DOI:
10.1039/D5RA01781E
(Paper)
RSC Adv., 2025,
15, 21666-21677
Identification of prochlorperazine dimaleate as a Sortase A inhibitor from FDA libraries for MRSA infection treatment†
Received
12th March 2025
, Accepted 12th June 2025
First published on 25th June 2025
Abstract
Staphylococcus aureus is acknowledged as an essential contributor to global disease burden, particularly with the emergence of methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA) strains. This study employs a systematic computational and experimental strategy to screen and validate FDA-approved drugs to target Sortase A, an essential enzyme involved in MRSA virulence. Herein, we have identified six molecules exhibiting antimicrobial potency against MRSA and reduced biofilm formation. Among the hits obtained, prochlorperazine dimaleate showed potent activity against MRSA while proving non-cytotoxic to hepatocellular Carcinoma (HepG2) cells at inhibitory concentration. Further, it also disrupts the membrane potential and creates pores inside the membrane of MRSA. In vivo thigh infection studies in mice showed that prochlorperazine dimaleate successfully reduced the MRSA infection load. Taken together, we herein report that prochlorperazine dimaleate attenuated the pathogenicity of S. aureus, thus reducing MRSA infection by directly targeting Sortase A. Therefore, prochlorperazine dimaleate can be utilized as an adjuvant therapy for treating MRSA infection.
1. Introduction
Staphylococcus aureus (S. aureus) is a highly concerning and rapidly spreading pathogen that causes soft tissue infection, bacteremia, pneumonia, endocarditis, osteomyelitis, toxic shock syndrome, and skin infection.1,2 Methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA) have already posed a hazard to human health. This is further exacerbated by the biofilm-forming nature of S. aureus, which exhibits a high propensity for resistance to conventional antibiotics, posing a significant challenge in treating wound infections. MRSA now accounts for more than 60% of all isolated S. aureus.3 Hence, there is a critical need for novel treatment approaches that address drug resistance while simultaneously decreasing bacterial infections.4,5 The pathogenicity of Staphylococcus aureus is closely linked to the virulence factors it secretes, encompassing surface proteins, toxins, and enzymes.6,7 These factors aid in bacterial adherence, tissue invasion and degradation, and evasion of host defense mechanisms. Surface-associated adhesins such as clumping factor A/clumping factor B (ClfA/ClfB), fibronectin-binding proteins (FnBPs), and collagen adhesin (CNA) are involved in the pathogenesis of S. aureusinfection.8–10 Sortase A (SrtA), a cysteine transpeptidase, covalently attaches crucial virulence factors of S. aureus to the bacterial surface, promotes bacterial adherence, facilitates biofilm formation, mediates host cell penetration, acquires vital nutrients from the host, and contributes to immune evasion and suppression.11–13 Further, SrtA stands out as a promising drug target due to its location on the extracellular side of the cell membrane, and an additional advantage is the absence of human homologs,14 suggesting that selective inhibitors of this enzyme should exhibit lower toxicity.15 Consequently, inhibiting this enzyme is anticipated to exert reduced selection pressure on developing resistance.16,17 Over the last decade, several natural products such as flavonoids,18 salvinolic acid,19 chalcone,20 peptide analogs, and synthetic small molecules have been reported as Sortase A inhibitors (Table S1†). However, potential toxicity and antimicrobial resistance are persistent concerns.
To address this, we have employed computational and experimental strategies to target Sortase A in MRSA. We have screened Food and Drug Administration (FDA)-approved drug libraries against Sortase A and identified 60 hit molecules. Phenotypic screening of the top virtual screening hits by antimicrobial assay identified six promising molecules against clinically isolated MRSA. Among these, prochlorperazine dimaleate exhibited potent antibacterial activity, biofilm eradication, membrane potential disruption, and was non-cytotoxic in the HepG2 cell line. Further, it also reduced the MRSA infection load in the thigh infection murine model. In summary, we have identified novel chemical scaffolds promising novel therapeutic agents for combating MRSA infections.
2. Experimental
2.1. Materials and methods
2.1.1. Bacterial culture and chemicals. The clinical isolate Staphylococcus aureus American Type Culture Collection 43300 (S. aureus ATCC 43300) was purchased from the American Type Culture Collection (Manassas, VA, USA). Conventional antibiotics, ciprofloxacin, gentamicin, vancomycin, and nisin, were purchased from Sigma-Aldrich. Mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride were purchased from Tokyo Chemical Industry (TCI chemicals). Cation-adjusted Muller–Hinton broth (CA-MHB) and Mueller–Hinton Broth (MHB) and Luria–Bertani medium (LB medium) were purchased from Becton Dickinson and Himedia, respectively. The human monocyte cell line Tohoku Hospital Pediatrics-1 (THP-1) and hepatocellular Carcinoma (HepG2) cells were maintained in Roswell Park Memorial Institute 1640 (RPMI-1640) and Dulbecco's Modified Eagle (DMEM) medium, respectively.
2.1.2. Cloning, expression, and purification of Sortase A. The gene encoding Sortase A, excluding the N-terminal transmembrane domain (N1–59), was amplified from Staphylococcus aureus genomic DNA via polymerase chain reaction (PCR) using the primers SrtA-F and SrtA-R.21 The amplified fragment was subsequently digested with XhoI and NdeI and inserted into the corresponding sites of the pET28a vector, generating the recombinant plasmid pET28a-SrtA. The pET28a-SrtA plasmid was introduced into E. coli BL21(DE3) cells using the heat shock method. Transformed cells were grown in LB medium supplemented with kanamycin (50 μg mL−1) at 37 °C with shaking at 200 rpm until the optical density at 600 nm (OD600) reached 0.6–0.8.22 Protein expression was induced by adding 0.5 mM IPTG (isopropyl-β-D-thiogalactopyranoside), and the culture was incubated overnight at 25 °C with shaking at 180 rpm. Following induction, the cells were harvested by centrifugation at 8000 rpm for 10 min at 4 °C to collect the supernatant. The cell lysate was sonicated on ice, and the soluble protein fraction was isolated by centrifugation. The protein was purified using immobilized metal affinity chromatography (IMAC) with a Ni-NTA resin (Qiagen). The lysate was loaded onto the column, followed by a series of washing steps and protein elution using buffers with increasing concentrations of imidazole. The eluted fractions were analyzed by SDS-PAGE, and those containing the target protein were pooled. The pooled fractions were subsequently desalted into a buffer containing 25 mM NaCl, 150 mM Tris, and 5% glycerol using PD-10 desalting columns (Cytiva).
2.1.3. Virtual screening of compounds in Sortase A. FDA-approved library drugs comprising 3040 molecules were prepared using the Ligand Prep Program in Flare™(V8) (Cresset).23–26 All calculations were run on a Windows 10 Pro 64-bit/Core i7-8700 CPU @ 3.20 GHz system. The molecules were recharged with hydrogen appropriately for pH 7.4 and stripped of salts. The 3D coordinates of all molecules were assigned with OpenMM using the AMBER force field for energy minimization. The NMR protein structure file of Sortase A attached to an inhibitor (PDB ID: 2KID) was obtained from the Protein Data Bank. The protein was prepared using the default Protein Preparation Program, wherein water molecules were removed while the terminal residues were capped with ACE or NME. The bound inhibitor was auto-extracted, and the active site with a 6 Å radius was identified and defined. Molecular docking of 3040 molecules was performed using virtual screening on the predefined grid. A maximum of 10 poses were obtained for each molecule in a single run.
