
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic adsorption and photocatalytic degradation of perfluorooctanoic acid in aqueous solution by a regenerable biochar-titania nanotube composite†
Yingjie Liu,
Dongjiao Lin,
Yang Yu,
Fei Wang,
Weizhao Yin,
Ying Liu,
Peilin Ye and
Yanyan Gong *
*
Guangdong Key Laboratory of Environmental Pollution and Health, School of Environment and Climate, Jinan University, Guangzhou 511443, China. E-mail: yanyangong@jnu.edu.cn
First published on 7th May 2025
Abstract
Perfluorooctanoic acid (PFOA), a recalcitrant perfluoroalkyl substance, presents escalating challenges for aquatic decontamination due to its extreme persistence and bioaccumulation. A biochar-titania nanotube (TNTs@biochar) combining the advantages of biochar and TNTs was synthesized for the first time via an alkaline hydrothermal approach and explored for the adsorption and photodegradation of PFOA in aqueous solution. Titania nanotubes interacted with biochar to form TNTs@biochar. The optimal composite was obtained at a biochar![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiO2 mass ratio of 1:1 and a calcination temperature of 550 °C. The composite efficiently adsorbed ∼99% of PFOA through hydrophobic and anion–π interactions and hydrogen bonding, concentrating PFOA on photoactive sites. The incorporation of biochar with TNTs enhanced light absorption in the 200–700 nm range, lowered the band gap energy to 3.10 eV, improved the formation rate and separation efficiency of e−–h+ pairs, and enhanced interfacial charge transfer, resulting in promoted photocatalytic activity. The degradation of pre-concentrated PFOA on TNTs@biochar reached up to 99%. The photodegradation also regenerated the composite, allowing for four successive adsorption–photodegradation cycles. Hydroxyl radical and h+-driven oxidation played a paramount part, leading to decarboxylation and C–F bond cleavage. The byproducts of the photodegradation demonstrated lower acute and chronic toxicity compared with PFOA. The composite exhibits synergistic adsorption and photocatalytic activity as well as offers efficiently and economically scalable solutions for PFOA-laden water remediation.
:TiO2 mass ratio of 1:1 and a calcination temperature of 550 °C. The composite efficiently adsorbed ∼99% of PFOA through hydrophobic and anion–π interactions and hydrogen bonding, concentrating PFOA on photoactive sites. The incorporation of biochar with TNTs enhanced light absorption in the 200–700 nm range, lowered the band gap energy to 3.10 eV, improved the formation rate and separation efficiency of e−–h+ pairs, and enhanced interfacial charge transfer, resulting in promoted photocatalytic activity. The degradation of pre-concentrated PFOA on TNTs@biochar reached up to 99%. The photodegradation also regenerated the composite, allowing for four successive adsorption–photodegradation cycles. Hydroxyl radical and h+-driven oxidation played a paramount part, leading to decarboxylation and C–F bond cleavage. The byproducts of the photodegradation demonstrated lower acute and chronic toxicity compared with PFOA. The composite exhibits synergistic adsorption and photocatalytic activity as well as offers efficiently and economically scalable solutions for PFOA-laden water remediation.
1 Introduction
Perfluorinated compounds (PFCs) are persistent organic substances in which all the hydrogens of the carbon chain are substituted by fluorine atoms. Owing to their extraordinary stability and surface active properties, PFCs have been extensively applied in industrial processes and consumer products. Perfluorooctanoic acid (PFOA) is one of the most universally studied long-chain PFC owing to its widespread distribution, environmental persistence, and bioaccumulative potential.1 It is mainly employed in the synthesis and fabrication of fluoropolymers and fluorine-based waterproof/oil-proof/antifouling finishing agents for fabrics and consumer products2 but results in water contamination. For instance, Pelch et al.3 reported a maximum PFOA concentration of 2100 ng L−1 in drinking water samples collected from 16 states in the United States. Stefano et al.4 found that the PFOA concentration in groundwater samples collected from southern Brazil reached up to 249 ng L−1. PFOA has been associated with various detrimental health effects, including liver and kidney disease, reproductive effects, a compromised immune system, and cancers.5 From the view of human health protection, PFOA was listed in Annex A of the Stockholm Convention in May 2019. The water quality limit of PFOA is stipulated to be 80 ng L−1 in drinking water according to the new edition of Standards for drinking water quality in China (GB 5749-2022) (State Administration for Market Regulation and Standardization Administration, 2022).Perfluorooctanoic acid is hardly biodegradable owing to the high C–F bond energy (531.5 kJ mol−1). Various physicochemical technologies have been explored, including adsorption, photocatalytic degradation, reverse osmosis, and nanofiltration. Adsorption is easily applied in practice owing to the low cost and simple operation.6 Adsorbents including activated carbon,7 graphene,8 ion exchange resins,9 mineral materials,10 organic framework materials,11 and biochar12 are capable of removing PFOA. Biochar is a carbon-rich and highly aromatic solid derived from pyrolysis and carbonization of biomass under oxygen-limited or anaerobic conditions, and is one of the most researched adsorbents. It has the advantages of a large specific surface area, high porosity, rich surface functional groups, thermal stability, and economic and environmental benefits.13 Zhang et al.14 revealed that the theoretical maximum adsorption capacity of acid-modified biochar derived from sludge for PFOA was 45.88 mg g−1. Yet, the adsorption does not degrade PFOA and the regeneration of the adsorbent is costly. To overcome these obstacles, composite materials combining both high adsorption capacity and reactivity have been developed to adsorb and catalytically degrade PFOA.
Photocatalytic degradation has been reported as a promising technique to decompose PFOA. Many efforts have been tried to develop photocatalysts, including In2O3,15 ZnO,16 and CeO2 (ref. 17) for photodegradation of PFOA. Titania nanotubes (TNTs) derived from TiO2 have great potential as a photocatalyst due to the large specific surface area, specific tubular structure, great ion exchange capacity, good photoelectron response, and high electron–hole separation efficiency.18 Chen et al.19 synthesized TNTs through a microwave hydrothermal method from TiO2 and found that 44% of PFOA (initial concentration = 50 mg L−1) was degraded by a 254-nm UV light at pH 4 with 0.25 g L−1 TNTs whereas the PFOA photodegradation was only 19% with TiO2. Liu et al.20 conceived an activated charcoal-supported TNTs composite (TNTs@AC) through a one-step hydrothermal approach. The composite absorbed 96.8% of phenanthrene in water within 180 min and completely degraded phenanthrene preconcentrated on the composite within 2 h under UV irradiation (365 nm, 1.42 mW cm−2). Li et al.21 reported a maximum PFOA adsorption capacity of 84.5 mg g−1 via Fe/TNTs@AC and a PFOA degradation percentage of 91.3% under UV light (254 nm, 21 mW cm−2). To the best of our knowledge, no studies have been reported about biochar-TNTs (TNTs@biochar). The preparation of TNTs@biochar is efficient and economically beneficial, and TNTs@biochar is expected to demonstrate the huge adsorption capacity of PFOA and great photocatalytic degradation of the adsorbed PFOA.
