
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular reductive radical-polar crossover-based acyl migration reactions of N-vinylimides with alkyl, silyl, and acyl radicals†
Yutao Jinga,
Li Zhang*d,
Li Qiu*a and
Yewen Fang *bc
*bc
aCollege of Chemistry and Chemical Engineering, Taiyuan University of Technology, No. 79 West Street Yingze, Taiyuan 030024, China. E-mail: qiuli@tyut.edu.cn
bSchool of Materials and Chemical Engineering, Ningbo University of Technology, No. 201 Fenghua Road, Ningbo 315211, China. E-mail: fang@nbut.edu.cn
cZhejiang Institute of Tianjin University, No. 201 Fenghua Road, Ningbo 315211, China
dCollege of Basic Science, Zhejiang Pharmaceutical University, No. 666 Siming Road, Ningbo 315500, China. E-mail: zhangl@zjpu.edu.cn
First published on 15th April 2025
Abstract
Herein, novel SET reduction-based N → C acyl migration protocols for the preparation of functionalized α-amino ketones were successfully developed. In addition to alkyl and silyl radicals, acyl radicals derived from dihydroquinazolinones or acyl oxime acetates could react with enamides to give various 1,4-diketones. These photocatalytic radical addition/acyl migration cascade reactions feature broad substrate scope, good functional group compatibility, and mild reaction conditions.
Due to the remarkable progress of photoredox catalysis, the generation of radicals under mild and environmentally benign reaction conditions has become especially facile. In addition to the facile formation of radicals, their reduction or oxidation could proceed efficiently via the single-electron-transfer (SET) pathway. Consequently, a range of radical-polar crossover (RPC) process-based transformations enabled by photoredox catalysis has been designed and realized.1 Recently, we employed a SET reduction-based formation of carbanion as a key strategy (Scheme 1), and a number of interesting and useful reactions were developed.2 Impressively, the SET reduction-based generation of carbanions could proceed efficiently under base-free conditions. Owing to the facile generation of radicals from various radical precursors, the SET reduction of radicals is a complementary and reliable approach to the well-documented formation of carbanions or their analogues via a two-electron pathway. Moreover, with the proper combination of photocatalyst and radical precursors, many reductive RPC processes do not require exogenous reductants because the photoexcited photocatalyst and the reduced photocatalyst could serve as the oxidant and reductant, respectively, in the same catalytic cycle.
 | ||
| Scheme 1 SET reduction-based formation of carbanion. | ||
Realizing this interesting and useful characteristic of photocatalysis, many efficient and robust transformations bearing a reductive RPC process have been developed and employed.3 Due to its good sustainability and efficiency, synthetic chemists will undoubtedly be interested to uncover more novel methods using redox-neutral photocatalyzed reductive RPC process as the key strategy.
More than 20 years ago, Hamada and co-workers demonstrated a base-mediated N → C acyl migration reaction producing α-amino ketones.4 According to their proposed mechanism, a base-generated carbanion neighboring the imide nitrogen was the key intermediate for this migration reaction. Recently, we realized the cyclopropanation of enamides with a halomethyl radical via an RPC process.2c Inspired by the formation of α-nitrogen carbanions from the corresponding radicals via SET reduction,2c,d redox-neutral photocatalyzed N → C acyl migration reactions were successfully developed via the reactions of N-vinylimides with alkylsilicates.5 Owing to the low, relatively uniform redox potentials, the bis-catecholatosilicates were attractive radical precursors for the formation of nonstabilized primary alkyl radicals, which could be involved in the expected migration reactions. However, because of the limited availability of organofunctional trialkoxysilanes, silicates are not a suitable choice for secondary and tertiary C-centered radicals as well as acyl radicals. In light of the importance of α-amino ketones serving as expedient precursors for β-aminoalcohols, which are useful for the preparation of pharmaceuticals and chiral auxiliaries, a modular approach to α-amino ketones via the N → C acyl migration reaction would be highly desirable (Scheme 2). Herein, with reductive RPC as the strategy, a modular access to functionalized α-amino ketones was generally developed via reactions of N-vinylimides with various radicals derived from dihydroquinazolinones (DHQs)6 or acyl oxime acetates.7 The decisive advantage of such a photo-driven strategy was the facile generation of α-imidocarbanion in the absence of a base and exogenous reductant under mild reaction conditions. Moreover, our method could significantly extend the applications of DHQs and acyl oxime acetates as radical precursors.
