
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew spirooxindole pyrrolidine/pyrrolizidine analogs: design, synthesis, and evaluation as an anticancer agent†
Narayanasamy Nivethaa,
Janarthanan Venkatesanb,
Dhanashree Muruganb,
Loganathan Rangasamy b,
Shu Pao Wuc and
Sivan Velmathi*a
b,
Shu Pao Wuc and
Sivan Velmathi*a
aOrganic and Polymer Synthesis Laboratory, Department of Chemistry, National Institute of Technology, Tiruchirappalli, 620015, Tamil Nadu, India. E-mail: velmathis@nitt.edu
bDrug Discovery Unit, Centre for Biomaterials, Cellular and Molecular Theragnostic (CBCMT), Vellore Institute of Technology, Vellore, 632014, Tamil Nadu, India
cDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu, 30010, Taiwan, Republic of China
First published on 19th May 2025
Abstract
A novel series of mesitylene-based spirooxindoles were synthesized via the multicomponent [3 + 2] cycloaddition reaction in a greener medium. Spectroscopic techniques such as 1H and 13C NMR and HRMS analysis were carried out for the structural elucidation of all the spirooxindole derivatives. The in vitro cytotoxicity properties of spirooxindole analogs 4/5(a–g) against the human lung (A549) cancer cell line exhibited encouraging outcomes. Of the fourteen synthesized spirooxindole analogs, seven compounds (4a, 4b, 4e, 4g, 5c, 5e, and 5f) showed greater potency towards the A549 lung cancer cell line. The cytotoxicity of the spirooxindole analogs was also investigated against a non-cancerous mouse embryonic fibroblast NIH-3T3 cell line. Compounds 5e and 5f, which exhibited better cytotoxic effect against the cancerous A549 cells (3.48 and 1.2 μM), appeared to be non-cytotoxic against the non-cancerous mouse embryonic fibroblast. Studies using Hoechst and acridine orange/ethidium bromide staining also demonstrated the apoptotic effect of the potent compounds, which decreased cell proliferation.
Introduction
Lung cancer poses a major threat to human health and is one of the most deadly and debilitating malignancies in the world.1,2 Nonetheless, the 5 year survival rate for lung cancer is less than 15%, and the death rate remains high.3 Over the past century, significant advancements in drug innovation regarding oral bioavailability and biological compatibility have been made; however, drug resistance has become a challenge for medicinal chemists. Consequently, pharmacologists and chemists are concentrating on the functional diversity of pharmacological leads,4 nano-formulation, and the advancement of drug delivery systems,5–7 with the objective of addressing current challenges. For this reason, it is crucial to find new therapeutic medications to treat lung cancer.Spiroheterocyclic hybrids with pyrrolidine ring systems have been documented as an important class of potentially bioactive compounds that are frequently constructed via [3 + 2] cycloaddition of azomethine ylide8–10 and possess a wide range of pharmacological activities.11–13 The spirooxindole scaffold is a unique structure made up of two fundamental subunits. Oxindole, the first subunit, has several functions and can interact with biological targets by hydrogen bonding as either donors or acceptors. A carbocyclic or heterocyclic moiety fused with an oxindole ring at the C-3 position makes up the second unit. In addition, hybridizing the spirooxindole nucleus with other moieties has led to the development of new molecules with enhanced anticancer activity profiles. Because of its facile synthesis and easily accessible reagents, azomethine ylide is one of the most useful reactive 1,3-dipoles in the [3 + 2] cycloaddition reaction.14,15 The most practical synthesis approach for functionalized spirooxindoles is the [3 + 2] cycloaddition of azomethine ylides with different dipolarophiles.
Mesitylene and its derivatives have gained huge attention in medicinal chemistry. Although mesitylene itself is not bioactive, its unique chemical structure providing a rigid, hydrophobic and electron-rich aromatic core makes it a valuable scaffold for drug design. The three methyl groups can act as points of substitution, enabling the attachment of bioactive moieties that interact with cancer cell lines. The advantages of mesitylene-based compounds are synthetic flexibility due to three reactive positions, lipophilicity aiding in cell membrane penetration and stability of the aromatic ring under physiological conditions.
Furthermore, the phenyl ring plays a crucial role in anticancer activity due to its chemical stability, planarity and ability to interact with biological targets. The presence and positions of substituents on the phenyl rings can affect how a drug molecule binds to a receptor, influencing its efficacy and selectivity. Substitution on the phenyl ring modifies biological activity like enhanced target selectivity, reduced toxicity and improved solubility and bioavailability. Generally, para-substitution enhances target affinity and creates hydrogen bond interactions with –OH or –NH2 groups whereas meta-substitution has the ability of tuning selectivity. In addition, the presence of –CH3 and –OCH3 groups modifies lipophilicity and membrane permeability. On the other hand, the presence of –Cl, –Br and –F groups increase metabolic stability and binding affinity. As a result, we have chosen mesitaldehyde and substituted acetophenones to form dipolarophiles or chalcones which undergo a cycloaddition reaction with isatin and secondary amino acids to form highly functionalized spirooxindole derivatives of interest.
Many natural spirooxindoles, such as spirotryprostatins A and B, exhibit remarkable anticancer properties.16 Coerulescine, horsfiline, and elacomine are examples of inhibitors of the mammalian cell cycle at the G2/M interphase.17 Among the several chemotherapeutic agents, MI-219,18 MI-888,19 and MI-773 (ref. 20) are some of the synthesized spirooxindole derivatives that are especially pertinent to the present study (Fig. 1). Barakat and colleagues reported a highly functionalized spirooxindole derivative and the p-bromophenyl arm (I) was identified as the most potent towards breast cancer cell lines (IC50 values of 15.49 ± 0.04 μM).21 Acharya's group designed and synthesized pyrrolizidine spirooxindole derivatives in which the compound with a p-chloro substituent (II) produced selective cytotoxicity against leukemia, and colon, prostate and renal cancer cell lines.22 Parasuraman's group developed a library of spirooxindole derivatives and examined their anticancer potential against the A549 cell line.23 The p-bromophenyl substituted spirooxindole derivative (III) showed substantial anticancer activity with IC50 values of 15.49 ± 0.04 μM. Barakat and co-workers reported a series of thiochromene based spirocyclic hybrids and tested them against the MCF-7 and MDA-MB231 breast cancer cell lines, with the compound with a fluoro substituent (IV) displaying remarkable activity in the series.24
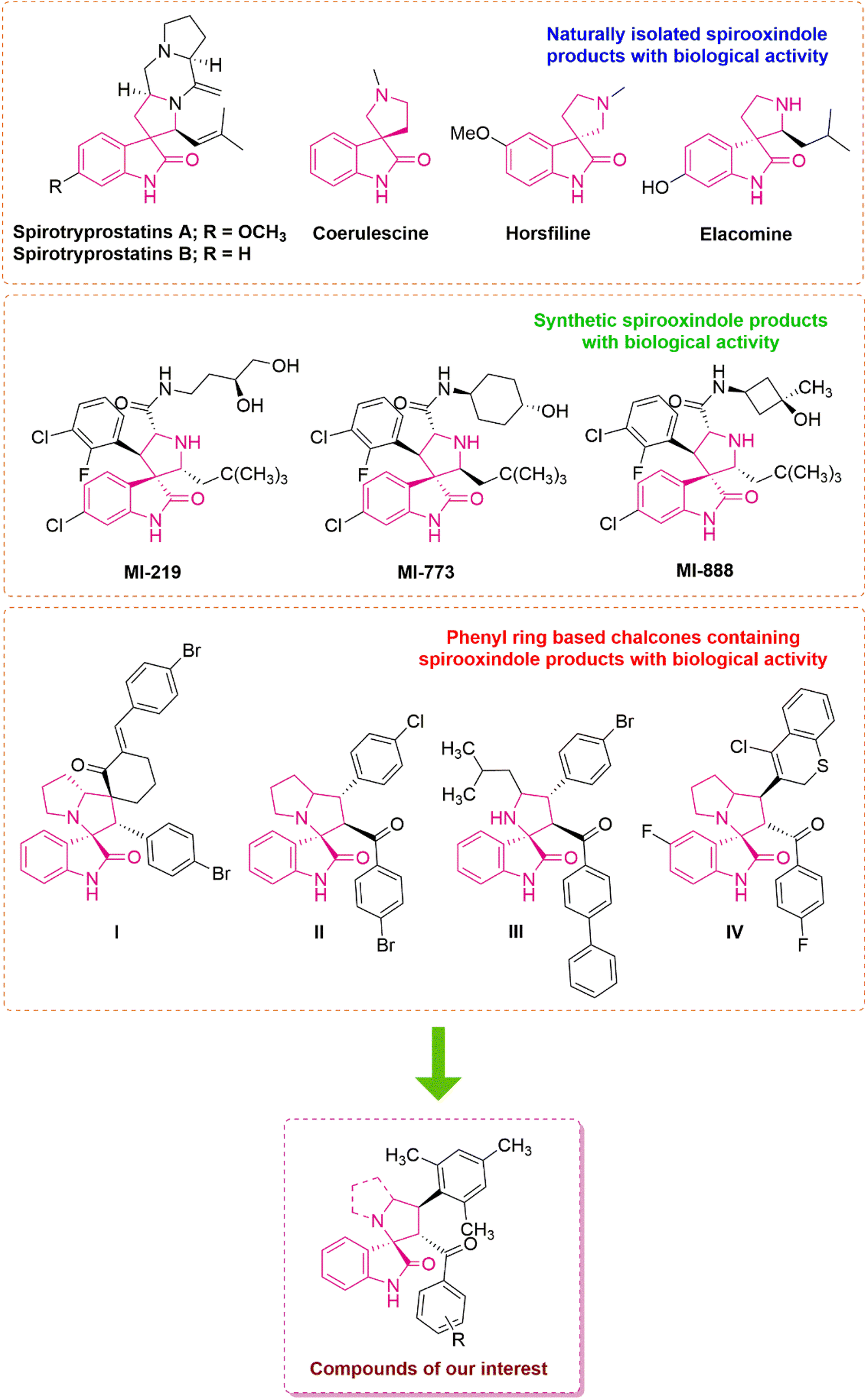 | ||
| Fig. 1 Representative examples of naturally occurring and synthesized biologically active spirooxindole derivatives and rationally designed compounds of interest. | ||
Motivated by these discoveries and as part of our ongoing research, we have rationally designed and synthesized a small combinatorial library of highly functionalized spirooxindoles in this work. Using a spectroscopic examination of the representative derivative, the absolute configuration was determined. Then, all derivatives were subjected to cytotoxicity screening against A549 cells to determine the selectivity using the MTT assay. Compared with cisplatin, the most promising derivatives in terms of potency and selectivity were assessed for their AO/EB and Hoechst staining tests. Additionally, the ability of the compound to reduce oxidative stress was examined by evaluating the intracellular reactive oxygen species of the most active compounds using 2′,7′-dichlorofluorescein diacetate.