To this end, the overall objective of this study was to prepare a regenerable TNTs@biochar and explore its adsorption and photodegradation ability of PFOA. The specific objectives were to: (1) synthesize and characterize TNTs@biochar; (2) investigate the effects of supporting material, biochar:TiO2 mass ratio, and calcination temperature on the sorption and photodegradation of PFOA via TNTs@biochar; (3) investigate the adsorption of TNTs@biochar, the subsequent destruction of PFOA, and reusability of TNTs@biochar; (4) clarify the underlying adsorption and photodegradation mechanisms; and (5) predict the toxicity of photodegradation products of PFOA in comparison with that of PFOA.
2 Materials and methods
2.1 Chemicals and reagents
All chemicals used in this study were of analytical grade or higher. The details are provided in Section S1 of the ESI.†2.2 Synthesis and characterization of TNTs@biochar
Biochar was synthesized following a previously reported approach.22 TNTs@biochar was prepared following a revised hydrothermal method.21 The details were provided in Section S2.† The details of the characterization tests were provided in Section S3.†2.3 Adsorption of PFOA by TNTs@biochar
Batch adsorption kinetic experiments of PFOA by TNTs@biochar (a biochar:TiO2 mass ratio of 1:1) were performed using 40 mL polypropylene (PP) tubes in duplicate at room temperature (23 ± 2 °C). The initial concentrations of PFOA and TNTs@biochar were set at 100 μg L−1 and 0.3 g L−1, respectively. The pH of the mixture was kept constant at 7.0 ± 0.3 using HCl and NaOH. The tubes were then mixed on an end-to-end rotator at 40 rpm. At predetermined times (i.e., 0, 0.083, 0.17, 0.5, 1, 2, 4, 8, 12, and 24 h), duplicate tubes were centrifuged at 4000 rpm for 2 min, and the supernatants were analyzed for PFOA.
Adsorption isotherms of PFOA by TNTs@biochar were carried out following the same procedure as in the kinetic tests. The experimental conditions were: initial PFOA = 0–2.0 mg L−1, TNTs@biochar = 0.3 g L−1, pH = 7.0 ± 0.3, and equilibrium time = 24 h. Following the sorption equilibrium experiments, desorption isotherms were conducted by replacing 95% of each supernatant with an equal volume of water, adjusting the mixture pH to 7.0 ± 0.3, and re-equilibrating the mixture for 24 h.
2.4 Photodegradation of TNTs@biochar-adsorbed PFOA
After establishing an adsorption equilibrium (initial PFOA = 100 μg L−1 and TNTs@biochar = 1.5 g L−1), the mixtures underwent centrifugation at 4000 rpm for 2 min, and 90% of the supernatant was discarded. The residual mixture was introduced into a quartz-covered Petri dish, and diluted with ultrapure water to reach a total volume of 10 mL. The pH was maintained at 7.0. Photodegradation was performed using a 254 nm UV source with an intensity of 30.0 mW cm−2. The reaction temperature was maintained at 23 ± 2 °C. Aliquots were collected at designated time intervals, centrifuged at 4000 rpm for 2 min, and the supernatants were subjected to PFOA quantification. The solids were extracted using methanol (40 mL, 70 °C, 4 h) followed by the analysis of PFOA and its degradation products. M2PFOA was applied as the recovery standard, and 90–103% mass recovery was achieved. To investigate the effects of light intensity on photodegradation, photodegradation experiments were conducted with light intensities varying from 16.4 to 57.0 mW cm−2.Scavenger tests were carried out to explore the roles of h+, ˙O2−, and ˙OH during the photodegradation of PFOA. The following scavengers were applied: KI for h+, ascorbic acid (AA) for ˙O2−, and isopropanol (IP) and tert-butanol (TB) for ˙OH. Electron spin resonance (ESR) assays were carried out to confirm the production of ˙OH and ˙O2− (Section S4†).
The acute and chronic toxicities of PFOA and its photodegradation products to green algae, daphnid, and fish were predicted using USEPA ECOSAR v1.11 software (Section S5†).
The reusability of the photo-regenerated composite was investigated by repeating adsorption and photodegradation experiments in four consecutive cycles.
2.5 Effects of TNTs@biochar dosage and pH
To evaluate the effect of TNTs@biochar dosage, adsorption experiments were conducted with a dosage of TNTs@biochar ranging from 0 to 1.5 g L−1 (initial PFOA = 100 μg L−1 and pH = 7.0 ± 0.3). The photodegradation tests were carried out with PFOA-laden TNTs@biochar at a dosage from 2.4 to 6.0 g L−1. To determine the impact of pH, sorption experiments were conducted with 0.3 g L−1 of TNTs@biochar and 100 μg L−1 of PFOA at pH values from 4.0 to 11.0. The photodegradation tests were carried out with pH varying from 5.0 to 9.0 (pH in the adsorption process = 7.0 ± 0.3, TNTs@biochar = 6.0 g L−1, and initial PFOA = 66.7 μg g−1).2.6 Analytical methods
Perfluorooctanoic acid, M2PFOA, and photodegradation byproducts analysis were performed using an ultra-high-performance liquid chromatography system (UHPLC, Shimadzu Corporation Inc., Kyoto, Japan) equipped with an AB-Sciex 5500 triple quadrupole mass spectrometry system (Applied Biosystems, Foster City, CA, USA). The methods are detailed in Section S6.† The detection limits of PFOA and M2PFOA were 100 ng L−1 and 0.3 ng L−1, respectively.3 Results and discussion
3.1 Characterization of TNTs@biochar
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of biochar, TNTs, and TNTs@biochar are shown in Fig. 1 to obtain a clear perspective on the surface morphology and microscopic structure. As shown in SEM images, biochar demonstrated a cylinder structure with loose pores on its surface (Fig. 1a) and TNTs presented as scattered irregular aggregates (Fig. 1b). Dispersed TNTs with slight agglomeration were observed on the surface of TNTs@biochar (Fig. 1c). As shown in TEM images, biochar showed a multi-layer sheet structure (Fig. 1d). TNTs exhibited uniformly hollow and open-ended tubes with an elongated tubular structure. They have an outer diameter of 10 nm and a length of 100 nm (Fig. 1e). The TEM image of TNTs@biochar revealed that the tubular TNTs presented as an interwoven network spreading all over the surface, indicating co-growth of TNTs and biochar particles.23 Free biochar nanoparticles were also observed in the composite, with particle sizes ranging from 10 to 30 nm (Fig. 1f). AFM was employed to explore the surface topography of TNTs@biochar (Fig. S1†). The average roughness was revealed to be 146 nm, demonstrating an obvious roughness. Enhanced surface roughness leads to a higher surface area, which is beneficial to adsorption and degradation of organic pollutants.24 The elemental compositions (Table S1†) revealed three major elements (C (62.35%), Ti (23.37%), and O (13.64%)) on the surface of TNTs@biochar. The fairly high carbon content indicated that TNTs were not simply coated on the surface of biochar; rather, some biochar nanoparticles were also coated on TNTs.21 | ||
| Fig. 1 SEM images (a–c) and TEM images (d–f) of biochar, TNTs, and TNTs@biochar. | ||
The BET specific surface areas of biochar, TNTs, and TNTs@biochar with various biochar:TiO2 mass ratios are compared in Table S2.† Based on the specific surface areas of biochar (295.18 m2 g−1) and TNTs (63.00 m2 g−1), the theoretical specific surface areas of TNTs@biochar with biochar:TiO2 mass ratios of 2:1, 1:1, and 1:2 were calculated to be 217.79, 179.09, and 140.39 m2 g−1, respectively, if they were combined without distortion. The measured specific surface areas were 339.90, 298.32, and 141.52 m2 g−1, respectively. Since they are higher than the theoretical values, this indicates that TNTs impregnation affected the pore structure or activated the pore opening on the surface of the TNTs@biochar.25 TNTs and biochar intermingled with each other to form TNTs@biochar. On the one hand, the impregnation of TNTs may narrow or block some interior pores of the biochar, favoring the adsorption of PFOA on the outer shell of TNTs@biochar and subsequently resulting in photodegradation. On the other hand, during the preparation of TNTs@biochar, the oxygen-containing functional groups may be removed and the biochar layer may be expanded, resulting in the increase of surface area.26 Moreover, the formation of nanosized biochar particles can lead to an increase in the specific surface area.