2.1.4. Fluorescence quenching assay. Various concentrations of compounds (0 to 128 μg mL−1) were added to Sortase A protein (500 nM mL−1) in reaction buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 10 mM CaCl2) and incubated at room temperature for 10 minutes.27 Subsequently, fluorescence intensity was measured using a microplate reader Cytation 5 (Agilent, USA) at excitation and emission wavelengths of 280 and 326 nm, respectively. The Ksv value was calculated using the previously reported study.
2.1.5. Sortase A activity assay by fluorescence resonance energy transfer (FRET) assay. Sortase A activity was assessed using a fluorescence resonance energy transfer (FRET) assay.28 A 200 μL reaction mixture was prepared, comprising Sortase A reaction buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 10 mM CaCl2), purified Sortase A protein (4 μM), and varying concentrations of test compounds (0 to 256 μg mL−1). The mixtures were incubated at 37 °C for 2 hours, followed by adding the fluorescent peptide substrate Abz-LPATG-Dap(DNP) (10 μM). The reaction was further incubated at 37 °C for 20 minutes. Fluorescence intensity was then recorded using a Cytation 5 microplate reader (Agilent, USA), with excitation and emission wavelengths set to 309 nm and 420 nm, respectively.
2.1.6. Determination of MIC and MBC. The micro broth dilution method was used to determine the minimum inhibitory concentration (MIC) of several compounds against S. aureus ATCC 43300. These compounds included mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride. The experiment adhered to the protocols outlined by the Clinical and Laboratory Standards Institute (CLSI).29 Briefly, S. aureus ATCC 43300 was cultured in Mueller–Hinton broth (MHB), and the bacterial cell suspensions were diluted up to 5 × 105 Colony-forming unit per milliliter (CFU mL−1).30 Bacterial culture was inoculated into 96-well plates containing serially diluted compounds and incubated at 37 °C for 18 hours.31 The MIC was calculated by noting the absence of observable growth at the lowest concentration tested. To determine minimum bactericidal concentration (MBC), 100 μL of suspension cultures from wells with concentrations above the MIC were spread onto Mueller–Hinton agar (MHA) plates.31 The plates were incubated for an additional 18 hours at 37 °C, and the presence or absence of bacterial colonies was evaluated. The lowest concentration of the compounds where no bacterial colonies were observed was considered the MBC.32 The MICs and MBCs were confirmed through biological replicates.
2.1.7. Time-kill kinetics. A time-kill kinetics experiment was carried out to assess the efficacy of compounds and ciprofloxacin against S. aureus ATCC 43300.33 Initially, S. aureus was cultured until reaching the exponential growth phase (OD600 = 0.5), corresponding to a bacterial concentration of 5 × 108 CFU mL−1.34 The culture was then diluted 1000-fold in CA-MHB and treated individually with the compounds and ciprofloxacin at 2 times the minimum inhibitory concentration (MIC). The samples were incubated at 37 °C with shaking at 200 rpm at distinct time periods (0, 2, 4, 8, 12, and 24 hours). 100 μL of the treated culture was disseminated on MHA plates. In addition, cultures without treatment and ciprofloxacin alone were included as untreated and antibiotic controls. The plates were then incubated for 24 hours at 37 °C. Following this incubation, viable bacterial colonies were enumerated, and the CFU mL−1 was determined. The data was analyzed using GraphPad Prism 8. The experiment was carried out in triplicate.
2.1.8. Biofilm growth inhibition assay. The compounds' efficiency in suppressing biofilm formation by S. aureus ATCC 43300 was demonstrated using crystal violet staining dye. S. aureus was cultured in tryptic soy broth (TSB) at 37 °C for 24 hours.31 The overnight culture was diluted in TSB (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100) with the 0.1% glucose supplement. This diluted culture was added to a flat-bottom 96-well microtiter plate, followed by compounds at MIC and 2 × MIC, and further incubation at 37 °C for 24 hours.32 Following the incubation, the planktonic cells were removed from the wells, and the wells were washed with 1 × PBS (pH 7.4).35 The microtiter plate was dried for 10 minutes, and biofilm was stained with 0.1% crystal violet for 10 minutes at 25 °C. The wells were washed with distilled water to remove any excess stain. To solubilize the stain, glacial acetic acid (33%) was added and incubated at room temperature for 30 minutes on an orbital shaker.36 The absorbance of the stained cells was measured at 600 nm with a plate reader, and the percentage of biofilm growth inhibition relative to the untreated control was calculated. The experiment was conducted in triplicate with three biological replicates, and the data were analyzed utilizing GraphPad Prism 8.
:100) with the 0.1% glucose supplement. This diluted culture was added to a flat-bottom 96-well microtiter plate, followed by compounds at MIC and 2 × MIC, and further incubation at 37 °C for 24 hours.32 Following the incubation, the planktonic cells were removed from the wells, and the wells were washed with 1 × PBS (pH 7.4).35 The microtiter plate was dried for 10 minutes, and biofilm was stained with 0.1% crystal violet for 10 minutes at 25 °C. The wells were washed with distilled water to remove any excess stain. To solubilize the stain, glacial acetic acid (33%) was added and incubated at room temperature for 30 minutes on an orbital shaker.36 The absorbance of the stained cells was measured at 600 nm with a plate reader, and the percentage of biofilm growth inhibition relative to the untreated control was calculated. The experiment was conducted in triplicate with three biological replicates, and the data were analyzed utilizing GraphPad Prism 8.
2.1.9. Membrane potential perturbation assay. The membrane potential perturbation of S. aureus ATCC 43300 was assessed using the fluorescent probe 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)], which is sensitive to changes in membrane potential.37 To initiate the experiment, S. aureus ATCC 43300 cells were centrifuged at 2400×g for 10 minutes at room temperature to remove the supernatant. Following centrifugation, the cells were resuspended in 1 × PBS and maintained at an OD600 1.0. Subsequently, the S. aureus ATCC 43300 cells were treated with 10 mM EDTA for 5 minutes, after which they were centrifuged again at 2400×g for 10 minutes to eliminate the EDTA. The S. aureus ATCC 43300 cells were again resuspended in resuspension buffer (10 mM glucose, 60 mM NaH2PO4, 130 mM NaCl, 5 mM KCl, 0.5 mM MgCl2, pH 7) and maintained OD600 = 1.0. To label cells with DiOC2(3), a 10 mM stock solution in DMSO was applied, resulting in a final concentration of 30 μM. The DiOC2(3)-loaded cells were transferred to a 96-well microplate.18 When investigating the effects of a compound, the cells and the compound were mixed homogeneously. After a 5-minute incubation at room temperature, the fluorescence of DiOC2(3) was monitored at 5-minute intervals for 30 minutes using excitation wavelength 450 nm and emission wavelength 670 nm.18 This experiment was conducted in triplicate, and the resulting data were analyzed using GraphPad Prism 8.