 | ||
| Scheme 2 Generation of α-amino ketone bearing a quaternary carbon center via acyl migration reaction. | ||
Our initial study of acyl migration reaction commenced with the reaction of N-acetyl-N-(1-phenylethenyl)acetamide 1a with pinacolone-derived dihydroquinazolinone 2a and the results are listed in Table 1. A simple survey of experimental parameters led us to identify the optimal reaction conditions (2 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6, 9 W blue LEDs irradiation, DMSO, rt, 24 h) (entry 1). As for the photocatalyst, the organic photocatalyst 4CzIPN proved ineffective for this transformation (entry 2). The reaction also gave a low yield using Ru(bpy)3Cl2 as the photocatalyst (entry 3). The subsequent screening of solvents indicated that both DMA and CH3CN are suitable solvents for this reaction (entries 4 and 5). Moreover, the lack of either [Ir] or light suggested the indispensable role of each in the desired reactivity (entries 6 and 7).
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield of 3a (%) |
| a Standard reaction conditions: a reaction mixture of 1a (0.2 mmol), 2a (0.4 mmol), [Ir] (2 mol%), and DMSO (6.0 mL) was irradiated with 9 W blue LEDs for 24 h at room temperature (r.t.) (cooling with a fan).b Yield of the isolated product 3a.c NMR analysis of crude reaction mixture.d The reaction was run in the dark. ND = not detected. | ||
| 1 | None | 76 |
| 2 | 4CzIPN instead of [Ir] | 20 |
| 3 | Ru(bpy)3Cl2 instead of [Ir] | 22 |
| 4 | DMA instead of DMSO | 66 |
| 5 | CH3CN instead of DMSO | 70 |
| 6c | Without photocatalyst | ND |
| 7cd | Without light | ND |

Having established the optimal conditions (entry 1, Table 1), we next set out to investigate the scope with respect to the N-vinylimides 1 using dihydroquinazolinone derivative 2a as the radical precursor. As shown in Table 2, N-vinylimides 1 having para-, meta-, or ortho-substituents were all eligible to forge the desired α-amino ketones 3b–3d in 31–66% yields. Clearly, steric variance on the phenyl ring had an influence on the reaction efficiency. Not surprisingly, substrates bearing a methoxy or methylenedioxy group on the phenyl ring were well tolerated, giving the corresponding products 3e–3g in good yields (64–79%). Moreover, enamides containing electron-withdrawing groups (–Cl and –Br) reacted smoothly to give the expected products 3h and 3i in 83% and 44% yields, respectively. In addition to the smooth reactions of monoaryl-substituted radical acceptors, enamides with biphenyl and naphthyl substituents could be efficiently transformed into the expected products 3j and 3k with good results. Interestingly, after the aryl substituent of enamides was replaced with a thiophene group, the yield of product 3l was 85%. Besides N-Boc-N-(1-phenylvinyl)acetamide 1m proved to be a viable substrate, giving 3m in 74% yield.

After demonstrating the scope of N-vinylimides, we next set out to investigate the generality of DHQs using N-acyl-protected enamide 1a as the model radical acceptor. As listed in Table 3, oxygen- or nitrogen-stabilized primary alkyl radicals were all readily accessed by the use of Me-substituted dihydroquinazolinones, giving the products 3n–3q in 66–86% yields. Interestingly, the products 3o and 3q could serve as the precursors of 1,3-aminoalcohol8 and 1,3-diamine,9 respectively. Moreover, stable 4-methoxybenzyl radical also gave the corresponding product 3r in 60% yield. Secondary oxygen-stabilized dimethoxymethyl radical10 generated from pyruvic aldehyde dimethyl acetal-derived dihydroquinazolinone participated in this transformation, forging 3s in 40% yield. Both acyclic and cyclic secondary alkyl radicals could react with 1a efficiently to produce the desired products 3t and 3u in good yields. The structure of 3t was further confirmed by single-crystal X-ray analysis (CCDC 2415008).11 Dihydroquinazolinone, derived from 1-adamantly methyl ketone, was also effective in delivering the expected 3v in 70% yield under these redox-neutral conditions. Again, this straightforward acyl migration reaction can be applied to N-Boc-N-(1-phenylvinyl)acetamide 1m, producing 3w in 79% yield.