The N2 adsorption–desorption isotherms and pore size distributions of biochar, TNTs, and TNTs@biochar with various biochar:TiO2 mass ratios are depicted in Fig. S2a.† Biochar demonstrated a type IV isotherm with a type H2 hysteresis loop, which was primary attributed to complex pore structure and capillary condensation.27 TNTs@biochar with biochar:TiO2 mass ratios of 2:1, 1:1, and 1:2 demonstrated a type II isotherm, reflecting unrestricted monolayer-multilayer adsorption. The type H3–hysteresis loop was related to the slit aperture structured by the accumulation of flake particles.28 The pore size distribution profile of biochar demonstrated a single peak at 3.8 nm (Fig. S2b†). Enlarged pore size distribution was observed for TNTs@biochar, peaking at 0.8, 1.4, 1.8, and 2.8 nm (Fig. S2c†). The preparation of TNTs@biochar may partially block the interior pores of biochar and produced nanosized carbon particles. The average pore diameter was decreased from 4.12 nm for biochar to 0.79 nm for TNTs@biochar.
The characteristic stretching frequencies of biochar, TNTs, and TNTs@biochar before and after PFOA adsorption were compared in Fig. 2a. For biochar, the peaks observed at 3433, 1627, 1554, 1093, and 879 cm−1 corresponded to the stretching vibrations of –OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, CC, C–O, and the aromatic C–H bending, respectively.29 For TNTs, the bands observed at 3433 and 1639 cm−1 were assigned to the stretching and deformation vibrations of the OH groups present at the TNTs surface and from adsorbed water, respectively.30 The peak observed at 2376 cm−1 corresponds to carbon dioxide adsorbed from air.31 The absorption band at 536 cm−1 corresponds to Ti–O vibration.30 Three peaks (3433 cm−1 for –OH, 1627 cm−1 for CO, and 528 cm−1 for the Ti–O functional group) were observed for TNTs@biochar. The Ti–O group shifted from 536 cm−1 for TNTs to 528 cm−1 for TNTs@biochar. The peaks of CC (1554 cm−1), C–O (1093 cm −1), and C–H (879 cm−1) for biochar were not observed for TNTs@biochar. Meanwhile, the peak intensities of –OH and Ti–O for TNTs@biochar were decreased compared with that of TNTs. All these changes were due to the combination of biochar and TNTs. The stretching intensities of –OH and CO for TNTs@biochar before calcination were decreased after calcination (TNTs@biochar), indicating the removal of negatively charged functional groups, which was beneficial to PFOA adsorption.32
O, CC, C–O, and the aromatic C–H bending, respectively.29 For TNTs, the bands observed at 3433 and 1639 cm−1 were assigned to the stretching and deformation vibrations of the OH groups present at the TNTs surface and from adsorbed water, respectively.30 The peak observed at 2376 cm−1 corresponds to carbon dioxide adsorbed from air.31 The absorption band at 536 cm−1 corresponds to Ti–O vibration.30 Three peaks (3433 cm−1 for –OH, 1627 cm−1 for CO, and 528 cm−1 for the Ti–O functional group) were observed for TNTs@biochar. The Ti–O group shifted from 536 cm−1 for TNTs to 528 cm−1 for TNTs@biochar. The peaks of CC (1554 cm−1), C–O (1093 cm −1), and C–H (879 cm−1) for biochar were not observed for TNTs@biochar. Meanwhile, the peak intensities of –OH and Ti–O for TNTs@biochar were decreased compared with that of TNTs. All these changes were due to the combination of biochar and TNTs. The stretching intensities of –OH and CO for TNTs@biochar before calcination were decreased after calcination (TNTs@biochar), indicating the removal of negatively charged functional groups, which was beneficial to PFOA adsorption.32
 | ||
| Fig. 2 (a) FTIR spectra and (b) XRD patterns of biochar, TNTs, and TNTs@biochar before and after calcination and PFOA-laden TNTs@biochar. | ||
Upon PFOA adsorption, similar absorption band characteristics were observed, namely, the –OH group (3433 cm−1), CO group (1627 cm−1), and Ti–O (520 cm−1). Yet, the Ti–O stretching band deviated from 528 cm−1 for TNTs@biochar to 520 cm−1 for PFOA–laden TNTs@biochar, and the peak intensity of the –OH group increased. These changes were attributed to the hydrophobic and anion–π interactions between TNTs@biochar and PFOA21 as well as the hydrogen bond formed between PFOA and the functional groups, e.g., –OH on TNTs@biochar.33
The XRD patterns of biochar, TNTs, TNTs@biochar before calcination, TNTs@biochar, and PFOA-laden TNTs@biochar are displayed in Fig. 2b. For biochar, the peak at 14.1° was assigned to the typical crystalline structure of the cellulose Iα (triclinic).34 For TNTs, the peaks at 25.3°, 37.8°, 48.0°, 53.8°, 55.1°, and 62.7° were assigned to the (101), (004), (200), (105), (211), and (204) crystal planes of anatase, respectively (JCPDS No. 83-2243).35 For TNTs@biochar, the broad peak at 14.1° of biochar disappeared, and the characteristic peaks of anatase at 25.3°, 48.0°, 53.8°, and 55.1° were still observed, confirming that the anatase crystal was covered on the biochar. The characteristic peak for biochar was not observed for TNTs@biochar, probably due to the formation of nanosized biochar particles coated on the TNTs. No diffraction peak of TNTs@biochar was observed before calcination. Evidently, titanate was transformed into anatase after calcination (TNTs@biochar).21 Upon PFOA adsorption, the XRD diffraction peaks remained the same, confirming that TNTs did not contribute to the adsorption of PFOA.
