAK inhibitors 12 and 13 based on deep understanding of the cocrystal structure of TAE226 and FAK. In this research work, they retained the pyrimidine ring to anchor the DFG short helix sequence of FAK hinge region, and oriented the 4-amino-N-methylbenzamide group toward the solvent region of FAK. This design made compounds 12 and 13 have extremely strong inhibitory activity against FAK, with IC50 value of 2.75 nm and 1.87 nm, respectively. Studies have shown that compounds 12 and 13 have potent antiproliferative activity against nine human cancer cell lines. In addition, 12 and 13 also showed good antiangiogenic effects. In 2022, Zhang et al.37 synthesized a compound 14 targeting FAK with 2,4-diaminopyrimidine as the backbone by using bromine atom instead of the common chlorine atom or trifluoromethyl group on the pyrimidine ring. The study showed that compound 14 had significant inhibitory activity against FAK (IC50 = 3.7 nM), and the inhibitory effect was higher than that of the positive drug GSK-2256098 (IC50 = 18 nM). In addition, compound 14 could significantly inhibit the tumor growth of S180 tumor bearing mice, and the tumor volume and weight were significantly reduced compared with PF-562271 as a positive control. They also successfullyprepared compound 14 containing radiolabeled 18F, which can specifically take up by tumor cells and can be used as a new tracer for tumor diagnosis and treatment. In 2023, Song et al.38 introduced cinnamic acid group into TAE226 for the first time and constructed a novel FAK inhibitor 15 (IC50 = 49 nM) based on 2,4-diaminopyrimidine skeleton. Studies have confirmed that compound 15 can effectively inhibit FAK related signals, including AKT/mTOR and MAPK pathway in MGC-803 cells. In addition, the molecular bonding experiments between 15 and FAK exhibited favorable binding properties. The oxygen atom of the carbonyl group in the o-aminobenzamide group of compound 15 and the chlorine atom on the pyrimidine ring formed hydrogen bonds and halogen bonds with the Asp564 residue of the FAK central kinase domain, respectively. The 3-position nitrogen atom and 2-position amino group of the pyrimidine ring formed two hydrogen bonds with the Cys502 residue of the FAK central kinase domain.
 | ||
| Fig. 4 FAK inhibitors based on 2,4-diaminopyrimidine core (continued). | ||
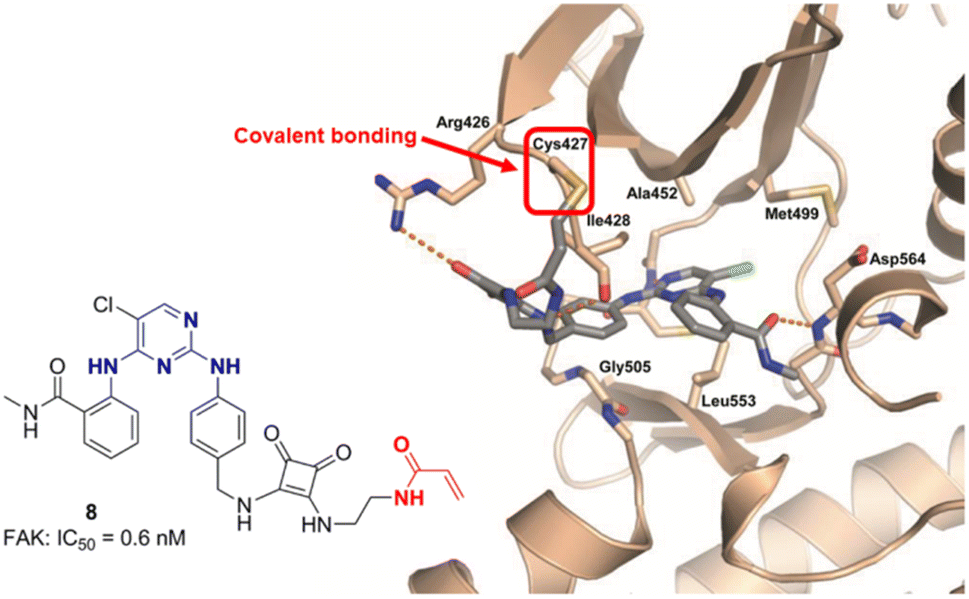 | ||
| Fig. 5 Crystal structure of FAK covalent inhibitor 8 with the kinase domain of FAK (PDB ID: 6GCX).32 | ||
As we all know, nitric oxide (NO) is a gas signaling molecule and has a wide range of effects in tumor biology.39,40 Relevant studies have shown that NO has the potential to regulate FAK signaling function. By inducing the release of elevated NO levels, it will have a synergistic inhibitory effect on FAK kinase catalytic activity and non-kinase functions.41 Phenylsulfonyl furazan is a NO release precursor. In 2023, Kang et al.42 introduced benzenesulfonyl furazan into the molecular design of FAK inhibitors and developed a nitric oxide releasing derivative 16 based on TAE226. The pyrimidine core structure of compound 16 linked to benzenesulfonyl furazan through long-chain fatty acids, and the IC50 for FAK was 29 nM. Studies have confirmed that compound 16 can induce a significant increase in the level of NO signal, exert the dual synergistic effect of NO release/FAK inhibition, and achieve potent tumor inhibition by regulating the kinase dependent and independent of FAK signaling pathway, which has made a beneficial exploration for the development of new FAK inhibitors. In 2025, Zhang et al. have prepared compound 17 through rational drug design strategy.43 Compound 17 is a very potent FAK inhibitor with IC50 value of 0.83 nM, which possesses a classic 2,4-diaminopyrimidine motif with one Br to replace Cl at pyrimidine ring, and the morpholine ring was replaced by 2-fluoroethoxyl group. Both in vitro and in vivo studies showed that 17 dramatically suppressed tumor cell viability, cancer stem cell activity, and cell migration in A549 and SKOV-3 cell lines with tumor inhibition rates of 59.15% and 57.9%, respectively.
The synthesis of TAE226 is from 2,4,5-trichloropyrimidine, 2-amino-N-methylbenzamide and 2-methoxy-4-morpholinoaniline in two steps (Fig. 6).44,45 2-Methoxy-4-morpholinoaniline was prepared from 4-fluoro-2-methoxy-1-nitrobenzene coupling with morpholine followed the reduction of nitro group under H2 atmosphere. 2,4,5-trichloropyrimidine is usually used as one starting compound due to the different reaction activities of two Cl atoms between two nitrogen atoms and adjacent to nitrogen on the pyrimidine ring. The synthesis of the other FAK inhibitors with a 2,4-diaminopyrimidine motif is similar to the preparation of TAE226. The structure–activity relationship (SAR) of FAK inhibitors containing 2,4-diaminopyrimidine backbone is illustrated on Fig. 7 by using TAE226 as a representative. The Cl atom on pyrimidine ring can be replaced by other electron-withdrawing groups, such as CF3, Br, NO2, or introduce a fused ring. The amide group of the phenyl ring on the left side can be replaced by a sulfonamide group, or connect with right side to form a macrocyclic structure. The elimination of methoxyl group of the phenyl ring on the right side has no decrease of FAK inhibitory activity. The morpholine ring does not interact with FAK, and its main function is to enhance the pharmacokinetic properties of TAE226, and it can be replaced by piperidine, piperazine, amide, ester group, or introduce a covalent warhead, for example, acrylamide group, to form a covalent FAK inhibitor. It can be seen from the above description that the molecular structure of FAK inhibitors with 2,4-diaminopyrimidine monocycle as backbone is mostly like TAE226. Without exception, there are halogen atoms (Cl, Br) or halogen containing groups (CF3) substituted at the 5-position of pyrimidine ring. The amino groups at the 2-position and 4-position of pyrimidine ring directly connect with benzene ring, ensuring that such molecules adopt a U-type structure binding to the central kinase domain of FAK.
 | ||
| Fig. 6 The synthesis of TAE226. | ||
 | ||
| Fig. 7 The structure–activity relationship (SAR) of FAK inhibitors containing 2,4-diaminopyrimidine backbone. | ||
 | ||
| Fig. 8 FAK inhibitors based on fused ring of 2,4-diaminopyrimidine. | ||
Cheng et al.46 designed and synthesized a novel FAK inhibitor 18 containing pyrrolo[2,3-d]pyrimidine structure. Molecular docking studies showed that the nitrogen atom in the newly introduced pyrrole ring has an additional hydrogen bonding interaction on the Glu500 residue of FAK hinge region, thereby enhancing the interaction between compound 18 and FAK.
Compound 18 exhibited potent FAK inhibitory activity with an IC50 value of 5.4 nM and good FAK selectivity among 26 kinase lineages. Compound 18 can inhibit the migration of A549 cells and induce their apoptosis, showing low toxicity to normal human cell line such as HK2 cells. Animal experiments confirmed that compound 18 has metabolic stability in mouse, rat and human liver microsomes, and the inhibitory effect on human cytochrome P450 is very weak, indicating that compound 18 has good druggability. The synthesis of 18 is starting from 2,4-dichloro-7H-pyrrolo[2,3-d]pyrimidine by protection of NH of pyrrole with TsCl followed coupling with (2-aminophenyl)dimethylphosphine oxide under basic conditions, and then, the Buchwald–Hartwig reaction between tert-butyl 4-(4-amino-3-methoxybenzoyl)piperazine-1-carboxylate and the above prepared intermediate was carried out to provide N-protected 18. After deprotection of N-Ts on pyrrole and N-Boc on piperazine, compound 18 was obtained in good yield (Fig. 9).
 | ||
| Fig. 9 The synthesis of compound 18. | ||
Huang et al.47 proposed a new pyrrolopyrimidine structure compound 19 (IC50 = 9.9 nM), which is characterized by 2,4-diaminopyrimidine formed by 2,7-disubstituted secondary amine cyclization as the core skeleton, and m-chlorobenzyl group is substituted on the pyrrole ring nitrogen, which is used to reduce the planar rigidity and enhance the interaction with FAK. Compound 19 could effectively induce apoptosis of A549 cells and retard the cell cycle at G2/M phase. Animal experiments showed its good pharmacokinetic properties and bioavailability. Yu et al.48 reported that the FAK inhibitor 20 with pyrrolo[2,3-d] pyrimidine as the core skeleton was connected to the aromatic ring by replacing the nitrogen atom at the 4-position of 2,4-diaminopyrimidine with an oxygen atom. The IC50 value of compound 20 for FAK was 1.9 nM, and its half-life was 4.08 hours at an oral dose of 20 mg kg−1, showing excellent drug metabolism parameters and good antitumor activity in vivo. The tumor growth inhibition rate (TGI) was 72.5% at a dose of 60 mg kg−1. Zhang et al.49 used Schrodinger software to carry out extensive simulation screening of the ATP binding pocket of FAK. Based on a FAK inhibitor PF-562271, which has entered the clinical phase I, compounds 21 and 22 were synthesized by cyclizing pyrrolo and pyrazolo-fused pyrimidine backbone, respectively. The IC50 for FAK was 55 nM and 206 nM, respectively. This cyclization strategy improved the occupancy of pyrimidine ring in the ATP binding pocket to a certain extent. The introduction of a halogen group at the 5- or 6-position of the pyrrole ring can improve the activity, and the compound 23 with a fluorine atom at the 5-position has a FAK inhibitory activity of 22 nM. Subsequently, the authors returned to a similar structure as TAE226, retained the pyrazolopyrimidine core skeleton, still used an o-aminobenzamide group at the 4-position of the pyrimidine ring, and introduced an m-aminobenzamide group at the 2-position of the pyrimidine ring to obtain compound 24, which showed excellent FAK inhibitory activity (IC50 = 12 nM). The IC50 values of compound 24 against YY8103 and SMMC7721 human hepatocellular carcinoma cells were 2.39 and 10.07 μM, respectively, which exceeded PF-562271. In addition to its excellent inhibitory activity, compound 24 also has good drug metabolism parameters, including a half-life (t1/2) of 3.65 hours (10 mg per kg oral), and showed significant in vivo antitumor activity (TGI = 78.6% at 30 mg per kg dose). The structure–activity relationship of FAK inhibitors containing fused pyrrole ring on 2,4-diaminopyrimidine is demonstrated on Fig. 10.
 | ||
| Fig. 10 The structure–activity relationship (SAR) of FAK inhibitors based on fused ring of 2,4-diaminopyrimidine. | ||
Gray et al.50 reported a series of novel FAK inhibitors based on a pyrazolo (or thiazol)-1,4-diaza-5-cycloheptanopyrimidine tricyclic structure. In this study, they used pyrazole (or thiazole) and 1,4-diaza-5-cycloheptanone to replace the o-aminobenzamide of TAE226, and replaced the morpholine ring with 4-methylpiperazine, and synthesized compounds 25 (BJG-02-063) and 26 (BJG-01-181), with inhibitory activities against FAK of 132 nM and 62.2 nM, respectively. The structure of compound 26 was further modified to produce better inhibitory activity. The hydrogen on the 4-position nitrogen atom of 1,4-diaza-5-cycloheptanone was replaced by methyl group to obtain compound 27 (BJG-03-025), which enhanced its FAK inhibitory activity to 22 nM. Compound 27 could significantly inhibit the proliferation of MDA-MB-231 cells (IC50 = 3.6 μM). The pharmacokinetics study in mice showed that the oral bioavailability of compound 27 was 18.3% (10 mg per kg dose), and the half-life of intravenous administration was 5.29 hours (2 mg per kg dose). Molecular docking studies revealed that the nitrogen atom of pyrrole ring and the oxygen atom of cycloheptanone formed hydrogen bonds with the Asp564 residue of FAK. The amino and nitrogen atoms of pyrimidine ring interacted with the oxygen atom on Cys502 residue, which was the key for this kind of tricyclic compounds to exert FAK inhibitory activity, showing its great potential in the treatment of gastric and breast cancer.