2.1.10. Membrane permeability assay. The membrane integrity of S. aureus ATCC 43300 was assessed by measuring the uptake of propidium iodide (PI) following exposure to the compounds.19 The bacterial culture was grown in MHB up to the exponential phase, washed twice, and the OD600nm of 0.5 was adjusted in 0.85% saline. Bacterial cells were incubated at 37 °C for an hour with compounds at MIC and 2 × MIC.38 Nisin (20 μM) has been used as a positive control. Next, we added fluorescent dye PI (30 μM) into the suspension culture and incubated it at room temperature for 20 minutes. Following incubation, the culture was centrifuged at 8000 rpm for 10 minutes, and the supernatant was carefully decanted. Subsequently, the pellet was reconstituted in 0.85% saline. The suspension cells were transferred into a black 96-well polystyrene plate. Fluorescence readings were taken at 5-minute intervals over 90 minutes, utilizing an excitation wavelength of 490 nm and an emission wavelength of 635 nm.39 The experiment was evaluated in triplicate. Data were analyzed by GraphPad Prism 8.
2.1.11. Macrophage invasion assay. In this study, the macrophage invasion assay was conducted using the THP-1 cell line, which is a type of monocyte.40 The THP-1 cells were grown in RPMI-1640 media supplemented with 1% (v/v) penicillin-streptomycin (Pen-Strep), and 10% (v/v) fetal bovine serum (FBS).41 To prepare the THP-1 cell line, phorbol 12-myristate 13-acetate (PMA) was added to the monocytes at a 25 ng mL−1 concentration. The cells were seeded at a density of 105 cells per well in 24-well plates and incubated at 37 °C with 5% CO2 for 24 hours.42 The THP-1 cell line was infected with S. aureus ATCC 43300 cells at a concentration of 5 × 108 CFU mL−1. During the infection, compounds were added at the MIC and 2 × MIC. The cells were then incubated for one hour. After the infection period, the infected THP-1 cell line was washed with 1 × PBS (pH 7.4) and treated with gentamicin (50 μg mL−1) for 30 minutes to eliminate any extracellular bacteria. The intracellular bacteria protected within the host cells were released by mild treatment with 0.1% saponin. The viable bacterial colonies were then counted by plating them on Mueller–Hinton agar (MHA). The experiment was conducted in triplicate, and the resulting data were analyzed using GraphPad Prism 8 software.
2.1.12. Hemolysis assay. A hemolysis assay was conducted using fresh rabbit blood.43 The blood was collected and washed with 1 × phosphate buffer saline (1× PBS) (pH 7.4).44 Red Blood Cells (RBCs) were resuspended in 1× PBS at 4% concentration (v/v).45 To perform the assay, 180 μL of the resuspended erythrocytes were added to flat-bottom 96-well plates. Next, 20 μL of compounds were added at MIC and 2× MIC concentrations. Triton X-100 (0.1%) was used as a positive control. The treated erythrocytes were then incubated at 37 °C for one hour. Following the incubation period, the test samples underwent centrifugation at 1000×g for 10 minutes, and the supernatant containing unlysed erythrocytes was carefully dispensed.46 The supernatant containing the released hemoglobin was collected, and its absorbance was measured at 540 nm using a spectrophotometer in a microplate reader.47 The percentage of hemoglobin released was calculated. The experiment was performed in triplicate, and the data were analyzed using GraphPad Prism 8 software.
2.1.13. Cytotoxicity assay. The cytotoxicity of compounds was evaluated using the MTT (3-(4, 5-dimethylthiazol-2yl)-2, 5-diphenyl tetrazolium bromide) assay in the HepG2 cell line.48 A flat-bottom 96-well plate was used, with 20000 HepG2 cells seeded in each well. The plate was then incubated at 37 °C with 5% CO2 for 24 hours.49 DMEM medium was removed, and the cells were treated with compounds at MIC and 2 × MIC, further incubated for 24 hours. Culture media was removed, and HepG2 cells were washed with 1 × PBS (pH 7.4) to eliminate residual media components.50 Next, the HepG2 cells were stained with MTT solution (5 mg mL−1) and incubated for 4 hours. Viable cells produced a purple color due to the formation of formazan crystals. After that, 50 μL of DMF (a solubilizing agent) was added to each well, incubating for 6 hours again, and measuring absorbance at 570 nm.51 The percentage of cell viability was calculated in triplicate.
2.1.14. Animal studies. The animal experiments were carried out at the IMTech Center for Animal Resources and Experimentation (iCARE) facility of the CSIR-Institute of Microbial Technology, adhering to the established ethical guidelines set by the Institutional Animal Ethics Committee (IAEC). The study involving animals followed a protocol that had received approval from the IAEC (approval IAEC/21/08).
2.1.14.1. Thigh infection in a neutropenic mouse. The effectiveness of prochlorperazine dimaleate was assessed in an in vivo model of thigh infection caused by S. aureus ATCC 43300.52 Female BALB/C mice (n = 4; 6–8 weeks old) were made neutropenic by receiving two doses of cyclophosphamide (Sigma-Aldrich) via intraperitoneal injection, with the first dose given 4 days before infection (150 mg kg−1 of body weight) and the second dose given one day prior (100 mg kg−1 of body weight).53 Following neutropenia induction, the mice were infected with S. aureus ATCC 43300 at a concentration of 5 × 105 CFU per mouse, administered intramuscularly into the right thigh muscles. Four hours after infection, each group of mice (n = 4) received an oral dose of prochlorperazine dimaleate and vancomycin 100 mg kg−1 of body weight, respectively. The remaining group of mice served as the vehicle control. After 24 hours of infection, the mice were sacrificed, and the right thigh muscles were dissected and preserved in 1 × PBS (pH 7.4). Subsequently, the muscles were homogenized, and the number of colony-forming units (CFUs) in the muscles was determined by spreading dilutions on MHA plates.
2.1.15. Statistical analysis. The experiments were performed multiple times using biological replicates, and each iteration consistently produced comparable outcomes. The data are depicted as mean values, and their corresponding standard deviations (SD) are provided. To evaluate the variances between groups, two-tailed unpaired t-tests were conducted utilizing GraphPad Prism 8.0.1 software. Statistical significance was assessed using p-values, with significance levels defined as follows:* indicating p ≤ 0.01, ** indicating p ≤ 0.001, *** indicating p ≤ 0.0001, and **** indicating p ≤ 0.0001.
3. Results
3.1. Identification of novel antimicrobial agents against MRSA
We performed molecular docking of FDA-approved drugs with Sortase A protein to identify novel inhibitors of S. aureus by drug-repurposing. This allows an accelerated drug development pathway for clinical use as the safety and pharmacological profiles of these molecules are already established. Additionally, this further strengthens the existing antimicrobial arsenal. Out of the 3040 molecules screened, 60 compounds were selected (Table S2†), based on binding scores and availability of the chemical molecules for further evaluation by in vitro phenotypic screening against MRSA.