The UV-vis-DRS spectra of TNTs, biochar, and TNTs@biochar are depicted in Fig. 3a. TNTs showed a significant absorption peak at 290 nm, whereas the peak red shifted to 310 nm for TNTs@biochar. TNTs@biochar demonstrated an enhanced absorption in the visible light region compared with TNTs, owing to the presence of biochar. Therefore, the composite can be excited to produce more e−–h+ pairs, resulting in higher photocatalytic activity.36 The optical band gaps of the samples were obtained via fitting the experimentally determined absorption coefficient to the Tauc equation37 (Fig. S3†). TNTs@biochar obtained a narrower band gap energy of 3.10 eV compared with 3.18 eV for the TNTs, which was probably caused by the introduction of biochar and the formation of Ti3+ in the surface disordered layer.38 The band gap energy of TNTs@biochar was narrower than other reported photocatalytic materials for PFOA removal; for instance, 3.34 eV for TiO2 quantum dots loaded sulfonated graphene aerogel39 and 3.12 eV for F-functionalized MOF with in situ-growth TiO2.40 The PL spectra (Fig. 3b) show a significant decrease of PL intensity for TNTs@biochar compared with that of TNTs, indicating lower recombination of e−–h+ on the composite. In the presence of TNTs@biochar, photogenerated electrons can be transferred from TNTs to biochar, enhancing the separation efficiency of e−-h+. On the other hand, biochar can absorb photoluminescence, leading to a quenching effect. Moreover, the maximum PL peak red-shifted from 508 nm to 512 nm, consistent with a wider absorption range of TNTs@biochar and might be attributed to the lower energy gap of TNTs@biochar.41 Fig. 3c compares the CV curves of TNTs and TNTs@biochar. The higher peak intensity of TNTs@biochar meant higher oxidation and reduction potentials, indicating that the introduction of biochar enhanced the oxidation and reduction properties of the composite.42 The CV integrated area for TNTs@biochar was larger than that for TNTs, indicating an enhanced charge storage capability.43 Electron transfer behavior and diffusion characteristics of TNTs and TNTs@biochar were compared by EIS analysis as shown in Nyquist plots (Fig. 3d). The incorporation of biochar increased the curvature of the arc compared with that of TNTs. The charge transfer resistance (Rct) was calculated by fitting with a Randles equivalent circuit (inset in Fig. 3d). The Rct value of TNTs@biochar (451 Ω) was significantly decreased compared with TNTs (2348 Ω). These changes indicated that the combination of biochar and TNTs led to a lower impedance, an enhanced number of e−–h+, and a faster interfacial charge transfer.42,44
 | ||
| Fig. 3 (a) UV-vis-DRS spectra of biochar, TNTs, and TNTs@biochar, and (b) PL spectra, (c) CV curves, and (d) EIS spectroscopy of TNTs and TNTs@biochar. | ||
3.2 Effects of preparation conditions of TNTs@biochar on PFOA sorption and photodegradation
:TiO2 mass ratio was increased from 1:2 to 2:1, the PFOA removal percentage was enhanced from 34.55% to 99.62%. The adsorption of PFOA was facilitated by the biochar phase via hydrophobic and anion–π interactions, as well as hydrogen bonding. With the increasing biochar:TiO2 mass ratio from 1:2 to 2:1, the specific surface area of the composite increased from 141.52 m2 g−1 to 339.90 m2 g−1 (Table S2†). Meanwhile, a decreased biochar:TiO2 mass ratio meant an increased TiO2:biochar mass ratio and some of the biochar sites were blocked due to the patching and/or blocking effects of TNTs, resulting in a decline in PFOA uptake.
 | ||
| Fig. 4 (a) Effects of biochar:TiO2 mass ratios on the adsorption of PFOA by TNTs@biochar. Experimental conditions: initial PFOA = 100 μg L−1, material dosage = 0.3 g L−1, solution volume = 40 mL, reaction time = 24 h, pH = 7.0 ± 0.3, and temperature = 23 ± 2 °C. (b) Effects of biochar:TiO2 mass ratios on photodegradation of pre-adsorbed PFOA by TNTs@biochar. Experimental conditions: initial pre-adsorbed PFOA quantity = 66.7 μg g−1, material dosage = 6 g L−1, solution volume = 10 mL, pH = 7.0 ± 0.3, temperature = 23 ± 2 °C, reaction time = 7 h, UV wavelength = 254 nm, and light intensity = 30.0 mW cm−2. | ||
Following the adsorption equilibrium (initial PFOA = 100 μg L−1 and sorbent = 1.5 g L−1, and 99% PFOA removal was reached (Fig. S6†)), the photodegradation effectiveness of pre-concentrated PFOA on TNTs@biochar at various biochar:TiO2 mass ratios (i.e., 66.7 μg PFOA per g sorbent) was compared after 7 h UV irradiation (Fig. 4b). Evidently, increasing the biochar:TiO2 mass ratio from 1:1 to 2:1, and further to 1:0 resulted in a sharp drop of the photodegradation percentage from 39.03% to 23.82%, and further to 16.49%. The abundant photoactive surface oxygen-containing functional groups such as –COOH,46 semiquinone,47 cyclopentadienyl, and phenoxy48 on the surface of biochar endowed it with a photocatalytic ability to degrade PFOA. TNTs have photoelectric responses under UV light. The combination of biochar with TNTs could affect PFOA photodegradation in the following ways: (1) biochar as a supporting material can decrease the aggregation of TNTs and increase the photocatalytic active sites;49 (2) the high specific surface area of biochar harvested the PFOA and concentrate them close to the active sites of TNTs; (3) the addition of biochar can promote the absorption of light (Fig. 3a); (4) biochar can transform photogenerated electrons to TNTs, facilitating the electron transfer from the active site to the target PFOA;50 (5) biochar has a large electron storage capacity and can serve as an electron scavenger, separating the photogenerated electrons and holes (Fig. 3b); and (6) the formation of a carbon–oxygen–titanium linkage between biochar and TNTs can narrow the band gap and extend the absorption band into the lower energy range (Fig. S3†). However, excess biochar may prevent light from reaching the photoactive sites of the composite.51 Considering both the adsorption capacity and photodegradation ability, the optimal biochar:TiO2 mass ratio was determined to be 1:1.
 | ||
| Fig. 5 (a) Effects of temperature of calcination on adsorption of PFOA by TNTs@biochar. Experimental conditions: initial PFOA = 100 μg L−1, material dosage = 0.3 g L−1, solution volume = 40 mL, reaction time = 24 h, pH = 7.0 ± 0.3, and temperature = 23 ± 2 °C. (b) Effects of the temperature of calcination on photodegradation of pre-adsorbed PFOA by TNTs@biochar. Experimental conditions: initial pre-adsorbed PFOA quantity = 66.7 μg g−1, material dosage = 6 g L−1, solution volume = 10 mL, pH = 7.0 ± 0.3, temperature = 23 ± 2 °C, reaction time = 7 h, UV wavelength = 254 nm, and light intensity = 30.0 mW cm−2. | ||
Following the adsorption equilibrium (initial PFOA = 100 μg L−1 and sorbent = 1.5 g L−1, and 99% PFOA removal was reached (Fig. S7†)), the photodegradation effectiveness of pre-concentrated PFOA on TNTs@biochar (i.e., 66.7 μg PFOA per g sorbent) prepared at various calcination temperatures was compared after 7 h UV irradiation (Fig. 5b). Evidently, improving the calcination temperature from 450 °C to 550 °C enhanced the photodegradation from 17.87% to 39.03%. Calcination temperature can induce phase transformation. Upon calcination at 550 °C, the titanate phase was converted into the anatase phase, which was more photoactive.53 Meanwhile, increasing the calcination temperature formed anatase with a larger crystal size and higher crystallinity.54 The calcination temperature of 550 °C was applied in the subsequent experiments.