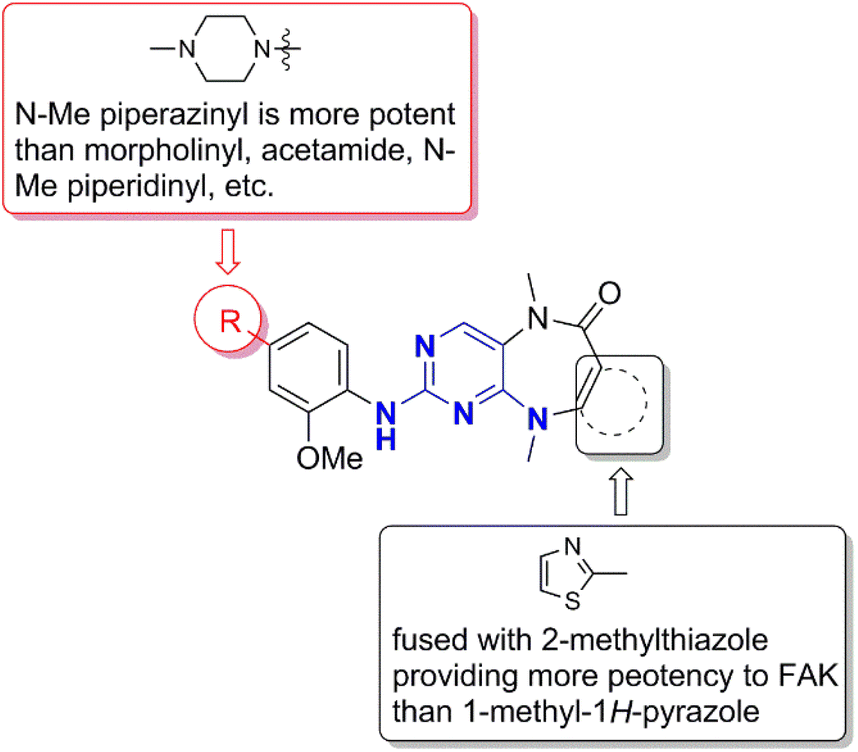
The synthesis of FAK inhibitors 25–27 with fused tricyclic structure started from ethyl 2-methylthiazole-4-carboxylate. Ethyl 2-methyl-5-(methylamino)thiazole-4-carboxylate was obtained from bromination, amination, deprotection and N-methylation. This above intermediate coupled with 2,4-dichloro-5-nitropyrimidine followed reductive cyclization to give tricyclic core, and then, the Buchwald–Hartwig reaction between the above tricyclic intermediate and 2-methoxy-4-(4-(4-methylpiperazin-1-yl)piperidin-1-yl)aniline was performed to give 27 (Fig. 11). The structure–activity relationship of these fused tricyclic FAK inhibitors is showed on Fig. 12. The fused tricyclic ring containing 2-methylthiazole is more potent than 1-methyl-1H-pyrazole, and N–Me piperazinyl is more potent than N–Me piperidinyl, morpholinyl, and acetamide on phenyl ring.
 | ||
| Fig. 11 The synthesis of compound 27. | ||
 | ||
| Fig. 12 The structure–activity relationship (SAR) of fused tricyclic FAK inhibitors. | ||
 | ||
| Fig. 13 FAK inhibitors based on 2-aminopyrimidine core. | ||
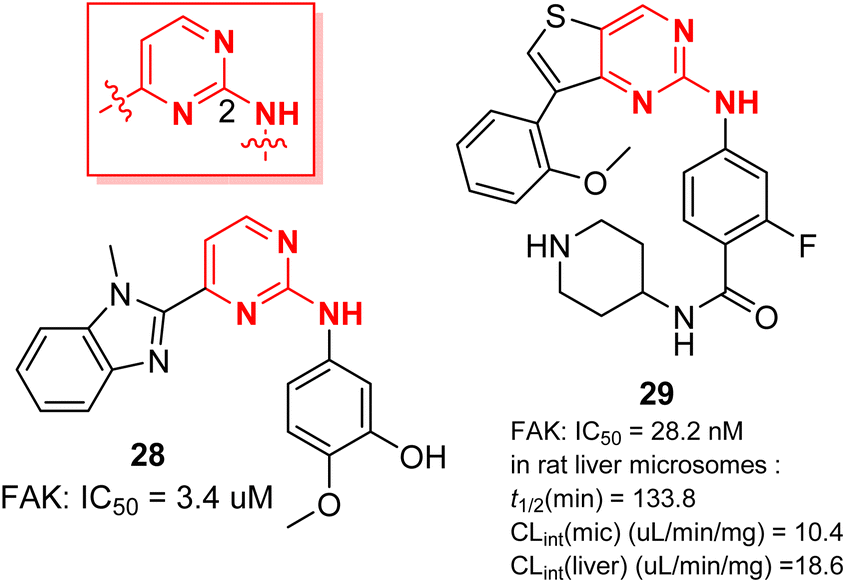
Kunick et al.51 have prepared compound 28, which has a unique 2-anilino-4-(benzimidazol-2-yl)-pyrimidine backbone. It is a multi-kinase inhibitor that can inhibit FAK, Aurora B, PLK1 and VEGFR2 (IC50 = 3.4, 6, 1.2, and 7.2 μM). Compound 28 showed good antiproliferative activity against many cancer cell lines from different sources, such as renal cancer, colon cancer and central nervous system cancer. Cheng et al.52 designed a class of thieno-2-aminopyrimidine derivatives to simulate the active conformation of 2,4-diaminopyrimidine derivatives, a classical FAK inhibitor, in which compound 29 indicated strong inhibition of FAK activity (IC50 = 28.2 nM). Compound 29 showed better efficacy than TAE226 in inhibiting the proliferation of U87MG, A549 and MDA-MB-231 cells. Further studies showed that compound 29 could induce apoptosis of MDA-MB-231 cells, retard the cell cycle at G0/G1 phase, and inhibit cell migration. In vitro studies showed that compound 29 had good metabolic stability in rat liver microsomes (t1/2 = 133.8 min).
2.2 FAK inhibitors based on triazine core
As bioelectronic isosteres of pyrimidines, 1,3,5-triazine and 1,2,4-triazine have also been used in the design and development of FAK inhibitors (Fig. 14). | ||
| Fig. 14 FAK inhibitors based on triazine core. | ||
Chen et al.53 designed and synthesized a new class of FAK inhibitors of diarylamino-1,3,5-triazine derivatives. The representative compound 30 showed moderate FAK inhibitory activity (IC50 = 0.4 μM) and could inhibit the expression of Y397 in HUVEC cells at low concentrations. The compound can effectively inhibit the generation of fibroblast growth factor receptor 2 (FGFR2) in blood vessels. Compared with TAE226, compound 30 has no 5-position chlorine atom on the triazinering, so it reduces the van der Waals interaction with Met499 on FAK to some extent. In addition, the 1,3,5-triazine ring in 30 has lower electron density and weaker ability to form hydrogen bonds with FAK than the 1,3-pyrimidine ring in TAE226, which may be the reason for the unsatisfactory inhibitory activity. In order to improve the inhibitory activity of compound 30, Chen et al.54 designed and synthesized imidazo[1,2-a]-[1,3,5]triazine derivative 31 which is a more selective inhibitor of FAK. It has no inhibitory activity against IGF-IR and Pyk2 at 1 μM, but its IC50 for FAK is 50 nM, and its selectivity for FAK is more than that of other 30 kinases. Compound 31 strongly inhibited FAK mediated autophosphorylation and cell proliferation in U87 MG, HCT-116, MDA-MB-231 and PC-3 cell lines, significantly induced apoptosis in HCT-116 cells at low concentrations, and retarded cell progression at the G2/M phase. Further studies indicated that 31 could inhibit the adhesion, migration and invasion of U87-MG cells to the matrix. Considering the importance of the pyrimidine ring and the chlorine atom at the 5-position of TAE226 to inhibit FAK, Chen et al.55 designed and synthesized a series of new compounds with 1,2,4-triazine skeleton. Among these compounds, the chlorine atom at the 6-position remained unchanged, compound 32 had weak inhibitory activity against FAK (IC50 = 0.23 μM). Molecule docking studies released that the van der Waals force between the 6-position chlorine atom in compound 32 and Met499 of FAK may not contribute much to its binding with FAK, and the hydrogen bonding between the triazine ring and its amino group and FAK hinge region is weak, affecting the inhibitory activity of 32 on FAK. Based on the previously identified ALK inhibitors and JAK2 (Janus kinase 2) inhibitors, scientists from Cephalon company developed a class of JAK2/FAK dual inhibitors based on pyrrolo[2,1-f]-[1,2,4] triazine skeleton, and compound 33 (ref. 56) can potently inhibit JAK2 (IC50 = 2.5 nM) and FAK (IC50 = 5 nM). Studies have shown that compound 33 has ideal pharmacokinetic characteristics and oral bioavailability. It can strongly inhibit pStat3 and reduce the phosphorylation level of FAK in vitro, and the inhibitory activity of 33 is not limited to JAK2 and FAK. At 1 μM concentration, it can inhibit 19 of 53 tumor related kinases with an inhibition rate of >90%.
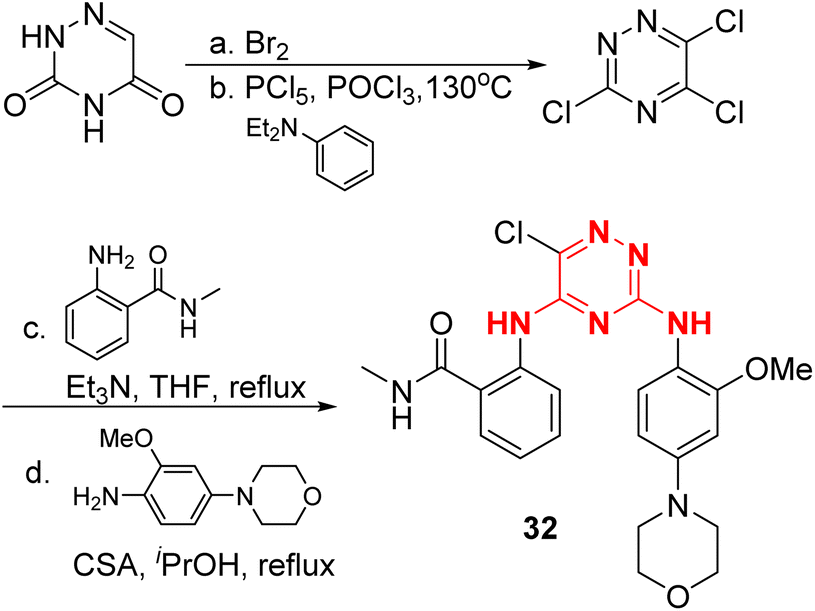
The preparation of compound 32 is listed on Fig. 15. 3,5,6-Trichloro-1,2,4-triazine was obtained from 1,2,4-triazine-3,5(2H,4H)-dione through chloration, and then coupling with the corresponding substituted anilines to provide compound 32, these two coupling steps are similar to the preparation of TAE226.
 | ||
| Fig. 15 The synthesis of compound 32. | ||
2.3 FAK inhibitors with other skeletons
In addition to the above FAK inhibitors with six membered heterocycles, such as pyrimidine and triazine as the core skeleton, some five membered N-containing heterocycles also appear in the structure of FAK inhibitors including 1,2,4-thiadiazole, 1,2,4-oxadiazole, 1,2,4-triazole, imidazole, pyrazole, etc. Compared with six membered N-containing heterocycles, these FAK inhibitors with five membered N-containing heterocycles generally have lower activity and the inhibition rate is generally at the μM level (Fig. 16).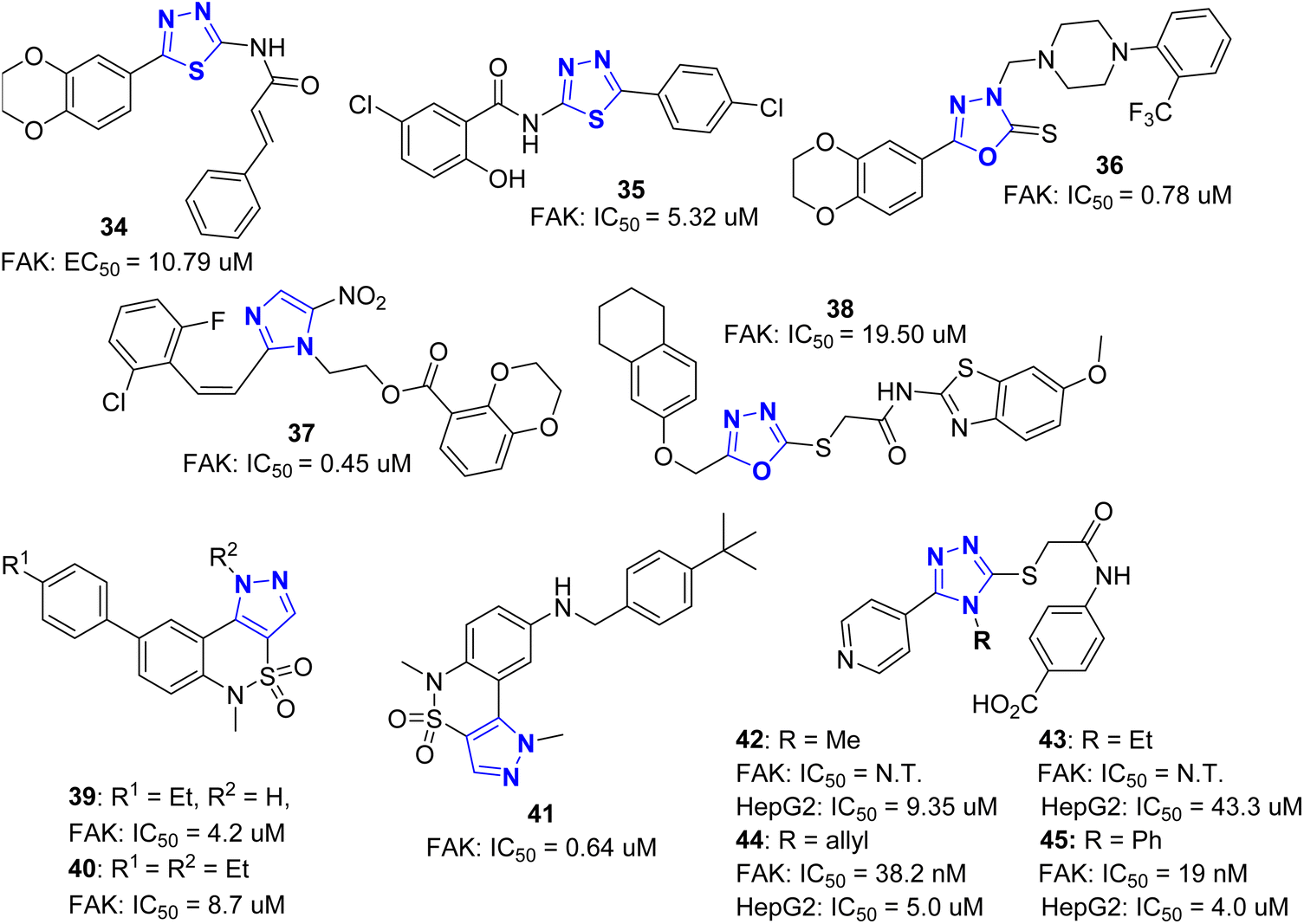 | ||
| Fig. 16 FAK inhibitors based on other N-containing five-member rings. | ||
Zhu et al.57,58 found a class of compounds 34 and 35 containing 1,2,4-thiadiazole, which can be used as FAK inhibitors. Docking studies proved that these two compounds extended into the ATP binding pocket through hydrogen bond π–σ interaction and π–cation interaction. Compound 34 inhibited FAK with an EC50 value of 10.79 μM and could induce apoptosis of HepG2 cells in a dose-dependent manner. The IC50 of compound 35 for FAK was 5.32 μM. Compounds 34 and 35 could effectively inhibit the growth of MCF-7 and B16–F10 cells with IC50 values of 0.45 and 0.31 μM, respectively. By replacing 1,2,4-thiadiazole in 34 and 35 with 1,2,4-oxadiazole-2(3H)-thione, they designed and synthesized a class of FAK inhibitors represented by compound 36.59 The IC50 of compound 36 for inhibition of FAK was 0.78 μM, and the IC50 for inhibition of the proliferation of HepG2 cells and SM1116 cells was 5.78 μM and 47.15 μM, respectively. They also reported a class of FAK inhibitors containing 2-styrene-5-nitroimidazole.60 The IC50 of representative compound 37 for inhibition of FAK was 0.45 μM, and it induced HeLa cell apoptosis in a dose-dependent manner. Similar to the research work of Zhu et al.,57,58 Altintop et al.61 designed and synthesized compound 38 with 1,2,4-oxadiazole as the framework of FAK inhibitor. Its IC50 for A549 and C6 cells were 9.33 μM and 4.63 μM, respectively. It also caused significant morphological changes in the above two types of cells and inhibited the expression of Caspase-3 in these two cell lines. The mechanism study revealed that it could effectively inhibit the activity of FAK and AKT, with IC50 of 19.50 μM and 2.60 μM, respectively. Docking studies showed that compound 38 could bind to the kinase domain in AKT and FAK. When bound to the FAK kinase domain, it can form π–π stacking and salt bridges with the Phe568 residue and π–cation interactions with the Lys457 residue. It is worth noting that the 6-methoxybenzothiazole group of 38 has a close relationship with the tight binding of FAK. These studies showed that compound 38 represents a new class of ATP competitive FAK inhibitors.
In addition to ATP competition for type I and type II kinase inhibitors, the development of allosteric kinase inhibitors62,63 has attracted more attention to medicinal chemists. Allosteric kinase inhibitors do not compete with ATP, so it is possible to overcome the side effects of poor selectivity of ATP competition and reduce drug resistance. Through high-throughput screening, Miki et al.64 found a new class of FAK inhibitors 39 and 40 containing 1,5-dihydropyrazolo[4,3-c][2,1] benzothiazine backbone, with IC50 of 8.7 μM and 4.2 μM for FAK, respectively. Compound 39 was pre-incubated with ATP for a long time, and no competitive binding with ATP was found. The crystal structure of compound 39 with the kinase domain of FAK (PDB ID: 4EBV) showed that the allosteric site involved in the induction of 39 was in the kinase domain and had polar and hydrophobic interactions with FAK. The pyrazole ring has π–cation interaction with the Arg550 side chain, one nitrogen atom in the pyrazole ring has hydrogen bonding with water molecules, and the other nitrogen atom has weak hydrogen bonding with the carboxylic acid group in the Asp604 side chain (Fig. 17). The introduction of ethyl on the pyrazole nitrogen of compound 39 produced compound 40, which was only two times weaker than 39 in inhibiting FAK, indicating that hydrogen bonding with water molecules is not important. Biological studies have shown that both 39 and 40 can bind to unphosphorylated FAK allosteric sites, but only 39 can target phosphorylated FAK. Docking studies showed that when bound to phosphorylated FAK, the nitrogen atom in the pyrazole ring of 39 has a hydrogen bonding interaction with Glu500, but in compound 40, the hydrogen on this nitrogen atom was replaced by ethyl, so phosphorylated FAK could not be formed. Interestingly, 40 showed higher FAK selectivity compared with compound 39, indicating that the pyrazole N-substitution of 39 significantly improved kinase selectivity. Based on the studies of 39 and 40, this research group synthesized N-substituted pyrazolo [4,3-c][2,1] benzothiazine derivative 41, which showed potent inhibitory activity against FAK (IC50 = 0.64 μM). The crystal structure shows that (PDB ID: 4I4F), the core structure of tricyclic compound 41 binds to FAK in the same way as compound 38 (Fig. 18). The amino group connected with 4-tert-butylbenzyl group in the core structure of compound 41 has hydrogen bonding with Gly563, while 4-tert-butyl group occupies the hydrophobic cavity that formed by Met475, Leu486, Met499 and Val484 residues of FAK.65
 | ||
| Fig. 17 Crystal structure of allosteric FAK inhibitor 39 complexed with the kinase domain of FAK (PDB ID: 4EBV).64 | ||
 | ||
| Fig. 18 Crystal structure of allosteric FAK inhibitor 41 complexed with the kinase domain of FAK (PDB ID: 4I4F).65 | ||
In 2021, Hayallah et al.66 reported novel FAK inhibitors (42–45) with 1,2,4-triazole structure. The structure–activity relationship (SAR) study of the substituents on the triazole ring showed that the 4-phenyl substituted compound 45 had higher FAK inhibitory activity (IC50 = 19.0 nM) than GSK-2256098 (IC50 = 22.1 nM). Compounds 44 and 45 showed significant inhibitory effects on Hep G2 and Hep 3B cells (IC50 of 4.72 and 3.78 μM for Hep G2 and 2.88 and 4.83 μM for Hep 3B).
2.4 FAK based dual inhibitors
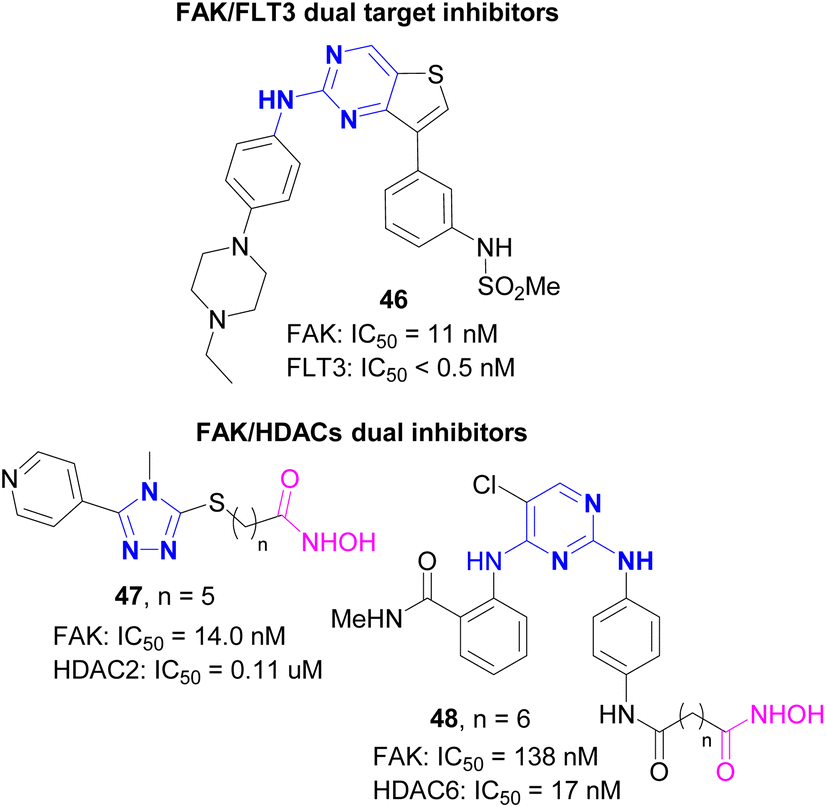
Compared with traditional single target inhibitors, dual target or multi-target inhibitors have the following advantages.67 Firstly, dual target or multi-target drugs can simultaneously regulate multiple tumor pathways to achieve the best therapeutic effect.68 Secondly, these drugs usually show lower side effects than drugs with two different targets at the same time.69 Studies have confirmed that dual target drugs can significantly reduce the generation of drug resistance in the treatment of fungal diseases.70,71 Therefore, the design and development of dual target inhibitors based on FAK is expected to become a new research trend in the field of cancer therapy.72FLT3 (FMS like tyrosine kinase 3, FMS like tyrosine kinase 3) belongs to the receptor tyrosine kinase III (RTK III) family. FLT3 can regulate the differentiation, proliferation and survival of hematopoietic progenitor cells and dendritic cells, and can activate the phosphorylation of downstream SHC1 and AKT1, involving the signaling cascade of mTOR, RAS and MAP kinases. In recent years, studies have confirmed that the activating mutation of FLT3 plays an important role in the occurrence and disease progression of acute myeloid leukemia (AML).73 In 2021, Sim et al.74 reported a novel dual target inhibitor 46 based on FAK, targeting both FAK and FLT3. Compound 46 was derived from the first fused ring FAK inhibitor reported by this research group.75,76 At the beginning of the study, they tried to use the benzenesulfonamide of PF-562271 (VS-6062) as a functional group entering the ATP binding pocket inside FAK. Subsequent SAR studies showed that the compound connected with the thiophene ring and the m-benzenesulfonamide group exhibited excellent inhibitory activity against FAK (IC50 = 7 nM). Compound 46 with 4-ethylpiperazine phenyl core finally screened and showed the best activity against FAK, even exceeding TAE226 and PF-562271. Kinase selectivity studies showed that compound 46 was highly specific among 334 kinases, with IC50 values of less than 0.5 nM for 15 of them including FLT3, and showed strong inhibitory effects on Ba/F3 cells carrying four FLT3 mutations. The in vivo activity study showed that 46 had significantly inhibitory effect on MV4-11 xenograft mice. This finding not only provides a new way for the study of FAK inhibitors and FLT3 mutation inhibitors, but also has potential in the change of AML treatment.
Histone deacetylase (HDAC) is a kind of protease, which plays an important role in the structural modification of chromosomes and the regulation of gene expression. Abnormal histone acetylation level is closely related to cancer and immune diseases, so HDAC is an important target for cancer treatment.77 HDAC inhibitors can inhibit the invasion and metastasis of tumor cells and antitumor angiogenesis, and play an important role in the field of antitumor drug research and development. Studies have confirmed that the combination of FAK inhibitors and HDAC inhibitors can show a synergistic effect, and they have a common regulatory function on the Hippo pathway. At the same time, targeting FAK and HDACs may synergistically improve the efficacy of cancer treatment. Generally, the structure of dual target inhibitors of FAK and HDACs contains three parts, the group interacting with the ATP binding pocket of FAK, the linker with a specific length, and the group chelating with zinc ions in HDAC (carboxylic acid, hydroxamic acid, 2-aminobenzamide are commonly used). In 2021, Mustafa et al.78 reported the first HDAC2 and FAK dual inhibitor 47. In this study, Mustafa used 5-pyridyl-1,2,4-triazole as the binding part with FAK, and introduced various substituents (methyl, ethyl, phenyl, allyl, etc.) on the N atom at the 4-position of the triazole ring. The inhibitory activities of representative compound 47 on HDAC2 and FAK were 0.09 μM and 12.59 nM, respectively, and it had significant inhibitory effects on a variety of tumor cells. In 2023, Song et al.79 reported a dual FAK and HDAC6 inhibitor 48 (MY-1259). Starting from TAE226, they used long-chain hydroxamic acid to replace the morpholine group of TAE226 to obtain compound 48. The results indicated that hydroxamic acid linked with six carbon atoms bearing the highest activity, with IC50 of 16 nM and 132 nM for HDAC6 and FAK, respectively. Compound 48 showed strong inhibitory effect on multiple tumor cells including gastric cancer, breast cancer and lung cancer. Compound 48, administered by intraperitoneal injection at a dose of 20 mg kg−1 for 21 days, had a tumor inhibitory effect of 53.54% on MGC-803 tumor bearing mice, which was significantly better than the combination of TAE226 and SAHA (Fig. 19).
 | ||
| Fig. 19 FAK/FLT3 and FAK/HDAC2 dual inhibitors. | ||
Epidermal growth factor receptor (EGFR) is a member of tyrosine kinase family and a key factor regulating intracellular signaling pathways such as PI3K, AKT and mTOR. EGFR plays a key role in tumor cell proliferation, migration, invasion and angiogenesis. EGFR expression increases in gastric cancer, breast cancer, bladder cancer, head and neck squamous cell carcinoma, so EGFR is a key target for anticancer drug development.80–82 Targeting FAK and EGFR can achieve dual inhibition of downstream signal transduction.83–85 In 2021, Abe et al.86 reported FAK/EGFR dual target inhibitors 50–53 with 2-arylquinoline as the core skeleton (Fig. 20). Starting from the lead compound 49, they carried out various substitutions on the substituents of the quinoline ring to obtain compounds 50–53. They found that when CF3 or Cl were present at the para-position of the benzene ring at the 2-position of quinoline, they had high inhibitory activity against FAK and EGFR, and compound 51 was the best, with IC50 of 14.25 and 20.15 nM for FAK and EGFR, respectively. Chen and co-workers87 have developed a new class of pyrimidine derivatives as potential dual FAK and EGFRT790M inhibitors based on a fragment-based drug design strategy. Among them, compounds 54 and 55 displayed strong inhibitory activities against FAK kinase (IC50 = 1.03, 3.05 nM) and EGFRT790M mutants (IC50 = 3.89, 7.13 nM), respectively. These compounds also exhibited strong inhibitory activity toward the three evaluated FAK-overexpressing PC cells (AsPC-1, BxPC-3, Panc-1) and two drug-resistant cancer cell lines (breast cancer MCF-7/adr cells and lung cancer H1975 cells) at concentrations below 6.94 μM. These compounds showed potent dual FAK and EGFRT790M inhibitors with a great potential for the treatment of hard to treat cancers.
 | ||
| Fig. 20 FAK/EGFR dual inhibitors. | ||
PLK1 (polo like kinase 1), a member of the polo like kinase family, is a crucial protein kinase that regulates cell mitosis.88,89 It can promote nuclear division and chromosome segregation in G2/M phase,90 and is closely related to tumorigenesis and progression. Recent studies have shown that simultaneously targeting FAK and PLK1 has a synergistic effect, probably by affecting p53 through affecting CDK2 in the MAPK pathway. Previous studies have also reported the importance of inhibiting FAK during PLK1 degradation.91,92 In 2023, Wu et al. reported a dual target inhibitor of FAK/PLK1 based on quinazolinone backbone through computer-aided drug design and structure–activity relationship (SAR) study.93 The results showed that compound 56 had a significant inhibitory effect on MCF-7, HepG2, SGC7901 and A375 cells. The IC50 for FAK and PLK1 were 90 and 81 nM, respectively, showing that quinazolinone can be used as an important framework for the design of FAK inhibitors (Fig. 21).
 | ||
| Fig. 21 FAK/PLK1 dual inhibitors. | ||
2.5 FAK-targeting protein–protein interactions (PPIs)
Protein–protein interaction (PPI) can regulate many physiological processes in organisms and can be used as an key point for the treatment of various diseases.94,95 Important physiological activities of life, such as cell signal transduction and regulation of material metabolism, protein synthesis and transport, DNA replication and transcription, are realized through PPI network. Changes in cellular microenvironment, epigenetic changes, gene mutations, etc., can interfere with PPI patterns or directly induce protein binding, forming tumor specific interactions, which can lead to the occurrence, development, invasion and metastasis of tumors. In recent years, significant progress has been achieved in the research and development of antitumor drugs targeting PPIs. Many drugs are in clinical trials. Venetoclax (ABT-199) has been approved to market for the treatment of p17 deficient chronic lymphocytic leukemia (CLL).96 More and more evidence shows that drugs targeting specific PPIs may have higher specificity and better efficacy than traditional anticancer drugs, providing new strategies and opportunities for treatment of specific tumors. As mentioned earlier, FAK protein interacts with MDM2, p53, IGF-1R, VEGFR-3 and other proteins, and plays an important role in the growth, proliferation, invasion and migration of tumor cells. Therefore, the development of blockers targeting the interaction between FAK and the above proteins can achieve the purpose of tumor inhibition (Fig. 22). | ||
| Fig. 22 Targeting FAK PPI blockers. | ||
MDM2 is an E3 ligase that plays a key role in mediating p53 degradation.97 There is an interaction between FERM domain of FAK protein and MDM2, which inactivates p53 ubiquitination through a kinase independent signaling pathway, resulting in slower apoptosis of tumor cells and promoting tumor cell growth and proliferation. Therefore, it is necessary to develop blockers targeting FAK/MDM2 interaction. Based on crystal structures of FAK and MDM2 proteins, Cance et al.98 first reported that FAK/MDM2 interaction blocker 57 can activate p53 in a dose-dependent manner. It induced apoptosis of liver, breast and colon cancer cells through molecular modeling and high-throughput virtual screening, confirming that simultaneous inhibition of FAK/MDM2 can effectively combat the growth and proliferation of cancer cells.
The FAK FERM domain can also directly interact with p53 and disrupt its pro-apoptotic function.99 Disrupting the binding of FAK to p53 may reactivate p53 to play a tumor suppressor role. Cance et al.100 found that compound 58 had potent inhibitory activity on colon cancer cell HCT116 by disrupting the scaffold function of FAK and blocking the interaction between FAK and p53 in cells, and its inhibitory activity was significantly enhanced when combined with doxorubicin or 5-fluorouracil. Studies have shown that FAK interacts with insulin-like growth factor receptor-1 (IGF-1R) and c-Met to promote the survival and growth of cancer cells, and blocking the interaction between FAK and c-Met or IGF-1R may be a new tumor treatment strategy.101,102 Compound 59 was obtained by Hochwald et al.103 through molecular modeling and high-throughput virtual screening. This compound can disrupt the interaction between FAK and IGF-1R and c-Met, reduce the phosphorylation of FAK and AKT in adenocarcinoma cells, inhibit the growth of a variety of cancer cells, and have no effect on normal cells. Compound 59 combined with gemcitabine can significantly enhance cancer cell apoptosis and significantly inhibit the growth of pancreatic tumor in vivo. In addition to the FERM domain, the FAT domain of FAK can also interact with a series of proteins, including vascular endothelial growth factor receptor-3 (VEGFR-3) and paxillin, which can promote cancer cell survival. Cance et al.104 found compound 60 by virtual docking using the FAK protein FAT domain crystal structure. Compound 60 decreased the phosphorylation level of FAK/VEGFR3 in a concentration-dependent manner, and had a significant inhibitory effect on some cancer cell lines. 60 could induce VEGFR3-dependent apoptosis of breast cancer cells. In vivo experiments showed that 60 effectively inhibited the overexpression of VEGFR-3 in tumor bearing mice and had a significant synergistic effect with doxorubicin. Compound 61 derived from structural modification of 60 had higher selectivity for FAK and VEGFR-3 overexpressing cancer cells than normal expressing cells.105
3. FAK inhibitors combined with other drugs in the treatment of tumors
Drug combination is very common in the treatment of tumor, diabetes and other aspects, and can play a good synergistic effect.106 Similarly, the combination of FAK inhibitors and other drugs can also play a synergistic role in tumor inhibition.In July 2019, Yang and others107 published a patent to disclose the method of combining FAK/ALK/ROS1 inhibitors with EGFR inhibitors to treat cancer. They combined one or more of FAK inhibitors (APG-2449), ALK inhibitors and ROS1 inhibitors with EGFR inhibitors (such as Osimertinib) and showed synergistic inhibitory effects on a variety of cancer cells in vitro and in vivo. In May 2021, VS-6766 broke the limitation of KRAS mutation status in patients.108 The results of the phase 1/2 frame trial showed that the ORR of the combination was 52%, that of KRAS mutant patients was 70%, and that of KRAS wild-type patients was 44%, showing a good therapeutic effect. In June 2021, Li and others109 applied for a patent for the combination of FAK inhibitor and KRAS inhibitor in the treatment of solid tumors. They verified the synergistic inhibitory effect of FAK inhibitor Ifebemtinib combined with KRAS G12C inhibitor (AMG510 or MRTX849) on a variety of cancer cells, especially on cancer cell lines carrying KRAS G12C mutation. They confirmed that the therapeutic effect of the combination was achieved through the synergistic anticancer effect of FAK-YAP signaling pathway.110 In August 2021, Inxmed Co. Ltd announced that FAK inhibitor Ifebemtinib (IN10018) combined with liposomal doxorubicin (PLD) for the treatment of platinum-resistant recurrent ovarian cancer. This combination had obtained the fast-track certification of the US FDA,111 and was recognized as a breakthrough therapeutic drug by the China National Drug Administration (NMPA) in April 2022. Studies have shown that Ifebemtinib can break the tumor defense mechanism and effectively overcome drug resistance and metastasis by overcoming the tumor stromal fibrosis barrier and regulating the tumor immunosuppressive microenvironment. At present, this combination treatment is being used for a variety of solid tumor indications that lack effective methods, including platinum-resistant ovarian cancer, melanoma, triple negative breast cancer, head and neck cancer, pancreatic cancer and so on. In October 2023, Inxmed Co. Ltd announced that Ifebemtinib combined with MEK inhibitor (Cobimetinib), PD-1 antibody (Toripalimab), KRAS G12C inhibitor (D-1553) and EGFR TKI inhibitor (Furmonertinib), respectively. These combinations showed great potential antitumor efficacy in clinical research to give significant survival benefit trend to patients.112 In May 2024, Chai et al.113 disclosed the use of FAK inhibitor GSK-2256098 or PF-573228 combined with autophagy inhibitor hydroxychloroquine to treat drug-resistant pancreatic cancer. Through cell and animal experiments, it was found that these combinations can significantly reduce the viability of pancreatic cancer cells and inhibit the growth of pancreatic tumors. FAK inhibitors can promote the efficacy of hydroxychloroquine chemotherapy and enhance the antitumor effect by reducing the density of matrix around pancreatic cancer cells. In September 2024, the US FDA granted Narmafotinib with Gemcitabine and Apatinib as a fast-track treatment for advanced pancreatic cancer.114 Among the 14 patients who received Narmafotinib combined with chemotherapy, the tumor of 6 patients significantly shrank, indicating that the combination had good efficacy in some patients. In the phase I clinical study, it showed good tolerance and safety. The strategy of small molecule FAK inhibitors combined with other targeted drugs in the treatment of cancer has been recognized by clinical research, providing new therapeutic opportunities and ways for patients with multidrug-resistant tumors.
4. FAK-targeting protacs
Protac (proteolysis targeting chimera, Protac), which is a protein degradation targeting chimera technology, has made great progress in the past 20 years. It is a method of targeted degradation of specific pathogenic proteins by using natural ubiquitin protease system with synthetic molecules.115 Protac molecule is heterobifunctional compound composed of E3 ligand, linker, and protein of interest ligand (POI). The most used E3 ligands are CRBN and VHL. The linkers are generally long-chain alkanes, ethers, etc., and POIs are usually small molecule kinase inhibitors. Due to its special targeting selectivity and anti-drug-resistance, protac has become a drug development pathway with great clinical application prospects.116,117 At present, the research of protacs targeting FAK has entered the clinical research stage.In 2018, Gray et al.118 designed and synthesized a multi kinase degradation compound 62 (TL12-186) by coupling FAK inhibitor with pomalidomide through ethylene glycol ether linker. The study found that 28 kinases including BTK, PTK2, FLT3, AurkA, AurkB, TEC, ULK1, ITK, CDK can be degraded by 62. Later, they prepared ALK degraders 63 and 64 based on the pyrimidine ALK inhibitor TAE684, and found that these compounds could not only induce ALK degradation, but also FAK, Aurora A, FER, rSK1 degradation.119 These studies demonstrate the flexibility of the development of FAK degraders. Also in 2018, Crews et al.120 reported that FAK selective degraders 65. They coupled the FAK inhibitor Defactinib with VHL ligand through ether linker to obtain compound 65. Its half degradation concentration DC50 of FAK was 3 nM, and the degradation rate of FAK was more than 99% at the concentration of 10 nM, reaching almost complete degradation of FAK. Compound 65 was more effective than Defactinib in inhibiting the migration and invasion of MDA-MB-231 cells (Fig. 23).
 | ||
| Fig. 23 FAK protacs 62–65. | ||
In 2019, scientists from Boehringer Ingelheim designed and synthesized FAK degraders 66 (BI-3663) and 67 (BI-0319).121 Studies have confirmed that 66 (Dmax = 95%) and 67 (Dmax = 80%) can specifically degrade FAK in 12 cell lines. However, 66 and 67 did not show higher antiproliferative activity than FAK inhibitors. Rao et al.122 connected the CRBN ligand pomalidomide through “click reaction” of FAK inhibitors VS-6062 or VS-6063 to prepare compound 68, which has a very potent degradation ability for FAK of five different cell lines, and the half degradation concentration DC50 is as low as 0.08 nM. It is worth noting that compound 68 could inhibit FAK autophosphorylation more effectively than VS-6062. Consistent with the findings of Boehringer Ingelheim scientists, FAK degraders did not show more potent antiproliferative activity than FAK inhibitors (Fig. 24).
 | ||
| Fig. 24 FAK protacs 66–68. | ||
In 2022, Cheng et al.123 reported that FAK degrader 69 based on TAE226 structure had significant effect in combating non-small cell lung cancer (NSCLC). They retained the pyrimidine backbone and o-aminobenzamide moiety of TAE226, only replaced the morpholine ring with a piperazine ring, and sequentially installed a linker and pomalidomide on the piperazine ring for the recruitment of E3. According to the structure–activity relationship study, compound 69 containing a linker of five carbon atoms showed excellent activity, reaching 86.4% of the degradation activity of FAK in A549 cells, and the IC50 for FAK was 14.9 nM. Cheng et al.124 also reported a FAK degrader 70 based on PF562271 (VS-6062). Compound 70, with an amide chain of 10 atoms as the linker, showed better antiproliferative activity and anti-invasion ability in A549 cells, with a degradation activity of 85% and an IC50 of 26.4 nM at a concentration of 10 nM. In addition, compound 70 has excellent stability with a plasma half-life t1/2 greater than 194.8 min (Fig. 25).
 | ||
| Fig. 25 FAK protacs 69 and 70. | ||
The synthesis of FAK protacs is listed on Fig. 26 by using 62 (TL12-186) as an example. The first two steps are similar to the preparation of TAE226. Following the deprotection of N-Boc group at piperazidine, the linker was installed by alkylation with 1-azido-2-(2-(2-bromoethoxy)ethoxy)ethane. After then, the azide group was reduced by PPh3 followed coupling with E3 ligand to give 62 in 33% overall yield.118 The synthesis of the other FAK-targeting protacs are similar to the preparation of 62 with different small molecular FAK inhibitors, linkers with proper length and E3 ligands.
 | ||
| Fig. 26 The synthesis of FAK protac 62. | ||
5. Clinical trials of FAK inhibitors
The research on FAK inhibitors and degraders is very active, and some small molecular FAK inhibitors and protacs targeting FAK have entered the clinical research stage.5.1 Clinical trials of small molecular FAK inhibitors
At present, there are nine small molecular FAK inhibitors to enter clinical research (Fig. 27)125 including CEP-37440, APG2449, etc. Among them, VS-6063 (Defactinib) and CT-707 (Conteltinib) have entered clinical trials at phase III stage.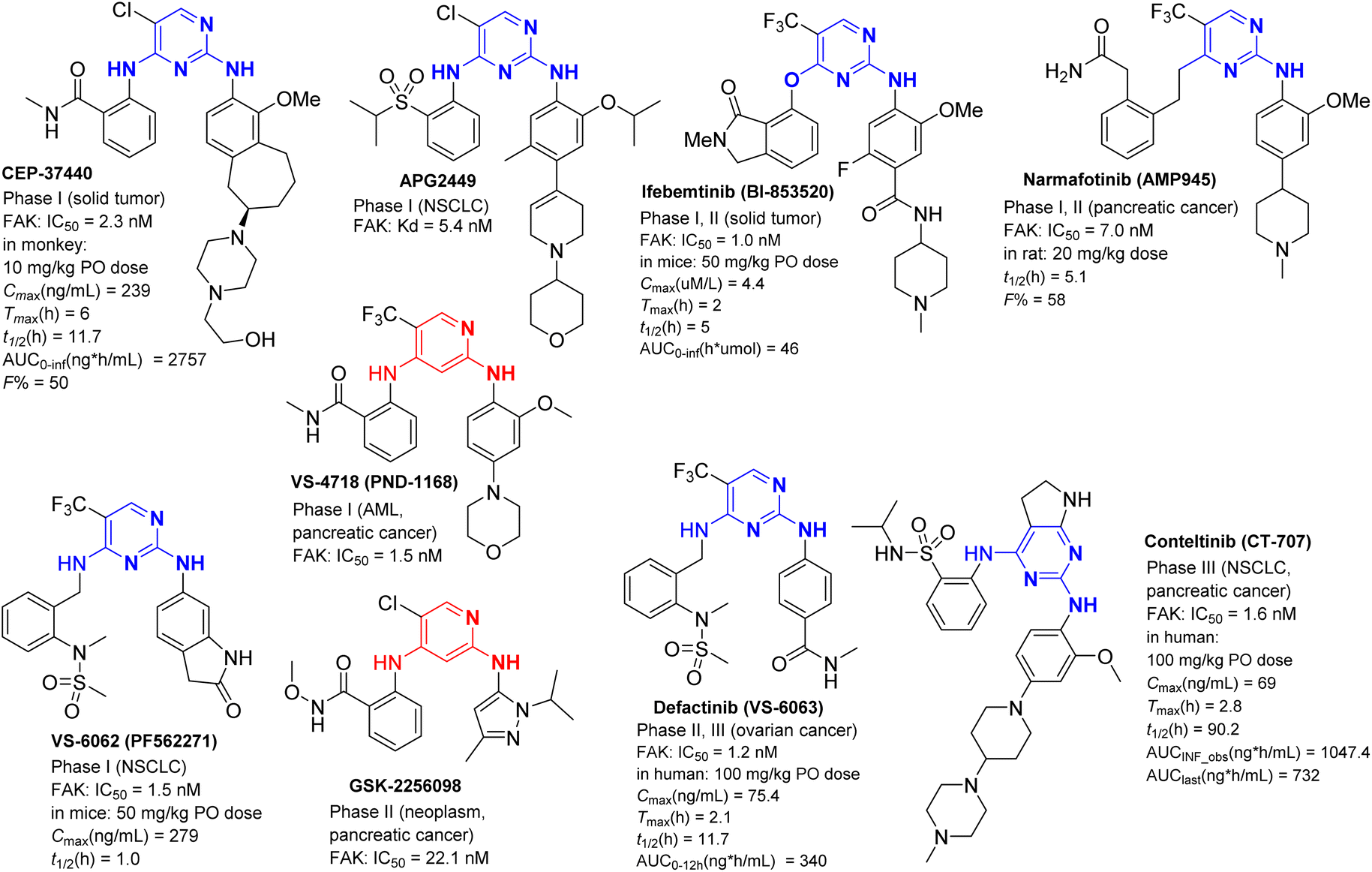 | ||
| Fig. 27 Small molecular FAK inhibitors in clinical trial. | ||
CEP-37440 is an oral effective dual inhibitor of FAK/ALK developed by Teva pharmaceutical company.126 The IC50 of FAK and ALK inhibition is 2.3 nM and 3.5 nm, respectively. It exerts the function of inhibiting tumor cell proliferation by blocking Tyr397 autophosphorylation of FAK. At present, it has completed clinical phase I research for the treatment of ALK positive non-small cell lung cancer (NSCLC) and inflammatory breast cancer (IBC). APG2449 is a new small molecule FAK/ALK/ROS1 triple tyrosine kinase inhibitor (TKI) with oral activity independently developed by Ascentage pharmaceutical.127 It is the first third-generation ALK inhibitor that has entered the clinical stage. It has excellent antitumor activity in a mouse model of non-small cell lung cancer (NSCLC) and is in clinical stage I. It shows potent inhibitory activity against NSCLC carrying ALK/ROS1 double mutations, providing a new treatment strategy for NSCLC patients resistant to the second-generation ALK inhibitor. Ifebemtinib (BI-853520 or IN10018),128 a FAK inhibitor with oral activity (IC50 = 1 nM) jointly developed by Boehringer Ingelheim and Inxmed, shows strong antiproliferative activity against a variety of solid tumors, including prostate cancer, and is currently in clinical phase I/II. Narmafotinib (AMP945)129 is a potent FAK inhibitor (IC50 = 7.0 nM) developed by Australian Amplia pharmaceutical company, which can inhibit the autophosphorylation of FAK protein Y397 in MDA-MB-231 cells and has low cytotoxicity. On September 25, 2024, the FDA of the United States granted it the fast-track qualification of the leading drug for the treatment of advanced pancreatic cancer. The clinical trial of Narmafotinib for the treatment of advanced pancreatic cancer is being carried out in Australia and South Korea, and is currently in clinical phase I/II. VS-4718 (PND-1168), which entered phase I clinical research, is a highly specific reversible FAK inhibitor with an IC50 of 1.5 nM, which can selectively promote tumor cell apoptosis and is used for the treatment of acute myeloid leukemia (AML) and pancreatic cancer. The structure of GSK-2256098 is very similar to TAE226, except that the CL on the pyrimidine ring of TAE226 is replaced by CF3.130 It is a selective FAK inhibitor that can inhibit the growth and proliferation of pancreatic cancer cells. It is currently in phase II clinical research for the treatment of pancreatic cancer and progressive meningioma.
VS-6062 (PF562271),131 VS-6063 (Defactinib)132 and VS-4718 (PND-1168)133 were FAK inhibitors developed by Pfizer in the early stage, and then Verastem was responsible for clinical research. VS-6062 is an effective and reversible ATP competitive FAK and PYK2 kinase inhibitor with IC50 of 1.5 nM and 13 nM, respectively. At present, phase I clinical research has ended for the treatment of non-small cell lung cancer (NSCLC). VS-6063 (Defactinib), which inhibits Tyr397 phosphorylation of FAK in a time- and dose-dependent manner, can also reduce the levels of AKT and YB-1 in taxane-resistant cell lines, and is now in clinical phase III. In March 2024, the US Food and Drug Administration (FDA) granted Avutometinib (VS-6766) alone or in combination with the FAK inhibitor VS-6063 (Defactinib) orphan drug qualification for the treatment of patients with KRAS mutated recurrent low-grade serous ovarian cancer, bringing new hope for the treatment of this refractory disease. In clinical trials, the efficacy of the combination of Defactinib and Avutometinib was better, 45% of patients had significantly reduced tumors, and the disease control rate was at least 90%. In KRAS mutant patients, the disease control rate of dual drug combination also reached 100%. In terms of safety, the most common treatment-related adverse events (occurring in more than 20% of patients) included nausea, diarrhea, elevated CPK, peripheral edema, vomiting, blurred vision, acne like dermatitis, fatigue, rash, dry skin, and anemia.
CT-707 (Conteltinib)134 is a first-in-class drug from China's Shouyao holdings. It is a multi-kinase inhibitor targeting FAK, ALK and Pyk2. The IC50 of FAK was 1.6 nM, which was used to treat ALK positive non-small cell lung cancer, including newly treated and Crizotinib resistant patients. The phase I clinical trial results of CT-707 for ALK positive non-small cell lung cancer patients showed that for newly treated patients, the objective remission rate of patients in the 450 mg dose group was 87.5%, and the disease control rate was as high as 100%. For Crizotinib resistant patients, the overall remission rate of patients in the 300 mg dose group was 83.3%, and the disease control rate was 100%. Among patients in the dose group above 450 mg, the median progression free survival was 13 months, and 58% of patients had remission lasting more than 11 months. The most common adverse reactions of CT-707 were gastrointestinal reactions and elevated transaminases, mostly grade 1–2, which showed good tolerability and safety. Multiple phase II and phase III clinical trials of CT-707 (ctr20200770, ctr20181770) are in progress. On October 22, 2024, CT-707 granules was accepted as a new drug by the State Drug Administration for the treatment of anaplastic lymphoma kinase (ALK)-positive locally advanced or metastatic non-small cell lung cancer (NSCLC).
5.2 Clinical trials of FAK protacs
Although the research of protacs is in full swing, no protac molecule has yet been approved for clinical use, as have the protacs targeting FAK. Up to now, three FAK protac molecules have entered clinical research, namely BI-3663, FAK degrader 1 and GSK215 (Fig. 28). BI-3663 is a highly selective protac molecule of PTK2/FAK (DC50 = 30 nM), which can degrade PTK2 with an IC50 of 18 nM. It is formed by linking PTK2/FAK inhibitor BI-4464 and CRBN E3 ligase ligand through long-chain ether. Protac FAK degrader 1 is a selective FAK degrader based on VHL E3 ligase ligand, with IC50 of 6.5 nM and DC50 of 3 nM. GSK215 is a protac molecule targeting FAK,135 which can induce rapid and persistent degradation of FAK and have a long-term effect on FAK levels, with a half degradation concentration of DC50 of 8.4 nM. GSK215 is designed with VS-4718 as a ligand targeting FAK protein and VHL as an E3 ligase ligand. The above three protacs targeting FAK are currently in phase I clinical research, and there are few published reports on their clinical experimental data. | ||
| Fig. 28 FAK protacs in clinical trial. | ||
6. Outlook and perspectives
FAK is a protein closely related to the proliferation, invasion and metastasis of tumor cells. Since its discovery in the 1990s, it has become one of the widely concerned and potential targets for antitumor drugs.136 Because of an important role of FAK in the proliferation and metabolism of normal cells, targeted FAK drugs may affect normal cells and even lead to serious adverse reactions. To enhance the curative effect and reduce the toxic and side effects, optimizing the design of FAK targeted drugs and achieving its excellent tumor targeting are essential for its clinical application. We outline the structural composition of FAK protein and highlight its key phosphorylation sites. We also summarize the drugs targeting FAK and highlight its backbone structure and molecular design.First, in the design of targeted FAK drugs, it is urgent to break through the classical 2,4-diaminopyrimidine core skeleton. 2,4-diaminopyrimidine is the traditional backbone of small molecule targeted drugs on the market and well developed. It competitively binds with ATP at most kinase sites. This backbone is bound to cause target mutations, multidrug resistance and off target effects. Therefore, it is necessary to search for small molecule FAK inhibitors with skeletal diversity. At present, five membered azole rings, fused heterocycles and linear frameworks also appear in the structure of FAK inhibitors,137 which is worthy of further exploration and development.
Second, the synergistic effect based on dual target or multi-target inhibitors of FAK can effectively improve the antitumor activity. Dual target or multi-target drugs located in the same signaling pathway or parallel pathways have attracted much attention in recent years. The dual- or multi-target small molecule drugs play a synergistic role, reduce the generation of drug resistance, and improve the therapeutic effect.138–141 However, there are few studies on dual target or multi-target inhibitors of FAK/SRC, FAK/FGFR, FAK/HER2/EGFR, FAK/ALK/ROS, FAK/AKT/mTOR. This situation leaves a broad space for the development of dual- or multi-target drugs based on FAK.
Third, the introduction of covalent structure in drug design has become a new direction for the development of FAK inhibitors.142 The antitumor drugs listed in recent years, such as Zanubrutinib targeting BTK, Sotorasib, Adagrasib, Garsorasib, and Fulzerasib targeting KRAS G12C mutation, are all covalent inhibitors. They have excellent performance in the treatment of refractory tumors such as drug resistance and easy relapse, which has inspired the “gold rush” wave of medicinal chemists to develop covalent drugs. The design and synthesis of FAK covalent inhibitors are also not lagging. Since the first irreversible covalent FAK inhibitor32 was reported in 2018, many FAK covalent inhibitors have been reported, but most of them use mature acrylamide as covalent warheads, and it is urgent to develop FAK inhibitors containing covalent warheads with other structures such as nitrile group.143,144
Fourth, the combination of FAK inhibitors and other antitumor drugs has great clinical application prospects. Many clinical trials have confirmed that the combination of FAK inhibitor and other antitumor drugs can play a synergistic effect. For example, the combination of Ifebemtinib (IN10018) and MEK inhibitor (Cobimetinib), PD-1 antibody (Toripalimab), KRAS G12C inhibitor (D-1553) and EGFR TKI inhibitor (Furmonertinib) described previously has shown excellent antitumor efficacy in clinical research and significantly increased the survival of patients.
Finally, the development of proximity induction strategy145,146 injects fresh vitality into the development of FAK targeted drugs. Proximity inducing strategy refers to the emerging drug design technology that drug molecules destroy the pathogenic target by using the ubiquitin proteasome system (UPS), autophagy, lysosomal degradation and other pathways possessed by the cell itself, or achieve the functional regulation of the target by narrowing the distance between the target and other functional bodies. Protac belongs to the proximity inducing strategy. FAK inhibitors bind to E3 ligase ligands such as CRBN or VHL through the linker to form protacs that target FAK for degradation, which has certain advantages. However, the inherent characteristics of protac molecules, such as difficulties in druggability, limit further application. This is also the main reason why no protac drug has been listed so far. Medicinal chemists are overcoming the lack of protac, and the first protac is likely to be approved in the very near future.
To sum up, although no drug targeting FAK has been approved for marketing, FAK inhibitors that have entered clinical research have achieved significant effects. For example, Defactinib (VS-6063) and Conteltinib (CT-707) have progressed to phase III clinical trials and have been used in the treatment of non-small cell lung cancer (NSCLC), ovarian cancer and pancreatic cancer, showing the great potential of FAK inhibitors in the fight against malignant tumors. It is believed that soon, targeted FAK drugs will become a breakthrough method for the treatment of human cancer.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Author contributions
S. K. Zhu and Q. Wu are the drafters of the overall framework of the article and are responsible for the content writing; G. X. Liu and X. Y. Wang were responsible for literature collation; P. G. Wang and A. Q. Geng directed the writing of the paper, checked and revised it.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Foundation of China (no. 21372259) for providing financial support for this work.Notes and references
- D. Ilic, Y. Furuta, S. Kanazawa, N. Takeda, K. Sobue, N. Nakatsuji, S. Nomura, J. Fujimoto, M. Okada and T. Yamamoto, Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice, Nature, 1995, 377, 539–544, DOI:10.1038/377539a0.
- Y. Lu and H. Sun, Progress in the development of small molecular inhibitors of focal adhesion kinase (FAK), J. Med. Chem., 2020, 63, 14382–14403, DOI:10.1021/acs.jmedchem.0c01248.
- X. Wang, N. Li, Y. Liu, J. Wu, Q. Liu, J. Niu, Y. Xu, C. Huang, S. Zhang and J. Song, Targeting focal adhesion kinase (FAK) in cancer therapy: A recent update on inhibitors and PROTAC degraders, Eur. J. Med. Chem., 2024, 276, 116678, DOI:10.1016/j.ejmech.2024.116678.
- M. Li, Y. Zhi, B. Liu and Q. Yao, Advancing strategies for proteolysis-targeting chimera design, J. Med. Chem., 2023, 66, 2308–2329, DOI:10.1021/acs.jmedchem.2c01555.
- X. Tan, Y. Yan, B. Song, S. Zhu, Q. Mei and K. Wu, Focal adhesion kinase: from biological functions to therapeutic strategies, Exp. Hematol. Oncol., 2023, 12, 83, DOI:10.1186/s40164-023-00446-7.
- S. Pomella, M. Cassandri, M. R. Braghini, F. Marampon, A. Alisi and R. Rota, New insights on the nuclear functions and targeting of FAK in cancer, Int. J. Mol. Sci., 2022, 23, 1998, DOI:10.3390/ijms23041998.
- P. Rosado, P. Lequerica-Fernandez, I. Pena, L. Alonso-Duran and J. C. de Vicente, In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression, Virchows Arch., 2012, 461, 163–168, DOI:10.1007/s00428-012-1283-2.
- T. Marlowe, A. Dementiev, S. Figel, A. Rivera, M. Flavin and W. Cance, High resolution crystal structure of the FAK FERM domain reveals new insights on the druggability of tyrosine 397 and the Src SH3 binding site, BMC Mol. Cell Biol., 2019, 20, 10, DOI:10.1186/s12860-019-0193-4.
- K. Brami-Cherrier, N. Gervasi, D. Arsenieva, K. Walkiewicz, M. Boutterin, A. Ortega, P. G. Leonard, B. Seantier, L. Gasmi, T. Bouceba, G. Kadare, J. Girault and S. T. Arold, FAK dimerization controls its kinase-dependent functions at focal adhesions, EMBO J., 2014, 33, 356–370, DOI:10.1002/embj.201386399.
- X. Zhao and J. Guan, Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis, Adv. Drug Delivery Rev., 2011, 63, 610–615, DOI:10.1016/j.addr.2010.11.001.
- Q. Fu, Y. Liu, Y. Fan, S. Hua, H. Qu, S. Dong, R. Li, M. Zhao, Y. Zhen, X. Yu, Y. Chen, R. Luo, R. Li, L. Li, X. Deng, W. Fang, Z. Liu and X. Song, Alpha-enolase promotes cell glycolysis, growth, migration, and invasion in non-small cell lung cancer through FAK-mediated PI3K/AKT pathway, J. Hematol. Oncol., 2015, 8, 22, DOI:10.1186/s13045-015-0117-5.
- D. Lietha, X. Cai, D. F. J. Ceccarelli, Y. Li, M. D. Schaller and M. J. Eck, Structural basis for the autoinhibition of focal adhesion kinase, Cell, 2007, 129, 1177–1187, DOI:10.1016/j.cell.2007.05.041.
- C. Lawson, S. Lim, S. Uryu, X. L. Chen, D. A. Calderwood and D. D. Schlaepfer, FAK promotes recruitment of talin to nascent adhesions to control cell motility, J. Cell Biol., 2012, 196, 223–232, DOI:10.1083/jcb.201108078.
- P. Mohanty and S. Bhatnagar, Structural basis of focal adhesion targeting domain-mediated signaling in cardiac hypertrophy, J. Recept. Signal Transduction Res., 2017, 37, 38–50, DOI:10.3109/10799893.2016.1155067.
- T. Liu, T. LaFortune, T. Honda, O. Ohmori, S. Hatakeyama, T. Meyer, D. Jackson, J. de Groot and W. K. A. Yung, Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferationin vitro andin vivo, Mol. Cancer Ther., 2007, 6, 1357–1367, DOI:10.1158/1535-7163.MCT-06-0476.
- D. Lietha and M. J. Eck, Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation, PLoS One, 2008, 3, e3800, DOI:10.1371/journal.pone.0003800.
- A. Schultze and W. Fiedler, Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer, Expert Opin. Invest. Drugs, 2010, 19, 777–788, DOI:10.1517/13543784.2010.489548.
- L. Wang, M. Ai, J. Yu, L. Jin, C. Wang, Z. Liu, X. Shu, Z. Tang, K. Liu, H. Luo, W. Guan, X. Sun and X. Ma, Structure-based modification of carbonyl-diphenylpyrimidines (Car-DPPYs) as a novel focal adhesion kinase (FAK) inhibitor against various stubborn cancer cells, Eur. J. Med. Chem., 2019, 172, 154–162, DOI:10.1016/j.ejmech.2019.04.004.
- C. Viegas-Junior, A. Danuello, V. Da Silva Bolzani, E. J. Barreiro and C. A. M. Fraga, Molecular hybridization: a useful tool in the design of new drug prototypes, Curr. Med. Chem., 2007, 14, 1829–1852, DOI:10.2174/092986707781058805.
- W. Huang, Y. Ding, Y. Miao, M. Liu, Y. Li and G. Yang, Synthesis and antitumor activity of novel dithiocarbamate substituted chromones, Eur. J. Med. Chem., 2009, 44, 3687–3696, DOI:10.1016/j.ejmech.2009.04.004.
- R. Li, X. Zhang, Q. Li, Z. Ge and R. Li, Novel EGFR inhibitors prepared by combination of dithiocarbamic acid esters and 4-anilinoquinazolines, Bioorg. Med. Chem. Lett., 2011, 21, 3637–3640, DOI:10.1016/j.bmcl.2011.04.096.
- M. Wei, J. Zhang, F. Ma, M. Li, J. Yu, W. Luo and X. Li, Synthesis and biological activities of dithiocarbamates containing 2(5H)-furanone-piperazine, Eur. J. Med. Chem., 2018, 155, 165–170, DOI:10.1016/j.ejmech.2018.05.056.
- Y. Su, R. Li, X. Ning, Z. Lin, X. Zhao, J. Zhou, J. Liu, Y. Jin and Y. Yin, Discovery of 2,4-diarylaminopyrimidine derivatives bearing dithiocarbamate moiety as novel FAK inhibitors with antitumor and anti-angiogenesis activities, Eur. J. Med. Chem., 2019, 177, 32–46, DOI:10.1016/j.ejmech.2019.05.048.
- K. M. Huttunen and J. Rautio, Prodrugs-an efficient way to breach delivery and targeting barriers, Curr. Top. Med. Chem., 2011, 11, 2265–2287, DOI:10.2174/156802611797183230.
- A. Kamiyama, M. Nakajima, L. Han, K. Wada, M. Mizutani, Y. Tabuchi, A. Kojima-Yuasa, I. Matsui-Yuasa, H. Suzuki, K. Fukuyama, B. Watanabe and J. Hiratake, Phosphonate-based irreversible inhibitors of human gamma-glutamyl transpeptidase (GGT), Bioorg. Med. Chem., 2016, 24, 5340–5352, DOI:10.1016/j.bmc.2016.08.050.
- H. Liu, B. Wu, Y. Ge, J. Huang, S. Song, C. Wang, J. Yao, K. Liu, Y. Li, Y. Li and X. Ma, Phosphamide-containing diphenylpyrimidine analogues (PA-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with enhanced activity against pancreatic cancer cell lines, Bioorg. Med. Chem., 2017, 25, 6313–6321, DOI:10.1016/j.bmc.2017.09.041.
- M. Qu, Z. Liu, D. Zhao, C. Wang, J. Zhang, Z. Tang, K. Liu, X. Shu, H. Yuan and X. Ma, Design, synthesis and biological evaluation of sulfonamide-substituted diphenylpyrimidine derivatives (Sul-DPPYs) as potent focal adhesion kinase (FAK) inhibitors with antitumor activity, Bioorg. Med. Chem., 2017, 25, 3989–3996, DOI:10.1016/j.bmc.2017.05.044.
- J. Farand, N. Mai, J. Chandrasekhar, Z. E. Newby, J. Van Veldhuizen, J. Loyer-Drew, C. Venkataramani, J. Guerrero, A. Kwok, N. Li, Y. Zherebina, S. Wilbert, J. Zablocki, G. Phillips, W. J. Watkins, R. Mourey and G. T. Notte, Selectivity switch between FAK and Pyk2: Macrocyclization of FAK inhibitors improves Pyk2 potency, Bioorg. Med. Chem. Lett., 2016, 26, 5926–5930, DOI:10.1016/j.bmcl.2016.10.092.
- X. Zheng, X. Li, L. Tian, B. Wu, J. Yu, C. Wang, X. Sun, X. Ma, L. Chen and Y. Li, Design, synthesis and activity evaluation of isopropylsulfonyl-substituted 2,4-diarylaminopyrimidine derivatives as FAK inhibitors for the potential treatment of pancreatic cancer, Eur. J. Med. Chem., 2022, 241, 114607, DOI:10.1016/j.ejmech.2022.114607.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Advances in covalent kinase inhibitors, Chem. Soc. Rev., 2020, 49, 2617–2687, 10.1039/c9cs00720b.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O'Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity, Nature, 2019, 575, 217–223, DOI:10.1038/s41586-019-1694-1.
- E. Yen-Pon, B. Li, M. Acebron-Garcia-de-Eulate, C. Tomkiewicz-Raulet, J. Dawson, D. Lietha, M. C. Frame, X. Coumoul, C. Garbay, M. Etheve-Quelquejeu and H. Chen, Structure-based sesign, synthesis, and characterization of the first irreversible inhibitor of focal adhesion kinase, ACS Chem. Biol., 2018, 13, 2067–2073, DOI:10.1021/acschembio.8b00250.
- B. Li, Y. Li, C. Tomkiewicz-Raulet, P. Dao, D. Lietha, E. Yen-Pon, Z. Du, X. Coumoul, C. Garbay, M. Etheve-Quelquejeu and H. Chen, Design, synthesis, and biological evaluation of covalent inhibitors of focal adhesion kinase (FAK) against human malignant glioblastoma, J. Med. Chem., 2020, 63, 12707–12724, DOI:10.1021/acs.jmedchem.0c01059.
- T. Chen, Y. Liu, M. Shi, M. Tang, W. Si, X. Yuan, Y. Wen and L. Chen, Design, synthesis, and biological evaluation of novel covalent inhibitors targeting focal adhesion kinase, Bioorg. Med. Chem. Lett., 2021, 54, 128433, DOI:10.1016/j.bmcl.2021.128433.
- T. Chen, Y. Liu, J. Liu, M. Tang, H. Huang, C. Bai, W. Si, T. Yang, X. Yuan, Y. Wen and L. Chen, Design, synthesis and biological evaluation of novel FAK inhibitors with better selectivity over IR than TAE226, Bioorg. Chem., 2022, 124, 105790, DOI:10.1016/j.bioorg.2022.105790.
- S. Wang, R. Zhang, H. Zhang, Y. Wang, D. Yang, Y. Zhao, G. Yan, G. Xu, H. Guan, Y. Zhou, D. Cui, T. Liu, Y. Li, S. Liao and M. Zhou, Design, synthesis, and biological evaluation of 2,4-diamino pyrimidine derivatives as potent FAK inhibitors with anti-cancer and anti-angiogenesis activities, Eur. J. Med. Chem., 2021, 222, 113573, DOI:10.1016/j.ejmech.2021.113573.
- Y. Li, Y. Qi, Y. Fang, H. Gao and H. Zhang, Design, synthesis, and biological evaluation of 4-arylamino pyrimidine derivatives as FAK inhibitors and tumor radiotracers, Mol. Pharm., 2022, 19, 2471–2482, DOI:10.1021/acs.molpharmaceut.2c00180.
- Y. Liu, L. Kong, N. Li, Y. Liu, M. Jia, Q. Liu, S. Zhang and J. Song, Design, synthesis and biological evaluation of novel 2,4-diaminopyrimidine cinnamyl derivatives as inhibitors of FAK with potent anti-gastric cancer activities, Bioorg. Chem., 2023, 141, 106895, DOI:10.1016/j.bioorg.2023.106895.
- Z. Huang, J. Fu and Y. Zhang, Nitric oxide donor-based cancer therapy: Advances and prospects, J. Med. Chem., 2017, 60, 7617–7635, DOI:10.1021/acs.jmedchem.6b01672.
- H. Cheng, L. Wang, M. Mollica, A. T. Re, S. Wu and L. Zuo, Nitric oxide in cancer metastasis, Cancer Lett., 2014, 353, 1–7, DOI:10.1016/j.canlet.2014.07.014.
- M. Chu, Y. Koshman, R. Iyengar, T. Kim, B. Russell and A. M. Samarel, Contractile activity regulates inducible nitric oxide synthase expression and NO(i) production in cardiomyocytes via a FAK-dependent signaling pathway, J. Signal Transduction, 2012, 2012, 473410, DOI:10.1155/2012/473410.
- J. Zhang, K. Xu, F. Yang, Y. Qiu, J. Li, J. Li, W. Wang, G. Tan, Z. Zou and F. Kang, Design, synthesis and evaluation of nitric oxide releasing derivatives of 2,4-diaminopyrimidine as novel FAK inhibitors for intervention of metastatic triple-negative breast cancer, Eur. J. Med. Chem., 2023, 250, 115192, DOI:10.1016/j.ejmech.2023.115192.
- Y. Li, Y. He, C. Zhang, L. Gan and H. Zhang, Discovery of potent focal adhesion kinase (FAK) inhibitor A8 with enhanced antitumor activity, Eur. J. Med. Chem., 2025, 291, 117593, DOI:10.1016/j.ejmech.2025.117593.
- X. Wang, A. Y. Sato, S. Marino, N. Akel, G. Boysen, A. G. Basnakian, T. M. Bellido and H. Li, Generation of BT-amide, a bone-targeted Pyk2 inhibitor, effective via oral administration, for the prevention of glucocorticoid induced bone loss, J. Med. Chem., 2024, 67, 20708–20720, DOI:10.1021/acs.jmedchem.4c02539.
- R. A. Ward, M. J. Anderton, S. Ashton, P. A. Bethel, M. Box, S. Butterworth, N. Colclough, C. G. Chorley, C. Chuaqui, D. A. E. Cross, L. A. Dakin, J. É. Debreczeni, C. Eberlein, M. R. V. Finlay, G. B. Hill, M. Grist, T. C. M. Klinowska, C. Lane, S. Martin, J. P. Orme, P. Smith, F. Wang and M. J. Waring, Structure- and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR), J. Med. Chem., 2013, 56, 7025–7048, DOI:10.1021/jm400822z.
- R. Wang, Y. Chen, X. Zhao, S. Yu, B. Yang, T. Wu, J. Guo, C. Hao, D. Zhao and M. Cheng, Design, synthesis and biological evaluation of novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potential FAK inhibitors and anticancer agents, Eur. J. Med. Chem., 2019, 183, 111716, DOI:10.1016/j.ejmech.2019.111716.
- S. Zeng, S. Yuan, Y. Zhang, J. Du, Y. Wu, Y. Chen, P. Zhu and W. Huang, Discovery of novel pyrrolo [2,3-d] pyrimidine derivatives as potent FAK inhibitors based on cyclization strategy, Bioorg. Chem., 2023, 139, 106713, DOI:10.1016/j.bioorg.2023.106713.
- W. Wei, Z. Feng, Z. Liu, X. Li, H. He, K. Ran, Y. Shi, Y. Zhu, T. Ye, C. Gao, N. Wang and L. Yu, Design, synthesis and biological evaluation of 7-((7H-pyrrolo[2,3-d]pyrimidin-4-yl)oxy)-2,3-dihydro-1H-inden-1-one derivatives as potent FAK inhibitors for the treatment of ovarian cancer, Eur. J. Med. Chem., 2022, 228, 113978, DOI:10.1016/j.ejmech.2021.113978.
- H. Tan, Y. Liu, C. Gong, J. Zhang, J. Huang and Q. Zhang, Synthesis and evaluation of FAK inhibitors with a 5-fluoro-7H-pyrrolo[2,3-d]pyrimidine scaffold as anti-hepatocellular carcinoma agents, Eur. J. Med. Chem., 2021, 223, 113670, DOI:10.1016/j.ejmech.2021.113670.
- B. J. Groendyke, B. Nabet, M. L. Mohardt, H. Zhang, K. Peng, E. Koide, C. R. Coffey, J. Che, D. A. Scott, A. J. Bass and N. S. Gray, Discovery of a pyrimidothiazolodiazepinone as a potent and selective focal adhesion kinase (FAK) inhibitor, ACS Med. Chem. Lett., 2021, 12, 30–38, DOI:10.1021/acsmedchemlett.0c00338.
- R. Determann, J. Dreher, K. Baumann, L. Preu, P. G. Jones, F. Totzke, C. Schachtele, M. H. G. Kubbutat and C. Kunick, 2-Anilino-4-(benzimidazol-2-yl)pyrimidines-a multikinase inhibitor scaffold with antiproliferative activity toward cancer cell lines, Eur. J. Med. Chem., 2012, 53, 254–263, DOI:10.1016/j.ejmech.2012.04.007.
- R. Wang, S. Yu, X. Zhao, Y. Chen, B. Yang, T. Wu, C. Hao, D. Zhao and M. Cheng, Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors, Eur. J. Med. Chem., 2020, 188, 112024, DOI:10.1016/j.ejmech.2019.112024.
- P. Dao, R. Jarray, J. Le Coq, D. Lietha, A. Loukaci, Y. Lepelletier, R. Hadj-Slimane, C. Garbay, F. Raynaud and H. Chen, Synthesis of novel diarylamino-1,3,5-triazine derivatives as FAK inhibitors with anti-angiogenic activity, Bioorg. Med. Chem. Lett., 2013, 23, 4552–4556, DOI:10.1016/j.bmcl.2013.06.038.
- P. Dao, N. Smith, C. Tomkiewicz-Raulet, E. Yen-Pon, M. Camacho-Artacho, D. Lietha, J. Herbeuval, X. Coumoul, C. Garbay and H. Chen, Design, synthesis, and evaluation of novel imidazo[1,2-a][1,3,5]triazines and their derivatives as focal adhesion kinase inhibitors with antitumor activity, J. Med. Chem., 2015, 58, 237–251, DOI:10.1021/jm500784e.
- P. Dao, D. Lietha, M. Etheve-Quelquejeu, C. Garbay and H. Chen, Synthesis of novel 1,2,4-triazine scaffold as FAK inhibitors with antitumor activity, Bioorg. Med. Chem. Lett., 2017, 27, 1727–1730, DOI:10.1016/j.bmcl.2017.02.072.
- C. A. Zificsak, D. E. Gingrich, H. J. Breslin, D. D. Dunn, K. L. Milkiewicz, J. P. Theroff, T. V. Thieu, T. L. Underiner, L. R. Weinberg, L. D. Aimone, M. S. Albom, J. L. Mason, L. Saville, J. Husten, T. S. Angeles, J. P. Finn, M. Jan, T. M. O'Kane, P. Dobrzanski and B. D. Dorsey, Optimization of a novel kinase inhibitor scaffold for the dual inhibition of JAK2 and FAK kinases, Bioorg. Med. Chem. Lett., 2012, 22, 133–137, DOI:10.1016/j.bmcl.2017.02.072.
- J. Sun, Y. Yang, W. Li, Y. Zhang, X. Wang, J. Tang and H. Zhu, Synthesis, biological evaluation and molecular docking studies of 1,3,4-thiadiazole derivatives containing 1,4-benzodioxan as potential antitumor agents, Bioorg. Med. Chem. Lett., 2011, 21, 6116–6121, DOI:10.1016/j.bmcl.2011.08.039.
- X. Yang, L. Xiang, X. Li, T. Zhao, H. Zhang, W. Zhou, X. Wang, H. Gong and H. Zhu, Synthesis, biological evaluation, and molecular docking studies of 1,3,4-thiadiazol-2-amide derivatives as novel anticancer agents, Bioorg. Med. Chem., 2012, 20, 2789–2795, DOI:10.1016/j.bmc.2012.03.040.
- J. Sun, S. Ren, X. Lu, J. Li, F. Shen, C. Xu and H. Zhu, Discovery of a series of 1,3,4-oxadiazole-2(3H)-thione derivatives containing piperazine skeleton as potential FAK inhibitors, Bioorg. Med. Chem., 2017, 25, 2593–2600, DOI:10.1016/j.bmc.2017.03.038.
- Y. Duan, Y. Yao, W. Huang, J. A. Makawana, S. B. Teraiya, N. J. Thumar, D. Tang, X. Tao, Z. Wang, A. Jiang and H. Zhu, Synthesis, biological evaluation, and molecular docking studies of novel 2-styryl-5-nitroimidazole derivatives containing 1,4-benzodioxan moiety as FAK inhibitors with anticancer activity, Bioorg. Med. Chem., 2014, 22, 2947–2954, DOI:10.1016/j.bmc.2014.04.005.
- M. D. Altintop, B. Sever, G. Akalin Ciftci, G. Turan-Zitouni, Z. A. Kaplancikli and A. Ozdemir, Design, synthesis, in vitro and in silico evaluation of a new series of oxadiazole-based anticancer agents as potential Akt and FAK inhibitors, Eur. J. Med. Chem., 2018, 155, 905–924, DOI:10.1016/j.ejmech.2018.06.049.
- J. F. Ohren, H. Chen, A. Pavlovsky, C. Whitehead, E. Zhang, P. Kuffa, C. Yan, P. McConnell, C. Spessard, C. Banotai, W. T. Mueller, A. Delaney, C. Omer, J. Sebolt-Leopold, D. T. Dudley, I. K. Leung, C. Flamme, J. Warmus, M. Kaufman, S. Barrett, H. Tecle and C. A. Hasemann, Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition, Nat. Struct. Mol. Biol., 2004, 11, 1192–1197, DOI:10.1038/nsmb859.
- C. W. Lindsley, Z. Zhao, W. H. Leister, R. G. Robinson, S. F. Barnett, D. Defeo-Jones, R. E. Jones, G. D. Hartman, J. R. Huff, H. E. Huber and M. E. Duggan, Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors, Bioorg. Med. Chem. Lett., 2005, 15, 761–764, DOI:10.1016/j.bmcl.2004.11.011.
- M. Iwatani, H. Iwata, A. Okabe, R. J. Skene, N. Tomita, Y. Hayashi, Y. Aramaki, D. J. Hosfield, A. Hori, A. Baba and H. Miki, Discovery and characterization of novel allosteric FAK inhibitors, Eur. J. Med. Chem., 2013, 61, 49–60, DOI:10.1016/j.ejmech.2012.06.035.
- N. Tomita, Y. Hayashi, S. Suzuki, Y. Oomori, Y. Aramaki, Y. Matsushita, M. Iwatani, H. Iwata, A. Okabe, Y. Awazu, O. Isono, R. J. Skene, D. J. Hosfield, H. Miki, T. Kawamoto, A. Hori and A. Baba, Structure-based discovery of cellular-active allosteric inhibitors of FAK, Bioorg. Med. Chem. Lett., 2013, 23, 1779–1785, DOI:10.1016/j.bmcl.2013.01.047.
- M. Mustafa, G. E. A. Abuo-Rahma, A. A. Abd El-Hafeez, E. R. Ahmed, D. Abdelhamid, P. Ghosh and A. M. Hayallah, Discovery of antiproliferative and anti-FAK inhibitory activity of 1,2,4-triazole derivatives containing acetamido carboxylic acid skeleton, Bioorg. Med. Chem. Lett., 2021, 40, 127965, DOI:10.1016/j.bmcl.2021.127965.
- H. Lu, L. Bai, Y. Zhou, Y. Lu, Z. Jiang and J. Shi, Recent study of dual HDAC/PARP inhibitor for the treatment of tumor, Curr. Top. Med. Chem., 2019, 19, 1041–1050, DOI:10.2174/1568026619666190603092407.
- W. Zhang, J. Huang, X. Tian, Y. Liu, M. Jia, W. Wang, C. Jin, J. Song and S. Zhang, A review of progress in o-aminobenzamide-based HDAC inhibitors with dual targeting capabilities for cancer therapy, Eur. J. Med. Chem., 2023, 259, 115673, DOI:10.1016/j.ejmech.2023.115673.
- F. Yun, C. Cheng, S. Ullah and Q. Yuan, Design, synthesis and biological evaluation of novel histone deacetylase1/2 (HDAC1/2) and cyclin-dependent Kinase2 (CDK2) dual inhibitors against malignant cancer, Eur. J. Med. Chem., 2020, 198, 112322, DOI:10.1016/j.ejmech.2020.112322.
- X. H. Makhoba, C. J. Viegas, R. A. Mosa, F. P. D. Viegas and O. J. Pooe, Potential impact of the multi-target drugapproach in the treatment of some complex diseases, Drug Des., Dev. Ther., 2020, 14, 3235–3249, DOI:10.2147/DDDT.S257494.
- A. Anighoro, J. Bajorath and G. Rastelli, Polypharmacology: challenges and opportunities in drug discovery, J. Med. Chem., 2014, 57, 7874–7887, DOI:10.1021/jm5006463.
- X. W, J. Wang, Q. Liang, R. Tong, J. Huang, X. Yang, Y. Xue, W. Wang, M. Sun and J. Shi, Recent progress on FAK inhibitors with dual targeting capabilities for cancer treatment, Biomed. Pharmacother., 2022, 151, 113116, DOI:10.1016/j.biopha.2022.113116.
- Z. Wang, J. Cai, J. Cheng, W. Yang, Y. Zhu, H. Li, T. Lu, Y. Chen and S. Lu, FLT3 inhibitors in acute myeloid leukemia: Challenges and recent developments in overcoming resistance, J. Med. Chem., 2021, 64, 2878–2900, DOI:10.1021/acs.jmedchem.0c01851.
- H. Cho, I. Shin, H. Yoon, E. Jeon, J. Lee, Y. Kim, S. Ryu, C. Song, N. H. Kwon, Y. Moon, S. Kim, N. D. Kim, H. G. Choi and T. Sim, Identification of thieno[3,2-d]pyrimidine derivatives as dual inhibitors of focal adhesion kinase and FMS-like tyrosine kinase 3, J. Med. Chem., 2021, 64, 11934–11957, DOI:10.1021/acs.jmedchem.1c00459.
- H. Choi, Z. Wang, W. Richmond, X. He, K. Yang, T. Jiang, T. Sim, D. Karanewsky, X. Gu, V. Zhou, Y. Liu, O. Ohmori, J. Caldwell, N. Gray and Y. He, Design and synthesis of 7H-pyrrolo[2,3-d]pyrimidines as focal adhesion kinase inhibitors. Part 1, Bioorg. Med. Chem. Lett., 2006, 16, 2173–2176, DOI:10.1016/j.bmcl.2006.01.053.
- H. Choi, Z. Wang, W. Richmond, X. He, K. Yang, T. Jiang, D. Karanewsky, X. Gu, V. Zhou, Y. Liu, J. Che, C. C. Lee, J. Caldwell, T. Kanazawa, I. Umemura, N. Matsuura, O. Ohmori, T. Honda, N. Gray and Y. He, Design and synthesis of 7H-pyrrolo[2,3-d]pyrimidines as focal adhesion kinase inhibitors. Part 2, Bioorg. Med. Chem. Lett., 2006, 16, 2689–2692, DOI:10.1016/j.bmcl.2006.02.032.
- J. C. Dawson, B. Serrels, A. Byron, M. T. Muir, A. Makda, A. Garcia-Munoz, A. von Kriegsheim, D. Lietha, N. O. Carragher and M. C. Frame, A synergistic anticancer FAK and HDAC inhibitor combination discovered by a novel chemical-genetic high-content phenotypic screen, Mol. Cancer Ther., 2020, 19, 637–649, DOI:10.1158/1535-7163.MCT-19-0330.
- M. Mustafa, A. A. Abd El-Hafeez, D. Abdelhamid, G. D. Katkar, Y. A. Mostafa, P. Ghosh, A. M. Hayallah and G. E. A. Abuo-Rahma, A first-in-class anticancer dual HDAC2/FAK inhibitors bearing hydroxamates/benzamides capped by pyridinyl-1,2,4-triazoles, Eur. J. Med. Chem., 2021, 222, 113569, DOI:10.1016/j.ejmech.2021.113569.
- J. Song, X. Liu, Y. Zhang, X. Tian, M. Deng, C. Huang and S. Zhang, The dual FAK-HDAC inhibitor MY-1259 displays potent activities in gastric cancers in vitro and in vivo, Bioorg. Chem., 2023, 131, 106328, DOI:10.1016/j.bioorg.2022.106328.
- C. Arienti, S. Pignatta and A. Tesei, Epidermal growthfactor receptor family and its role in gastric cancer, Front. Oncol., 2019, 9, 1308, DOI:10.3389/fonc.2019.01308.
- X. Hu, Q. Xun, T. Zhang, S. Zhu, Q. Li, L. Tong, M. Lai, T. Huang, C. Yun, H. Xie, K. Ding and X. Lu, 2-Oxo-3,4-dihydropyrimido[4,5-d] pyrimidines as new reversible inhibitors of EGFR C797S(Cys797 to Ser797)mutant, Chin. Chem. Lett., 2020, 31, 1281–1287 CrossRef CAS.
- N. Lyu, B. Pedersen, E. Shklovskaya, H. Rizos, M. P. Molloy and Y. Wang, SERS characterization of colorectal cancer cell surface markers upon anti-EGFR treatment, Exploration, 2022, 2, 20210176, DOI:10.1002/EXP.20210176.
- Q. Luo, S. Zhou, W. Pan, J. Sun, L. Yang, L. Zhang, M. Qiu and D. Yang, A multi-kinase inhibitor APG-2449 enhances the antitumor effect of ibrutinib in esophageal squamous cell carcinoma via EGFR/FAK pathway inhibition, Biochem. Pharmacol., 2021, 183, 114318, DOI:10.1016/j.bcp.2020.114318.
- M. Ai, C. Wang, Z. Tang, K. Liu, X. Sun, T. Ma, Y. Li, X. Ma, L. Li and L. Chen, Design and synthesis of diphenylpyrimidine derivatives (DPPYs) as potential dual EGFR T790M and FAK inhibitors against a diverse range of cancer cell lines, Bioorg. Chem., 2020, 94, 103408, DOI:10.1016/j.bioorg.2019.103408.
- I. Eke and N. Cordes, Dual targeting of EGFR and focal adhesion kinase in 3D grown HNSCC cell cultures, Radiother. Oncol., 2011, 99, 279–286, DOI:10.1016/j.radonc.2011.06.006.
- M. M. Elbadawi, W. M. Eldehna, A. A. Abd El-Hafeez, W. R. Somaa, A. Albohy, S. T. Al-Rashood, K. K. Agama, E. B. Elkaeed, P. Ghosh, Y. Pommier and M. Abe, 2-Arylquinolines as novel anticancer agents with dual EGFR/FAK kinase inhibitory activity: synthesis, biological evaluation, and molecular modelling insights, J. Enzym. Inhib. Med. Chem., 2022, 37, 349–372, DOI:10.1080/14756366.2021.2015344.
- M. Ai, C. Wang, Z. Tang, K. Liu, X. Sun, T. Ma, Y. Li, X. Ma, L. Li and L. Chen, Design and synthesis of diphenylpyrimidine derivatives (DPPYs) as potential dual EGFR T790M and FAK inhibitors against a diverse range of cancer cell lines, Bioorg. Chem., 2020, 94, 103408, DOI:10.1016/j.bioorg.2019.103408.
- A. Schuldt, Cell division: a timely exit for PLK1, Nat. Rev. Mol. Cell Biol., 2013, 14, 194–195, DOI:10.1038/nrm3570.
- Z. Liu, Q. Sun and X. Wang, PLK1, A potential target for cancer therapy, Transl. Oncol., 2017, 10, 22–32, DOI:10.1016/j.tranon.2016.10.003.
- S. Iliaki, R. Beyaert and I. S. Afonina, Polo-like kinase 1 (PLK1) signaling in cancer and beyond, Biochem. Pharmacol., 2021, 193, 114747, DOI:10.1016/j.bcp.2021.114747.
- D. K. Gupta, J. Du, S. A. Kamranvar and S. Johansson, Tension-induced cytokinetic abscission in human fibroblasts, Oncotarget, 2018, 9, 8999–9009, DOI:10.18632/oncotarget.24016.
- S. A. Kamranvar, D. K. Gupta, Y. Huang, R. K. Gupta and S. Johansson, Integrin signaling via FAK-Src controls cytokinetic abscission by decelerating PLK1 degradation and subsequent recruitment of CEP55 at the midbody, Oncotarget, 2016, 7, 30820–30830, DOI:10.18632/oncotarget.9003.
- J. Sun, Z. Fang, Y. Tao, Y. Zhang, Y. Zhang, H. Sun, Y. Zhou and Y. Wu, Design, synthesis and antitumor activity of FAK/PLK1 dual Inhibitors with quinazolinone as the skeleton, Chem. Biodivers., 2023, 20, e202300146, DOI:10.1002/cbdv.202300146.
- S. Zhang, J. Lou, Y. Li, F. Zhou, Z. Yan, X. Lu and Y. Zhao, Recent progress and clinical development of inhibitors that block MDM4/p53 protein-protein interactions, J. Med. Chem., 2021, 64, 10621–10640, DOI:10.1021/acs.jmedchem.1c00940.
- Z. Wang, W. Huang, K. Zhou, X. Ren and K. Ding, Targeting the non-catalytic functions: a new paradigm for kinase drug discovery?, J. Med. Chem., 2022, 65, 1735–1748, DOI:10.1021/acs.jmedchem.1c01978.
- A. J. Souers, J. D. Leverson, E. R. Boghaert, S. L. Ackler, N. D. Catron, J. Chen, B. D. Dayton, H. Ding, S. H. Enschede, W. J. Fairbrother, D. C. S. Huang, S. G. Hymowitz, S. Jin, S. L. Khaw, P. J. Kovar, L. T. Lam, J. Lee, H. L. Maecker, K. C. Marsh, K. D. Mason, M. J. Mitten, P. M. Nimmer, A. Oleksijew, C. H. Park, C. Park, D. C. Phillips, A. W. Roberts, D. Sampath, J. F. Seymour, M. L. Smith, G. M. Sullivan, S. K. Tahir, C. Tse, M. D. Wendt, Y. Xiao, J. C. Xue, H. Zhang, R. A. Humerickhouse, S. H. Rosenberg and S. W. Elmore, ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets, Nat. Med., 2013, 19, 202–208, DOI:10.1038/nm.3048.
- S. Wang, Y. Zhao, A. Aguilar, D. Bernard and C. Y. Yang, Targeting the MDM2-p53 protein-protein interaction for new cancer therapy: Progress and challenges, Cold Spring Harb. Perspect. Med., 2017, 7, a26245, DOI:10.1101/cshperspect.a026245.
- V. M. Golubovskaya, N. L. Palma, M. Zheng, B. Ho, A. Magis, D. Ostrov and W. G. Cance, A small-molecule inhibitor, 5'-O-tritylthymidine, targets FAK and MDM-2 interaction, and blocks breast and colon tumorigenesis in vivo, Anti-Cancer Agents Med. Chem., 2013, 13, 532–545, DOI:10.2174/1871520611313040002.
- V. M. Golubovskaya, R. Finch and W. G. Cance, Direct interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53, J. Biol. Chem., 2005, 280, 25008–25021, DOI:10.1074/jbc.M414172200.
- V. M. Golubovskaya, B. Ho, M. Zheng, A. Magis, D. Ostrov, C. Morrison and W. G. Cance, Disruption of focal adhesion kinase and p53 interaction with small molecule compound R2 reactivated p53 and blocked tumor growth, BMC Cancer, 2013, 13, 342, DOI:10.1186/1471-2407-13-342.
- S. Fukami, D. Tomioka, Y. Murakami, T. Honda and S. Hatakeyama, Pharmacological profiling of a dual FAK/IGF-1R kinase inhibitor TAE226 in cellular and in vivo tumor models, BMC Res. Notes, 2019, 12, 347, DOI:10.1186/s13104-019-4389-7.
- D. A. Ucar, L. H. Dang and S. N. Hochwald, Focal adhesion kinase signaling and function in pancreatic cancer, Front. Biosci., 2011, 3, 750–756, DOI:10.2741/e283.