Results and discussion
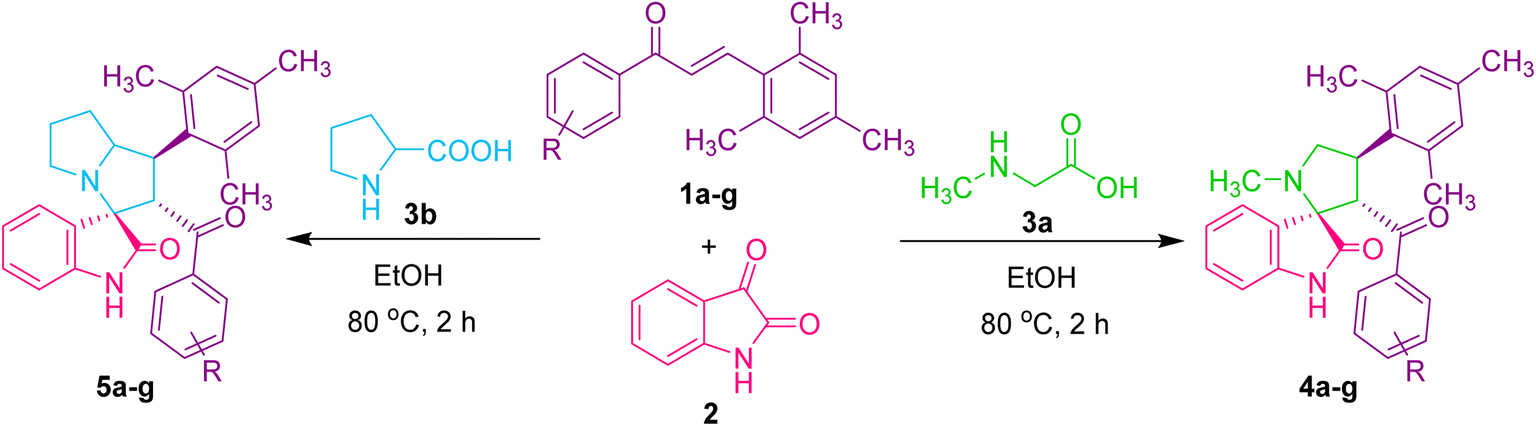
We present a one-pot multicomponent 1,3-dipolar azomethine ylide cycloaddition reaction to form the mesitylene appended novel spirooxindole pyrrolidine derivative to continue our research on the synthesis of novel biologically significant heterocyclic compounds, particularly spirooxindoles.The focus of the current study is 1,3-dipolar cycloaddition of azomethine ylides produced in situ through the decarboxylative condensation of substituted (E)-3-mesityl-1-phenylprop-2-en-1-one 1 to isatin 2 and sarcosine 3a in ethanol, affording novel spirooxindole pyrrolidines 4 (Scheme 1).
 | ||
| Scheme 1 Model reaction. | ||
The reaction was carried out under various conditions, and the outcomes of the reaction are summarized in Table 1. As evidenced from the results, no desired products were observed when the reaction was agitated in water at room temperature and refluxed for 24 h, respectively (Table 1, entries 1 and 2). Several solvents, including CHCl3, CH3CN, DMF, MeOH, EtOH, H2O/MeOH, and H2O/EtOH combinations were used in the process under refluxing conditions to increase the product yields (Table 1, entries 3–9). Protic solvents produced the product in good to outstanding yields, while aprotic solvents produced lesser yields. Remarkably, ethanol was found to be the best solvent that produced a high yield in a short amount of time.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temperature (°C) | Time (h) | Yieldb (%) |
| a Reaction condition: 1a (1.0 mmol), 2 (1.0 mmol) and 3a (1.0 mmol) in solvent 10 mL.b Isolated yield.c RT = room temperature.d NR = no reaction. | ||||
| 1 | H2O | RTc | 24 | NRd |
| 2 | H2O | 100 | 24 | NR |
| 3 | CHCl3 | 63 | 12 | 44 |
| 4 | CH3CN | 84 | 12 | 55 |
| 5 | DMF | 155 | 12 | 50 |
| 6 | MeOH | 67 | 2 | 60 |
| 7 | EtOH | 80 | 2 | 73 |
| 8 | H2O/MeOH | 80 | 2 | 63 |
| 9 | H2O/EtOH | 80 | 2 | 66 |

Under the optimized reaction conditions, a series of 3′-benzoyl-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one derivatives 4(a–g) were synthesized by reacting an equimolar mixture of dipolarophiles 1(a–g), isatin 2 and sarcosine 3 in ethanol under reflux for 2 h. After completion of the reaction (TLC), the reaction mixture was poured into ice-cold water, and the resulting solid was filtered off and purified by column chromatography to obtain pure spirooxindole pyrrolidine derivatives 4(a–g) in 69–88% yields (Table 2). To further investigate the potential of this method, another series of (1′R,2′S,3R)-2′-benzoyl-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one derivatives 5(a–g) were synthesized by reacting an equimolar mixture of substituted dipolarophiles 1(a–g), isatin 2 and L-proline 3b in ethanol under reflux for 2 h (Table 2). It was discovered that dipolarophiles with both electron-donating and electron-withdrawing substituents have good reaction rates. Dipolarophiles containing electron-donating substituents produced products in comparatively lower yields than dipolarophiles with electron-withdrawing substituents, as seen from the results displayed in Table 2. Likewise, superior product yields were noted when substituents were absent in the dipolarophile.
The structure of new spirooxindole pyrrolidine derivatives produced by 1,3-dipolar cycloaddition of azomethine ylide was elucidated with the help of IR, 1H NMR, 13C NMR, and mass data as illustrated for compound 4a. In the IR spectrum, the absorption bands at 3160 cm−1 correspond to the NH group present in product 4a. The absorption bands in 1712 and 1679 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching frequency of ketone and amide groups, respectively. In the 1H NMR spectrum, the singlets at δ 1.96 and 2.62 confirmed the presence of –CH3 protons of mesitylene, respectively. A singlet at δ 2.11 shows the presence of –NCH3 protons of the pyrrolidine ring. A doublet and triplet at δ 3.26 and 3.43 for two protons showed the presence of pyrrolidine ring –CH2 group (H-5). A triplet in the region of δ 4.93 corresponds to –CH protons (H-4 and H-3) of the pyrrolidine ring. The peaks in the range of δ 6.46–7.46 are attributed to 11 aromatic protons. The NH proton of the oxindole ring appeared as a singlet at δ 10.32. In the 13C NMR spectrum, the peaks at δ 20.70 and 21.17 correspond to methyl carbons of the mesitylene ring. The signal at δ 35.15 corresponds to –NCH3 carbon. The peaks in the range of δ 39.37, 57.60, and 58.57 are attributed to C-4, C-5, and C-3 carbons of the pyrrolidine ring. A peak appeared at δ 74.65, confirming the spiro carbon of spirooxindole pyrrolidine in compound 4a. Aromatic carbons resonated in the region of δ 109.73–142.04. The peaks at δ 178.21 and 197.82 confirmed the presence of two carbonyl groups. A distinguishing peak observed at m/z: 425.2269 in the high-resolution mass spectrum corresponds to the [M + H]+ ion of product 4a.