3.3 Adsorption and desorption of PFOA by TNTs@biochar
The adsorption of PFOA via TNTs@biochar depicted a rapid initial rate during the first 0.5 h and then slowed down until it reached equilibrium at 8 h (Fig. 6a). Upon equilibrium, 86.05% of PFOA was removed. Readily accessible adsorption sites were occupied first. The pseudo-first-order (eqn (S1)†), pseudo-second-order (eqn (S2)†), external mass transfer (eqn (S3)†), and intraparticle diffusion models (eqn (S4)) (Section S7†) were applied to interpret the data. The pseudo-second-order kinetic model better fitted the sorption kinetic data (R2 = 0.989) than the pseudo-first-order kinetic model (R2 = 0.807) (Table S3†), indicating that chemisorption involving valence forces through sharing or exchanging electrons between TNTs@biochar and PFOA was the potential rate-controlling step during adsorption.55 The equilibrium PFOA uptake calculated from the pseudo-second-order kinetic model (250.58 μg g−1) was comparable to that experimentally obtained (276.23 μg g−1). The external mass transfer model (R2 = 0.972) outperformed the intraparticle diffusion model (R2 = 0.926 for the first stage and R2 = 0.908 for the second stage) (Table S3†), indicating that the adsorption was external mass transfer limited. The kf value was 3.02 × 10−4 cm s−1. | ||
| Fig. 6 (a) PFOA adsorption kinetics on TNTs@biochar; (b) pseudo-first-order, (c) pseudo-second-order, (d) external mass transfer, and (e) intraparticle diffusion models applied for simulating PFOA adsorption kinetics. Experimental conditions: initial PFOA = 100 μg L−1, material dosage = 0.3 g L−1, solution volume = 40 mL, reaction time = 24 h, pH = 7.0 ± 0.3, and temperature = 23 ± 2 °C. | ||
Fig. 7a displays the adsorption isotherms of PFOA by TNTs@biochar. The classical Langmuir (eqn (S9)†), Freundlich (eqn (S10)†), and dual-mode (eqn (S11)†) models (Section S8†) are employed to fit the data. Table S4† presents the best-fitted sorption isotherm model parameters. The Freundlich isotherm model (R2 = 0.982) best simulated the experimental data with a 1/n value of 0.364 ± 0.025, revealing that the adsorption sites of TNTs@biochar were energetically heterogeneous and the PFOA adsorption by TNTs@biochar was favorable.56 The lack of a clear plateau (Fig. 7a) suggested that multi-layer adsorption occurs.
 | ||
| Fig. 7 (a) Adsorption isotherms for PFOA on TNTs@biochar and (b) adsorption and desorption isotherms of PFOA on TNTs@biochar. Experimental conditions: initial PFOA = 0–2.0 mg L−1, material dosage = 0.3 g L−1, solution volume = 40 mL, reaction time = 24 h, pH = 7.0 ± 0.3, and temperature = 23 ± 2 °C. | ||
Fig. S5† compares the zeta potential of TNTs@biochar, biochar, and TNTs. The introduction of biochar suppressed the negative surface potential of TNTs and elevated the pHpzc value from 1.76 for TNTs to 3.12 for TNTs@biochar. Under the experimental pH of 7.0, TNTs@biochar was negatively charged and PFOA existed in an anionic form, with electrostatic repulsion existing between PFOA and TNTs@biochar. TNTs itself did not remove PFOA. The introduction of biochar during the preparation of TNTs@biochar induced adsorption sites for PFOA. FTIR analysis (Fig. 2a) demonstrated that TNTs@biochar effectively removed PFOA from aqueous solutions via hydrophobic interactions, anion–π interactions, and hydrogen bonding.
The desorption isotherm of PFOA from TNTs@biochar is shown in Fig. 7b. Evidently, PFOA adsorption on TNTs@biochar was irreversible during desorption. The hysteresis was probably attributed to the hydrogen bonding between TNTs@biochar and PFOA, which was not susceptible to desorption.57 Moreover, the porous structure of biochar may swell during adsorption and collapse during desorption, contributing to the desorption hysteresis.58 It should be noted that the sorption hysteresis is beneficial to the subsequent photodegradation of pre-concentrated PFOA by TNTs@biochar.
3.4 Photodegradation of pre-concentrated PFOA
 | ||
| Fig. 8 (a) Photodegradation kinetics of pre-adsorbed PFOA on TNTs@biochar; (b) pseudo-first-order and (c) retarded first-order kinetics models applied for fitting degradation data. Experimental conditions: initial pre-adsorbed PFOA quantity = 66.7 μg g−1, material dosage = 6 g L−1, solution volume = 10 mL, pH = 7.0 ± 0.3, temperature = 23 ± 2 °C, reaction time = 8 h, UV wavelength = 254 nm, and light intensity = 30.0 mW cm−2. | ||
Increasing the light intensity significantly enhanced the PFOA photodegradation. As the light intensity was increased from 16.4 to 30 mW cm−2, the PFOA degradation was improved from 7.84% to 38.97%. As the light intensity was further increased to 48.3 and 57.0 mW cm−2, the PFOA degradation was elevated to 80% and 99%, respectively. The enhancement was attributed to a faster generation rate of holes and more production of oxygen-containing radicals (e.g., ˙OH),59 which contributed to the photodegradation of PFOA (Section 3.4.2).