We determined the minimum inhibitory concentration and minimum bactericidal concentration (MIC and MBC) of 60 compounds against the S. aureus ATCC 43300 strain. Among them, prochlorperazine dimaleate, loperamide hydrochloride, and chlorpromazine hydrochloride displayed the lowest MIC activity of 32 μg mL−1. Whereas, mequitazine, loperamide hydrochloride, and amlodipine besylate exhibited MICs of 64 μg mL−1. Furthermore, we determined the MBC of these compounds. Triflupromazine hydrochloride, prochlorperazine dimaleate, and chlorpromazine hydrochloride showed the lowest bactericidal activity of 64 μg mL−1. On the other hand, mequitazine, loperamide hydrochloride, and amlodipine besylate demonstrated the lowest bactericidal activity of 128 μg mL−1 (Table 1).
Table 1 The MIC and MBC values of mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride against S. aureus ATCC 43300
| Compounds |
S. aureus ATCC 43300 |
| MIC (μg mL−1) |
MBC (μg mL−1) |
| Mequitazine |
64 |
128 |
| Triflupromazine hydrochloride |
32 |
64 |
| Prochlorperazine dimaleate |
32 |
64 |
| Loperamide hydrochloride |
64 |
128 |
| Amlodipine besylate |
64 |
128 |
| Chlorpromazine hydrochloride |
32 |
64 |
We further explored the binding interaction of these drugs exhibiting potent activity against MRSA with Sortase A enzyme. The binding fingerprints of top most potent molecules are shown in Fig. 1. All the examined drug molecules showed strong binding at their active site, ranging from −5.9 to −7.67 kcal mol−1 (Fig. S1 and Table S3†). The primary amino acids involved in forming hydrogen bonds with the drug molecules include Val108, Asp112, Glu113, Gln114, Trp136, and Arg139.
 |
| | Fig. 1 The 2D- and 3D-visualization of protein–ligand interactions depicting the spatial arrangement of key amino acids involved in hydrogen bonding and hydrophobic interactions with the compounds. (A–B) triflupromazine hydrochloride, (C–D) prochlorperazine dimaleate, and (E–F) chlorpromazine hydrochloride. | |
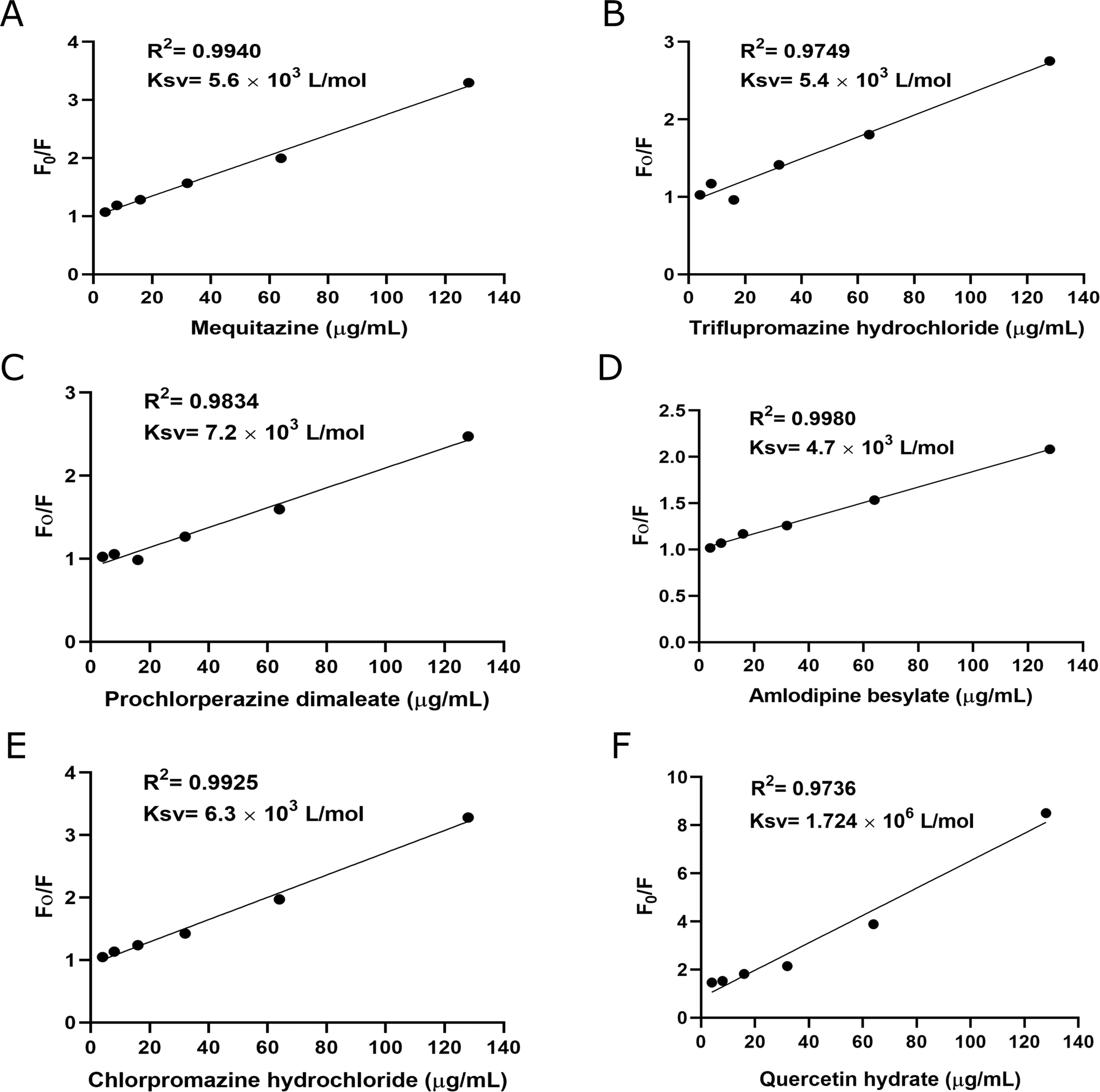
3.2. Fluorescence quenching assay
A fluorescence quenching assay was used to characterize the binding affinity of Sortase A with the compounds. Sortase A exhibits intrinsic fluorescence due to aromatic amino acids, tryptophan, tyrosine, and phenylalanine. Treatment of Sortase A with these compounds resulted in a dose-dependent quenching of its fluorescence intensity, except loperamide hydrochloride Fig. 2. The binding constants (Ksv) for mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, amlodipine besylate, and chlorpromazine hydrochloride with Sortase A were found to be 5.6 × 103 L mol−1, 5.4 × 103 L mol−1, 7.2 × 103 L mol−1, 4.7 × 103 L mol−1, and 6.3 × 103 L mol−1, respectively. Quercetin hydrate has been used as positive control. These results indicate that these compounds significantly interact with Sortase A.
 |
| | Fig. 2 Binding affinities between compounds (A) mequitazine, (B) triflupromazine hydrochloride, (C) prochlorperazine dimaleate, (D) amlodipine besylate, (E) chlorpromazine hydrochloride, and (F) quercetin hydrate) and Sortase A were determined by fluorescence quenching assay. Affinity constant Ksv was calculated by plotting Stern–Volmer Sortase A quenching. Data is presented as mean ± SD (n = 3). | |
3.3. Sortase A activity assay
We determined the percent inhibition of compounds (mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, chlorpromazine hydrochloride) against Sortase A protein. Sortase A activity was evaluated using the fluorescent substrate peptide Abz-LPATG-Dap(DNP). The enzyme cleaves the peptide specifically between threonine (T) and glycine (G), disrupting fluorescence resonance energy transfer (FRET) and leading to an increase in fluorescence intensity. When incubated with Sortase A, all tested compounds, except loperamide hydrochloride, demonstrated inhibitory activity against the enzyme (Table 2).
