
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBio-inspired benzimidazole-functionalized manganese terpyridine complexes for electrochemical CO2 reduction†
Runqi Mo,
Rui Li,
Ping Zhang ,
Ying Xiong and
Lin Chen*
,
Ying Xiong and
Lin Chen*
State Key Laboratory of Environment-Friendly Energy Material, School of Materials and Chemistry, Southwest University of Science and Technology, Mianyang 621010, P. R. China. E-mail: chenlin101101@aliyun.com
First published on 18th July 2025
Abstract
Inspired by the active site of carbon monoxide dehydrogenase (CODH), where a pair of amino acids facilitating hydrogen bonding manages the reversible interconversion of CO and CO2 with high efficiency, we developed a family of manganese terpyridine derivatives (1–4), in which a benzimidazole moiety functions as a proton relay to assist the CO2 reduction reaction (CO2RR). To regulate the position of the proton donor, the benzimidazole moiety was introduced into the framework by two approaches, and the pKa of the proton relay was adjusted by methylation of the benzimidazole moiety. We found that all such designs led to a sharp corruption in the activity of electrochemical CO2 reduction compared with that of our previously reported analogues. The corruption was ascribed to the pKa of the benzimidazole moiety, which resulted in inefficient proton exchange.
Introduction
In the past decade, extensive efforts have been devoted for exploring highly efficient catalysts for the CO2 reduction reaction (CO2RR).1 Owing to their rich electronic and structural tunability, transition-metal-based molecules have been utilized to systematically investigate CO2RR mechanisms.2 Mono-nuclear metal complexes, such as chromium,3 manganese,4–9 iron,10–12 cobalt,13 nickel,14 ruthenium,15 and rhenium,16,17 have exhibited activity in the electrochemical CO2RR. In the [NiFe] carbon monoxide dehydrogenase (CODH) active site (Scheme 1),18 a redox active nickel center, an iron(II) ion acting as a Lewis acid, and a pair of amino acids facilitating hydrogen bonding manage the reversible interconversion of CO and CO2 with high efficiency. The second-sphere histidine and lysine residues in Ni–Fe CODH are positioned to stabilize the CO2–metal adduct and assist in the C–O bond cleavage.18 Inspired by the active site of CODH, installing a proton relay inside the second coordination sphere of catalysts has proven to be instrumental in improving the catalysis of the CO2RR.9,19–21 Previously, we systematically investigated a series of manganese terpyridine complexes (M1–M4) bearing a proximal proton relay (Scheme 1). M1 exhibits significantly higher selectivity than its bipyridine analogues.22 Both catalysts (M2 and M3) exhibit high selectivity for CO2-to-CO conversion, and the isomer with a proximal amide (M3) shows a rate ca. two orders of magnitude higher than that of the isomer with a distal amide.23 Furthermore, Mn terpyridine derivatives with urea groups (M4) functioning as multipoint hydrogen-bonding hangman accelerate the reaction rate of CO2 reduction to ca. 360 s−1.24 These studies confirm the key influence of a proximal proton relay in improving the CO2RR. However, much improvement is still needed to optimize the synergism between the proton relay and the Mn terpyridine framework. | ||
| Scheme 1 Active site of Ni–Fe CO dehydrogenase inspires the design of a series of manganese terpyridine dicarbonyl catalytic active sites. The catalytic active sites in the red dashed box are studied in this work. | ||
To better mimic to the structure of the natural enzyme CODH, in this study, a benzimidazole moiety was introduced into the second coordination sphere of the Mn terpyridine framework to assist the CO2RR (Scheme 1, the red dashed box). As shown in Scheme 2, the benzimidazole moiety was incorporated into the framework by two approaches. One approach involves attaching the benzimidazole moiety to the pyridine without a spacer, and the other involves separating the benzimidazole moiety and pyridine with a phenyl spacer. The distance between the proton donor and CO2 adduct could be adjusted by rotating the benzimidazole moiety or phenyl (Scheme S10†). In addition, the benzimidazole moiety was methylated to regulate the pKa of the proton relay. Herein, the electrochemical reduction of CO2 using this series of complexes was studied.