O stretching frequency of ketone and amide groups, respectively. In the 1H NMR spectrum, the singlets at δ 1.96 and 2.62 confirmed the presence of –CH3 protons of mesitylene, respectively. A singlet at δ 2.11 shows the presence of –NCH3 protons of the pyrrolidine ring. A doublet and triplet at δ 3.26 and 3.43 for two protons showed the presence of pyrrolidine ring –CH2 group (H-5). A triplet in the region of δ 4.93 corresponds to –CH protons (H-4 and H-3) of the pyrrolidine ring. The peaks in the range of δ 6.46–7.46 are attributed to 11 aromatic protons. The NH proton of the oxindole ring appeared as a singlet at δ 10.32. In the 13C NMR spectrum, the peaks at δ 20.70 and 21.17 correspond to methyl carbons of the mesitylene ring. The signal at δ 35.15 corresponds to –NCH3 carbon. The peaks in the range of δ 39.37, 57.60, and 58.57 are attributed to C-4, C-5, and C-3 carbons of the pyrrolidine ring. A peak appeared at δ 74.65, confirming the spiro carbon of spirooxindole pyrrolidine in compound 4a. Aromatic carbons resonated in the region of δ 109.73–142.04. The peaks at δ 178.21 and 197.82 confirmed the presence of two carbonyl groups. A distinguishing peak observed at m/z: 425.2269 in the high-resolution mass spectrum corresponds to the [M + H]+ ion of product 4a.
As a representative case, the strong peak in the IR spectrum of compound 5a appeared at 3190 cm−1, corresponding to –NH stretching, and the sharp peak in 1723 and 1678 cm−1 corresponds to CO stretching. In the 1H NMR spectrum, the multiplets in the region of δ 1.57–1.63 and 1.67–1.73 correspond to H-6 protons of the pyrrolizidine ring. The H-7 protons appeared as a multiplet at δ 1.80–1.89. A singlet at δ 2.12 is attributed to –CH3 protons. A multiplet at δ 2.36–2.39 corresponds to H-8 protons of the pyrrolizidine ring. The singlets at δ 2.60 and 2.66 confirmed the presence of –CH3 protons. The multiplet, triplet, and doublet at δ 4.04–4.10, 4.39, and 5.34 correspond to methine protons (H-4, H-3, and H-5) of the pyrrolizidine ring, respectively. The 11 aromatic protons appeared in the range of δ 6.45–7.47. A singlet at δ 10.15 confirmed the presence of the –NH group of product 5a. In the 13C NMR spectrum, the peaks in the region of δ 20.70, 21.27, and 21.86 are attributed to –CH3 carbons of the mesitylene ring. The peaks resonating at δ 28.00, 32.32, 46.88, 47.25, 61.47, and 69.72 confirmed the presence of C-7, C-6, C-4, C-8, C-3, and C-5 carbons of the pyrrolizidine ring of compound 5a. A peak observed at δ 73.83 corresponds to the spiro carbon (C-2). Aromatic carbons appeared between the region of δ 109.99–142.39. The two carbonyl groups resonated at δ 179.54 and 197.81, respectively. A distinguishing peak observed at m/z: 451.2424 in the HRMS for the [M + H]+ ion further confirms product 5a.
A plausible reaction pathway to validate the formation of 3′-benzoyl-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one 4 is depicted in Scheme 2. The reaction of isatin 2 and sarcosine 3a by dehydration and successive decarboxylation furnishes the azomethine ylide or 1,3-dipole. The azomethine ylide probably undergoes cycloaddition with dipolarophile 1a via path A, furnishing 4. The regioselectivity observed in the reaction may be explained by the fact that the electron-rich carbon of the 1,3-dipole prefers to add over the electron-deficient carbon of the α,β-unsaturated moiety of the dipolarophile 1a, which is more encouraging due to the presence of a secondary orbital interaction (SOI)25 which is not in path B. The other possible regioisomer 4′ was not observed in the reaction.
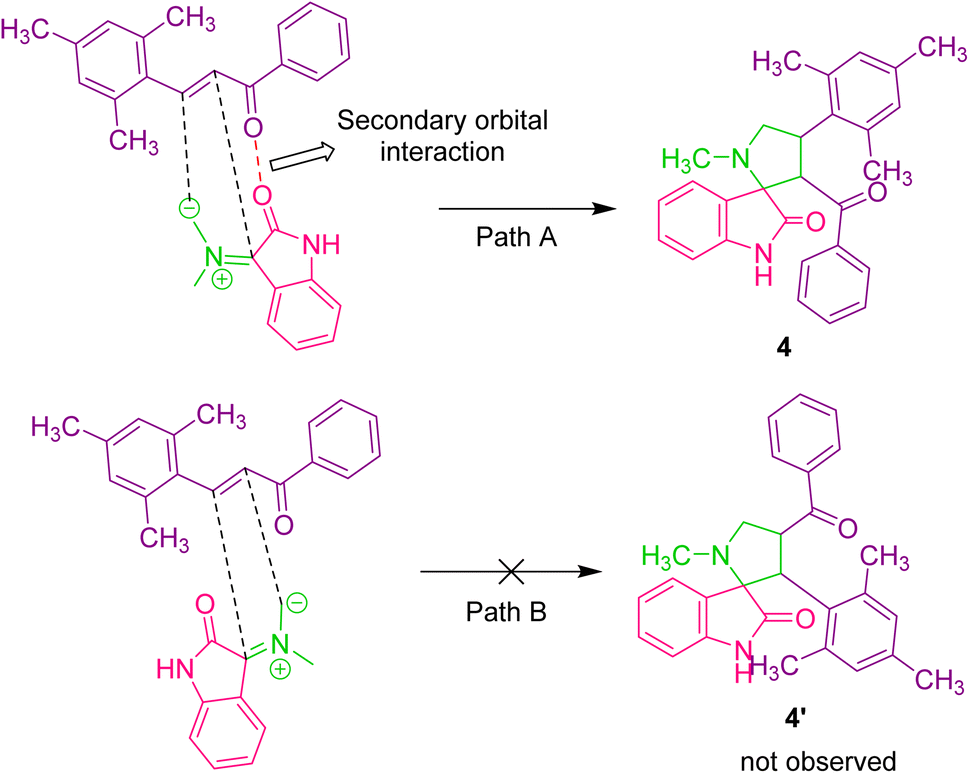 | ||
| Scheme 2 Plausible reaction mechanism for the formation of spirooxindole pyrrolidine. | ||
Cytotoxicity studies
| Compounds | IC50a at 24 h (μM) | IC50a at 48 h (μM) |
|---|---|---|
| a Data are mean% ± SD% of each triplicate. | ||
| 4a | 6.6025 ± 0.108 | 3.814 ± 0.02 |
| 4b | 9.023 ± 2.44 | 3.22 ± 0.17 |
| 4c | >100 | 10.26 ± 2.73 |
| 4d | 33.53 ± 0.34 | 14.49 ± 3.18 |
| 4f | >100 | >100 |
| 4e | 6.929 ± 0.271 | 5.5 ± 0.5 |
| 4g | 9.40 ± 0.611 | 3.5 ± 0.13 |
| 5a | 28.90 ± 12.7 | 10.76 ± 1.9 |
| 5b | 64.39 ± 9.8 | 17.39 ± 1.82 |
| 5c | 12.90 ± 2.28 | 8.242 ± 0.34 |
| 5d | 55.61 ± 0.18 | 13.45 ± 1.27 |
| 5e | 15.65 ± 0.063 | 3.48 ± 1.32 |
| 5f | 16.915 ± 3.92 | 1.2 ± 0.412 |
| 5g | 11.06 ± 8.9 | 7.8 ± 2.3 |
| Cisplatin | >50 | 22.35 ± 0.64 |
Compounds 4a, 4b, 4e, 4g, 5c, 5e, and 5f with better cytotoxic effects identified through MTT assays have been evaluated for the AO/EB and Hoechst staining assay and the plots were displayed in Fig. S5 and S6, ESI.† A higher number of early and late apoptotic cells were observed upon treatment with compounds 5f and 5e in correlation with their lower IC50. The control group of A549 cells (control), in Fig. 2, displays uniform cell shape and strong green fluorescence, whereas the cells treated with 5e, 5f, and cisplatin represent distinct changes as an indication of apoptosis and cell death, such as swelling, cytoplasmic disintegration and rounding of cells within 24 and 48 h, as shown in Fig. 2–5. These studies represent the potency of compounds 5f, 5e, 4a, 4b, 4e, 4g, and 5c as potential cytotoxic agents against A549 cells, which follows the order 5f > 5e > 4a > 4b > 5c > 4e > 4g > cisplatin.