Table 2 The percent inhibition of mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, amlodipine besylate, chlorpromazine hydrochloride and quercetin hydrate against Sortase A
| Compounds |
S. aureus ATCC 43300 Sortase A |
| Concentration (μg mL−1) |
% inhibition |
| Mequitazine |
128 |
64 |
| Triflupromazine hydrochloride |
128 |
48 |
| Prochlorperazine dimaleate |
128 |
47 |
| Amlodipine besylate |
128 |
57 |
| Chlorpromazine hydrochloride |
128 |
49 |
| Quercetin hydrate |
128 |
73 |
3.4. Time-dependent eradication of MRSA by potent compounds
We evaluated these compounds' effectiveness in killing S. aureus ATCC 43300 strains over time. Mequitazine and amlodipine besylate effectively eliminated the bacteria within 12–24 hours and 4–24 hours, respectively. Similarly, loperamide hydrochloride and chlorpromazine hydrochloride eliminated the bacteria within 8–12 hours, and re-growth of the bacteria was observed after 24 hours in the presence of these compounds. All four of these compounds (mequitazine, amlodipine besylate, loperamide hydrochloride, and chlorpromazine hydrochloride) exhibited a ≥3 log reduction in colony-forming units (CFU) from the initial inoculum. These results indicated that the above four drugs exhibit bactericidal characteristics. On the other hand, triflupromazine hydrochloride and prochlorperazine dimaleate achieved a ∼2 log reduction in CFU within 2 to 4 hours. However, re-growth of the bacteria was observed after 24 hours in the presence of these compounds Fig. 3. This suggests that although these two compounds initially reduced the bacterial population, they were ineffective in completely eradicating S. aureus ATCC 43300. In conclusion, the findings highlight the efficient bactericidal activity of mequitazine, amlodipine besylate, loperamide hydrochloride, and chlorpromazine hydrochloride against S. aureus ATCC 43300. These compounds demonstrated rapid and sustained killing of the bacteria, making them promising candidates for further investigation in combating S. aureus infections.
 |
| | Fig. 3 Time-dependent killing of S. aureus ATCC 43300 at 2 × MIC of compounds ciprofloxacin, mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride. The data points depict the mean ± SD (n = 3). | |
3.5. The identified drugs successfully inhibit the MRSA biofilm
The effectiveness of these compounds in inhibiting biofilm formation by the S. aureus ATCC 43300 strain was investigated. When used at MIC and 2 × MIC, the compounds successfully inhibited biofilm formation in S. aureus ATCC 43300 cells. When the compounds mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride were employed at MIC, they significantly inhibited the biofilm growth by 95%, 19%, 67%, 95%, 40%, and 93%, respectively. Whereas, at 2 × MIC, all these compounds inhibit 94% of the biofilm growth Fig. 4. Since biofilms provide a protective shield for pathogenic bacteria from antibiotic treatments and host immune responses, this association often leads to chronic infections. The present study's compounds can eliminate MRSA biofilm formation and are promising candidates for novel drug development.
 |
| | Fig. 4 The biofilm growth inhibition activity of six compounds, mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride, was evaluated against S. aureus ATCC 43300 at MIC and 2× MIC. The data is presented as mean ± SD (n = 3). Statistical significance is denoted as follows:*, P < 0.1;**, P < 0.01; ***, P < 0.001; and ****, P < 0.0001. | |
3.6. Membrane potential disruption of MRSA by drugs
We determined the membrane potential perturbation of the S. aureus ATCC 43300 strain. Gram-positive bacteria have negatively charged structures on their outer surface, such as teichoic acid and polysaccharide components in the cytoplasmic membrane.54 The negative potential is crucial for several membrane complex systems, bacterial cell division and proliferation, signaling, and antibiotic susceptibility.55–59 All these compounds, except loperamide hydrochloride, successfully disrupted the membrane potential at their MIC and 2 × MIC. While loperamide hydrochloride showed significant reduction only at 2 × MIC (Fig. 5), A proton ionophore, Carbonyl cyanide m-chlorophenyl hydrazone (CCCP), 1 mg mL−1, has been used as a positive control. CCCP can shift the membrane potential to ∼ zero mV (proton Nernst potential in the current scenario). These findings indicate that the compounds can interfere with the membrane potential of S. aureus ATCC 43300, disrupting its normal function.
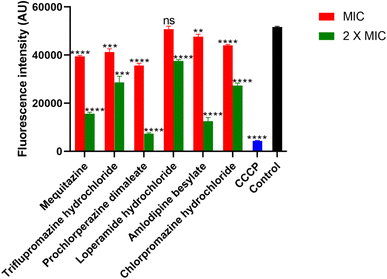 |
| | Fig. 5 Measurement of S. aureus ATCC 43300 membrane potential perturbation after being treated with compounds at MIC and 2 × MIC. CCCP (carbonyl cyanide 3-chlorophenylhydrazone) was the positive control, while untreated cells were the negative control. The data is presented as mean ± SD (n = 3), ns, non-significant; **, P ≤ 0.001; ***, P ≤ 0.0001; and ****, P < 0.0001. | |
3.7. The drugs compromise the MRSA cell membrane integrity
We investigated the membrane disruption capability of compounds by performing a membrane permeability assay. In this scenario, propidium iodide dye (PI) permeates the bacterial cell membrane and binds to double-stranded DNA only when the compounds damage the membrane, which otherwise remains impermeable to the dye.60,61 Nisin was used as a positive control due to its ability to create pores in the cell membrane.62,63 We observed that the fluorescence intensity of PI was increased over time after being treated with these compounds, which can be attributed to membrane permeabilization Fig. 6. Therefore, the results suggest that the compounds have the capability to permeabilize the cell membrane of S. aureus ATCC 43300.
 |
| | Fig. 6 Fluorescence intensity of PI was measured over the period after the addition of MIC and 2 × MIC compounds. Nisin (20 μM) and untreated cells have been used as positive and negative controls, respectively. The data is presented as mean ± SD (n = 3). aP = 0.01; and bP < 0.0001. | |
3.8. Assessment of macrophage invasion by MRSA
The invasion of S. aureus ATCC 43300 in macrophage (THP1) was determined in the presence of drugs under investigation at MIC and 2 × MIC. The results reveal that the compounds significantly reduce S. aureus ATCC 43300 invasion at MIC and 2 × MIC. Prochlorperazine dimaleate successfully reduced MRSA invasion of macrophages at 2 × MIC Fig. 7.
