
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneral one-step access to unsymmetric propargylic acetals via alcohol functionalization with allenyl ethers†
Ai Long,
Di Li,
Bo Yan,
Yue Yuan,
Haoqi Liu,
Lan Yang,
Yang Chen* and
Shuzhong He *
*
School of Pharmaceutical Sciences, Guizhou Engineering Laboratory for Synthetic Drugs, Guizhou University, Guiyang, Guizhou 550025, China. E-mail: szhe@gzu.edu.cn
First published on 15th April 2025
Abstract
We report a straightforward and robust protocol for synthesizing unsymmetric propargylic acetals through single-step alkoxypropargylation of aliphatic alcohols. This method employs allenyl ethers and hypervalent iodine reagents to achieve direct functionalization under mild conditions, producing 26 distinct acetals in 60–94% yields. The reaction's broad substrate compatibility accommodates diverse hydroxyl-containing molecules, offering a versatile and scalable platform for installing alkynyl moieties in complex molecular architectures.
Introduction
Propargylic acetals have emerged as versatile synthetic building blocks, demonstrating remarkable utility across click chemistry and natural product synthesis. For instance, Fevig and Pinto employed 3,3-diethoxypropyne in azide–alkyne click chemistry to construct serine protease factor Xa (FXa) inhibitors (Scheme 1).1 Wang and coworkers utilized the identical 3,3-diethoxypropyne acetal to access O-GlcNAc transferase (OGT) inhibitors.2 In the total synthesis of indoxamycins, Liang designed an unsymmetrical propargylic acetal precursor to enable the pivotal Pauson-Khand cycloaddition step.3 Similarly, Hu and Hong developed a synthetic strategy for assembling scabrolide A's tricyclic core via cascade radical cyclization using propargylic acetal-based chemistry.4 | ||
| Scheme 1 Applications of propargylic acetals. | ||
Several synthetic methods have been developed for propargylic acetal construction (Scheme 2). Forsyth's seminal work pioneered a two-step protocol for unsymmetrical propargylic acetals through bromoacetalization of allenyl ether substrates followed by β-hydride elimination.5 This methodology was subsequently adopted in the aforementioned total syntheses. In 2014, Zhou reported a Cu(I)-catalyzed coupling strategy between terminal alkynes and oxadiazolines, achieving unsymmetric propargyl acetals via dialkoxycarbene pathway.6 Wang's 2019 investigation demonstrated a decarboxylative cross-coupling of 2,2-diethoxyacetic acid with alkynyl bromides for streamlined propargylic acetal assembly.7 However, in contrast to Forsyth's direct alcohol functionalization protocol, these contemporary approaches necessitate elaborate precursors while proving incompatible with terminal alkyne generation.
 | ||
| Scheme 2 Synthesis strategy of unsymmetric propargylic acetals. | ||
A related class of compounds, N,O-propargylic acetals, was recently accessed by Yu through PhI(OAc)2-mediated oxidation of allenamides, generating propargylic iminium ions that undergo 1,2-addition with alcohols.8 Building upon our expertise in allenyl ether chemistry,9,10 we now report a one-step synthesis of unsymmetric propargylic acetals via hypervalent iodine oxidation of allenyl ethers followed by alcohol trapping.11 This method eliminates the need for pre-functionalized intermediates and enables direct alcohol functionalization under mild conditions.