 | ||
| Fig. 9 (a) Effects of various scavengers on photodegradation of PFOA pre-sorbed on TNTs@biochar and (b) electron spin resonance (ESR) spectra of TNTs@biochar. Scavengers experimental conditions: initial PFOA pre-adsorption quantity = 66.7 μg g−1, material dosage = 6 g L−1, solution volume = 10 mL, pH = 7.0 ± 0.3, temperature = 23 ± 2 °C, UV wavelength = 254 nm, light intensity = 30.0 mW cm−2. KI, tert-butanol (TB)/isopropanol (IP)/ascorbic acid (AA) concentration = 2 mM and reaction time = 7 h. | ||
The intermediates and products after 7 h of PFOA photodegradation were analyzed and summarized in Table S6.† Perfluoroheptanoic acid (PFHpA), perfluorohexanoic acid (PFHxA), perfluoropentanoic acid (PFPeA), and perfluorobutyric acid (PFBA) were detected. Accordingly, the photocatalytic degradation process of pre-concentrated PFOA by TNTs@biochar is proposed:
| C7F15COO− + TNTs@biochar → C7F15COO−/TNTs@biochar | (1) |
| TNTs@biochar + hv → e− (CB) + h+ (VB) | (2) |
| h+ (VB) + H2O → ˙OH + H+ | (3) |
| h+ (VB) + OH−→ ˙OH | (4) |
| C7F15COO− + h+ (VB) → C7F15COO˙ | (5) |
| C7F15COO˙ → ˙C7F15 + CO2 | (6) |
| ˙C7F15 + ˙OH → C7F15OH | (7) |
| C7F15OH → C6F13COF + H+ + F− | (8) |
| C6F13COF + H2O → C6F13COO− + 2H+ + F− | (9) |
| C6F13COO− + h+ (VB)/˙OH/H2O → C5F11COO− + 2F− + CO2 + 3H+ → … → F− + CO2 | (10) |
Fig. 10 exhibits the photocatalytic degradation mechanisms. First, PFOA is adsorbed on the TNTs@biochar via hydrophobic and anion–π interactions as well as hydrogen bonding (eqn (1)). Second, electrons (e−, conduction band) and holes (h+, valence band) are produced under UV irradiation (eqn (2)).60 The photogenerated h+ reacts with H2O and OH− to produce ˙OH radicals (eqn (3) and (4)).61 Third, C7F15COO− is oxidized by the photogenerated h+ to produce C7F15COO˙ (eqn (5)). C7F15COO˙ underwent a Kolbe decarboxylation reaction to produce ˙C7F15 and CO2 (eqn (6)). The resultant ˙C7F15 further reacts with ˙OH to generate highly unstable C7F15OH (eqn (7)), which leads to the cleavage of a C–F bond and the release of F− (eqn (8)). The C6F13COF intermediate conveniently reacts with H2O to produce C6F13COO− (eqn (9)).62 The shorter-chain C6F13COO− experiences the same decarboxylation/defluorination cycle and each cycle eliminates one carbon and two fluorine atoms (eqn (10)).63
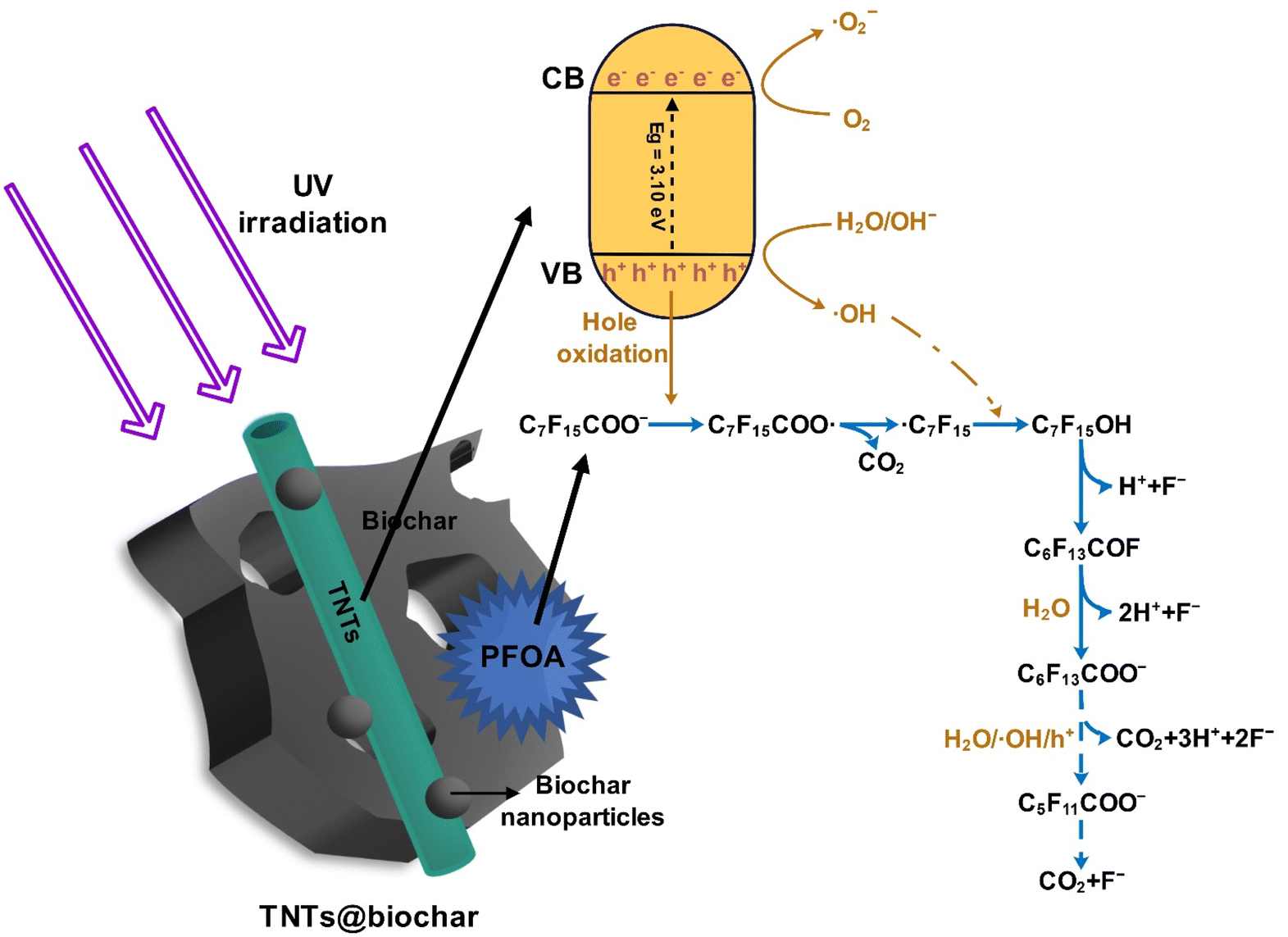 | ||
| Fig. 10 Conceptualized illustration of photocatalytic reaction mechanisms of PFOA by TNTs@biochar. | ||
3.5 Effects of TNTs@biochar dosage and pH on PFOA sorption and photodegradation
Fig. S8† compares the adsorption and photodegradation of PFOA via TNTs@biochar at various dosages. Clearly, increasing the dosage from 0.25 to 0.6 g L−1 significantly enhanced the sorption from 29.02% to >99% (Fig. S8a†). Higher dosage provided more sorption sites for PFOA. As the composite dosage was increased from 2.4 to 4.8 g L−1, the photodegradation of pre-concentrated PFOA declined from 39.13% to 27.61% (Fig. S8b†). The lower degradation was attributed to reduced light penetration caused by an enhanced shading effect of denser suspended composites.64 Yet, the photodegradation reached 38.97% as the dosage was further increased to 6.0 g L−1. Although the light penetration was diminished at higher dosage, the gain in the amount of holes and ˙OH produced with increasing dosage outweighs the loss in light penetration.Fig. S9a† shows effects of pH on PFOA sorption by TNTs@biochar. The sorption rose sharply from 25.26% to 85.92% as the pH decreased from 11.0 to 7.0, and then remained at 99% from pH 4.0 to 6.0. As the pH increases, the electrostatic repulsion between TNTs@biochar and PFOA was enhanced (Fig. S5†). On the other hand, more OH− competed with PFOA anions for adsorption sites. Fig. S9b† compares the photodegradation of PFOA pre-concentrated on TNTs@biochar at various pH values. After 7 h UV irradiation, PFOA degradation decreased from 42.0% at pH 5.0 to 39.0% at pH 7.0 and further to 27.3% at pH 9.0. Higher pH was less favorable for the interaction between the photoactive sites of TNTs@biochar and PFOA. In addition, alkaline conditions hindered the generation of holes and radicals,65 which may restrain the degradation of PFOA (Section 3.4.2). Excessive OH− competed with PFOA for photogenerated holes,21 impeding direct h+-driven oxidation of PFOA (Section 3.4.2).