As an extension, with 1a as the radical acceptor, spiro dihydroquinazolinone 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12 successfully underwent the expected ring-opening/radical addition/acyl migration cascade reaction to give 5 in 54% yield (Scheme 3a). Of note, the acyl migration reaction is highly dependent on the electron properties of radical acceptors.5 Using enamide-bearing pyridine moiety 6 as the radical acceptor, the Giese-type reaction instead of acyl migration reaction was observed, yielding 7 in 40% yield (Scheme 3b). Similarly, only the Giese-type reaction product 9 was isolated for the reaction of 2a with N-Boc-N-acetyldehydroalanine methyl ester 8 (Scheme 3c).
12 successfully underwent the expected ring-opening/radical addition/acyl migration cascade reaction to give 5 in 54% yield (Scheme 3a). Of note, the acyl migration reaction is highly dependent on the electron properties of radical acceptors.5 Using enamide-bearing pyridine moiety 6 as the radical acceptor, the Giese-type reaction instead of acyl migration reaction was observed, yielding 7 in 40% yield (Scheme 3b). Similarly, only the Giese-type reaction product 9 was isolated for the reaction of 2a with N-Boc-N-acetyldehydroalanine methyl ester 8 (Scheme 3c).
 | ||
| Scheme 3 Acyl migration and Giese-type reactions. | ||
To investigate the generality of this method, we focused on the acyl migration reactions of enamides with silyl and acyl radicals instead of alkyl radicals (Table 4). Interestingly, under the optimized reaction conditions in combination with K2CO3 (2.0 equiv.), using 2-silylated dihydroquinazolinone 10a as the silyl radical precursor,13 a range of N,N-diacetylimidostyrenes bearing various functional groups underwent acyl migrations to give 11a–11d in 36–45% yields. Furthermore, pyridine-containing enamide was also applied, producing 11e in moderate yields. In addition, the benzoyl radical generated from 2-benzoyl-2-methyl-2,3-dihydroquinazolin-4(1H)-one 10b readily reacted with various N-acetyl-N-(1-arylvinyl)acetamides 1, furnishing the expected products 12a–12d in moderate to good yields ranging from 35 to 65% yields. Moreover, enamide bearing a heteroaryl unit, thiophene, was tolerated here, affording product 12e in 37% yield. In addition to employing 2-benzyl-2-phenylbenzothiazoline 10c as a benzoyl radical source,14 4-methyl-subsituted benzoyl radical derived from 10d also reacted with 1a to give product 12f in 42% yield in the presence of a stoichiometric amount of Cs2CO3. Of note, compared to the broad substrate scope of 2,2-disubstituted DHQs for the formation of alkyl radicals, the generality of 2-acyl-substituted DHQs and benzothiazolines remains an issue.15

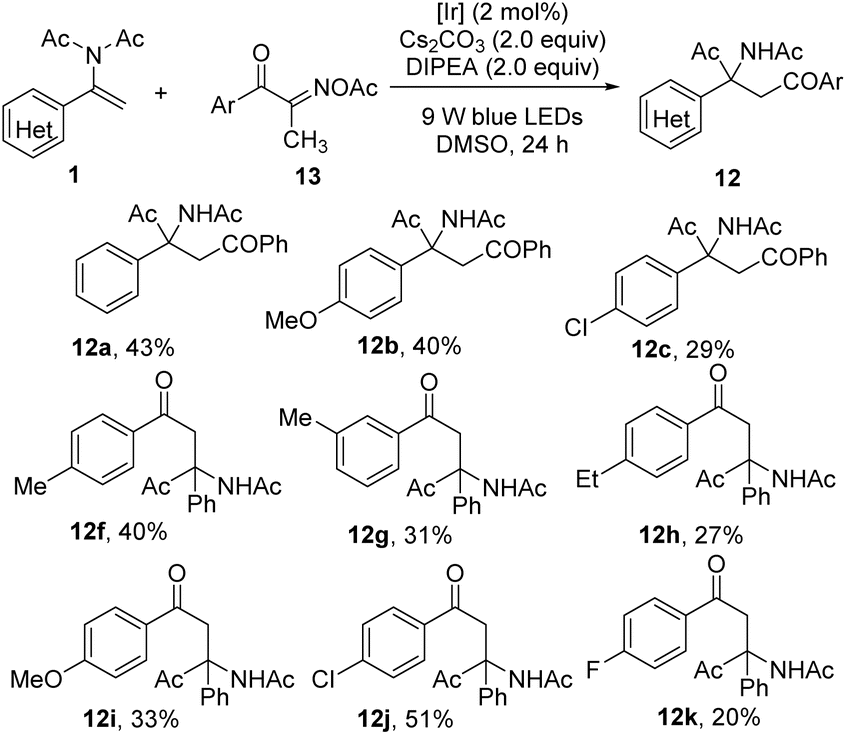