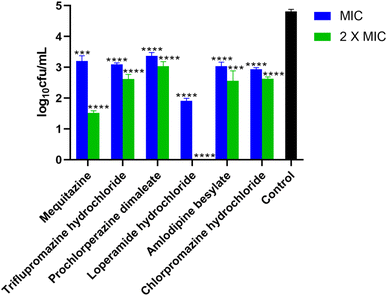 |
| | Fig. 7 Macrophage (THP1) invasion assay of S. aureus ATCC 43300 strains was treated with compounds at MIC, and 2 × MIC. The data is presented as mean ± SD (n = 3). ***, P ≤ 0.0001; and ****, P < 0.0001. | |
3.9. Toxicity assessment in rabbit blood
We examined the hemolytic effects of these compounds in rabbit blood. A hemolytic value below 10% indicates non-hemolytic activity, while a value above 25% indicates hemolytic activity.64 We treated the rabbit blood with the compounds at their MIC and 2 × MIC. Remarkably, we observed that all the compounds demonstrated a percentage of hemolysis lower than 10% Fig. 8. Based on our investigation, the compounds mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride have shown acceptable safety profiles in relation to hemolytic activity when tested on rabbit blood. Therefore, these findings suggest that these compounds exhibit a favorable safety profile and can be potentially used for adjuvant therapy.
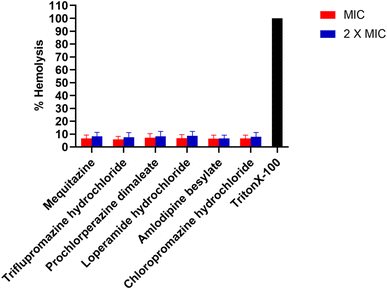 |
| | Fig. 8 Hemolysis of rabbit erythrocytes treated with compounds, mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride. Triton X-100 served as the positive control in the experiment. The data is presented as mean ± SD (n = 3). ****, P <0.0001. | |
3.10. Cytotoxicity assessment in HepG2 cell line
Cytotoxicity in the HepG2 cell line was assessed utilizing the MTT assay. When tested at their respective minimum inhibitory concentration levels, mequitazine, triflupromazine hydrochloride, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride demonstrated a percentage viability of 30%, 63%, 84%, 60%, and 28%. Prochlorperazine dimaleate exhibited a percentage cell viability of 86% at MIC and approximately 50% at 2 × MIC. These results indicate that prochlorperazine dimaleate exhibits low cytotoxicity towards mammalian cells at MIC and 2 × MIC Fig. 9. Hence, these findings suggest that prochlorperazine dimaleate demonstrates a promising safety profile and may have potential applications in biomedicine.
 |
| | Fig. 9 The cytotoxicity of compounds was evaluated in the mammalian HepG2 cell line in an in vitro assay. The data is presented as mean ± SD (n = 3). **, P < 0.01, ***, P < 0.001,and ****, P < 0.0001. | |
3.11. Animal studies
3.11.1. In vivo thigh infection assay in mice. The drug prochlorperazine dimaleate has shown potent activity against MRSA in vitro and is non-cytotoxic in nature.65 Therefore, prochlorperazine dimaleate was selected to evaluate in vivo efficacy in the murine thigh infection model. The experiment was conducted on BALB/C mice to assess the effectiveness of prochlorperazine dimaleate in addressing a thigh infection induced by S. aureus ATCC 43300. The mice were orally administered 100 mg kg−1 of prochlorperazine dimaleate. Vancomycin (100 mg kg−1) was used as a positive control. After treatment with the drugs, the bacterial count of MRSA was reduced by approximately 1 log colony forming unit per milliliter (CFU mL−1) in both vancomycin and prochlorperazine dimaleate treated mice as compared to the control group that received no treatment Fig. 10. Hence, prochlorperazine dimaleate exhibited effective tissue penetration and demonstrated efficacy against MRSA, in contrast to the vehicle group. These findings underscore the therapeutic effectiveness of this chemical scaffold, suggesting its potential as a promising candidate for combating methicillin-resistant S. aureus.
 |
| | Fig. 10 The in vivo efficacy of prochlorperazine dimaleate was evaluated in the thigh of each group of BALB/C mice (n = 4). One BALB/C died during the experiment in the case of prochlorperazine dimaleate treatment group (n = 3). The right thigh of the mice was infected with S. aureus ATCC 43300. After 4 hours post-infection, the mice were orally dosed with vancomycin or prochlorperazine dimaleate. The vehicle group received oral treatment with sunflower oil. After 24 hours post-infection, CFU mL−1 was quantified in triplicate for each mouse. The data were presented as mean ± SD. *, P = < 0.05, and **, P = < 0.01. | |
4. Discussion
This study identifies FDA-approved drugs from diverse chemical classes as potent inhibitors of Staphylococcus aureus Sortase A, offering promising candidates against multidrug-resistant MRSA. By integrating computational screening with functional validation, we demonstrate that repurposed neuropharmacologic agents-including phenothiazines (triflupromazine, prochlorperazine, chlorpromazine), mequitazine, amlodipine, and loperamide-target SrtA's catalytic domain with submicromolar affinity. Their established clinical usage underscores the feasibility of drug repurposing for infectious diseases and raises the prospect of rapid clinical translation. These compounds expand the chemotherapeutic arsenal against multidrug-resistant MRSA by exploiting non-growth-dependent antivirulence mechanisms.
Sortase A anchors virulence-associated surface proteins to the bacterial cell wall, thereby enhancing MRSA's capacity to colonize and persist in host tissues. The identified compounds demonstrated high-affinity binding to the Sortase A active site, as confirmed by molecular docking, fluorescence quenching, and enzyme inhibition assays. All molecules, except loperamide hydrochloride, inhibited Sortase A activity in vitro, supporting their mechanism-based antimicrobial potential. Among the six candidates, mequitazine and amlodipine besylate exhibited bactericidal activity, achieving ≥3 − log reduction in MRSA colony-forming units, while the remaining compounds displayed bacteriostatic effects. This differentiation is critical for tailoring therapeutic strategies, especially in life-threatening infections requiring rapid bacterial clearance.
In addition to antimicrobial activity, these compounds also inhibited MRSA biofilm formation, a major contributor to chronic and device-associated infections. Several hits notably compromised the extracellular matrix, a hallmark of biofilm resilience, suggesting their potential in addressing recalcitrant infections that evade standard antibiotic therapy. Mechanistically, five of the six compounds induced membrane depolarization in MRSA, collapsing the proton gradient critical for ATP generation and nutrient uptake. This disruption likely underpins their antibacterial efficacy. Furthermore, increased membrane permeability was observed, consistent with irreversible damage and bacteriolysis.
The ability of these compounds to reduce intracellular MRSA burden in macrophage (THP-1) models suggests additional utility in eradicating bacterial reservoirs that contribute to persistence and relapse. This is particularly relevant given the emerging recognition of S. aureus's capacity for transient intracellular survival.
Importantly, the compounds exhibited minimal hemolytic activity (<10%), indicating low risk of erythrocyte toxicity. Cytotoxicity profiling in HepG2 cells confirmed favorable safety margins, with prochlorperazine dimaleate demonstrating high cell viability even at 2 × MIC concentrations. In vivo validation in a neutropenic thigh infection model confirmed that prochlorperazine dimaleate significantly reduced MRSA burden in murine muscle tissue, showing efficacy comparable to vancomycin. These findings highlight its translational potential as a repurposed anti-MRSA agent with a favorable safety and pharmacological profile.