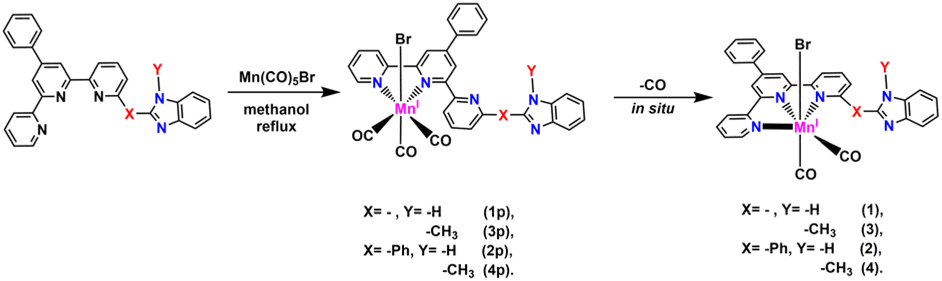 | ||
| Scheme 2 Preparation of manganese catalysts 1–4. | ||
Results and discussion
Synthesis and characterization
Precursors 1p–4p were obtained according to our previously reported methods (Schemes 2 and S1–S8†) by refluxing Mn(CO)5Br and the corresponding ligands L1–L4 in methanol, respectively.25 All of the complexes were characterized by high-resolution mass spectroscopic (HR-MS), 1H NMR, FTIR and elemental analyses (Fig. S1–S13†). The configurations of 1p–4p are hypothesized to resemble those of the analogues reported previously,23 in which the terpyridine serves as a bidentate ligand, the pyridyl proximal to the benzimidazole moiety is uncoordinated (Scheme 2), and bromide is located at an apex, opposite the axial carbonyl. IR and HR-MS indicate that the Mn(I) centre of these complexes is coordinated with three CO ligands in a facial mode.7 The 1H NMR spectroscopy of these complexes indicates a mixture of atropisomers (the NH or N–CH3 moieties parallel and antiparallel to the axial plane).21In situ conversion of the coordination mode from k2-N,N′,N′′-tpy (k2-tpy) to k3-N,N′,N′′-tpy (k3-tpy)
As reported in our previous studies,22,23 the target catalysts 1–4 can be in situ obtained decarbonylating of precursors 1p–4p promoted by an electric field (ca. −1.5 V vs. Fc+/0). The switch in the coordination mode is very complicated. As reported by Kubiak et al., the k2-mode reductive intermediates in situ transform to the corresponding k3 mode.7 Our previous studies found that the switch in the coordination mode always occurs before the reduction reaction.22–25 In this study, the switch process was monitored by infrared spectroelectrochemistry (IR-SEC) performed in a self-designed thin-layer specular reflectance cell under Ar.As shown in Fig. S14–S17† (red line), precursors 1p–4p studied herein almost inherit the features reported previously and in situ convert to k3-mode complexes 1–4 (IR absorption at around 1949, 1925, 1878 and 1853 cm−1) with the help of an electric field before the reduction reaction. Subsequently, at a more negative applied potential, the reduction of 1p–4p results in a mixture of [X2−–Br−] and [Xp2−–Br−] (X = 1–4). Herein, [X2−–Br−] may be obtained from the reduction of X or decarbonylating of [Xp2−–Br−]. Observably, [Xp2−–Br−] cannot totally convert to [X2−–Br−] in the timescale of the IR scan. Among all these complexes, 2p possesses the largest ratio of [X2−–Br−]/[Xp2−–Br−] in the intensity of IR absorption, indicating the higher stability of [22−–Br−] compared to the other [X2−–Br−]. In addition, the two lower-intensity absorption bands at around 1983 and 1892 cm−1 for 1p and 3p are assigned to the [Mn0-bipyridine]2 dimer22 (Fig. S14 and S16†), while the IR signal at 1983 and 1892 cm−1 cannot be found for 2p and 4p with a phenyl linkage (Fig. S15 and S17†), indicating that the bulky ligand eliminates this dimerization.