Considering the synthetic importance of 1,4-diketones and their unique biological activities,16 we decided to develop a simple and straightforward method for their access based on the acyl migration of enamides with acyl radicals. Fortunately, with N,N-diisopropylethylamine (DIPEA) as the reductant and Cs2CO3 as the base, we found easily accessible acyl oxime esters could serve as general and efficient acyl radical precursors enabled by photocatalysis (Table 5).7,17 The para-position of N,N-diacetylimidostyrenes with electron-rich and electron-deficient groups proceeded smoothly, affording 1,4-diketones 12b and 12c in moderate yields. In addition to benzoyl radicals, aryl acyl radicals bearing substituents (p-Me, m-Me, p-Et, p-OMe, p-Cl, p-F) on the aromatic ring at different positions or with different electronic properties can uniformly undergo expected cascade reactions, delivering the desired 1,4-diketones (12f–12k, 20–51% yields). Note that in contrast to the oxidative generation of acyl radicals from 2-acyl-substituted DHQs, acyl oxime esters serve as precursors of acyl radicals under reductive conditions. In this regard, the current reductive method is complementary to the above-mentioned oxidative SET approaches (see the ESI† for details).18
| a Reaction conditions: a reaction mixture of 1 (0.2 mmol), 13 (0.4 mmol), [Ir] (2 mol%), Cs2CO3 (0.4 mmol), DIPEA (0.4 mmol) and DMSO (6.0 mL) was irradiated with 9 W blue LEDs for 24 h at room temperature (rt) (cooling with a fan). Isolated yields. |
|---|
 |
In summary, with reductive RPC as the strategy, we have described a new protocol that enables modular access to functionalized α-amino ketones through photoredox catalysis. Using DHQs or acyl oxime acetates as the radical precursors, a variety of N-vinylimides bearing various functional groups could undergo smooth radical addition/acyl migration cascade reactions under mild conditions. Both SET oxidation and photoreduction pathways for the generation of acyl radicals are compatible with the following catalytic cascade reaction for the preparation of 1,4-diketones. Expectedly, the modular preparation of functionalized α-amino ketones bearing quaternary carbon centers would be feasible using radical addition/acyl migration cascade reactions as the strategy under mild reaction conditions.19
Data availability
The data supporting this study have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22271168) and the Specialty Fund of Zhejiang Institute of Tianjin University (No. ZITJU2024-ZYHT003).Notes and references
- (a) R. J. Wiles and G. A. Molander, Isr. J. Chem., 2020, 60, 281 CrossRef CAS PubMed; (b) M. Liu, X. Ouyang, C. Xuan and C. Shu, Org. Chem. Front., 2024, 11, 895 RSC.
- (a) T. Guo, L. Zhang, X. Liu, Y. Fang, X. Jin, Y. Yang, Y. Li, B. Chen and M. Ouyang, Adv. Synth. Catal., 2018, 360, 4459 CrossRef CAS; (b) W. Luo, Y. Yang, Y. Fang, X. Zhang, X. Jin, G. Zhao, L. Zhang, Y. Li, W. Zhou, T. Xia and B. Chen, Adv. Synth. Catal., 2019, 361, 4215 CrossRef CAS; (c) L. Huai, L. Zhang, Z. Wang, H. Wu and Y. Fang, Org. Chem. Front., 2023, 10, 1245 RSC; (d) X. Jin and L. Zhang, Org. Biomol. Chem., 2022, 20, 5377 RSC.