Results and discussion
Inspired by Yu's iminium ion strategy,8 we hypothesized that allenyl ethers could undergo analogous oxidation to generate propargylic oxonium ions, which might be trapped by alcohols to form acetals (Scheme 2). Initial studies focused on optimizing the oxidation of phenyl allenyl ether (1a) in methanol. Screening of oxidants revealed PhI(OAc)2 as optimal, delivering the methyl acetal 2a in 89% yield within 5 minutes at room temperature (Table 1).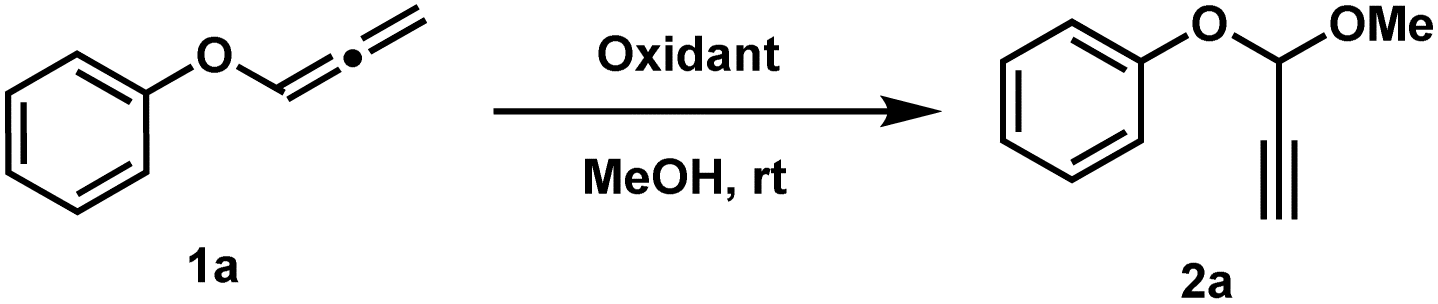
With optimized conditions (1.2 equiv. PhI(OAc)2, 0.3 M in MeOH), diverse allenyl ethers were evaluated. As summarized in Table 2, a wide range of aryl allenyl ethers proved effective in this transformation, delivering the corresponding acetals in good yields. Three para-substituted phenyl allenyl ethers—bearing –CF3 (2h), –Me (2g), and –OMe (2i) groups—exhibited moderately reduced yields (60%, 60%, and 62%, respectively). In contrast, methoxy-substituted substrates at meta (2j, 91%) and ortho (2k, 83%) positions maintained high efficiency. The protocol extended successfully to aliphatic and N-heterocyclic-containing allenyl ethers (2l–2p), providing acetals in 69–86% yields. To validate scalability, gram-scale reactions of both 2d and 2i proceeded efficiently (94% and 64% yield, respectively), confirming the protocol's robustness.

Given our research goal of developing a general alcohol functionalization method, we proceeded to introduce various alcohols into the reaction system. Initial attempts employed isopropanol as both solvent and nucleophile in the reaction of benzyl allenyl ether with PhI(OAc)2. However, this system exclusively produced the O-acetoxy acetal 4 rather than the anticipated O-isopropyl product 3a (Scheme 3). Structural analysis of 4 revealed interception of the in situ generated oxonium ion by the oxidant's acetate counterion during the 1,2-addition step. This outcome was attributed to the steric bulk and low nucleophilicity of isopropanol, which proved insufficient to compete with the acetate anion.
 | ||
| Scheme 3 Synthetic Attempts toward propargyl isopropyl acetal. | ||
This observation prompted investigation of oxidants bearing less nucleophilic counterions. Furthermore, to accommodate solid and complex alcohol substrates incompatible with solvent roles, we sought to develop non-solvent-dependent conditions. Replacement of PhI(OAc)2 with PhI(OCOCF3)2 in isopropanol successfully afforded 3a in 68% yield, validating the counterion strategy. Subsequent solvent optimization identified no conversion in THF, partial success in ethyl acetate at elevated temperatures, and optimal performance in CH2Cl2 (84% yield at rt) (Table 3).