 | ||
| Fig. 2 Acridine orange/ethidium bromide (AO/EB) stained A549 cell line with untreated control (A and B), treated with compounds 4a (C and D), 4b (E and F), 4e (G and H), 4g (I and J), and cisplatin positive control (K and L) at 24 h and 48 h. White arrows indicate normal cells; yellow arrows indicate nuclear disintegration, chromatin condensation, and fragmentation, which are various stages of apoptosis; blue arrows indicate dead cells; and green arrows indicate swelled cells characteristic of necrosis. Scale = 150 μm, magnification = 20×. | ||
 | ||
| Fig. 3 Acridine orange/ethidium bromide (AO/EB) stained A549 cell line with untreated control (A and B), treated with compounds 5c (C and D), 5e (E and F), 5f (G and H), and cisplatin positive control (I and J) at 24 h and 48 h. White arrows indicate normal cells; yellow arrows indicate nuclear disintegration, chromatin condensation, and fragmentation, which are various stages of apoptosis; blue arrows indicate dead cells; and green arrows indicate swelled cells characteristic of necrosis. Scale = 150 μm, magnification = 20×. | ||
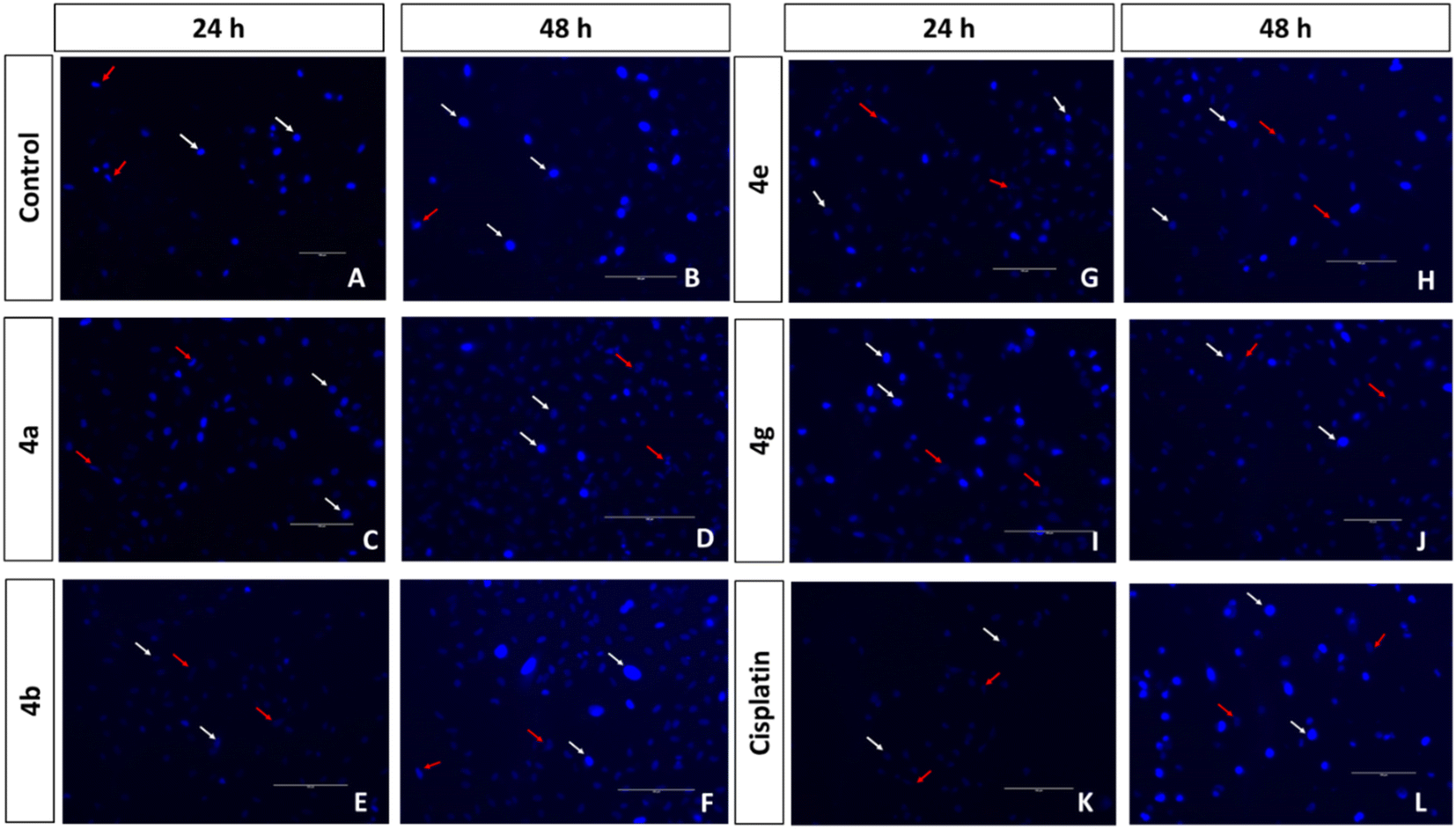 | ||
| Fig. 4 Fluorescent images of Hoechst 33258 stained A549 cell line with untreated control (A and B), treated with compounds 4a (C and D), 4b (E and F), 4e (G and H), 4g (I and J), and cisplatin positive control (K and L) at 24 h and 48 h. White arrows indicate normal cells and red arrows indicate nucleus disintegrated dead cells. Scale = 150 μm, magnification = 20×. | ||
 | ||
| Fig. 5 Fluorescent images of Hoechst 33258 stained A549 cell line with untreated control (A and B), treated with compounds 5c (C and D), 5e (E and F), 5f (G and H), and cisplatin positive control (I and J) at 24 h and 48 h. White arrows indicate normal cells and red arrows indicate nucleus disintegrated dead cells (scale = 150 μm, magnification = 20×). | ||
 | ||
| Fig. 6 ROS produced by 4a, 4b, 4e, 4g, 5c, 5e, 5f, and cisplatin at 0, 2, 4, 6, 8, 12 and 24 h. The concentrations used for the studies are their respective IC50 values. Data are mean% ± SD% of each triplicate. | ||
| Compound | IC50a at 24 h (μM) | IC50a at 48 h (μM) |
|---|---|---|
| a Data are mean% ± SD% of each triplicate. | ||
| 4a | >100 | 25.40 ± 3.74 |
| 4b | >100 | 32.82 ± 5.95 |
| 4c | >100 | >100 |
| 4d | >100 | >100 |
| 4e | >100 | >100 |
| 4f | >100 | >100 |
| 4g | >100 | 75.28 ± 0.94 |
| 5a | >100 | 71.97 ± 4.45 |
| 5b | >100 | 69.91 ± 1.30 |
| 5c | >100 | 72.09 ± 0.95 |
| 5d | >100 | 42.86 ± 1.51 |
| 5e | >100 | >100 |
| 5f | >100 | >100 |
| 5g | >100 | >100 |
| Cisplatin | >50 | 24.40 ± 6.18 |
Conclusions
We synthesized a novel series of mesitylene-based spirooxindole derivatives via a cost-effective one-pot multicomponent cycloaddition reaction and evaluated their anticancer activity against A549 lung cancer cell lines. Seven compounds (4a, 4b, 4e, 4g, 5c, 5e and 5f) exhibited IC50 values of less than 10 μM in A549 cells. Notably, 5f outperformed cisplatin, showing that the compound was potent and selective against the A549 lung cancer cell line. The AO/EB and Hoechst staining assays indicated that compound 5f induces apoptosis. Compound 5f is non-cytotoxic towards non-cancerous mouse embryonic fibroblast NIH-3T3 cells and selective towards A549 cells. Overall, we found that the viability of cancer cells decreased while that of non-cancer cells remained intact during incubation with the synthesized compound. These findings suggested that the synthesized compound may be used as a therapeutic agent for cancer chemotherapy.Materials and methods
General
All chemicals and solvents were purchased from commercial suppliers and used as such. Reaction progresses were monitored by TLC using Merck silica gel 60 F254 plates. Melting points were recorded using the Veego (VMP-DS) apparatus in open capillary tubes. 1H and 13C NMR spectra were run on an Agilent 400 MHz spectrometer in DMSO-d6 as a solvent. Chemical shifts are given in parts per million (ppm), and the coupling constants (J) are given in Hertz (Hz). IR spectra were recorded on a Bruker Alpha II ATR mode spectrometer. An Agilent QTOF G6545 spectrometer was used to obtain high-resolution mass spectra. The A549 and NIH-3T3 cell lines were procured from the National Centre for Cell Science (NCCS), Pune, India. The reagents used for the in vitro experiments such as Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (RM10432), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), cell culture grade DMSO, acridine orange and ethidium bromide were procured from Himedia. Hoechst 33258 was obtained from Thermo Fisher Scientific. 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) was procured from Sigma.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 v/v) to afford the spirooxindole pyrrolidines 4(a–g)/pyrrolizidines 5(a–g) in good yields.
:20 v/v) to afford the spirooxindole pyrrolidines 4(a–g)/pyrrolizidines 5(a–g) in good yields.
3′-Benzoyl-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (4a). Beige solid. Yield: 73%. Mp. 156–158 °C. IR: νmax (KBr, cm−1): 3160 (NH), 1712 (C
O), 1679 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.96 (CH3) (s, 3H), 2.11 (N–CH3) (s, 3H), 2.62 (CH3) (s, 6H), 3.26 (H-5) (d, 1H, J = 8.4 Hz), 3.43 (H-5) (t, 1H, J = 8.2 Hz), 4.93 (H-3 and H-4) (t, 2H, J = 10.0 Hz), 6.46 (d, 1H, J = 7.6 Hz, Ar–H), 6.76 (s, 2H, Ar–H), 6.89–6.93 (m, 1H, Ar–H), 7.01–7.05 (m, 1H, Ar–H), 7.12 (d, 1H, J = 7.6 Hz, Ar–H), 7.27 (t, 2H, J = 7.6 Hz, Ar–H), 7.31–7.33 (m, 2H, Ar–H), 7.42–7.46 (m, 1H, Ar–H), 10.32 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.70 (CH3), 21.17 (CH3), 35.15 (N–CH3), 39.37 (C-4), 57.60 (C-5), 58.57 (C-3), 74.65 (C-2), 109.73, 121.94, 126.32, 126.85, 127.79, 128.82, 129.36, 133.55, 133.88, 135.54, 137.17, 137.43, 142.04, 178.21 (CO), 197.82 (CO). HRMS (ESI) m/z [M + H]+: calcd for C28H29N2O2: 425.2229, found: 425.2269.
3′-(4-Fluorobenzoyl)-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (4b). White solid. Yield: 88%. Mp. 195–197 °C. IR: νmax (KBr, cm−1): 3153 (NH), 1715 (C
O), 1684 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.93 (CH3) (s, 3H), 2.10 (N–CH3) (s, 3H), 2.60 (CH3) (s, 6H), 3.27 (H-5) (d, 1H, J = 7.2 Hz), 3.39 (H-5) (t, 1H, J = 7.6 Hz), 4.87–4.85 (H-3 and H-4) (m, 2H), 6.46 (d, 1H, J = 7.6 Hz, Ar–H), 6.75 (m, 1H, Ar–H), 6.88–6.92 (m, 1H, Ar–H), 7.01–7.05 (m, 1H, Ar–H), 7.09 (t, 3H, J = 8.6 Hz, Ar–H), 7.37–7.34 (m, 3H, Ar–H), 10.32 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.75 (CH3), 21.20 (CH3), 35.24 (N–CH3), 39.14 (C-4), 57.75 (C-5), 58.92 (C-3), 74.77 (C-2), 109.85, 115.94 (d, JCF = 22.0 Hz), 122.06, 126.22, 126.85, 129.52, 130.40, 130.83 (d, JCF = 9.4 Hz), 135.98, 135.63, 137.50, 142.05, 178.18 (CO), 196.56 (CO). HRMS (ESI) m/z [M + H]+: calcd for C28H28FN2O2: 443.2135, found: 443.2169.
3′-(4-Chlorobenzoyl)-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (4c). White solid. Yield: 85%. Mp. 193–195 °C. IR: νmax (KBr, cm−1): 3177 (NH), 1715 (C
O), 1690 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.93 (CH3) (s, 3H), 2.10 (N–CH3) (s, 3H), 2.60 (CH3) (s, 6H), 3.25 (H-5) (d, 1H, J = 8.0 Hz), 3.38 (H-5) (t, 1H, J = 5.4 Hz), 4.86 (H-3 and H-4) (t, 2H, J = 5.6 Hz), 6.46 (d, 1H, J = 7.6 Hz, Ar–H), 6.75 (s, 2H, Ar–H), 6.88–6.92 (m, 1H, Ar–H), 7.01–7.05 (m, 1H, Ar–H), 7.10 (d, 1H, J = 7.2 Hz, Ar–H), 7.26 (d, 2H, J = 8.4 Hz, Ar–H), 7.33 (d, 2H, J = 8.8 Hz, Ar–H), 10.32 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 21.17 (CH3), 21.62 (CH3), 35.64 (N–CH3), 39.52 (C-4), 58.19 (C-5), 59.46 (C-3), 75.15 (C-2), 110.30, 122.51, 126.57, 127.25, 129.42, 130.00, 130.12, 134.36, 136.07, 136.32, 137.92, 138.91, 142.48, 178.53 (CO), 197.52 (CO). HRMS (ESI) m/z [M + H]+: calcd for C28H28ClN2O2: 459.1839, found: 459.1879.
3′-(4-Bromobenzoyl)-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (4d). Yellow solid. Yield: 80%. Mp. 169–170 °C. IR: νmax (KBr, cm−1): 3181 (NH), 1715 (C
O), 1690 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.95 (CH3) (s, 3H), 2.12 (N–CH3) (s, 3H), 2.62 (CH3) (s, 6H), 3.27 (H-5) (d, 1H, J = 7.2 Hz), 3.41 (H-5) (t, 1H, J = 6.0 Hz), 4.87–4.89 (H-3 and H-4) (m, 2H), 6.49 (d, 1H, J = 7.6 Hz, Ar–H), 6.77 (s, 2H, Ar–H), 6.90–6.94 (m, 1H, Ar–H), 7.04–7.08 (m, 1H, Ar–H), 7.11 (d, 1H, J = 7.6 Hz, Ar–H), 7.20 (d, 2H, J = 8.4 Hz, Ar–H), 7.49 (d, 2H, J = 8.4 Hz, Ar–H), 10.33 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.76 (CH3), 21.21 (CH3), 35.23 (N–CH3), 39.10 (C-4), 57.77 (C-5), 59.01 (C-3), 74.73 (C-2), 109.89, 122.10, 126.14, 126.84, 127.73, 129.59, 129.78, 130.41, 131.96, 133.94, 135.66, 136.22, 137.50, 142.06, 178.09 (CO), 197.31 (CO). HRMS (ESI) m/z [M + H]+: calcd for C28H28BrN2O2: 503.1334, found: 503.1371.
4′-Mesityl-1′-methyl-3′-(4-methylbenzoyl)spiro[indoline-3,2′-pyrrolidin]-2-one (4e). Pale yellow solid. Yield: 78%. Mp. 103–105 °C. IR: νmax (KBr, cm−1): 3180 (NH), 1712 (C
O), 1694 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.96 (CH3) (s, 3H), 2.11 (N–CH3) (s, 3H), 2.23 (CH3) (s, 3H), 2.61 (CH3) (s, 6H), 3.28 (H-5) (d, 1H, J = 8.0 Hz), 3.39–3.43 (H-5) (m, 1H), 4.89–4.91 (H-3 and H-4) (m, 2H), 6.49 (d, 1H, J = 7.6 Hz, Ar–H), 6.75 (s, 2H, Ar–H), 6.91 (t, 1H, J = 7.4 Hz, Ar–H), 7.04 (t, 1H, J = 7.2 Hz, Ar–H), 7.08 (d, 2H, J = 8.0 Hz, Ar–H), 7.14 (d, 1H, J = 7.2 Hz, Ar–H), 7.27 (d, 1H, J = 8.0 Hz, Ar–H), 10.33 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.69 (CH3), 21.15 (CH3), 21.48 (CH3), 35.17 (N–CH3), 39.56 (C-4), 57.62 (C-5), 58.30 (C-3), 74.79 (C-2), 109.74, 121.90, 126.31, 126.96, 127.98, 129.32, 129.41, 130.35, 134.00, 134.74, 135.51, 137.40, 142.00, 143.95, 178.19 (CO), 197.06 (CO). HRMS (ESI) m/z [M + H]+: calcd for C29H31N2O2: 439.2386, found: 439.2423.
3′-(3-Chlorobenzoyl)-4′-mesityl-1′-methylspiro[indoline-3,2′-pyrrolidin]-2-one (4f). White solid. Yield: 80%. Mp. 124–126 °C. IR: νmax (KBr, cm−1): 3151 (NH), 1712 (C
O), 1691 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.96 (CH3) (s, 3H), 2.12 (N–CH3) (s, 3H), 2.62 (CH3) (s, 6H), 3.27 (H-5) (d, 1H, J = 7.2 Hz), 3.41 (H-5) (t, 1H, J = 6.0 Hz), 4.85–4.88 (H-3 and H-4) (m, 2H), 6.47 (d, 1H, J = 7.6 Hz, Ar–H), 6.77 (s, 2H, Ar–H), 6.93 (t, 1H, J = 7.4 Hz, Ar–H), 7.06 (t, 1H, J = 7.8 Hz, Ar–H), 7.10–7.12 (m, 2H, Ar–H), 7.26 (d, 1H, Ar–H), 7.32 (t, 1H, J = 7.8 Hz, Ar–H), 7.51 (d, 1H, J = 8.4 Hz, Ar–H), 10.37 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.70 (CH3), 21.16 (CH3), 35.12 (N–CH3), 39.10 (C-4), 57.67 (C-5), 59.26 (C-3), 74.56 (C-2), 109.80, 122.06, 126.11, 126.40, 126.68, 127.29, 129.51, 130.37, 130.76, 133.16, 133.69, 133.76, 135.60, 137.46, 138.93, 142.09, 178.07 (CO), 197.13 (CO). HRMS (ESI) m/z [M + H]+: calcd for C28H28ClN2O2: 459.1839, found: 459.1878.
4′-Mesityl-1′-methyl-3′-(3-methylbenzoyl)spiro[indoline-3,2′-pyrrolidin]-2-one (4g). Off-white solid. Yield: 69%. Mp. 155–157 °C. IR: νmax (KBr, cm−1): 3147 (NH), 1712 (C
O), 1676 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.96 (CH3) (s, 3H), 2.111 (N–CH3) (s, 3H), 2.17 (CH3) (s, 3H), 2.62 (CH3) (s, 6H), 3.28 (H-5) (d, 1H, J = 8.0 Hz), 3.42 (H-5) (t, 1H, J = 8.6 Hz), 4.87–4.92 (H-3 and H-4) (m, 2H), 6.47 (d, 1H, J = 7.6 Hz, Ar–H), 6.76 (s, 2H, Ar–H), 6.90 (t, 1H, J = 7.2 Hz, Ar–H), 7.01–7.05 (m, 1H, Ar–H), 7.09–7.12 (m, 2H, Ar–H), 7.15 (d, 2H, J = 4.8 Hz, Ar–H), 7.24–7.25 (m, 1H, Ar–H), 10.34 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.73 (CH3), 21.23 (CH3), 35.19 (N–CH3), 57.62 (C-5), 58.74 (C-3), 74.72 (C-2), 109.75, 121.96, 125.10, 126.44, 126.87, 128.33, 128.71, 129.34, 130.38, 133.98, 134.14, 137.26, 137.48, 138.12, 142.14, 178.31 (CO), 197.92 (CO). HRMS (ESI) m/z [M + H]+: calcd for C29H31N2O2: 439.2386, found: 439.2423.
2′-Benzoyl-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5a). Beige solid. Yield: 80%. Mp. 170–173 °C. IR: νmax (KBr, cm−1): 3190 (NH), 1723 (C
O), 1678 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.57–1.63 (H-6) (m, 1H), 1.67–1.73 (H-6) (m, 1H), 1.80–1.89 (H-7) (m, 2H), 2.12 (CH3) (s, 3H), 2.36–2.39 (H-8) (m, 2H), 2.60 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.04–4.10 (H-4) (m, 1H), 4.39 (H-3) (t, 1H, J = 10.6 Hz), 5.34 (H-5) (d, 1H, J = 12.0 Hz), 6.45 (d, 1H, J = 7.6 Hz, Ar–H), 6.78 (d, 2H, J = 10.0 Hz, Ar–H), 6.96 (t, 1H, J = 7.2 Hz, Ar–H), 7.05–7.10 (m, 1H, Ar–H), 7.15 (d, 1H, J = 7.6 Hz, Ar–H), 7.26 (d, 4H, J = 4.4 Hz, Ar–H), 7.43–7.47 (m, 1H, Ar–H), 10.15 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.70 (CH3), 21.27 (CH3), 21.86 (CH3), 28.00 (C-7), 32.32 (C-6), 46.88 (C-4), 47.25 (C-8), 61.47 (C-3), 69.72 (C-5), 73.83 (C-2), 109.99, 121.66, 127.55, 127.82, 128.78, 129.53, 129.62, 131.47, 133.49, 135.57, 137.13, 138.18, 142.39, 179.54 (CO), 197.81 (CO). HRMS (ESI) m/z [M + H]+: calcd for C30H31N2O2: 451.2386, found: 451.2424.
2′-(4-Fluorobenzoyl)-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5b). Off-white solid. Yield: 90%. Mp. 186–188 °C. IR: νmax (KBr, cm−1): 3185 (NH), 1715 (C
O), 1678 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.63 (H-6) (m, 1H), 1.67–1.73 (H-6) (m, 1H), 1.78–1.91 (H-7) (m, 1H), 2.12 (CH3) (s, 3H), 2.35–2.39 (H-8) (m, 2H), 2.59 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.03–4.09 (H-4) (m, 1H), 4.37 (H-3) (t, 1H, J = 10.4 Hz), 5.29 (H-5) (d, 1H, J = 11.6 Hz), 6.47 (d, 1H, J = 7.6 Hz, Ar–H), 6.78 (d, 2H, J = 8.8 Hz, Ar–H), 6.97 (t, 1H, J = 7.6 Hz, Ar–H), 7.07–7.15 (m, 4H, Ar–H), 7.29–7.32 (m, 2H, Ar–H), 10.17 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.69 (CH3), 21.26 (CH3), 21.84 (CH3), 28.05 (C-7), 32.38 (C-6), 46.72 (C-4), 47.24 (C-8), 61.80 (C-3), 69.83 (C-5), 73.88 (C-2), 110.04, 115.84 (d, JCF = 14.6 Hz), 121.74, 125.34, 127.48, 129.52, 129.71, 130.76 (d, JCF = 6.4 Hz), 131.48, 132.54, 133.87, 135.59, 136.23, 138.17, 142.35, 164.35, 166.02, 179.51 (CO), 196.51 (CO). HRMS (ESI) m/z [M + H]+: calcd for C30H30FN2O2: 469.2291, found: 469.2331.
2′-(4-Chlorobenzoyl)-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5c). White solid. Yield: 87%. Mp. 164–166 °C. IR: νmax (KBr, cm−1): 3193 (NH), 1720 (C
O), 1681 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.63 (H-6) (m, 1H), 1.66–1.73 (H-6) (m, 1H), 1.81–1.89 (H-7) (m, 2H), 2.12 (CH3) (s, 3H), 2.34–2.39 (H-8) (m, 2H), 2.59 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.03–4.09 (H-4) (m, 1H), 4.36 (H-3) (t, 1H, J = 10.4 Hz), 5.28 (H-5) (d, 1H, J = 11.6 Hz), 6.48 (d, 1H, J = 7.6 Hz, Ar–H), 6.78 (d, 2H, J = 8.4 Hz, Ar–H), 6.95–6.99 (m, 1H, Ar–H), 7.07–7.12 (m, 1H, Ar–H), 7.14 (d, 1H, J = 7.2 Hz, Ar–H), 7.21 (d, 2H, J = 8.8 Hz, Ar–H), 7.34 (d, 2H, J = 8.8 Hz, Ar–H), 10.18 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.70 (CH3), 21.27 (CH3), 21.84 (CH3), 28.05 (C-7), 32.39 (C-6), 46.65 (C-4), 47.22 (C-8), 61.92 (C-3), 69.87 (C-5), 73.84 (C-2), 110.08, 121.78, 125.29, 127.46, 128.91, 129.53, 129.63, 129.77, 131.48, 132.50, 135.62, 135.85, 136.23, 138.16, 138.36, 142.36, 179.43 (CO), 197.05 (CO). HRMS (ESI) m/z [M + H]+: calcd for C30H30ClN2O2: 485.1996, found: 485.2026.
2′-(4-Bromobenzoyl)-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5d). Yellow solid. Yield: 83%. Mp. 178–180 °C. IR: νmax (KBr, cm−1): 3193 (NH), 1719 (C
O), 1682 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.62 (H-6) (m, 1H), 1.66–1.73 (H-6) (m, 1H), 1.81–1.89 (H-7) (m, 2H), 2.12 (CH3) (s, 3H), 2.34–2.39 (H-8) (m, 2H), 2.59 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.03–4.09 (H-4) (m, 1H), 4.36 (H-3) (t, 1H, J = 10.4 Hz), 5.28 (H-5) (d, 1H, J = 11.6 Hz), 6.48 (d, 1H, J = 7.6 Hz, Ar–H), 6.78 (d, 2H, J = 8.8 Hz, Ar–H), 6.97 (t, 1H, J = 7.4 Hz, Ar–H), 7.09 (d, 1H, J = 7.6 Hz, Ar–H), 7.13 (d, 3H, J = 8.8 Hz, Ar–H), 7.49 (d, 2H, J = 8.4 Hz, Ar–H), 10.19 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.70 (CH3), 21.27 (CH3), 21.84 (CH3), 28.05 (C-7), 32.38 (C-6), 46.65 (C-4), 47.22 (C-8), 61.87 (C-3), 69.86 (C-5), 73.83 (C-2), 110.08, 121.78, 125.28, 127.46, 127.58, 129.53, 129.72, 129.77, 131.48, 131.85, 132.49, 135.62, 136.17, 136.23, 138.16, 142.36, 179.41 (CO), 197.24 (CO). HRMS (ESI) m/z [M + H]+: calcd for C30H30BrN2O2: 529.1491, found: 529.1527.
1′-Mesityl-2′-(4-methylbenzoyl)-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5e). Off-white solid. Yield: 80%. Mp. 160–162 °C. IR: νmax (KBr, cm−1): 3188 (NH), 1720 (C
O), 1676 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.63 (H-6) (m, 1H), 1.67–1.71 (H-6) (m, 1H), 1.79–1.88 (H-7) (m, 2H), 2.11 (CH3) (s, 3H), 2.24 (CH3) (s, 3H), 2.35–2.38 (H-8) (m, 2H), 2.59 (CH3) (s, 3H), 2.65 (CH3) (s, 3H), 4.04–4.09 (H-4) (m, 1H), 4.39 (H-3) (t, 1H, J = 10.6 Hz), 5.32 (H-5) (d, 1H, J = 11.6 Hz), 6.48 (d, 1H, J = 7.6 Hz, Ar–H), 6.77 (d, 2H, J = 10.4 Hz, Ar–H), 6.94–6.98 (m, 1H, Ar–H), 7.05–7.09 (m, 3H, Ar–H), 7.16 (d, 1H, J = 7.2 Hz, Ar–H), 7.22 (d, 2H, J = 8.4 Hz, Ar–H), 10.17 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.69 (CH3), 21.28 (CH3), 21.49 (CH3), 21.85 (CH3), 28.02 (C-7), 32.30 (C-6), 46.98 (C-4), 47.23 (C-8), 61.10 (C-3), 69.65 (C-5), 73.92 (C-2), 109.99, 121.62, 125.49, 127.63, 128.02, 129.39, 129.52, 129.55, 131.45, 132.61, 134.66, 135.54, 136.20, 138.17, 142.35, 143.88, 179.56 (CO), 197.00 (CO). HRMS (ESI) m/z [M + H]+: calcd for C31H33N2O2: 465.2542, found: 465.2575.
2′-(3-Chlorobenzoyl)-1′-mesityl-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5f). White solid. Yield: 85%. Mp. 144–146 °C. IR: νmax (KBr, cm−1): 3180 (NH), 1711 (C
O), 1679 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.62 (H-6) (m, 1H), 1.66–1.73 (H-6) (m, 1H), 1.80–1.89 (H-7) (m, 2H), 2.13 (CH3) (s, 3H), 2.35–2.39 (H-8) (m, 2H), 2.60 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.03–4.09 (H-4) (m, 1H), 4.35 (H-3) (t, 1H, J = 10.4 Hz), 5.26 (H-5) (d, 1H, J = 11.6 Hz), 6.47 (d, 1H, J = 7.6 Hz, Ar–H), 6.79 (d, 2H, J = 8.8 Hz, Ar–H), 6.98 (t, 1H, J = 7.0 Hz, Ar–H), 7.03 (t, 1H, J = 1.6 Hz, Ar–H), 7.09–7.14 (m, 2H, Ar–H), 7.20 (d, 1H, J = 8.0 Hz, Ar–H), 7.31 (t, 1H, J = 8.0 Hz, Ar–H), 7.51–7.53 (m, 1H, Ar–H), 10.20 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.53 (CH3), 21.10 (CH3), 21.67 (CH3), 27.82 (C-7), 32.18 (C-6), 46.48 (C-4), 47.10 (C-8), 62.06 (C-3), 69.72 (C-5), 73.59 (C-2), 109.89, 121.66, 125.10, 126.26, 127.11, 127.23, 129.36, 129.61, 130.56, 131.33, 132.28, 132.94, 133.50, 135.47, 136.10, 137.99, 138.79, 142.30, 179.28 (CO), 197.04 (CO). HRMS (ESI) m/z [M + H]+: calcd for C30H30ClN2O2: 485.1996, found: 485.2029.
1′-Mesityl-2′-(3-methylbenzoyl)-1′,2′,5′,6′,7′,7a′-hexahydrospiro[indoline-3,3′-pyrrolizin]-2-one (5g). Pale yellow solid. Yield: 70%. Mp. 106–108 °C. IR: νmax (KBr, cm−1): 3190 (NH), 1713 (C
O), 1680 (CO). 1H NMR (400 MHz, DMSO-d6) δH: 1.56–1.62 (H-6) (m, 1H), 1.66–1.73 (H-6) (m, 1H), 1.81–1.88 (H-7) (m, 2H), 2.12 (CH3) (s, 3H), 2.17 (CH3) (s, 3H), 2.36–2.39 (H-8) (m, 2H), 2.59 (CH3) (s, 3H), 2.66 (CH3) (s, 3H), 4.04–4.09 (H-4) (m, 1H), 4.38 (H-3) (t, 1H, J = 10.4 Hz), 5.31 (H-5) (d, 1H, J = 12.0 Hz), 6.46 (d, 1H, J = 7.6 Hz, Ar–H), 6.78 (d, 2H, J = 11.2 Hz, Ar–H), 6.96 (t, 1H, J = 7.4 Hz, Ar–H), 7.02 (s, 1H, Ar–H), 7.09 (t, 2H, J = 6.2 Hz, Ar–H), 7.15 (t, 2H, J = 7.0 Hz, Ar–H), 7.25 (d, 1H, J = 7.2 Hz, Ar–H), 10.15 (NH) (s, 1H). 13C NMR (100 MHz, DMSO-d6) δC: 20.69 (CH3), 21.21 (CH3), 21.29 (CH3), 21.85 (CH3), 27.97 (C-7), 32.30 (C-6), 46.94 (C-4), 47.27 (C-8), 61.59 (C-3), 69.69 (C-5), 73.84 (C-2), 109.95, 121.63, 125.07, 125.50, 128.29, 128.62, 129.53, 131.46, 132.65, 134.00, 135.54, 137.20, 138.02, 142.45, 179.59 (CO), 197.89 (CO). HRMS (ESI) m/z [M + H]+: calcd for C31H33N2O2: 465.2542, found: 465.2573.
In vitro studies
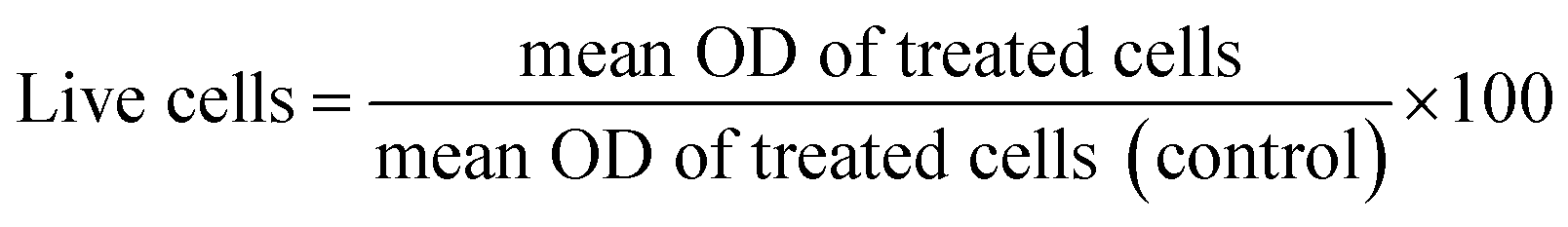
IC50 values were calculated using dose–response inhibition curves in GraphPad Prism.
Intracellular ROS generation. Intracellular ROS activity was detected using a ROS indicator, 2′,7′-dichlorofluorescein diacetate (DCFH-DA), a cell-permeable non-fluorescent compound. DCFH-DA is deacetylated by esterase inside the cell and then oxidized by ROS to DCF. A549 cells (3.0 × 104 cells per mL) were seeded in 24 well plates for 24 h. Later, selective compounds at IC50 concentrations (24 h) and 30 μL of 100 μM H2O2 were added to each well. Sixty μL of 10 μM DCFH-DA was added to each well and incubated in the dark for 10 min. Then, fluorescence intensity was measured immediately under a fluorescence multi-plate reader (TECAN INFINITE M Plex, Switzerland) with excitation at 488 nm and emission at 510 nm. The readings were measured at an interval time of 0 h, 2 h, 4 h, 6 h, 8 h, 12 h and 24 h. The results were analyzed through OriginLab software by plotting fold change vs. time.
Cytotoxicity in NIH 3T3 cells. Cytotoxicity of the synthesized compounds was evaluated in NIH 3T3 cells through an MTT assay. The cells (5 × 103 cells per well in 200 μL of media) were seeded into a 96 well plate and allowed to grow for 24 h at 37 °C and 5% CO2. Cells were treated with increasing concentrations of compounds for 24 h and 48 h. The study was conducted in triplicate. DMSO and media were used as a negative control, and cisplatin was used as a standard drug, respectively. After 24 h and 48 h, 20 μL of MTT reagent (5 mg mL−1 in sterile distilled water) was added to each well, and plates were wrapped with aluminium foil and incubated for 4 h at 37 °C. The purple formazan product was dissolved by the addition of 100 μL DMSO to each well. The absorbance was measured at 595 nm using a microplate reader (TECAN INFINITE M Plex, Switzerland). Data were collected for three replicates each and used to calculate the mean. The percentage of live cells was calculated from this data using the following formula:
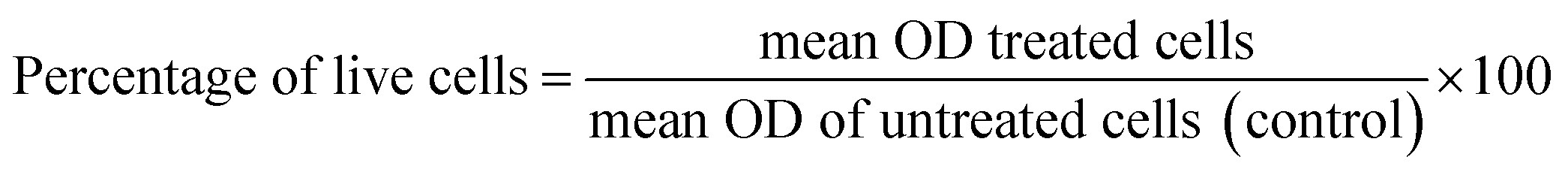
IC50 values were calculated using dose–response inhibition curves in GraphPad Prism.
Data availability
All data supporting the findings of this research are available in the main article and the ESI.†Conflicts of interest
The authors declare no conflicts of interest.References
- A. Leiter, R. R. Veluswamy and J. P. Wisnivesky, Nat. Rev. Clin. Oncol., 2023, 20, 624–639 CrossRef.
- Z. Wang, J. Kim, P. Zhang, J. M. G. Achi, Y. Jiang and L. Rong, Cell Insight, 2022, 1, 100015 CrossRef PubMed.
- L. Yan, J. Ma, Y. Zhu, J. Zan, Z. Wang, L. Ling, Q. Li, J. Lv, S. Qi, Y. Cao, Y. Liu, L. Cao, Y. Zhang, Z. Qi and L. Nie, J. Cell. Biochem., 2018, 119, 3989 CrossRef CAS.
- Y. M. A. Aziz, G. Lotfy, M. M. Said, E. S. H. El-Ashry, E. S. H. El Tamany, S. M. Soliman, M. M. Abu-Serie, M. Teleb, S. Yousuf, L. R. Domingo and A. Barakat, Front. Chem., 2021, 9, 735236 CrossRef PubMed.
- A. M. Omer, M. S. Ahmed, G. M. El-Subruiti, R. E. Khalifa and A. S. Eltaweil, Pharmaceutics, 2021, 13, 338 CrossRef CAS PubMed.
- M. Hosny, M. Fawzy, A. M. Abdelfatah, E. F. Fawzy and A. S. Eltaweil, Adv. Powder Technol., 2021, 32, 3220–3233 CrossRef CAS.
- A. M. Omer, T. M. Tamer, R. E. Khalifa, A. S. Eltaweil, M. M. Agwa, S. Sabra, M. S. Abd-Elmonem, M. S. Mohy-Eldin and Z. M. Ziora, Polymers, 2021, 13, 2428 CrossRef CAS.
- H. L. Teng, H. Huang and C. J. Wang, Chem, 2012, 18, 12614–12618 CrossRef CAS PubMed.
- K. R. Braun, T. H. E. Freysoldt and F. Wierschem, Chem. Soc. Rev., 2005, 34, 507–516 RSC.
- H. Pellissier, Tetrahedron, 2007, 63, 3235 CrossRef CAS.
- (a) N. Lashgari and G. M. Ziarani, ARKIVOC, 2012, 1, 277–320 Search PubMed; (b) B. Yu, D. Q. Yu and H. M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS; (c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS.
- (a) C. Marti and E. M. Carreira, Eur. J. Org Chem., 2003, 2003, 2209–2219 CrossRef; (b) G. Bhaskar, Y. Arun, C. Balachandran, C. Saikumar and P. T. Perumal, Eur. J. Med. Chem., 2012, 51, 79–91 CrossRef CAS; (c) M. Ghadi, A. Taheri and A. Abbasi, Tetrahedron, 2010, 66, 6744–6748 CrossRef.
- (a) A. Thangamani, Eur. J. Med. Chem., 2010, 45, 6120–6126 CrossRef CAS; (b) A. I. Almansour, R. S. Kumar, N. Arumugam, A. Basiri, Y. Kia, M. A. Ali, M. Farooq and V. Murugaiyah, Molecules, 2015, 20, 2296–2309 CrossRef.
- (a) S. J. Tang, X. Zhang, J. Y. Sun, D. W. Niu and J. J. Chruma, Chem. Rev., 2018, 118, 10393–10457 CrossRef CAS; (b) A. Shaabani, A. H. Rezayan and A. Sarvary, Mol. Diversity, 2011, 15, 41–68 CrossRef CAS PubMed; (c) N. Kielland and R. Lavilla, Synthesis of Heterocycles via Multicomponent Reactions II, Topics in Heterocyclic Chemistry, Springer, Berlin, Heidelberg, 2010, pp. 127–168 Search PubMed.
- (a) Y. Liu, H. Y. Hu, X. Wang, S. J. Zhi, Y. Kan and C. Wang, J. Org. Chem., 2017, 82, 4194–4202 CrossRef CAS; (b) Z. M. Zhang, B. Xu, S. Xu, H. H. Wu and J. L. Zhang, Angew. Chem., Int. Ed., 2016, 55, 6324–6328 CrossRef CAS PubMed; (c) E. Conde, I. Rivilla, A. Larumbe and F. P. Cossio, J. Org. Chem., 2015, 80, 11755–11767 CrossRef CAS; (d) J. Mancebo-Aracil, C. Najera and J. M. Sansano, Org. Biomol. Chem., 2013, 11, 662–675 RSC.
- C. B. Cui, H. Kakeya and H. Osada, Tetrahedron, 1996, 52, 12651–12666 CrossRef CAS.
- C. B. Cui, H. Kakeya, G. Okada, R. Onose and H. Osada, J. Antibiot., 1996, 49, 527–533 CrossRef CAS.
- M. Zheng, J. Yang, X. Xu, J. T. Sebolt, S. Wang and Y. Sun, Anticancer Res., 2010, 30, 3321–3331 CAS.
- Y. Zhao, S. Yu, W. Sun, L. Liu, J. Lu, D. McEachern, S. Shargary, D. Bernard, X. Li, T. Zhao, P. Zou, D. Sun and S. Wang, J. Med. Chem., 2013, 56, 5553–5561 CrossRef CAS PubMed.
- J. Lu, S. Guan, Y. Zhao, Y. Yu, Y. Wang, Y. Shi, X. Mao, K. L. Yang, W. Sun, X. Xu, J. S. Yi, T. Yang, J. Yang and J. G. Nuchtern, Oncotarget, 2016, 7, 82757–82769 CrossRef.
- G. Lotfy, E. S. H. E. Ashry, M. M. Said, E. S. E. Tamany, Y. M. A. Aziz, A. Al-Dhfyan, A. M. Al-Majid and A. Barakat, J. Photochem. Photobiol., B, 2018, 180, 98–108 CrossRef CAS.
- R. Ghosh, J. B. Vitor, E. Mendes, A. Paulo and P. C. Acharya, ACS Omega, 2020, 5, 27332–27343 CrossRef CAS PubMed.
- M. Sivanandhan, S. Ragupathy, A. Thangamani and A. Parasuraman, Mol. Diversity, 2024 DOI:10.1007/s11030-024-10974-x.
- A. Barakat, M. S. Islam, M. Ali, A. M. Al-Majid, S. Alshahrani, A. S. Alamary, S. Yousuf and M. I. Choudhary, Symmetry, 2021, 13, 1426 CrossRef CAS.
- (a) R. T. Pardasani, P. Pardasani, V. Chaturvedi, S. K. Yadav, A. Saxena and I. Sharma, Heteroat. Chem., 2003, 14, 36–41 CrossRef CAS; (b) N. V. Lakshmi, P. Thirumurugan and P. T. Perumal, Tetrahedron Lett., 2010, 51, 1064–1068 CrossRef CAS; (c) J. Li, J. Wang, Z. Xu and S. Zhu, ACS Comb. Sci., 2014, 16, 506–512 CrossRef CAS.
- B. Yu, D.-Q. Yu and H.-M. Liu, Eur. J. Med. Chem., 2015, 97, 673–698 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01632k |
| This journal is © The Royal Society of Chemistry 2025 |