3.6 Reusability of TNTs@biochar
Biochar-titania nanotube composite was repeatedly applied for four consecutive adsorption–photodegradation cycles. As shown in Fig. S10,† the PFOA adsorption was maintained at >99%, and the PFOA degradation also remained constant. The PFOA-laden TNTs@biochar can be regenerated via photodegradation, and the resultant composite can be reutilized in several cycles. Notably, the composite demonstrated remarkable cost advantages through sustainable biomass utilization, energy efficiency, and material reusability.4 Conclusion
A novel adsorptive photocatalyst TNTs@biochar was synthesized through an alkaline hydrothermal approach using low-cost biochar and TiO2, and investigated for the removal of PFOA in water. The composite exhibited synergistic adsorption and photocatalytic activities and was able to effectively degrade PFOA in water via a concentration and destroy strategy. TNTs@biochar adsorbed PFOA through hydrophobic and anion–π interactions as well as hydrogen bonding. The incorporation of biochar with TNTs improved photocatalytic activity, leading to an efficient degradation of pre-concentrated PFOA under UV irradiation via direct h+-driven oxidation and ˙OH. The generated shorter-chain PFCs demonstrated lower toxicity. Increasing the light intensity enhanced the degradation. A lower pH favored the adsorption and photodegradation of PFOA. TNTs@biochar demonstrated good reusability after four adsorption–photodegradation cycles. Further studies can focus on the sunlight-induced photocatalytic degradation of concentrated PFOA in various water matrices for practical application, the quantification of photodegradation products, and the development toxicity of PFOA and photodegradation products.Data availability
Besides the main manuscript, the data supporting this article have been included as part of the ESI.† Data will be available upon request.Author contributions
Data curation, formal analysis, investigation, methodology, validation, writing – original draft, and writing – review & editing was performed by Yingjie Liu. Methodology, investigation, and data curation were performed by Dongjiao Lin. Methodology, formal analysis, and writing – review & editing were carried out by Yang Yu, Fei Wang, and Weizhao Yin. Formal analysis was performed by Ying Liu and Peilin Ye. Conceptualization, methodology, resources, writing – review & editing, supervision, project administration, and funding acquisition were performed by Yanyan Gong. All the authors read and approved the final manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors gratefully acknowledge the partial financial support from Guangzhou Basic and Applied Basic Research Foundation (2025A04J0991), Natural Science Foundation of China (42177186), and Guangdong Key Laboratory of Environmental Pollution and Health (2016B030301005).References
- Y. Zhang, T. Ding, Z. Huang, H. Liang, S. Du, J. Zhang and H. Li, Chemosphere, 2023, 339, 139537 CrossRef CAS.
- D. Du, Y. Lu, Q. Li, Y. Zhou, T. Cao, H. Cui and G. Han, Sci. Total Environ., 2023, 867, 161507 CrossRef CAS PubMed.
- K. E. Pelch, T. McKnight and A. Reade, Sci. Total Environ., 2023, 876, 162978 CrossRef CAS.
- P. H. P. Stefano, A. Roisenberg, R. D'Anna Acayaba, A. P. Roque, D. R. Bandoria, A. Soares and C. C. Montagner, Environ. Sci. Pollut. Res., 2023, 30, 6159–6169 CrossRef CAS PubMed.
- K. Li, P. Gao, P. Xiang, X. Zhang, X. Cui and L. Q. Ma, Environ. Int., 2017, 99, 43–54 CrossRef CAS PubMed.
- M. T. Baig and A. Kayan, Sep. Sci. Technol., 2023, 58, 862–883 CrossRef CAS.
- J. Yuan, S. Mortazavian, E. Passeport and R. Hofmann, Sci. Total Environ., 2022, 156406 CrossRef CAS.
- S. S. Elanchezhiyan, S. M. Prabhu, J. Han, Y. M. Kim, Y. Yoon and C. M. Park, Appl. Surf. Sci., 2020, 528, 146579 CrossRef CAS.
- S. E. Woodard, J. Berry and B. Newman, Rem, 2017, 27, 19–27 Search PubMed.
- H. Wan, R. Mills, K. Qu, J. C. Hower, M. A. Mottaleb, D. Bhattacharyya and Z. Xu, Chem. Eng. J., 2021, 433, 133271 CrossRef.
- L. Hakim Mohd Azmi, D. R. Williams and B. P. Ladewig, Chemosphere, 2020, 262, 128072 CrossRef.
- D. Zhang, Q. He, M. Wang, W. Zhang and Y. Liang, Environ. Technol., 2021, 42, 1798–1809 CrossRef CAS PubMed.
- J. Wang and S. Wang, J. Cleaner Prod., 2019, 227, 1002–1022 CrossRef CAS.
- Y. Zhang, X. Tan, R. Lu, Y. Tang, H. Qie, Z. Huang, J. Zhao, J. Cui, W. Yang and A. Lin, ACS ES&T Water, 2023, 3, 817–826 Search PubMed.
- Y. Wu, Y. Li, C. Fang and C. Li, ChemCatChem, 2019, 11, 2297–2303 CrossRef CAS.
- D. Wu, X. Li, Y. Tang, P. Lu, W. Chen, X. Xu and L. Li, Chemosphere, 2017, 180, 247–252 CrossRef CAS.
- C. Fu, X. Xu, C. Zheng, X. Liu, D. Zhao and W. Qiu, Environ. Geochem. Health, 2022, 44, 2943–2953 CrossRef CAS PubMed.
- P. Zhang, Z. Mo, Y. Wang, L. Han, C. Zhang, G. Zhao and Z. Li, RSC Adv., 2016, 6, 39348–39355 RSC.
- Y. Chen, S. Lo and J. Kuo, Water Res., 2011, 45, 4131–4140 CrossRef CAS PubMed.
- W. Liu, Z. Cai, X. Zhao, T. Wang, F. Li and D. Zhao, Environ. Sci. Technol., 2016, 50, 11174–11183 CrossRef CAS PubMed.
- F. Li, Z. Wei, K. He, L. Blaney, X. Cheng, T. Xu, W. Liu and D. Zhao, Water Res., 2020, 185, 116219 CrossRef CAS.
- H. Lyu, J. Tang, Y. Huang, L. Gai, E. Y. Zeng, K. Liber and Y. Gong, Chem. Eng. J., 2017, 322, 516–524 CrossRef CAS.
- J.-M. A. Juve, F. Li, Y. Zhu, W. Liu, L. D. Ottosen, D. Zhao and Z. Wei, Chemosphere, 2022, 300, 134495 CrossRef.
- N. Popa and M. Visa, Mater. Chem. Phys., 2021, 258, 123927 CrossRef CAS.