Electrochemical properties
The cyclic voltammograms (CVs) of all complexes were tested in CH3CN (all potentials are reported against ferrocene, Fc+/0, ca. 0.63 V vs. NHE). As shown in Fig. 1a, complex 1p features two successive irreversible reduction peaks at around −1.5 V and one reversible couple at −2.0 V. The former can be assigned to the successive reduction on the Mn center and the ligand.7 The latter is ascribed to the redox of the intermediate formed by intramolecular proton transfer from the benzimidazole to reductive Mn center following the two successive reduction events.9,21,22 The intramolecular proton transfer leads to the irreversibility of the former reduction events. By contrast, the methylation of benzimidazole results in the reversible redox couple for complex 3p and the lack of the Mn–H intermediate.9,21,22 The difference between complexes 2p and 4p lies in the methylation on the benzimidazole moiety. The difference exhibits negligible influence on the initial states of 2p and 4p as well as the k3 modes 2 and 4 (Fig. S18†). The significant contrast between CVs of 2p and 4p may be ascribed to the intramolecular electron transfer following the double reduction of 2. The reduction waves, consistent with those of 4, are assigned to the successive reduction of 2, and the third new peak is assigned to reduction on the in situ-formed Mn hydride. This difference soundly supports the transformation in the coordination modes for 2p and 4p before the reduction because the k3 mode shortens the proton transfer distance between the proton donor and reductive Mn center. In addition, the Mn hydride is responsible for the strong response to phenol with 2p, which is discussed in the next section. | ||
| Fig. 1 Cyclic voltammograms of 0.5 mM complexes 1p–4p (a) under Ar and (b) under CO2 in the presence of 1 M phenol. (c) Dependence of the catalytic current (icat) on the square root of the concentration of phenol. (d) Catalytic Tafel plots of CO2-to-CO conversion showing the relationship between the TOF and driving force (using foot-of-the-wave analysis), compared to previously reported electrocatalysts Mn(bpy-tBu)(CO)3Br, Mn(bpy-CONHMe)(CO)3Br, and Mn(tpy-NHCO-Ph)(CO)3Br. Scan rate = 100 mV s−1, 0.1 M nBu4NPF6 in CH3CN, glassy carbon working electrode, Ag+/Ag reference electrode, and Pt wire counter electrode. | ||
The differential pulse voltammogram (DPV) tests of 1p–4p are consistent with the reduction events in CVs (Fig. S14–S17†). Furthermore, the cathodic and anodic peak currents (ip) of complexes 1p–4p show linear correlations with the square root of the scan rate (v1/2) from 0.025 to 10 V s−1 under Ar (Fig. S19–S22†), consistent with diffusion-controlled processes described by the Randles–Sevcik equation.13 The electrochemical property parameters of complexes 1p–4p are summarized in Table 1.
| Catalyst | Eθcat (V) | ηa (V) | icat/ipb | TOFCVc (s−1) | TONCPE | FEtotal (%) | FECOd (%) |
|---|---|---|---|---|---|---|---|
a Derived from catalytic half-wave potentials 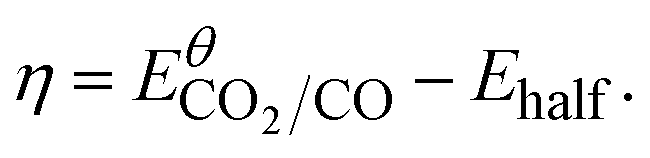 b Obtained at ca. −1.80 V.c Derived using the equations reported in the ESI file.d FE for CO is reported as an average of three CPE experiments at −1.85 V. b Obtained at ca. −1.80 V.c Derived using the equations reported in the ESI file.d FE for CO is reported as an average of three CPE experiments at −1.85 V. |
|||||||
| 1p | −1.58 | 0.28 | 8.47 | 11.31 | 0.05 | 97.8% ± 5% | 80.8% ± 5% |
| 2p | −1.74 | 0.39 | 17.72 | 81.25 | 4.50 | 100.2% ± 5% | 61.1% ± 5% |
| 3p | −1.55 | 0.24 | 5.67 | 22.44 | 4.75 | 99.2% ± 5% | 62.3% ± 5% |
| 4p | −1.66 | 0.31 | 4.11 | 8.48 | 0.86 | 99.0% ± 5% | 77.3% ± 5% |
After the addition of phenol, 1p–4p exhibit different responses to protons. The response to protons follows the order of 2p >> 1p > 3p >> 4p (Fig. S23–S26†). As shown in Fig. S23,† 1p catalyzes the proton reduction to hydrogen at −2.0 V, which is based on the redox couple of the Mn–H hydride. The third reversible redox event for complex 2p matches well with the reduction of phenol, leading to a strong increase in the current as the concentration of phenol. Complex 3p mediates the conversion of the proton to hydrogen with an obviously lower overpotential, but the amplitude of the catalytic current is low. Complex 4p exhibits a weak response to phenol following the second reduction peak. According to our previous studies, in the in situ-formed k3 mode, the benzimidazole moiety in 2 features the best position and pKa to assist the proton transfer to the Mn center. The configuration and performance of 2 are very similar to those of our previously reported analogue with a carboxylic acid proton relay.25 By contrast, the distance between the proton relay and Mn center in complex 3 is significantly longer than that in complex 2.22
After saturating the test solution with CO2 in the presence of 1 M phenol, complexes 1p–4p exhibit varied responses (icat/ip) to CO2. Among these complexes, 2p exhibits the strongest response to CO2 (Fig. 1b). As shown in Fig. S24,† CO2 leads to a sharp increase in the current at ca. 100 mV, more positive than that for proton reduction, corresponding to the reduction of CO2 to CO. The catalytic wave of complex 2p can be divided into two sections at the catalytic current plateau (Fig. 1b). The first section is assigned to the CO2 reduction, while the other section is ascribed to the reduction of protons, similar to that in the absence of CO2 (Fig. S24†). The catalytic half-wave potential for 2p is ca. −1.75 V, corresponding to an overpotential of ca. 400 mV  10 By contrast, complexes 3p and 4p feature lower overpotentials (ca. 250 mV), while the catalytic current becomes observably lower. The catalytic activity for CO2 reduction of complex 1p is a little stronger than that of 3p and 4p.
10 By contrast, complexes 3p and 4p feature lower overpotentials (ca. 250 mV), while the catalytic current becomes observably lower. The catalytic activity for CO2 reduction of complex 1p is a little stronger than that of 3p and 4p.
To be consistent with the previous Mn polypyridine analogues, external phenol is essential for the CO2-to-CO conversion using 1p–4p.16 As shown in Fig. 1c, the icat/ip values of 1p–4p show a linear dependence on the square root of the concentration of phenol, indicating a first-order dependence on the concentration (Fig. 1c and S27–S30†).26 Kinetic studies found the current response (icat/ip) of complexes 1p–4p all exhibit linear dependence on the catalyst concentration (Fig. S31–S34†). Among these complexes, 2p is the most sensitive to the concentrations of the catalyst and phenol. However, as the scan rate increases (Fig. S35–S38†), the conversion in the coordination mode (k2 to k3) cannot keep pace with the electrochemical reduction reaction; therefore, the catalytic contribution of the k2 mode increases at a higher scan rate.23
To compare the CO2 reduction activity of 1p–4p and other catalysts, the TOFmax values of 1p–4p were calculated by foot-of-the-wave analysis (FOWA) (Fig. 1d and S39†)26 under general conditions (in the presence of 1 M phenol, under CO2 saturation, and at a scan rate of 100 mV s−1), and the plateau current of the first section was adopted for the calculation. The state-of-the-art benchmarking of performances clearly revealed that these new Mn catalysts exhibit worse performances than the previous analogues in terms of the overpotential and catalytic rate (Fig. 1d). The corruption is ascribed to the pKa of the benzimidazole moiety, which results in the inefficiency of the proton exchange. The acidity of the benzimidazole is stronger than that of amide and urea moieties, which is favorable for the transfer of protons from the proton relay to CO2 adduct but is adverse for regaining protons from the bulk solution. The passable performance of the benzimidazole moiety in the artificial catalyst compared to that in CODH is ascribed to the alkalinity of the catalytic center as well as the CO2 adduct.
Controlled potential electrolysis (CPE) experiments (Fig. S40†) of all complexes in a CO2 atmosphere in the presence of 1.0 M phenol at varied potentials were carried out in a custom-designed cell (Fig. S41†). An average current of around 0.5 mA cm−2 is observed throughout the electrolysis period of 1 h under an applied potential of −1.85 V. CVs after electrolysis show a slight decrease in the amplitude of the catalytic current (Fig. S42–S44†), consistent with the observation of a decrease in the catalytic current during CPE. The formation of catalytically active nanoparticles is excluded owing to the negligible current observed after a “rinse test” following CPE (Fig. S45†). Therefore, the current decay is ascribed to the decomposition of the catalyst. The analysis of the gas mixture in the headspace of the working compartment by gas chromatography (GC) confirms CO and H2 as the main products. The analysis of the liquid phase by ion chromatography (IC) detects amounts of formic acid for complexes 1p–4p. Control experiments show significantly lower current densities and a negligible amount of CO (or H2 and formic acid) in the absence of catalysts (Fig. S40†). Isotope labelling experiments using 13CO2 confirm that CO is formed via the reduction of CO2 (Fig. S46†). The electrocatalytic properties of 1p–4p are summarized in Tables 1 and S1.† Using product analysis under varied electrolysis potentials, we found that the selectivity of products becomes corrupted as the potential decreases, which is consistent with the observations in CVs. At a more negative scan potential, the contribution of proton reduction becomes strong.
Conclusions
Inspired by the active site of carbon monoxide dehydrogenase (CODH), we integrate the benzimidazole moiety into the manganese terpyridine frame as a proton relay by two approaches. It is a pity that the performance of the benzimidazole moiety in the manganese terpyridine catalyst for the electrochemical CO2RR is worse than that in CODH. The poor activity of this series of catalysts is because the benzimidazole moiety disrupts the acid–base equilibrium between the proton relay and bulk solution as well as the metal center, reducing the proton exchange speed between the bulk solution and metal center. The results here clearly advise that ongoing studies in the design of CO2RR catalysts should pay more attention to the balance among the functional fragments of catalysts rather than solely focusing on the resemblance to the structure of natural enzymes. The consideration of the findings presented here in future catalyst designs may lead to more-efficient catalysts.Data availability
All data supporting this study, including experimental procedures, spectroscopic analyses, 1H-NMR data and electro chemical details, are available within the article and its ESI.†Author contributions
L. C. designed the research. R. Q. M., R. L., P. Z. performed electrochemistry, synthesis, and analyzed the data. Y. X. and L. C. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Chinese National Natural Science Foundation (U20A20125, 22088102, 22172018 and 21403132), the State Key Laboratory of Environment-Friendly Energy Materials Research on Independent Subjects (20fksy15, 21fksy18), and the State Key Laboratory of Fine Chemicals, Dalian University of Technology (KF2214).Notes and references
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119, 2752–2875 CrossRef CAS PubMed.
- N. Elgrishi, M. B. Chambers, X. Wang and M. Fontecave, Chem. Soc. Rev., 2017, 46, 761–796 RSC.