Among the phenothiazine scaffold, mequitazine exhibits weaker antimicrobial potency. This decreased activity may be attributed to the presence of bridgehead ring system, which decreases the molecule lipophilicity, and thus may hinder its penetration into the peptidoglycan layer of MRSA (Fig. S2†). Advancing our understanding of the structure-activity relationship (SAR) of SrtA inhibitors and the enzyme structural features will enable the development of potent antibiotic agents against MRSA.
5. Conclusion
In conclusion, docking studies together with experimental evaluation suggest that mequitazine, triflupromazine hydrochloride, prochlorperazine dimaleate, loperamide hydrochloride, amlodipine besylate, and chlorpromazine hydrochloride are potential novel lead molecules that bind with Sortase A and exhibit antibacterial activity against the S. aureus ATCC 43300 strain. The prochloroperazine showed comparative in vivo potency against S. aureus. However, it is essential to acknowledge the constraints in the present study. The antimicrobial compounds evaluated in the present study, apart from prochloperazine dimaleate, displayed toxic potential in mammalian cells. Hence, the identification of toxicophores and rational scaffold optimization may be carried out to enrich the antimicrobial arsenal against MRSA. These molecules have remarkable potential for future medicinal chemistry and represent the lead candidates in adjuvant therapy for fatal MRSA infection.
Data availability
The data supporting this article have been included in the ESI.†
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
The authors express their gratitude to the CSIR-Institute of Microbial Technology for the financial support (projects: OLP0136 and OLP0186). A. K. is grateful to the University Grants Commission (UGC) for awarding the Senior Research Fellowship (SRF). S. C. is thankful to the Council of Scientific and Industrial Research (CSIR) for awarding the Senior Research Fellowship (SRF).
References
- F. D. Lowy, N. Engl. J. Med., 1998, 339, 520–532 CrossRef CAS PubMed.
- F. D. Lowy, N. Engl. J. Med., 2011, 364, 1987–1990 CrossRef CAS PubMed.
- Z. Bai, M. Chen, Q. Lin, Y. Ye, H. Fan, K. Wen, J. Zeng, D. Huang, W. Mo, Y. Lei and Z. Liao, Front. Cell Dev. Biol., 2021, 9, 629681 CrossRef PubMed.
- A. Kali, Pharmacogn. Rev., 2015, 9, 29–34 CrossRef CAS.
- S. S. Boswihi and E. E. Udo, Current Medicine Research and Practice, 2018, 8, 18–24 CrossRef.
- M. S. Smeltzer, Trends Microbiol., 2016, 24, 681–682 CrossRef CAS.
- D. Oliveira, A. Borges and M. Simões, Toxins, 2018, 10, 252 CrossRef PubMed.
- J. M. Patti and M. Höök, Curr. Opin. Cell Biol., 1994, 6, 752–758 CrossRef CAS PubMed.
- Y.-A. Que, P. François, J.-A. Haefliger, J.-M. Entenza, P. Vaudaux and P. Moreillon, Infect. Immun., 2001, 69, 6296–6302 CrossRef CAS PubMed.
- R. Heying, J. van de Gevel, Y.-A. Que, P. Moreillon and H. Beekhuizen, Thromb. Haemostasis, 2007, 97, 617–626 CrossRef CAS.
- T. Spirig, E. M. Weiner and R. T. Clubb, Mol. Microbiol., 2011, 82, 1044–1059 CrossRef CAS PubMed.
- L. A. Marraffini, A. C. DeDent and O. Schneewind, Microbiol. Mol. Biol. Rev., 2006, 70, 192–221 CrossRef CAS PubMed.
- S. Cascioferro, M. Totsika and D. Schillaci, Microb. Pathog., 2014, 77, 105–112 CrossRef CAS.
- S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science (New York, N.Y.), 1999, 285, 760–763 CrossRef CAS PubMed.
- S. Cascioferro, D. Raffa, B. Maggio, M. V. Raimondi, D. Schillaci and G. Daidone, J. Med. Chem., 2015, 58, 9108–9123 CrossRef CAS PubMed.
- K. R. V. Thappeta, L. N. Zhao, C. E. Nge, S. Crasta, C. Y. Leong, V. Ng, Y. Kanagasundaram, H. Fan and S. B. Ng, Int. J. Mol. Sci., 2020, 21, 8601 CrossRef CAS.
- S. Alharthi, S. E. Alavi, P. M. Moyle and Z. M. Ziora, Drug discovery today, 2021, 26, 2164–2172 CrossRef CAS.
- M. Chamlagain, J. Hu, R. V. Sionov and D. Steinberg, Front. Microbiol., 2024, 15, 1333274 CrossRef PubMed.
- R. Boudjemaa, C. Cabriel, F. Dubois-Brissonnet, N. Bourg, G. Dupuis, A. Gruss, S. Lévêque-Fort, R. Briandet, M. P. Fontaine-Aupart and K. Steenkeste, Antimicrob. Agents Chemother., 2018, 62, e00023 CrossRef CAS PubMed.
- G. Wang, Y. Gao, H. Wang, X. Niu and J. Wang, Front. Cell. Infect. Microbiol., 2018, 8, 418 CrossRef CAS PubMed.
- Y. Zong, T. W. Bice, H. Ton-That, O. Schneewind and S. V. Narayana, J. Biol. Chem., 2004, 279, 31383–31389 CrossRef CAS.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 CrossRef PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, J. Chem. Inf. Model., 2006, 46, 665–676 CrossRef CAS PubMed.
- M. R. Bauer and M. D. Mackey, J. Med. Chem., 2019, 62, 3036–3050 CrossRef CAS.
- M. Kuhn, S. Firth-Clark, P. Tosco, A. Mey, M. Mackey and J. Michel, J. Chem. Inf. Model., 2020, 60, 3120–3130 CrossRef CAS PubMed.
- T. Ginex, E. Madruga, A. Martinez and C. Gil, Front. Pharmacol., 2023, 14, 1244317 CrossRef CAS PubMed.
- D. Mu, Y. Luan, L. Wang, Z. Gao, P. Yang, S. Jing, Y. Wang, H. Xiang, T. Wang and D. Wang, Emerging Microbes Infect., 2020, 9, 169–179 CrossRef CAS.
- X. Wang, J. L. Chen, G. Otting and X. C. Su, Sci. Rep., 2018, 8, 16371 CrossRef PubMed.
- J. S. Lewis Ii, Clinical and Laboratory Standards Institute, Performance Standards for Antimicrobial Susceptibility Testing, Clinical and Laboratory Standards Institute, 33rd edn, 2023 Search PubMed.
- M. A. T. Blaskovich, K. A. Hansford, Y. Gong, M. S. Butler, C. Muldoon, J. X. Huang, S. Ramu, A. B. Silva, M. Cheng, A. M. Kavanagh, Z. Ziora, R. Premraj, F. Lindahl, T. A. Bradford, J. C. Lee, T. Karoli, R. Pelingon, D. J. Edwards, M. Amado, A. G. Elliott, W. Phetsang, N. H. Daud, J. E. Deecke, H. E. Sidjabat, S. Ramaologa, J. Zuegg, J. R. Betley, A. P. G. Beevers, R. A. G. Smith, J. A. Roberts, D. L. Paterson and M. A. Cooper, Nat. Commun., 2018, 9, 22 CrossRef PubMed.