- D. A. Ucar, A. T. Magis, D. He, N. J. Lawrence, S. M. Sebti, E. Kurenova, M. Zajac-Kaye, J. Zhang and S. N. Hochwald, Inhibiting the interaction of c-Met and IGF-1R with FAK effectively reduces growth of pancreatic cancer cells in vitro and in vivo, Anti-Cancer Agents Med. Chem., 2013, 13, 595–602, DOI:10.2174/1871520611313040009.
- E. V. Kurenova, D. L. Hunt, D. He, A. T. Magis, D. A. Ostrov and W. G. Cance, Small molecule chloropyramine hydrochloride (C4) targets the binding site of focal adhesion kinase and vascular endothelial growth factor receptor 3 and suppresses breast cancer growth in vivo, J. Med. Chem., 2009, 52, 4716–4724, DOI:10.1021/jm900159g.
- P. N. Gogate, M. Ethirajan, E. V. Kurenova, A. T. Magis, R. K. Pandey and W. G. Cance, Design, synthesis, and biological evaluation of novel FAK scaffold inhibitors targeting the FAK-VEGFR3 protein-protein interaction, Eur. J. Med. Chem., 2014, 80, 154–166, DOI:10.1016/j.ejmech.2014.04.041.
- L. Shen, B. Wang, S. Wang, S. Ji, M. Fu, S. Wang, W. Hou, X. Dai and H. Liu, Combination therapy and dual-target inhibitors based on LSD1: New emerging tools in cancer therapy, J. Med. Chem., 2024, 67, 922–951, DOI:10.1021/acs.jmedchem.3c02133.
- Suzhou Ascentage Pharma Inc, The combination of FAK/ALK/ROS1 and EGFR inhibitors for treatment of cancer, CN Pat., CN201910659457.6, China, 2019 Search PubMed.
- J. C. Dawson, A. Serrels, D. G. Stupack, D. D. Schlaepfer and M. C. Frame, Targeting FAK in anticancer combination therapies, Nat. Rev. Cancer, 2021, 21, 313–324, DOI:10.1038/s41568-021-00340-6.
- Chengdu Hinova Pharma Inc, A combination drug for the prevention and/or treatment of solid tumors, CN Pat., CN202410377819.3, China, 2024 Search PubMed.
- B. Zhang, Y. Zhang, J. Zhang, P. Liu, B. Jiao, Z. Wang and R. Ren, Focal adhesion kinase (FAK) inhibition synergizes with KRAS G12C inhibitors in treating cancer through the regulation of the FAK-YAP signaling, Adv. Sci., 2021, 8, e2100250, DOI:10.1002/advs.202100250.
- InxMed (Shanghai) Co. Ltd, https://clinicaltrials.gov/studyNCT06014528.
- InxMed (Shanghai) Co. Ltd. https://clinicaltrials.gov/study/NCT05830539, https://clinicaltrials.gov/study/NCT06166836, https://clinicaltrials.gov/study/NCT05994131.
- Southwest Hospital, Use of FAK inhibitor combined with hydroxychloroquine in preparation of anti-pancreatic cancer drugs, CN Pat., CN202410575969.5, China, 2024 Search PubMed.
- C. Burns, T. Cock, A. Bishop, N. Kruger, S. McCormack, S. Ananda, M. Harris, W. L. Joubert, J. W. Kim, C. Lee, W. J. Lee, L. R. Lipton, A. Nagrial, D. Oh, J. O. Park, N. Pavlakis and J. Lickliter, Phase 1b/2a of narmafotinib (AMP945) in combination with gemcitabine and nab-paclitaxel in first-line patients with advanced pancreatic cancer (ACCENT trial): Interim analysis, J. Clin. Oncol., 2024, 42, 1 Search PubMed.
- K. M. Sakamoto, K. B. Kim, A. Kumagai, F. Mercurio, C. M. Crews and R. J. Deshaies, Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559, DOI:10.1073/pnas.141230798.
- X. Wang, Z. Qin, N. Li, M. Jia, Q. Liu, Y. Bai, J. Song, S. Yuan and S. Zhang, Annual review of PROTAC degraders as anticancer agents in 2022, Eur. J. Med. Chem., 2024, 267, 116166, DOI:10.1016/j.ejmech.2024.116166.
- K. Li and C. M. Crews, PROTACs: past, present and future, Chem. Soc. Rev., 2022, 51, 5214–5236, 10.1039/d2cs00193d.
- H. Huang, D. Dobrovolsky, J. Paulk, G. Yang, E. L. Weisberg, Z. M. Doctor, D. L. Buckley, J. Cho, E. Ko, J. Jang, K. Shi, H. G. Choi, J. D. Griffin, Y. Li, S. P. Treon, E. S. Fischer, J. E. Bradner, L. Tan and N. S. Gray, A chemoproteomic approach to query the degradable kinome using a multi-kinase degrader, Cell Chem. Biol., 2018, 25, 88–99, DOI:10.1016/j.chembiol.2017.10.005.
- C. E. Powell, Y. Gao, L. Tan, K. A. Donovan, R. P. Nowak, A. Loehr, M. Bahcall, E. S. Fischer, P. A. Janne, R. E. George and N. S. Gray, Chemically induced degradation of anaplastic lymphoma kinase (ALK), J. Med. Chem., 2018, 61, 4249–4255, DOI:10.1021/acs.jmedchem.7b01655.
- P. M. Cromm, K. T. G. Samarasinghe, J. Hines and C. M. Crews, Addressing kinase-independent functions of FAK via PROTAC-mediated degradation, J. Am. Chem. Soc., 2018, 140, 17019–17026, DOI:10.1021/jacs.8b08008.
- J. Popow, H. Arnhof, G. Bader, H. Berger, A. Ciulli, D. Covini, C. Dank, T. Gmaschitz, P. Greb, J. Karolyi-Ozguer, M. Koegl, D. B. McConnell, M. Pearson, M. Rieger, J. Rinnenthal, V. Roessler, A. Schrenk, M. Spina, S. Steurer, N. Trainor, E. Traxler, C. Wieshofer, A. Zoephel and P. Ettmayer, Highly selective PTK2 proteolysis targeting chimeras to probe focal adhesion kinase scaffolding functions, J. Med. Chem., 2019, 62, 2508–2520, DOI:10.1021/acs.jmedchem.8b01826.
- H. Gao, Y. Wu, Y. Sun, Y. Yang, G. Zhou and Y. Rao, Design, synthesis, and evaluation of highly potent FAK-targeting PROTACs, ACS Med. Chem. Lett., 2020, 11, 1855–1862, DOI:10.1021/acsmedchemlett.9b00372.
- Y. Sun, R. Wang, Y. Sun, L. Wang, Y. Xue, J. Wang, T. Wu, W. Yin, Q. Qin, Y. Sun, D. Zhao and M. Cheng, Identification of novel and potent PROTACs targeting FAK for non-small cell lung cancer: Design, synthesis, and biological study, Eur. J. Med. Chem., 2022, 237, 114373, DOI:10.1016/j.ejmech.2022.114373.
- Q. Qin, R. Wang, Q. Fu, G. Zhang, T. Wu, N. Liu, R. Lv, W. Yin, Y. Sun, Y. Sun, D. Zhao and M. Cheng, Design, synthesis, and biological evaluation of potent FAK-degrading PROTACs, J. Enzym. Inhib. Med. Chem., 2022, 37, 2241–2255, DOI:10.1080/14756366.2022.2100886.
- P. A. Quispe, M. J. Lavecchia and I. E. León, Focal adhesion kinase inhibitors in the treatment of solid tumors: Preclinical and clinical evidence, Drug Discovery Today, 2022, 27, 664–674, DOI:10.1016/j.drudis.2021.11.025.
- I. Salem, M. Alsalahi, I. Chervoneva, L. D. Aburto, S. Addya, G. R. Ott, B. A. Ruggeri, M. Cristofanilli and S. V. Fernandez, The effects of CEP-37440, an inhibitor of focal adhesion kinase, in vitro and in vivo on inflammatory breast cancer cells, Breast Cancer Res., 2016, 18, 37, DOI:10.1186/s13058-016-0694-4.
- D. D. Fang, R. Tao, G. Wang, Y. Li, K. Zhang, C. Xu, G. Zhai, Q. Wang, J. Wang, C. Tang, P. Min, D. Xiong, J. Chen, S. Wang, D. Yang and Y. Zhai, Discovery of a novel ALK/ROS1/FAK inhibitor, APG-2449, in preclinical non-small cell lung cancer and ovarian cancer models, BMC Cancer, 2022, 22, 752, DOI:10.1186/s12885-022-09799-4.
- U. A. Hirt, I. C. Waizenegger, N. Schweifer, C. Haslinger, D. Gerlach, J. Braunger, U. Weyer-Czernilofsky, H. Stadtmuller, I. Sapountzis, G. Bader, A. Zoephel, B. Bister, A. Baum, J. Quant, N. Kraut, P. Garin-Chesa and G. R. Adolf, Efficacy of the highly selective focal adhesion kinase inhibitor BI 853520 in adenocarcinoma xenograft models is linked to a mesenchymal tumor phenotype, Oncogenesis, 2018, 7, 21, DOI:10.1038/s41389-018-0032-z.
- I. Street, M. D. Sylva, K. Lackovic, D. Ganame and M. Devlin, Abstract LB-308: Combination of CTx-0294945 a highly selective inhibitor of focal adhesion kinase with bevacizumab in pre-clinical models of breast cancer, Cancer Res., 2012, 72, 308, DOI:10.1158/1538-7445.am2012-lb-308.
- J. Zhang, D. He, M. Zajac-Kaye and S. N. Hochwald, A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells, Cell Cycle, 2014, 13, 3143–3149, DOI:10.4161/15384101.2014.949550.
- W. G. Roberts, E. Ung, P. Whalen, B. Cooper, C. Hulford, C. Autry, D. Richter, E. Emerson, J. Lin, J. Kath, K. Coleman, L. Yao, L. Martinez-Alsina, M. Lorenzen, M. Berliner, M. Luzzio, N. Patel, E. Schmitt, S. LaGreca, J. Jani, M. Wessel, E. Marr, M. Griffor and F. Vajdos, Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271, Cancer Res., 2008, 68, 1935–1944, DOI:10.1158/0008-5472.CAN-07-5155.
- Y. Kang, W. Hu, C. Ivan, H. J. Dalton, T. Miyake, C. V. Pecot, B. Zand, T. Liu, J. Huang, N. B. Jennings, R. Rupaimoole, M. Taylor, S. Pradeep, S. Y. Wu, C. Lu, Y. Wen, J. Huang, J. Liu and A. K. Sood, Role of focal adhesion kinase in regulating YB-1-mediated paclitaxel resistance in ovarian cancer, J. Natl. Cancer Inst., 2013, 105, 1485–1495, DOI:10.1093/jnci/djt210.
- I. Tanjoni, C. Walsh, S. Uryu, A. Tomar, J. Nam, A. Mielgo, S. Lim, C. Liang, M. Koenig, C. Sun, N. Patel, C. Kwok, G. McMahon, D. G. Stupack and D. D. Schlaepfer, PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments, Cancer Biol. Ther., 2010, 9, 764–777, DOI:10.4161/cbt.9.10.11434.
- D. Wang, Y. Chen, Z. Chen, F. Yan, X. Dai, M. Ying, J. Cao, J. Ma, P. Luo, Y. Han, Y. Peng, Y. Sun, H. Zhang, Q. He, B. Yang and H. Zhu, CT-707, a Novel FAK inhibitor, synergizes with Cabozantinib to suppress hepatocellular carcinoma by blocking Cabozantinib-induced FAK activation, Mol. Cancer Ther., 2016, 15, 2916–2925, DOI:10.1158/1535-7163.MCT-16-0282.
- R. P. Law, J. Nunes, C. Chung, M. Bantscheff, K. Buda, H. Dai, J. P. Evans, A. Flinders, D. Klimaszewska, A. J. Lewis, M. Muelbaier, P. Scott-Stevens, P. Stacey, C. J. Tame, G. F. Watt, N. Zinn, M. A. Queisser, J. D. Harling and A. B. Benowitz, Discovery and characterisation of highly cooperative FAK-degrading PROTACs, Angew. Chem., Int. Ed., 2021, 60, 23327–23334, DOI:10.1002/anie.202109237.
- F. J. Sulzmaier, C. Jean and D. D. Schlaepfer, FAK in cancer: mechanistic findings and clinical applications, Nat. Rev. Cancer, 2014, 14, 598–610, DOI:10.1038/nrc3792.
- Y. Sun, Z. Gao, R. Wang, G. Zhang, T. Wu, W. Yin, Y. Sun, Q. Qin, D. Zhao and M. Cheng, Design, synthesis, and biological evaluation of diaminopyrimidine derivatives as novel focal adhesion kinase inhibitors, RSC Med. Chem., 2023, 14, 2301–2314, 10.1039/D3MD00324H.
- J. Huang, L. Chen, J. Wu, D. Ai, J. Zhang, T. Chen and L. Wang, Targeting the PI3K/AKT/mTOR signaling pathway in the treatment of human diseases: Current status,trends, and 2olutions, J. Med. Chem., 2022, 65, 16033–16061, DOI:10.1021/acs.jmedchem.2c01070.
- T. Liu, Y. Wan, Y. Xiao, C. Xia and G. Duan, Dual-target inhibitors based on HDACs: Novel antitumor agents for cancer therapy, J. Med. Chem., 2020, 63, 8977–9002, DOI:10.1021/acs.jmedchem.0c00491.
- W. Zhang, L. Xiao, D. Li, Y. Hu and W. Yu, New strategies for responding to SARS-CoV-2: The present and future of dual-target drugs, J. Med. Chem., 2024, 67, 11522–11542, DOI:10.1021/acs.jmedchem.4c00384.
- X. Li, X. Li, F. Liu, S. Li and D. Shi, Rational multitargeted drug design strategy from the perspective of a medicinal chemist, J. Med. Chem., 2021, 64, 10581–10605, DOI:10.1021/acs.jmedchem.1c00683.
- Y. Zhou, H. Yu, A. C. Vind, L. Kong, Y. Liu, X. Song, Z. Tu, C. Yun, J. B. Smaill, Q. Zhang, K. Ding, S. Bekker-Jensen and X. Lu, Rational design of covalent kinase inhibitors by an integrated computational workflow (Kin-Cov), J. Med. Chem., 2023, 66, 7405–7420, DOI:10.1021/acs.jmedchem.3c00088.
- M. Gehringer and S. A. Laufer, Emerging and re-emerging warheads for targeted covalent inhibitors: Applications in medicinal chemistry and chemical biology, J. Med. Chem., 2019, 62, 5673–5724, DOI:10.1021/acs.jmedchem.8b01153.
- V. Bonatto, R. F. Lameiro, F. R. Rocho, J. Lameira, A. Leitao and C. A. Montanari, Nitriles: an attractive approach to the development of covalent inhibitors, RSC Med. Chem., 2023, 14, 201–217, 10.1039/d2md00204c.
- G. E. Winter, Extrapolating lessons from targeted protein degradation to other proximity-inducing drugs, ACS Chem. Biol., 2024, 19, 2089–2102, DOI:10.1021/acschembio.4c00191.
- Q. Zhang, J. Yu, Q. You and L. Wang, Modulating phosphorylation by proximity-inducing modalities for cancer therapy, J. Med. Chem., 2024, 67, 21695–21716, DOI:10.1021/acs.jmedchem.4c02624.
| This journal is © The Royal Society of Chemistry 2025 |