- S. L. Hooe, J. M. Dressel, D. A. Dickie and C. W. Machan, ACS Catal., 2020, 10, 1146–1151 CrossRef CAS.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- J. A. Barrett, C. J. Miller and C. P. Kubiak, Trends Chem., 2021, 3, 176–187 CrossRef CAS.
- M. L. Clark, P. L. Cheung, M. Lessio, E. A. Carter and C. P. Kubiak, ACS Catal., 2018, 8, 2021–2029 CrossRef CAS.
- C. W. Machan and C. P. Kubiak, Dalton Trans., 2016, 45, 17179–17186 RSC.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- M. H. Rønne, D. Cho, M. R. Madsen, J. B. Jakobsen, S. Eom, É. Escoudé, H. C. D. Hammershøj, D. U. Nielsen, S. U. Pedersen, M.-H. Baik, T. Skrydstrup and K. Daasbjerg, J. Am. Chem. Soc., 2020, 142, 4265–4275 CrossRef PubMed.
- C. Costentin, S. Drouet, M. Robert and J. M. Saveant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- E. M. Nichols, J. S. Derrick, S. K. Nistanaki, P. T. Smith and C. J. Chang, Chem. Sci., 2018, 9, 2952–2960 RSC.
- P. Gotico, B. Boitrel, R. Guillot, M. Sircoglou, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, Angew. Chem., Int. Ed., 2019, 58, 4504–4509 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pecaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- D. Hong, T. Kawanishi, Y. Tsukakoshi, H. Kotani, T. Ishizuka and T. Kojima, J. Am. Chem. Soc., 2019, 141, 20309–20317 CrossRef CAS PubMed.
- B. A. Johnson, S. Maji, H. Agarwala, T. A. White, E. Mijangos and S. Ott, Angew. Chem., Int. Ed., 2016, 55, 1825–1829 CrossRef CAS PubMed.
- C. Riplinger, M. D. Sampson, A. M. Ritzmann, C. P. Kubiak and E. A. Carter, J. Am. Chem. Soc., 2014, 136, 16285–16298 CrossRef CAS PubMed.
- J. M. Smieja, E. E. Benson, B. Kumar, K. A. Grice, C. S. Seu, A. J. M. Miller, J. M. Mayer and C. P. Kubiak, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15646–15650 CrossRef CAS PubMed.
- J. H. Jeoung and H. Dobbek, Science, 2007, 318, 1461–1464 CrossRef CAS PubMed.
- J. B. Jakobsen, M. H. Rønne, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2021, 60, 9174–9179 CrossRef CAS PubMed.
- J. M. Saveant, Angew. Chem., Int. Ed., 2019, 58, 2125–2128 CrossRef CAS PubMed.
- J. Agarwal, T. W. Shaw, H. F. Schaefer and A. B. Bocarsly, Inorg. Chem., 2015, 54, 5285–5294 CrossRef CAS PubMed.
- S. He, Y. Qing, P. Zhang, Y. Xiong, Q. Wu, Y. Zhang, L. Chen, F. Huang and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202216082 CrossRef CAS PubMed.
- Y. Qing, Q. Wu, S. He, P. Zhang, Y. Xiong, Y. Zhang, F. Huang, F. Li and L. Chen, Dalton Trans., 2023, 52, 14309–14313 RSC.
- M. Li, F. Huang, P. Zhang, Y. Xiong, Y. Zhang, F. Li and L. Chen, Chem.–Eur. J., 2024, 30, e202304218 CrossRef CAS PubMed.
- Q. Wu, M. Li, S. He, Y. Xiong, P. Zhang, H. Huang, L. Chen, F. Huang and F. Li, Chem. Commun., 2022, 58, 5128–5131 RSC.
- K. J. Lee, N. Elgrishi, B. Kandemir and J. L. Dempsey, Nat. Rev. Chem., 2017, 1, 0039 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01621e |
| This journal is © The Royal Society of Chemistry 2025 |