|
|||||
|---|---|---|---|---|---|
| Entrya | Oxidant (1.2 equiv.) | Additive | T (°C) | Solvent (0.3 M) | Yieldb (%) |
| a All reactions were carried out with 1.50 mmol 1l, 5.0 mL solvent, 1.80 mmol oxidant, 2.25 mmol i-PrOH.b Isolated yields.c All additive were 2.0 equivalent. | |||||
| 1 | PhI(OCOCF3)2 | — | 25 °C | i-PrOH | 68 |
| 2 | PhI(OCOCF3)2 | — | 50 °C | THF | No reaction |
| 3 | PhI(OCOCF3)2 | — | 50 °C | EtOAc | 41 |
| 4 | PhI(OCOCF3)2 | — | 25 °C | EtOAc | No reaction |
| 5 | PhI(OCOCF3)2 | — | 25 °C | CH2Cl2 | 84 |
| 6c | PhI(OCOCF3)2 | DIPEA | 25 °C | CH2Cl2 | 13 |
| 7c | PhI(OCOCF3)2 | Pyridine | 25 °C | CH2Cl2 | 32 |
| 8c | PhI(OCOCF3)2 | NaHCO3 | 25 °C | CH2Cl2 | 36 |

As shown in Table 4, The refined protocol (PhI(OCOCF3)2, CH2Cl2, rt) demonstrated broad applicability with alcohols bearing diverse functional groups, including bromo (3d, 60%), trifluoromethyl (3e, 63%), cyclobutyl (3f, 85%), and alkyne (3h, 78%) moieties. Notably, solid alcohols such as L-menthol (3i, 67%)—which cannot serve as reaction solvents due to their physical state at room temperature—were successfully functionalized under these homogeneous conditions. The protocol's scalability was further demonstrated through gram-scale reactions, which maintained efficiency (75% yield for 3b, 84% for 3f). Phenolic substrates universally failed under standard conditions, presumably due to hypervalent iodine-mediated dearomatization pathways. A singular exception was observed with 2-hydroxybenzyl allenyl ether, where intramolecular cyclization afforded cyclic acetal 6 (85% yield, Scheme 4).
| a The reactions were carried out with 1l (100 mg, 0.68 mmol, 1.0 equiv.), PIFA (353 mg, 0.82 mmol, 1.2 equiv.), and the corresponding alcohol (1.03 mmol, 1.5 equiv.) in CH2Cl2 (2.28 mL, 0.3 M) at room temperature for 5–30 minutes.b Isolated yields.c Yield of a 3.0 g scale reaction.d Yield of a 2.5 g scale reaction. |
|---|
 |
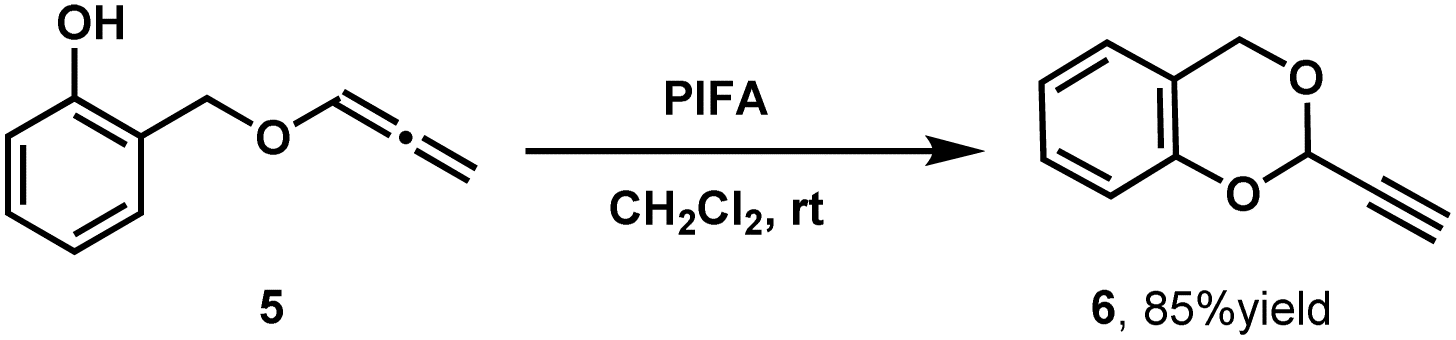 | ||
| Scheme 4 Intramolecular cyclization of 2-hydroxybenzyl allenyl ether. | ||
A plausible reaction mechanism, consistent with prior reports,8 is outlined in Scheme 5. Initial oxidation of the electron-rich alkene generates iodonium intermediate 7, which undergoes oxygen-assisted ring-opening to oxonium ion 8. When PhI(OAc)2 is employed, the acetate counterion competes with sterically hindered or weakly nucleophilic alcohols in the subsequent 1,2-addition step. This pathway, proceeding intermolecularly or intramolecularly, culminates in β-H elimination to yield O-acetoxy acetal 4. In contrast, PhI(OCOCF3)2—bearing a less nucleophilic trifluoroacetate counterion—shifts the equilibrium to favor alcohol addition. Subsequent β-H elimination from this intermediate delivers the desired propargylic acetal.