- (a) L. Pitzer, J. L. Schwarz and F. Glorius, Chem. Sci., 2019, 10, 8285 RSC; (b) K. Donabauer and B. König, Acc. Chem. Res., 2021, 54, 242 CrossRef CAS PubMed.
- O. Hara, M. Ito and Y. Hamada, Tetrahedron Lett., 1998, 39, 5537 CrossRef CAS.
- L. Huai, L. Zhang, Z. Wang and Y. Fang, Org. Chem. Front., 2024, 11, 2344 RSC.
- S. Miñoza, I. L. Librando and H.-H. Liao, Synlett, 2024, 35, 1072 CrossRef.
- (a) Y. Bao, Z.-J. Song, J.-L. Dai, S. Yan, Y. Zhang, J.-Y. Wang and G. Li, Chin. J. Chem., 2024, 42, 1399 CrossRef CAS; (b) H. Shi, H. Dong and C. Wang, Org. Chem. Front., 2023, 10, 5843 RSC.
- S. M. Lait, D. A. Rankic and B. A. Keay, Chem. Rev., 2007, 107, 767 CrossRef CAS PubMed.
- X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557–10566 RSC.
- T. Guo, L. Zhang, Y. Fang, X. Jin, Y. Li, R. Li, X. Li, W. Cen, X. Liu and Z. Tian, Adv. Synth. Catal., 2018, 360, 1352 CrossRef CAS.
- CCDC 2415008 (3t) contains the supplementary crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre.
- H.-J. Miao, J.-H. Zhang, W. Li, W. Yang, H. Xin, P. Gao, X.-H. Duan and L.-N. Guo, Chem. Sci., 2024, 15, 8993 RSC.
- T. Uchikura, H. Nakamura, H. Sakai and T. Akiyama, Chem.–Eur. J., 2023, 29, e202301090 CrossRef CAS PubMed.
- L. Li, S. Guo, Q. Wang and J. Zhu, Org. Lett., 2019, 21, 5462 CrossRef CAS PubMed.
- (a) Z.-N. Tsai, L.-Y. Li, A. S. Paculba, S. Miñoza, Y.-T. Tsao, P.-S. Lin and H.-H. Liao, Chem. - Asian J., 2024, 19, e202301004 CrossRef CAS PubMed; (b) X.-Y. Lv, R. Abrams and R. Martin, Nat. Commun., 2022, 13, 2394 CrossRef CAS PubMed.
- Y.-Y. Cheng, J.-X. Yu, T. Lei, H.-Y. Hou, B. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 26822 CrossRef CAS PubMed.
- C. Ai, T. Wang, Y. Bao, S. Yan, Y. Zhang and J.-Y. Wang, Org. Biomol. Chem., 2024, 22, 9197 RSC.
- (a) Y. Zhang, T. Zhu, Y. Lin, X. Wei, X. Xie, R. Lin, Z. Zhang, W. Fang, J.-J. Zhang, Y. Zhang, M.-Y. Hu, L. Cai and Z. Chen, Org. Biomol. Chem., 2024, 22, 5561 RSC; (b) L. Zheng, P.-J. Xia, Q.-L. Zhao, Y.-E. Qian, W.-N. Jiang, H.-Y. Xiang and H. Yang, J. Org. Chem., 2020, 85, 11989 CrossRef CAS PubMed.
- (a) Y. Zhu and J. Zhang, Macromol. Rapid Commun., 2024, 45, 2300695 CrossRef CAS PubMed; (b) Y. Zhu, D. Zhu, Y. Chen, Q. Yan, C.-Y. Liu, K. Ling, Y. Liu, D. Lee, X. Wu, T. P. Senftle and R. Verduzco, Chem. Sci., 2021, 12, 16092 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and compound characterisation data. CCDC 2415008. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra01542a |
| This journal is © The Royal Society of Chemistry 2025 |