- M. A. Rahman, D. Lamb, M. M. Rahman, M. M. Bahar, P. Sanderson, S. Abbasi, A. F. Bari and R. Naidu, J. Hazard. Mater., 2021, 409, 124488 CrossRef CAS PubMed.
- S. Wang and J. Wang, J. Hazard. Mater., 2021, 418, 126309 CrossRef CAS PubMed.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramırez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- K. Sun, J. Tang, Y. Gong and H. Zhang, Environ. Sci. Pollut. Res., 2015, 22, 16640–16651 CrossRef CAS.
- Y. Juan and Q. qiang, Environ. Sci. Technol., 2009, 43, 3385–3390 CrossRef PubMed.
- V. R. Djokić, A. D. Marinković, O. Ersen, P. S. Uskoković, R. D. Petrović, V. R. Radmilović and D. T. Janaćković, Ceram. Int., 2014, 40, 4009–4018 CrossRef.
- M. Triki, H. Tanazefti and H. Kochkar, J. Colloid Interface Sci., 2017, 493, 77–84 CrossRef CAS.
- X. Lei, Q. Lian, X. Zhang, T. Wang, M. Gee, W. Holmes, S. Jin, S. K. Ponnusamy, D. D. Gang and M. E. Zappi, Chemosphere, 2022, 308, 136379 CrossRef CAS.
- Y. Wu, L. Qi and G. Chen, J. Cleaner Prod., 2022, 340, 130742 CrossRef CAS.
- Z. Ma, Y. Yang, Q. Ma, H. Zhou, X. Luo, X. Liu and S. Wang, J. Anal. Appl. Pyrolysis, 2017, 127, 350–359 CrossRef CAS.
- M. Dhayal, R. Kapoor, P. G. Sistla, R. R. Pandey, S. Kar, K. K. Saini and G. Pande, J. Mater. Sci. Eng., C, 2014, 37, 99–107 CrossRef CAS PubMed.
- S. G. Kumar and K. K. Rao, Appl. Surf. Sci., 2017, 391, 124–148 CrossRef CAS.
- Ł. Haryński, A. Olejnik, K. Grochowska and K. Siuzdak, Opt. Mater., 2022, 127, 112205 CrossRef.
- X. Feng, X. Li, B. Su and J. Ma, Colloids Surf., A, 2022, 648, 129114 CrossRef CAS.
- C. Zhu, J. Xu, S. Song, J. Wang, Y. Li, R. Liu and Y. Shen, Sci. Total Environ., 2020, 698, 134275 CrossRef CAS.
- Z. Kong, L. Lu, C. Zhu, J. Xu, Q. Fang, R. Liu and Y. Shen, Sep. Purif. Technol., 2022, 297, 121449 CrossRef CAS.
- Z. Chen, S. Zhang, X. Wang, N. Mi, M. Zhang, G. Zeng, H. Dong, J. Liu, B. Wu and S. Wei, Environ. Sci. Technol., 2023, 57, 10438–10447 CrossRef CAS PubMed.
- J. Liu, M. Xu, T. Zhang, X. Chu, K. Shi and J. Li, Environ. Sci. Pollut. Res., 2023, 30, 9738–9748 CrossRef CAS PubMed.
- S. Sardar, T. Munawar, F. Mukhtar, M. S. Nadeem, S. A. Khan, M. Koc, S. Manzoor, M. N. Ashiq and F. Iqbal, Mater. Sci. Eng., B, 2023, 288, 116151 CrossRef CAS.
- Y. Ma, Z. Zhao, C. Zhang, Y. Zhang, C. Zhang, J. Li, M. Xu and H. Ma, Chem. Eng. J., 2023, 475, 146303 CrossRef CAS.
- Y. Zhou, M. Xu, D. Huang, L. Xu, M. Yu, Y. Zhu and J. Niu, Sci. Total Environ., 2021, 757, 143719 CrossRef CAS.
- C. Chen and C. T. Jafvert, Carbon, 2011, 49, 5099–5106 CrossRef CAS.
- D. Wu, F. Li, Q. Chen, M. Wu, W. Duan, Q. Zhao, B. Pan and B. Xing, Chemosphere, 2020, 256, 127082 CrossRef CAS PubMed.
- P. Devi, U. Das and A. K. Dalai, Sci. Total Environ., 2016, 571, 643–657 CrossRef CAS.
- P. Lisowski, J. C. Colmenares, O. Mašek, W. Lisowski, D. Lisovytskiy, A. Kamińska and D. Łomot, ACS Sustainable Chem. Eng., 2017, 5, 6274–6287 CrossRef CAS.
- M. Ahmaruzzaman, Mater. Res. Bull., 2021, 140, 111262 CrossRef CAS.
- Y. Zhu, T. Xu and D. Zhao, Sci. Total Environ., 2022, 853, 158573 CrossRef CAS PubMed.
- H. Huang, Z. Niu, R. Shi, J. Tang, L. Lv, J. Wang and Y. Fan, Bioresour. Technol., 2020, 306, 123096 CrossRef CAS.
- K. L. Schulte, P. A. DeSario and K. A. Gray, Appl. Catal., B, 2010, 97, 354–360 CrossRef CAS.
- F. He, F. Ma, J. Li, T. Li and G. Li, Ceram. Int., 2014, 40, 6441–6446 CrossRef CAS.
- R. Xie, L. Zhou, A. E. Smith, C. B. Almquist, J. A. Berberich and N. D. Danielson, J. Hazard. Mater., 2022, 431, 128521 CrossRef CAS PubMed.
- H. M. Jang, S. Yoo, Y.-K. Choi, S. Park and E. Kan, Bioresour. Technol., 2018, 259, 24–31 CrossRef CAS PubMed.
- Y. Jiang, P. Tan, X. Liu and L. Sun, Acc. Chem. Res., 2021, 55, 75–86 CrossRef PubMed.
- R. Hameed, C. Lei and D. Lin, Environ. Sci. Pollut. Res., 2020, 27, 18412–18422 CrossRef CAS.
- S. Liu, E. Véron, S. Lotfi, K. Fischer, A. Schulze and A. I. Schäfer, J. Hazard. Mater., 2023, 447, 130832 CrossRef CAS.
- W. Yu, L. Zhao, F. Chen, H. Zhang and L. H. Guo, J. Phys. Chem. Lett., 2019, 10, 3024–3028 CrossRef CAS.
- H. Ji, P. Du, D. Zhao, S. Li, F. Sun, E. C. Duin and W. Liu, Appl. Catal., B, 2020, 263, 118357 CrossRef CAS.
- N. Duinslaeger and J. Radjenovic, Water Res., 2022, 213, 118148 CrossRef CAS PubMed.
- R. Shan, L. Lu, J. Gu, Y. Zhang, H. Yuan, Y. Chen and B. Luo, Mater. Sci. Semicond. Process., 2020, 114, 105088 CrossRef CAS.
- N. Ahmadpour, M. H. Sayadi and S. Homaeigohar, RSC Adv., 2020, 10, 29808–29820 RSC.
- Y. Zhu, H. Ji, K. He, L. Blaney, T. Xu and D. Zhao, Water Res., 2022, 220, 118650 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01700a |
| This journal is © The Royal Society of Chemistry 2025 |