 | ||
| Scheme 5 Possible reaction mechanism. | ||
Conclusions
We have developed a novel method for the preparation of unsymmetric propargylic acetals through direct functionalization of alcohols. This approach involves a hypervalent iodine-mediated oxidation of allenyl ethers followed by 1,2-addition of alcohols under mild conditions, proceeding efficiently at room temperature and reaching completion within half an hour. The reaction demonstrates remarkable compatibility with sterically bulky alcohols and solid substrates, consistently delivering corresponding acetals in synthetically useful yields. As a scalable strategy for converting hydroxyl groups into alkynyl functionalities, this methodology offers broad utility in complex molecule synthesis and chemical biology applications.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as potential conflicts of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 22061008, 22361008, 82473814) and Guizhou Provincial Science and Technology Projects (Grant No. ZK [2023]-097).References
- J. M. Fevig, D. J. Pinto, Q. Han, M. L. Quan, J. R. Pruitt, I. C. Jacobson, R. A. Galemmo, Jr, S. Wang, M. J. Orwat, L. L. Bostrom, R. M. Knabb, P. C. Wong, P. Y. S. Lam and R. R. Wexler, Bioorg. Med. Chem. Lett., 2001, 11, 641 CrossRef CAS PubMed.
- Y. Wang, J. Zhu and L. Zhang, J. Med. Chem., 2017, 60, 263 CrossRef CAS PubMed.
- N. Hu, C. Dong, C. Zhang and G. Liang, Angew. Chem., Int. Ed., 2019, 58, 6659 CrossRef CAS PubMed.
- C. Peng, Q. Guo, G. Xu, X. Hong and P. Hu, J. Am. Chem. Soc., 2024, 146, 14422 CrossRef CAS PubMed.
- C. J. Forsyth and J. Clardy, J. Am. Chem. Soc., 1990, 112, 3497 CrossRef CAS.
- T. Xiao, P. Zhang, Y. Xie, J. Wang and L. Zhou, Org. Biomol. Chem., 2014, 12, 6215 RSC.
- X. L. Lyu, S. Huang, H. Song, Y. Liu and Q. Wang, RSC Adv., 2019, 9, 36213 RSC.
- R. Huang, P. Xu, W. Wang, G. Peng and H. Yu, Tetrahedron Lett., 2020, 61, 151753 CrossRef CAS.
- S. He, R. P. Hsung, W. R. Presser, Z. Ma and B. J. Haugen, Org. Lett., 2014, 16, 2180 CrossRef CAS PubMed.
- X. Huang, W. Thor, X. Feng, L. Kang, M. Yang, C. Lee, Y. Cheng and S. He, Org. Chem. Front., 2020, 7, 255 RSC.
- S. He, A. Long and T. Long, Green preparation of asymmetric alkynyl acetal, Chinese Patent, No. CN118702554, September 27, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01533b |
| This journal is © The Royal Society of Chemistry 2025 |