- E. V. K. Ledger and A. M. Edwards, mBio, 2023, 14, e0355822 Search PubMed.
- W. Liang, X. F. Liu, J. Huang, D. M. Zhu, J. Li and J. Zhang, BMC Infect. Dis., 2011, 11, 109 CrossRef CAS.
- G. A. McKay, S. Beaulieu, F. F. Arhin, A. Belley, I. Sarmiento, T. Parr, Jr. and G. Moeck, J. Antimicrob. Chemother., 2009, 63, 1191–1199 CrossRef CAS PubMed.
- A. Ogunsile, N. Songnaka, S. Sawatdee, M. Lertcanawanichakul, S. Krobthong, Y. Yingchutrakul, J. Uchiyama and A. Atipairin, PeerJ, 2023, 11, e16143 CrossRef PubMed.
- G. A. McKay, S. Beaulieu, F. F. Arhin, A. Belley, I. Sarmiento, T. Parr, Jr. and G. Moeck, J. Antimicrob. Chemother., 2009, 63, 1191–1199 CrossRef CAS PubMed.
- A. Ogunsile, N. Songnaka, S. Sawatdee, M. Lertcanawanichakul, S. Krobthong, Y. Yingchutrakul, J. Uchiyama and A. Atipairin, PeerJ, 2023, 11, e16143 CrossRef PubMed.
- M. A. Hudson, D. A. Siegele and S. W. Lockless, Antimicrob. Agents Chemother., 2020, 64, e00910 CrossRef CAS PubMed.
- H. Cao, Y. Qiao, X. Liu, T. Lu, T. Cui, F. Meng and P. K. Chu, Acta Biomater., 2013, 9, 5100–5110 CrossRef CAS PubMed.
- A. Masood, N. Ahmed, M. F. M. Razip Wee, A. Patra, E. Mahmoudi and K. S. Siow, Polymers, 2023, 15, 307 CrossRef CAS PubMed.
- N. P. Kalia, P. Mahajan, R. Mehra, A. Nargotra, J. P. Sharma, S. Koul and I. A. Khan, J. Antimicrob. Chemother., 2012, 67, 2401–2408 CrossRef CAS PubMed.
- L. Kovacs, R. A. Davis, T. Ganguly, R. Chammas and J. L. Sutcliffe, Biomed. Pharmacother., 2022, 145, 112469 CrossRef CAS PubMed.
- F. Richter, L. Martin, K. Leer, E. Moek, F. Hausig, J. C. Brendel and A. Traeger, J. Mater. Chem. B, 2020, 8, 5026–5041 RSC.
- F. Tramer, T. Da Ros and S. Passamonti, Methods Mol. Biol., 2012, 926, 203–217 CrossRef CAS.
- C. Xu, R. Song, P. Lu, J. Chen, Y. Zhou, G. Shen, M. Jiang and W. Zhang, Int. J. Nanomed., 2020, 15, 65–80 CrossRef CAS PubMed.
- M. Jangra, M. Kaur, R. Tambat, R. Rana, S. K. Maurya, N. Khatri, A. Ghafur and H. Nandanwar, Antimicrob. Agents Chemother., 2019, 63, 00338 CrossRef PubMed.
- S. Sreelatha, N. Kumar and S. Rajani, Front. Microbiol., 2022, 13, 1085113 CrossRef PubMed.
- S. Afrasiabi, A. Partoazar and N. Chiniforush, Sci. Rep., 2023, 13, 11552 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pognan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef PubMed.
- D. Seleem, B. Benso, J. Noguti, V. Pardi and R. M. Murata, PloS one, 2016, 11, e0157188 CrossRef PubMed.
- A. A. Javia, J. Maharaja Sayajirao Univ. Baroda, 2022 Search PubMed.
- M. V. Abdul Jaleel, Petroleum ether extract of Ophiorrhiza eriantha Wight induces apoptosis in human breast cancer MCF-7 cell line, J. Appl. Pharm. Sci., 2022, 12, 134–142 Search PubMed.
- P. A. Smith, M. F. T. Koehler, H. S. Girgis, D. Yan, Y. Chen, Y. Chen, J. J. Crawford, M. R. Durk, R. I. Higuchi, J. Kang, J. Murray, P. Paraselli, S. Park, W. Phung, J. G. Quinn, T. C. Roberts, L. Rougé, J. B. Schwarz, E. Skippington, J. Wai, M. Xu, Z. Yu, H. Zhang, M. W. Tan and C. E. Heise, Nature, 2018, 561, 189–194 CrossRef CAS PubMed.
- X. Zhang, J. Cao, Y. Pei, J. Zhang and Q. Wang, Oncol. Lett., 2016, 11, 3465–3470 CrossRef CAS PubMed.
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS PubMed.
- H. Strahl and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12281–12286 CrossRef CAS PubMed.
- J. P. Stratford, C. L. A. Edwards, M. J. Ghanshyam, D. Malyshev, M. A. Delise, Y. Hayashi and M. Asally, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9552–9557 CrossRef CAS PubMed.
- A. Prindle, J. Liu, M. Asally, S. Ly, J. Garcia-Ojalvo and G. M. Süel, Nature, 2015, 527, 59–63 CrossRef CAS PubMed.
- H. Zhang, Y. Pan, L. Hu, M. A. Hudson, K. S. Hofstetter, Z. Xu, M. Rong, Z. Wang, B. V. V. Prasad, S. W. Lockless, W. Chiu and M. Zhou, Nat. Commun., 2020, 11, 547 CrossRef CAS PubMed.
- C. Y. Yang, M. Bialecka-Fornal, C. Weatherwax, J. W. Larkin, A. Prindle, J. Liu, J. Garcia-Ojalvo and G. M. Süel, Cell Syst., 2020, 10, 417–423.e413 CrossRef CAS PubMed.
- A. Müller, M. Wenzel, H. Strahl, F. Grein, T. N. V. Saaki, B. Kohl, T. Siersma, J. E. Bandow, H. G. Sahl, T. Schneider and L. W. Hamoen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7077–e7086 CrossRef PubMed.
- S. M. Stocks, Cytom. J. Int. Soc. Anal. Cytol., 2004, 61, 189–195 CrossRef CAS PubMed.
- T. Schneider and H. G. Sahl, Curr. Opin. Invest. Drugs, 2010, 11, 157–164 CAS.
- E. Breukink and B. de Kruijff, Nat. Rev. Drug Discov., 2006, 5, 321–332 CrossRef CAS PubMed.
- I. Greco, N. Molchanova, E. Holmedal, H. Jenssen, B. D. Hummel, J. L. Watts, J. Håkansson, P. R. Hansen and J. Svenson, Sci. Rep., 2020, 10, 13206 CrossRef CAS PubMed.
- W. Wirth, R. Goesswald and W. Vater, Arch. Int. Pharmacodyn. Ther., 1959, 123, 78–114 CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab